

Neuroinflammation following immune activation in the central nervous system (CNS) leads to neuronal loss in specific areas of the CNS resulting in neurodegenerative disorders. Thus, fine tuning the immune responses within the brain is essential, because most of the brain diseases are associated with chronic inflammation, aberrant microglia activation that follows inappropriate T cell activation and polarization. Brain's immune cells, microglia, are sensitized to extrinsic and intrinsic stimuli mediating neuroinflammation contributing to the removal of pathogen/injurious stimuli and initiate healing process in the CNS. However, dysregulation of the inflammatory response leads to bystander tissue damage. During sterile and infectious acute brain injuries chronic inflammation exacerbates neurodegeneration and contributes to pathogenesis of Alzheimer's disease, Parkinson's disease, Huntington's disease, multiple sclerosis, depression, sickness behaviour, diminished cognition and memory. Neuroinflammation in the CNS can be mediated by many of the factors including pattern/danger-associated molecular patterns (PAMPs and DAMPs) are recognised by the various classes of pattern recognition receptors (PRRs). Among the group of PRRs, the toll-like receptor-4 (TLR4) is crucially involved in neuropathology. Microglia are the only cell type in resting CNS that express TLR4 mediating neuronal damage (Lehnardt et al., 2003). Its upregulation is reported in neuronal injuries and neurodegenerative diseases while the deficiency protects against neurodegeneration and shows increased survival of neurons. Mutation in the TLR4 decreases microglial activation and preserves cognitive functions in mouse model of Alzheimer's disease.
Role of TLR4 in neurodegeneration: Decades of research indicates involvement of TLR4 in many inflammatory diseases like sepsis, influenza, cancer, diabetes, atherosclerosis and neurodegenerative diseases; suggesting its manipulation (inhibition/antagonism) to have a huge clinical value. TLR4 recognizes bacterial lipopolysaccharide (LPS) and other endogenous molecules like HMGB-1, HSP 70, HSP 60, β-amyloid and β-synuclein, followed by activation of innate and adaptive immune system. Its antagonism has shown promising results in preclinical study of sepsis and has been demonstrated for its safety efficacy (Barochia et al., 2011). Preclinical studies using Eritoran tetrasodium (TLR4 antagonist) suggests that TLR4 antagonism inhibits LPS induced production of tumor necrosis factor alpha (TNF-α), interleukin-1 beta (IL-β) and IL-6, suggesting it as a strategy to limit excessive inflammation associated with TLR4 activation. Phase I and II clinical trials showed that Eritoran inhibits sepsis at lower endotoxin levels while in phase III clinical trial, it did not significantly reduce mortality of severe sepsis patients. Possibly the drug failed because (i) LPS may not be the only pathogenic molecule; (ii) differences in the response against Gram-negative and Gram-positive bacterial sepsis; (iii) other treatments interfering with interaction of drug and TLR4 (Opal et al., 2013). There are a variety of other microbial and endogenous host-derived ligands (in addition to LPS) that are recognized by a number of PRRs and can activate mitogen-activated protein kinases (MAPKs), nuclear factor-kappa B (NF-κB)-dependent and TLR4-independent signaling (Barochia et al., 2011). Thus, common intermediary molecules like MAPKs/NF-κB in the inflammatory cascade may be targeted for better clinical benefits. In our recent paper (Gaikwad and Agrawal-Rajput, 2015), we have demonstrated a detailed molecular mechanism of TLR4 induced damage of neurons and strategies to confer bidirectional neuroprotection (Figure 1).
Figure 1.
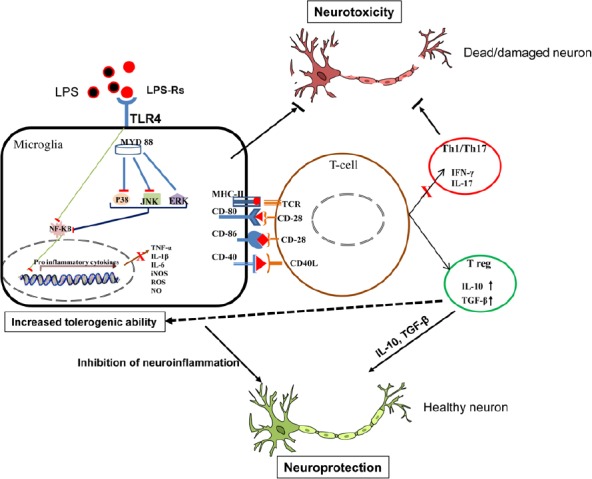
Schematic representation of TLR4 antagonism mediated bidirectional neuroprotection.
LPS stimulation results in increased microglial TLR4 activation following phosphorylation of MAPKs (JNK, ERK and p38) and NF-κB which enhances inflammatory mediators (TNF-α, IL-1β, IL-6, NO and ROS) causing damage or death of neurons. TLR4 antagonist (LPS-Rs) decreases TLR4 induced neuroinflammation and neurotoxicity. The antagonism also promotes neurosupportive Treg cells priming with decrease in Th1/Th17 cytokines. The immunosuppressive Treg cytokines (IL-10) confers tolerogenicity to microglia. ERK: Extracellular regulated protein kinases; IFN-γ: interferon gamma; IL-1β: interleukin-1 beta; iNOS: inducible nitric oxide synthase; JNK: c-Jun N-terminal kinases; LPS: lipopolysaccharide; MAPKs: mitogen-activated protein kinases; MHC-II: major histocompatibility complex class II; MYD 88: myeloid differentiation primary response 88; NF-κB: nuclear factor-kappa B; NO: nitric oxide; ROS: reactive oxygen species; TGF-β: transforming growth factor beta; TLR4: toll-like receptor-4; TNF-α: tumor necrosis factor alpha.
