

Abstract
It has been shown that the TSH receptor (TSHR) couples to a number of different signaling pathways, although the Gs-cAMP pathway has been considered primary. Here, we measured the effects of TSH on bone marker mRNA and protein expression in preosteoblast-like U2OS cells stably expressing TSHRs. We determined which signaling cascades are involved in the regulation of IL-11, osteopontin (OPN), and alkaline phosphatase (ALPL). We demonstrated that TSH-induced up-regulation of IL-11 is primarily mediated via the Gs pathway as IL-11 was up-regulated by forskolin (FSK), an adenylyl cyclase activator, and inhibited by protein kinase A inhibitor H-89 and by silencing of Gαs by small interfering RNA. OPN levels were not affected by FSK, but its up-regulation was inhibited by TSHR/Gi-uncoupling by pertussis toxin. Pertussis toxin decreased p38 MAPK kinase phosphorylation, and a p38 inhibitor and small interfering RNA knockdown of p38α inhibited OPN induction by TSH. Up-regulation of ALPL expression required high doses of TSH (EC50 = 395nM), whereas low doses (EC50 = 19nM) were inhibitory. FSK-stimulated cAMP production decreased basal ALPL expression, whereas protein kinase A inhibition by H-89 and silencing of Gαs increased basal levels of ALPL. Knockdown of Gαq/11 and a protein kinase C inhibitor decreased TSH-stimulated up-regulation of ALPL, whereas a protein kinase C activator increased ALPL levels. A MAPK inhibitor and silencing of ERK1/2 inhibited TSH-stimulated ALPL expression. We conclude that TSH regulates expression of different bone markers via distinct signaling pathways.
Bone is a dynamic tissue being continuously degraded by osteoclasts and renewed by osteoblasts, whose functions are regulated by various hormones and cytokines (1–3). Bone homeostasis represents a balance between these 2 opposing processes. Once this balance is disrupted, however, metabolic bone diseases such as osteoporosis and osteopetrosis occur. Recently evidence has emerged showing TSH receptor (TSHR) expression in bone and an antiosteoporotic action of TSH that was independent of thyroid hormones. Absence of the TSHR in the global Tshr−/− mouse caused high-turnover osteoporosis (4). Activation of TSHR in osteoclasts attenuated osteoclastogenesis and prevented bone resorption. TSH displayed osteoprotective effects in rodents by preventing bone loss after ovariectomy (5, 6). Hyperthyroid Tshr−/− mice completely devoid of TSH signaling displayed higher levels of bone resorption and bone loss compared with hyperthyroid wild-type mice (7). TSH has also been shown to induce osteoblastogenesis in embryonic stem cell cultures (8). And patients with hyperthyroidism displayed strong correlations between low TSH and high bone turnover, reduced bone mineral density, osteoporosis, and increase in fracture risk (9).
We and others have shown that the TSHR stimulates a number of different cell signal transduction pathways. It activates several G proteins including Gαs that stimulates the cAMP-protein kinase A (PKA) pathway and Gαq/11 that stimulates phospholipase C-mediated generation of diacylglycerol and inositol-1,4,5-trisphosphate that activates the calcium and protein kinase C (PKC) pathways (10–12). We have also shown that TSH causes translocation of both β-arrestin-1 and β-arrestin-2 to the plasma membrane (13). Knockdown of β-arrestin-1 inhibited TSH-stimulated up-regulation of osteopontin (OPN) secretion demonstrating that TSHR-mediated signaling plays a role in osteoblastogenesis and is in part caused by activatory signals mediated by β-arrestin-1 (13).
Oligomerization, a common feature of G protein-coupled receptors, has also been demonstrated for the TSHR. Oligomerization of TSHR has consequences for functional features like expression, ligand binding, signal induction, and conversion (14). Allen et al (11) demonstrated that binding of 2 TSH molecules to a TSHR homodimer is necessary to activate both Gs and Gq/11. In this study, we were interested in gaining insight into which signaling cascades are predominantly involved in TSHR-mediated effects on osteoblast differentiation and activity that result in its bone osteoprotective properties.
We used pathway specific activators and inhibitors and small interfering RNA (siRNA) silencing to demonstrate that distinct TSHR-mediated pathways regulate osteoblast markers, IL-11, secreted phosphoprotein 1 or OPN, and alkaline phosphatase (ALPL) in U2OS-TSHR cells.
Materials and Methods
Materials
U0126, H-89, and LY2228820 were purchased from Selleckchem; GF109203X (GFX) and phorbol 12, 13-dibutyrate from Enzo Life Sciences. Pertussis toxin (PTX) was obtained from Invitrogen; forskolin (FSK) from Sigma-Aldrich. Bovine TSH was from Sigma-Aldrich. M22 thyroid-stimulating antibodies (TSAbs) were from KRONUS (ID 83669; Star). Thyrostimulin was kindly provided by ZymoGenetics, Inc. Tissue-nonspecific anti-ALPL antibody was from Abcam, Inc (diluted 1:2000). The antibody to β-actin was from Sigma-Aldrich. RDye 680 or 800 secondary antibodies were from Li-Cor. GNAS, GNAQ, GNA11, GNAO, GNAI1–3, MAPK1, MAPK3, and MAPK14 ON-TARGET plus SMART pool human siRNAs or ON-TARGET plus nontargeting pool siRNA (scrambled) were from Thermo Fisher Scientific, Inc.
Cell culture
The generation of a U2OS cell line stably expressing TSHRs (U2OS-TSHR) was described previously (13). Cells were cultured in Eagle's MEM (EMEM) (ATCC) supplemented with 10% fetal bovine serum (FBS) (Sigma-Aldrich), 50-U/mL penicillin and 50-μg/mL streptomycin (Life Technologies, Inc), and hygromycin (250 μg/mL) (Life Technologies, Inc) as a selection marker. Cells were cultured at 37°C in a humidified 5% CO2 incubator. Experiments were performed in serum-free conditions: FBS was replaced with 0.1% BSA 24 hours before experiments.
Measurement of ERK1/2 and p38 MAPK phosphorylation by Luminex assay
U2OS-TSHR cells (1.5 × 105 cells/well) were seeded into 12-well plates in EMEM containing 10% FBS. Twenty-four hours before the experiment, media were changed to serum-free 0.1% BSA-containing EMEM. Cells were incubated for 15 minutes with TSH. Lysates were prepared using the Bio-Plex Pro cell signaling kit (Bio-Rad Laboratories) according to the manufacturer's directions, and lysate volume was adjusted so that all samples had equal protein concentrations, which were determined with a BCA assay (Pierce). Total and phosphorylated ERK1/2 and p38 MAPK were measured using a magnetic bead-based assay (Bio-Rad Laboratories). Assays were run as per manufacturer's instructions with 50-μL cell lysate sample in duplicates or triplicates, overnight incubation with shaking (18 h, at room temperature, 400 rpm), and using a handheld magnetic washer. Data were acquired on a Bio-Plex MAGPIX multiplex reader (Bio-Rad Laboratories) using the total and phospho-ERK1/2 (T185/Y187) and phospho-p38 MAPK (T180/Y182) sets (Bio-Rad Laboratories) according to the manufacturer's directions. Data were analyzed with Bio-Plex Manager 6.1 software (Bio-Rad Laboratories).
Transfection of U2OS-TSHR cells with siRNA
U2OS-TSHR cells were seeded into 12-well plates at 0.9 × 105 cells per well. After 24 hours, the cells were transfected with ON-TARGET plus SMART pool human siRNAs or ON-TARGET plus nontargeting pool siRNA (Scr) using DharmaFECT 1 transfection reagent according to the manufacturer's instructions (Thermo Fisher Scientific, Inc). Twenty-four hours after transfection, cells were incubated in 0.1% BSA-containing EMEM with or without TSH for 5 days.
Quantitative real-time PCR
Total RNA was purified using RNeasy Mini kits (QIAGEN, Inc). First strand cDNA was prepared using a High Capacity cDNA Reverse Transcription kit (Applied Biosystems). RT-PCR was performed in 25-μL reactions using cDNA prepared from 100 ng or less of total RNA and TaqMan Universal PCR Master Mix (Applied Biosystems). Levels of mRNA were measured using primers and probes from Applied Biosystems. Quantitative RT-PCR results were normalized to GAPDH to correct for differences in RNA input.
Immunoblotting
U2OS-TSHR cells (1.5 × 105 cells/well) were seeded into 12-well plates in EMEM containing 10% FBS. Twenty-four hours before the experiment, media were changed to 0.1% BSA-containing EMEM. Cells were stimulated with 100-mU/mL TSH for 5 days. Cells were washed once with cold PBS and lysed by addition of 1% sodium dodecyl sulfate sample buffer. Lysates were boiled for 10 minutes, and 40 μL of cell lysate were electrophoresed on a standard 10% sodium dodecyl sulfate-polyacrylamide gel (Bio-Rad Laboratories). After electrophoresis, proteins were transferred to nitrocellulose membranes (Bio-Rad Laboratories) and subsequently probed with the indicated primary antibodies and appropriate secondary antibodies conjugated with infrared fluorophores for detection and quantification on the Odyssey Infrared Imaging System (Li-Cor).
IL-11 and OPN secretion measurement by ELISA
U2OS-TSHR cells were seeded into 12-well plates at 1.5 × 105 cells per well. Cells were incubated with or without 100-mU/mL TSH for 5 days in 0.1% BSA-containing EMEM. Cell culture supernatants were used to determine IL-11 and OPN secretion levels by ELISA (Human IL11 and OPN Quantikine ELISA kits; R&D Systems, Inc) according to the manufacturer's instructions.
Measurement of cAMP production
U2OS-TSHR cells 1.0 × 105 cells per well were seeded into 24-well plates. Twenty-four hours before the experiment, media were changed to 0.1% BSA-containing EMEM. Cells were incubated for 15 minutes with increasing concentrations of TSH in 0.1% BSA-containing EMEM at 37°C. After aspiration of the incubation medium, cells were lysed with lysis buffer of the cAMP-Screen Direct System (Applied Biosystems), and cAMP content was determined as described by the manufacturer. The chemiluminescence signal was measured in a VICTOR3 V 1420 Multilabel Counter (PerkinElmer). Data analysis was performed with GraphPad Prism 6 for Windows (GraphPad Software).
Measurement of inositol monophosphate (IP1) production
U2OS-TSHR cells 1.0 × 105 cells per well were seeded into 24-well plates. Twenty-four hours before the experiment, media were changed to 0.1% BSA-containing EMEM. TSH stimulation was measured as IP1 production in cells incubated at 37°C for 15 minutes in 0.1% BSA-containing EMEM with increasing concentrations of TSH and 50mM LiCl to inhibit IP1 breakdown by blocking IP1 phosphatases. IP1 production was stopped by addition 0.05-mL lysis buffer (IP-One ELISA kit; CIS Bio International). IP1 content in the samples was determined according to the manufacturer's protocol. Optical density was measured in SpectraMax Plus384 (Molecular Devices). Data analysis was performed with GraphPad Prism 6 for Windows (GraphPad Software).
Results
TSH up-regulates expression of osteoblast genes in U2OS-TSHR cells
We used human preosteoblast-like osteosarcoma-derived U2OS cells engineered to stably overexpress TSHR as a model system to investigate TSH effects in bone (13). Treatment with 100-mU/mL bTSH resulted in up-regulation of bone markers associated with osteoblast activity such as IL-11, OPN, and ALPL at the mRNA and protein levels (Figure 1). IL-11 and OPN secreted proteins were measured by ELISA. ALPL protein was measured by immunoblotting (Supplemental Figure 1). We noted that the kinetics of induction were different for the 3 genes. For example, TSH-stimulated IL-11 mRNA up-regulation was rapid and reached a near maximum after 4 hours of stimulation (not shown), whereas the increases in OPN and ALPL mRNAs were much slower requiring 72 and 24 hours, respectively, for up-regulation. These observations were consistent with the idea that up-regulation of these genes were controlled by different TSHR-mediated signaling pathways.
Figure 1.

Time course demonstrating TSH-mediated up-regulation of mRNA and protein expression of IL-11, OPN, and ALPL. U2OS-TSHR cells were treated with 100-mU/mL bTSH in EMEM supplemented with 0.1% BSA. Cell lysates were analyzed by RT-qPCR. ALPL was detected by Western blotting using tissue-nonspecific anti-ALPL antibodies. IL-11 and OPN secretion were measured by ELISAs in cell culture medium. The data are from 3 independent experiments with duplicate samples and are presented as the mean ± SEM.
Next, we tested whether alternative activators of TSHR elicited responses similar to TSH effects on gene expression. TSAbs are present in Graves' disease. They bind TSHR and cause excess secretion of thyroid hormones. Recently, thyrostimulin, an ancestral glycoprotein hormone with high affinity for the TSHR, has been shown to regulate osteoblastic bone formation during early skeletal development (15). We used M22, a thyroid-stimulating human monoclonal autoantibody, and thyrostimulin to show that both agonists exert effects similar to those of TSH in up-regulation of IL-11, OPN, and ALPL (Figure 2).
Figure 2.

M22 and thyrostimulin exert effects similar to TSH on up-regulation of IL-11, OPN, and ALPL. U2OS-TSHR cells were treated with 300-ng/mL M22 and 200-ng/mL thyrostimulin in EMEM supplemented with 0.1% BSA for 5 days. Cell lysates were analyzed by RT-qPCR. The data are from 3 independent experiments with duplicate samples and are presented as the mean ± SEM.
TSHR-mediated up-regulation of IL-11 is primarily mediated by the Gs-cAMP-PKA signaling pathway
We first studied the Gαs-cAMP-PKA pathway, because it is traditionally considered the primary signaling pathway for TSHR. We compared the effects of TSH with those of the adenylyl cyclase activator FSK and found that FSK also up-regulated IL-11 5.6-fold over control (Figure 3A). PKA inhibitor H-89 abolished TSH-stimulated IL-11 mRNA expression and secreted IL-11 (Figure 3B). Finally, silencing Gαs by siRNA (average reduction of GNAS mRNA of 96%) (data not shown) lowered basal IL-11 mRNA expression by 72% and inhibited TSH-mediated up-regulation of IL-11 mRNA to levels below basal (Figure 3C). These data demonstrate that regulation of IL-11 expression by TSH is primarily regulated by the Gs-cAMP pathway.
Figure 3.

Effects of adenylyl cyclase activator FSK, PKA inhibitor H-89, and Gαs siRNA knockdown on IL-11 levels. U2OS-TSHR cells were treated with 100-mU/mL bTSH, 10μM FSK, or 10μM H-89 in 0.1% BSA EMEM. GNAS siRNA was used to silence Gαs. After 5 days, cells were lysed and IL-11 expression was measured by RT-qPCR. IL-11 secretion was measured by ELISA in cell culture medium. Effects of FSK (A), H-89 (B), and siRNA knockdown of Gαs (C). The bars represent the mean ± SEM of 3 experiments with at least 2 biological replicates.
TSHR-mediated up-regulation of OPN is primarily mediated by the Gi-p38 MAPK kinase signaling pathway
We studied up-regulation of OPN and found that it was not regulated by the Gs pathway as FSK had no effect on OPN (data not shown). However, we found that PTX, which inhibits receptor activation of Go/i proteins, inhibited TSH-mediated up-regulation of OPN by 91.5% (Figure 4A). After silencing of GNAO and GNAI1–3 levels by siRNA with 63%–88% knockdown efficiency (data not shown), we observed a 46.2% decrease of OPN expression pointing out that Gαi, but not Gαo, plays a role in TSH up-regulation of OPN (Figure 4B). Activation of p38 MAPK kinase by TSHR has been shown to be cAMP dependent in CHO-hTSHR and the rat thyroid cell line FRTL-5 (16). It has also been known that in vascular smooth muscle cells, phosphorylation of p38 by oxidized low-density lipoprotein was mediated via PTX-sensitive G proteins (17). Hence, we studied effects of cAMP and PTX on p38 MAPK activation and found that it was cAMP-independent as FSK had no significant effect (data not shown), but PTX inhibited p38 phosphorylation by 47.6% (Figure 5A). Next, we established that down-regulation of p38α MAPK14 by siRNA (knockdown efficiency 79%) (data not shown) reduced TSH effect on OPN by 74.3% (Figure 5B). In addition, an inhibitor of p38 activation, LY2228820, reduced TSH-stimulated up-regulation of OPN mRNA by 98.2% and secreted protein by 93% (Figure 5C). These results indicate that Gi and p38 MAPK kinase signaling are important regulators of TSH-stimulated OPN expression.
Figure 4.

Gαi, but not Gαo, is required for OPN up-regulation by TSH. U2OS-TSHR cells were pretreated with PTX overnight (A), GNAO and GNAI1–3 siRNAs were used to silence Gαo and Gαi1–3 for 24 hours (B) before 5-day treatment with 100-mU/mL bTSH. OPN mRNA expression was determined by RT-qPCR. The bars represent the mean ± SEM of 3 experiments with at least 2 biological replicates.
Figure 5.

p38 MAPK kinase mediates TSH-stimulated up-regulation of OPN. U2OS-TSHR cells were pretreated with 100-ng/mL PTX overnight before 15-minute treatment with 100-mU/mL bTSH. Activation of p38 MAPK was determined by Luminex Multiplex assay (A). MAPK14 siRNA was used to silence p38α 24 hours before 5-day treatment with 100-mU/mL bTSH (B). Cells were treated with 100-mU/mL bTSH with or without 1μM LY2228820. After 5 days, OPN expression (C) was determined by RT-qPCR. OPN secretion was determined by ELISA in cell culture medium. The bars represent the mean ± SEM of 3 experiments with at least 2 biological replicates.
TSHR-mediated induction of ALPL expression is biphasic with inhibition via Gs-cAMP-PKA and up-regulation predominantly by the Gq/11-PKC-ERK1/2 signaling pathway
Our preliminary experiments measuring the TSH dose response of ALPL expression showed that it was biphasic with lower doses of TSH causing down-regulation of ALPL and higher doses stimulating up-regulation (Figure 6). We had previously shown that the EC50 for TSH activation of Gs was much lower than that for activation of Gq/11 (11). We postulated that the down-regulation of ALPL might be caused by the Gs pathway and the up-regulation by the Gq/11 pathway. As hypothesized, the EC50 for stimulation of cAMP production was similar to that of down-regulation of ALPL (Figure 6A), whereas the EC50 for stimulation of IP1, a product of a signaling molecule in the Gq/11 pathway, was similar to that of up-regulation of ALPL (Figure 6B). In support of the idea that down-regulation of ALPL expression was mediated by the Gs-cAMP-PKA pathway, we observed 45% down-regulation of basal ALPL levels by FSK (Figure 7) and conversely, both inhibition of PKA by H-89 and GNAS siRNA knockdown increased basal levels of ALPL expression approximately 4- and 12-fold, respectively (Figure 7).
Figure 6.

TSH dose-response curves reveal biphasic effects on ALPL mRNA. A, Low TSH doses (shaded) are inhibitory for ALPL expression and correlate with stimulation of cAMP production. ALPL expression was determined by RT-qPCR after 5-day treatment with increasing doses of bTSH. cAMP production was measured by ELISA after 15 minutes stimulation with increasing doses of bTSH. Data are representative of 3 independent experiments with duplicate samples, and values are expressed in mean ± SEM. B, High TSH doses (shaded) are activatory for ALPL expression and correlate with stimulation of IP1 production. ALPL expression was determined by RT-qPCR after 5 days of treatment with increasing doses of bTSH. IP1 was measured by ELISA after 15 minutes of stimulation with increasing doses of bTSH in the presence of 50mM LiCl. Data are representative of 3 independent experiments with duplicate samples, and values are expressed in mean ± SEM.
Figure 7.
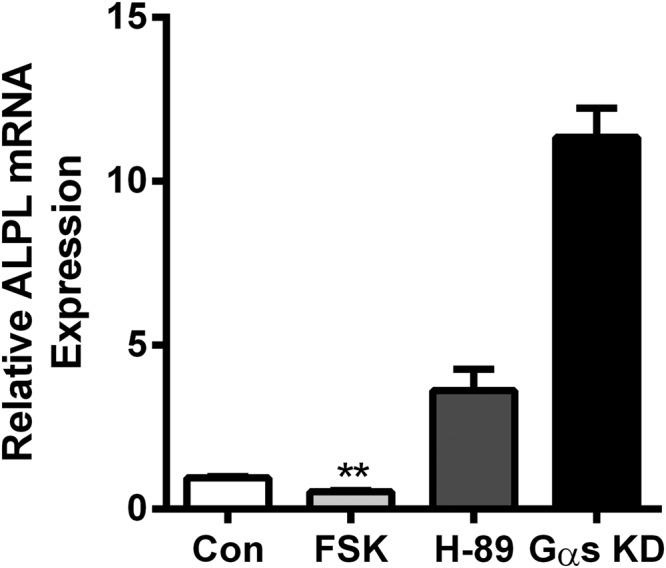
Increase of intracellular cAMP by FSK decreases nonstimulated ALPL levels, whereas H-89 and Gαs knockdown markedly increase it. U2OS-TSHR cells were treated with 10μM FSK or H-89. GNAS siRNA was used to knockdown Gαs. After 5 days, levels of ALPL expression were determined by RT-qPCR. The data are from 3 independent experiments with duplicate samples, and are presented as the mean ± SEM.
We next determined whether the Gq/11 pathway mediated up-regulation of ALPL by higher doses of TSH. We knocked down GNAQ and GNA11 (100% and 88% knockdown efficiency, respectively) (data not shown) using siRNA and concluded that Gαq/11 signaling is involved in TSH-mediated up-regulation of ALPL as we observed a 87.4% reduction of the TSH effect (Figure 8A). Because it had been previously shown that ALPL activity was modulated by PTH in periodontal ligament cells via a PKC-dependent pathway (18), we explored a role for PKC in ALPL up-regulation by TSH in our system. We used the PKC inhibitor GFX and demonstrated that ALPL up-regulation by TSH was 81.5% lower than control in GFX-treated cells (Figure 8B) and conversely, the PKC activator phorbol 12, 13-dibutyrate treatment increased basal ALPL expression level 4.7-fold (Figure 8C).
Figure 8.

Gαq/11 signaling is involved in TSH-stimulated ALPL up-regulation. GNAQ and GNA11 siRNAs were used to knockdown Gαq/11 (A). U2OS-TSHR cells were treated with 100-mU/mL bTSH with or without 2μM GFX, a PKC inhibitor (B), or phorbol 12, 13-dibutyrate, a PKC activator (C). ALPL expression was measured by RT-qPCR after 5 days. The data are from 3 independent experiments with duplicate samples and are presented as the mean ± SEM.
PKC downstream signaling has been reported to activate the mitogen-activated protein kinases ERK1/2 (19, 20). We previously showed the time course of TSHR-mediated ERK1/2 phosphorylation in U2OS-TSHR cells (13). Here, we show that the dose response to TSH of ERK1/2 phosphorylation is similar to that of up-regulation of ALPL (Figure 9A). Next, we used the MEK inhibitor U0126 to determine whether ERK1/2 phosphorylation affected TSH-induced levels of ALPL. U0126 resulted in 75% inhibition of the TSH response (Figure 9B). Silencing MAPK3/MAPK1 genes encoding for ERK1/2 (knockdown efficiency 94% and 82%, respectively) (data not shown), resulted in a 64.2% inhibition of TSH-up-regulated ALPL (Figure 9C). Combined, these findings indicate that the Gαq/11-PKC-ERK1/2 signaling cascade is important for induction of ALPL expression by TSH.
Figure 9.

ERK1/2 mediates TSH-stimulated up-regulation of ALPL. TSH activates ERK1/2 at high doses (shaded) (A). Total and phospho-ERK1/2 were analyzed by Luminex Multiplex assay after 15 minutes of stimulation of U2OS-TSHR cells with increasing doses of bTSH. The data are from 3 independent experiments with duplicate samples and are presented as the mean ± SEM. U2OS-TSHR cells were treated with 100-mU/mL bTSH with or without 1μM U0126 (B) for 5 days. MAPK3 and MAPK1 siRNAs were used to knockdown genes encoding for ERK1/2 (C) 24 hours before 5-day treatment with bTSH. ALPL expression was measured by RT-qPCR after 5 days. The data are from 3 independent experiments with duplicate samples and are presented as the mean ± SEM.
Discussion
Understanding molecular interactions involved in signal transduction is a key goal in pharmacology and toxicology. With a greater understanding of the multidimensional nature of GPCR signaling has come the appreciation that signal transduction is far more complex and context dependent than previously imagined (21). A common attribute linked to GPCRs is the ability to couple to more than one G protein subclass (22). TSHR exhibits promiscuous G protein coupling; it appears to signal primarily through Gs, but also through interaction with Gq/11, G12/13, Go/i (10) as well as with β-arrestins, with evidence that coupling to β-arrestin-1 is involved in activatory signaling and to β-arrestin-2 in receptor inactivation (13). We have shown previously that TSHR activation by TSH leads to up-regulation of osteoblast markers ALPL, OPN, and receptor activator of nuclear factor kappa-B ligand in U2OS-TSHR cells (13). We determined that β-arrestin-1 plays a key role in downstream signaling events leading to induction of OPN and receptor activator of nuclear factor kappa-B ligand expression. However, the contribution of specific G proteins to TSH-mediated up-regulation of osteoblast genes was not determined. Here, we showed that TSHR couples to at least 3 different G proteins to induce expression of IL-11, OPN, and ALPL in U2OS-TSHR cells.
TSH treatment up-regulated all 3 osteoblast markers under study, IL-11, ALPL, and OPN, in U2OS-TSHR cells. However, only up-regulation of IL-11, which is required for normal bone turnover (23), is primarily mediated via the cAMP pathway. Of note, we provided evidence that cAMP inhibits expression of ALPL, an enzyme that plays an important role in osteoid formation and bone mineralization, and has no effect on the expression of the organic component of bone, OPN. In contrast, we demonstrated that the induction of ALPL primarily occurs via the Gq/11-ERK1/2 pathway. The steep curve for the activatory part of the ALPL biphasic dose response with a Hill slope of 2.8 is consistent with positive cooperative interaction and multisite binding. This corresponds to our previous findings showing that TSHR coupling to phosphoinositide signaling is dependent on binding 2 molecules of TSH to a TSHR homodimer, causing a conformational change allowing coupling to Gq/11 (11). Lastly, we showed that OPN up-regulation requires Gi-p38 MAPK activation and, as shown previously, β-arrestin-1 (13).
The Gs-cAMP pathway has been considered to be the primary pathway of TSHR signal transduction based largely on data derived from studies of thyroid cells. In thyrocytes, TSH-mediated cAMP accumulation is a positive modulator of thyroid cell differentiation (24, 25), and many of the effects of TSH can be simulated by the direct activation of the cAMP pathway by FSK, cAMP analogs, or cholera toxin. However, these findings do not exclude the possibility that other G protein-dependent pathways are involved also. Indeed multiple pathways appear to be involved in TSHR signaling in U2OS-TSHR cells. The mediation of TSHR signaling by different pathways in thyrocytes and preosteoblast-like cells may be dependent on the levels of TSHR and G proteins in these cells. For example, TSHR abundance is much lower in bone compared with thyroid and this likely influences G protein coupling. Differences such as these could explain the differences in the pathways stimulated in thyrocytes vs preosteoblasts and the relative importance of non-cAMP mediated TSH signal transduction in preosteoblasts.
In our study, the EC50s for OPN and the activatory part of ALPL dose responses are approximately 400nM (or 20 mU/mL), which are somewhat higher than normal physiological concentration of TSH in blood (0.3–5 mU/mL). However, it is not possible to predict how these in vitro TSH concentrations would translate in an in vivo system. Moreover, this is true also for other TSHR agonists, such as TSAbs in Graves' patients and perhaps paracrine secreted thyrostimulin, a more potent ligand of TSHR than TSH in in vitro systems (26). Basset et al (15) have recently identified the expression of both subunits of thyrostimulin, Gpa2 and Gpb5, in the newborn mouse skeleton as well as primary osteoblasts and osteoclasts. Despite the fact that the authors could not detect either cAMP response to thyrostimulin or phosphorylation of protein kinase B, ERK, or p38 MAPK, juvenile thyrostimulin-deficient mice had increased bone volume and mineralization as a result of increased osteoblastic bone formation. They hypothesized that thyrostimulin may exert paracrine actions in bone by regulating osteoblastic bone formation during early skeletal development. Thyrostimulin has also been demonstrated to act as a paracrine regulator in mammalian ovary (27).
Another receptor that plays an important role in bone biology is PTH1 receptor (PTH1R). PTH1R is a primary regulator of mineral ion homeostasis and is expressed in osteoblasts. PTH1R's principal pathway is via Gs and activation of cAMP-PKA signaling process (28). However, it has also been shown to couple to Gq/11, G12/13, and Go/i as well as promote translocation of both β-arrestin-1 and β-arrestin-2 and activation of ERK1/2 (29). Moreover, the findings obtained using a β-arrestin signaling biased agonist suggested that the β-arrestin-dependent pathway selectively contributes to anabolic bone formation and does not stimulate bone resorption (30). Nonselective PTH-mediated osteoblast-osteoclast coupling is cAMP-dependent and sustained cAMP signaling in the absence of β-arrestin interaction promotes bone turnover (31, 32). Thus, TSHR and PTH1R use similar signal transduction pathways to regulate preosteoblast gene expression. Another feature shared by TSHR and PTH1R is the ability to promote bone formation when administered intermittently. Intermittent administration of PTH(1–34) led to bone formation, whereas continuous administration paradoxically led to bone resorption (29–31). Similarly, intermittently administered TSH restored ovariectomy-induced bone loss and improved bone strength in rats (6).
If TSH exerts predominant actions to promote osteoblastogenesis and/or inhibit bone turnover, a question arises as to why individuals with Graves' hyperthyroidism are susceptible to osteoporosis and fracture (33). It would be predicted that high concentrations of circulating TSAbs in Graves' disease should protect patients from bone loss (34). However, the cumulative effect of the Graves' disease environment is most likely caused by the elevated levels of T3 and T4, which directly stimulate excessive bone resorption (35).
In conclusion, we demonstrated that TSH-mediated up-regulation of IL-11, ALPL, and OPN occurs via distinct G protein-coupled signaling pathways. Development of functionally biased agonists of TSHR, where one signaling pathway may be favored over another, may allow the development of drugs that selectively modulate only the therapeutically relevant physiological functions (36) as has been accomplished for PTH1R. Our findings suggest that the development of TSHR agonists that are biased towards the β-arrestin-1 and Gq/11-ERK1/2 pathways could be useful to treat osteoporosis.
Acknowledgments
This work was supported by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health Grant Z01 DK011006.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- ALPL
- alkaline phosphatase
- EMEM
- Eagle's MEM
- FBS
- fetal bovine serum
- FSK
- forskolin
- GFX
- GF109203X
- GNA
- guanine nucleotide-binding protein (G protein), subunit alpha
- IP1
- inositol monophosphate
- OPN
- osteopontin
- PKA
- protein kinase A
- PKC
- protein kinase C
- PTH1R
- PTH1 receptor
- PTX
- pertussis toxin
- siRNA
- small interfering RNA
- TSAb
- thyroid-stimulating antibody
- TSHR
- TSH receptor.
References
- 1. Phan TC, Xu J, Zheng MH. Interaction between osteoblast and osteoclast: impact in bone disease. Histol Histopathol. 2004;19(4):1325–1344. [DOI] [PubMed] [Google Scholar]
- 2. Martin T, Gooi JH, Sims NA. Molecular mechanisms in coupling of bone formation to resorption. Crit Rev Eukaryot Gene Expr. 2009;19(1):73–88. [DOI] [PubMed] [Google Scholar]
- 3. Matsuo K, Irie N. Osteoclast-osteoblast communication. Arch Biochem Biophys. 2008;473(2):201–209. [DOI] [PubMed] [Google Scholar]
- 4. Abe E, Marians RC, Yu W, et al. TSH is a negative regulator of skeletal remodeling. Cell. 2003;115(2):151–162. [DOI] [PubMed] [Google Scholar]
- 5. Sampath TK, Simic P, Sendak R, et al. Thyroid-stimulating hormone restores bone volume, microarchitecture, and strength in aged ovariectomized rats. J Bone Miner Res. 2007;22(6):849–859. [DOI] [PubMed] [Google Scholar]
- 6. Sun L, Vukicevic S, Baliram R, et al. Intermittent recombinant TSH injections prevent ovariectomy-induced bone loss. Proc Natl Acad Sci USA. 2008;105(11):4289–4294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Baliram R, Sun L, Cao J, et al. Hyperthyroid-associated osteoporosis is exacerbated by the loss of TSH signaling. J Clin Invest. 2012;122(10):3737–3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Baliram R, Latif R, Berkowitz J, et al. Thyroid-stimulating hormone induces a Wnt-dependent, feed-forward loop for osteoblastogenesis in embryonic stem cell cultures. Proc Natl Acad Sci USA. 2011;108(39):16277–16282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Reddy PA, Harinarayan CV, Sachan A, Suresh V, Rajagopal G. Bone disease in thyrotoxicosis. Indian J Med Res. 2012;135(3):277–286. [PMC free article] [PubMed] [Google Scholar]
- 10. Latif R, Morshed SA, Zaidi M, Davies TF. The thyroid-stimulating hormone receptor: impact of thyroid-stimulating hormone and thyroid-stimulating hormone receptor antibodies on multimerization, cleavage, and signaling. Endocrinol Metab Clin North Am. 2009;38(2):319–341, viii. [DOI] [PubMed] [Google Scholar]
- 11. Allen MD, Neumann S, Gershengorn MC. Occupancy of both sites on the thyrotropin (TSH) receptor dimer is necessary for phosphoinositide signaling. FASEB J. 2011;25(10):3687–3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Boutin A, Allen MD, Geras-Raaka E, Huang W, Neumann S, Gershengorn MC. Thyrotropin receptor stimulates internalization-independent persistent phosphoinositide signaling. Mol Pharmacol. 2011;80(2):240–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Boutin A, Eliseeva E, Gershengorn MC, Neumann S. β-Arrestin-1 mediates thyrotropin-enhanced osteoblast differentiation. FASEB J. 2014;28(8):3446–3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kleinau G, Neumann S, Grüters A, Krude H, Biebermann H. Novel insights on thyroid-stimulating hormone receptor signal transduction. Endocr Rev. 2013;34(5):691–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bassett JH, van der Spek A, Logan JG, et al. Thyrostimulin regulates osteoblastic bone formation during early skeletal development. Endocrinology. 2015;156(9):3098–3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pomerance M, Abdullah HB, Kamerji S, Correze C, Blondeau JP. Thyroid-stimulating hormone and cyclic AMP activate p38 mitogen-activated protein kinase cascade. Involvement of protein kinase A, rac1, and reactive oxygen species. J Biol Chem. 2000;275(51):40539–40546. [DOI] [PubMed] [Google Scholar]
- 17. Jing Q, Xin SM, Cheng ZJ, et al. Activation of p38 mitogen-activated protein kinase by oxidized LDL in vascular smooth muscle cells: mediation via pertussis toxin-sensitive G proteins and association with oxidized LDL-induced cytotoxicity. Circ Res. 1999;84(7):831–839. [DOI] [PubMed] [Google Scholar]
- 18. Wolf M, Jäger A, Abuduwali N, Götz W, Lossdörfer S. Continuous PTH modulates alkaline phosphatase activity in human PDL cells via protein kinase C dependent pathways in vitro. Ann Anat. 2013;195(5):455–460. [DOI] [PubMed] [Google Scholar]
- 19. Naor Z, Benard O, Seger R. Activation of MAPK cascades by G-protein-coupled receptors: the case of gonadotropin-releasing hormone receptor. Trends Endocrinol Metab. 2000;11(3):91–99. [DOI] [PubMed] [Google Scholar]
- 20. May LT, Hill SJ. ERK phosphorylation: spatial and temporal regulation by G protein-coupled receptors. Int J Biochem Cell Biol. 2008;40(10):2013–2017. [DOI] [PubMed] [Google Scholar]
- 21. Neubig RR, Enna SJ. Pharmacology of G Protein Coupled Receptors. San Diego, CA: Academic Press; 2011. [DOI] [PubMed] [Google Scholar]
- 22. Galandrin S, Bouvier M. Distinct signaling profiles of β1 and β2 adrenergic receptor ligands toward adenylyl cyclase and mitogen-activated protein kinase reveals the pluridimensionality of efficacy. Mol Pharmacol. 2006;70(5):1575–1584. [DOI] [PubMed] [Google Scholar]
- 23. Sims NA, Jenkins BJ, Nakamura A, et al. Interleukin-11 receptor signaling is required for normal bone remodeling. J Bone Miner Res. 2005;20(7):1093–1102. [DOI] [PubMed] [Google Scholar]
- 24. Roger PP, Christophe D, Dumont JE, Pirson I. The dog thyroid primary culture system: a model of the regulation of function, growth and differentiation expression by cAMP and other well-defined signaling cascades. Eur J Endocrinol. 1997;137(6):579–598. [DOI] [PubMed] [Google Scholar]
- 25. Tramontano D, Moses AC, Veneziani BM, Ingbar SH. Adenosine 3′,5′-monophosphate mediates both the mitogenic effect of thyrotropin and its ability to amplify the response to insulin-like growth factor I in FRTL5 cells. Endocrinology. 1988;122(1):127–132. [DOI] [PubMed] [Google Scholar]
- 26. Nakabayashi K, Matsumi H, Bhalla A, et al. Thyrostimulin, a heterodimer of two new human glycoprotein hormone subunits, activates the thyroid-stimulating hormone receptor. J Clin Invest. 109(11):1445–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sun SC, Hsu PJ, Wu FJ, Li SH, Lu CH, Luo CW. Thyrostimulin, but not thyroid-stimulating hormone (TSH), acts as a paracrine regulator to activate the TSH receptor in mammalian ovary. J Biol Chem. 2010;285(6):3758–3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sinha P, Aarnisalo P, Chubb R, et al. Loss of Gsα in the postnatal skeleton leads to low bone mass and a blunted response to anabolic parathyroid hormone therapy. J Biol Chem. 2016;291(4):1631–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gesty-Palmer D, Chen M, Reiter E, et al. Distinct β-arrestin- and G protein-dependent pathways for parathyroid hormone receptor-stimulated ERK1/2 activation. J Biol Chem. 2006;281(16):10856–10864. [DOI] [PubMed] [Google Scholar]
- 30. Gesty-Palmer D, Flannery P, Yuan L, et al. A β-arrestin-biased agonist of the parathyroid hormone receptor (PTH1R) promotes bone formation independent of G protein activation. Sci Transl Med. 2009;1(1):1ra1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bisello A, Chorev M, Rosenblatt M, Monticelli L, Mierke DF, Ferrari SL. Selective ligand-induced stabilization of active and desensitized parathyroid hormone type 1 receptor conformations. J Biol Chem. 2002;277(41):38524–38530. [DOI] [PubMed] [Google Scholar]
- 32. Ferrari SL, Pierroz DD, Glatt V, et al. Bone response to intermittent parathyroid hormone is altered in mice null for β-Arrestin2. Endocrinology. 2005;146(4):1854–1862. [DOI] [PubMed] [Google Scholar]
- 33. Vestergaard P, Mosekilde L. Hyperthyroidism, bone mineral, and fracture risk–a meta-analysis. Thyroid. 2003;13(6):585–593. [DOI] [PubMed] [Google Scholar]
- 34. Brassill MJ, Williams GR. Advances in our understanding of hyperthyroidism-associated bone loss. Eur Endocrinol. 2009;4:103–106. [Google Scholar]
- 35. Mundy GR, Shapiro JL, Bandelin JG, Canalis EM, Raisz LG. Direct stimulation of bone resorption by thyroid hormones. J Clin Invest. 58(3):529–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sivertsen B, Holliday N, Madsen AN, Holst B. Functionally biased signalling properties of 7TM receptors - opportunities for drug development for the ghrelin receptor. Br J Pharmacol. 2013;170(7):1349–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]