Neuroinflammation: defence or offence? Neuroinflammation following CNS insults dysregulates homeostasis; microglia play a critical role in restoring homeostasis and remodeling of the active neuronal circuits through removal of apoptotic cells and improvement of synaptic plasticity. Microglia-derived cytokines, neurotransmitters, proteolytic enzymes and neurotrophins can influence the differentiation and survival of neurons, astrocytes and oligodendrocytes. We demonstrate that microglia are rapidly activated following TLR4 stimulation as demonstrated by phagocytic phenotype and massive increase in potent proinflammatory cytokines like TNF-α, IL-1β and IL-6. During CNS pathophysiology, microglia are induced by pattern recognition receptors (PRRs) agonists followed by activation of MAPKs pathways. They have a critical role in regulating cell proliferation, differentiation, survival and death; thus implicated in the pathogenesis of brain health. MAPK family consist of ERK, p38 and JNK. MAPKs play equally important roles in both immune cells and neurons, in microglia they mediate secretion of inflammatory cytokines while in neurons, inflammatory cytokines TNF-α and IL-1β activate the MAPKs. The MAPK pathways in brain are closely associated with cellular response to inflammation providing, degeneration and repair and their manipulation provide benefits. MAPK activation promotes production of inflammatory cytokines such as IL-1β, TNF-α, IL-6 and nitric oxide causing significant neuronal damage. In contrast, IL-4 promotes production of anti-inflammatory cytokines through AKT/PI3K and FOXO1, STAT3 and IRF4 and dictates benefits during CNS pathology (Varnum and Ikezu, 2012). They facilitate CNS recovery through clearance of cellular debris, resolution of inflammation and release plethora of trophic factors through PI3/AKT and MAPK pathways. However, fine tuning the microglial TLR4 activation plays a key role in immune surveillance or neuroinflammatory propagation which needs to be tightly controlled for therapeutic benefits. The failure to prevent overactivation leads to aberrant phagocytic activity resulting in potential loss of the healthy neurons (Neher et al., 2011). In our recent paper using LPS, as an exemplary ligand, for TLR4 activation we have demonstrated that JNK/p38 MAPKs and p65-NF-κB participate in mediating LPS induced neuroinflammation. Moreover, LPS-Rs through TLR4 antagonism attenuated phosphorylation of JNK/p38 MAPKs and p65-NF-κB accompanied by decreased inflammatory cytokines by microglia (Gaikwad and Agrawal-Rajput, 2015). Report by De Paola et al. (2012) also demonstrates TLR4 antagonism to display neuroprotection in a mouse model of motor neuron degeneration. Our study thus adds a new dimension to the fact that aberrant and persistent activation of JNK/p38 potentiates chronic inflammation causing a failure to initiate repair or remodelling. TLR4 antagonism attenuated phosphorylation of JNK/p38 may help to re-instigate the CNS healing. Additionally, the MAPK pathways have been reported for their regenerative actions. During regeneration, reduction in ERK signalling is necessary; however, stem cell factor (SCF) induced protection and survival of neurons is mediated by ERK-PI3K-NF-κB. The available reports thus suggest both protective (enhanced survival, neurite growth) and pathological role (cell death) of these signaling cascades (Walker et al., 2013). With vast and contradictory role of MAPKs in neuronal survival and damage, researcher should precisely select the targets for manipulation of these signals to achieve protection, homeostasis, pluripotency, regeneration and/or transdifferentiation for better CNS functions.
Regulatory T cells in the brain homeostasis: implications of TLR4 antagonism: The inflammatory network generated through microglial TLR4 and downstream MAPKs contributes to induce neuronal damage and their subsequent apoptosis and clearance. Using microglia-neuron co-culture system, we have demonstrated that TLR4 activation in the microglia is essential to mediate neuronal cell death. LPS-Rs protect microglia-mediated neuronal morphology, and decreased Bax/Bcl2 apoptotic pathway conferring neuroprotective functions. The TLR4 silencing confirms the essentiality of the receptor for neuroinflammatory damage. Following neuronal apoptosis microglia play an active role in clearance of the dead and/or dying neurons through phagocytosis. Microglial execute phagocytic activity and its inhibition is sufficient to prevent inflammatory neurodegenerative processes (Neher et al., 2011). We for the first time have provided a novel mechanism of phagocytic inhibition of microglia through TLR4 antagonism that may provide clinical benefits.
The activation or tolerogenic state of the innate immune cells orchestrates the T cell response. Microglia, as an antigen presenting cell, dictates the adaptive immune functions through infiltration of leucocytes in the brain and exacerbating/resolving the problem. Microglia-mediated neuroinflammatory milieu and the surface molecules would prime the T cell subsets, Th1 and Th17, to enhance neuroinflammatory burden. The immunosuppressive Tregs provides neurosupportive microenvironment through secretion of glia-derived neurotrophic factor (GDNF), brain-derived neurotrophic factor (BDNF) and insulin-like growth factor (IGF-1) (Gonzalez and Pacheco, 2014). A mechanistic study by Zhou et al. (2006) suggests microglial TLR4 is necessary for leukocyte recruitment in the brain. We, in our paper, have demonstrated that TLR4 activated microglia primed the T cells to release interferon gamma (IFN-γ) and IL-17, the key Th1 and Th17 cytokines respectively. However, the tolerogenic state of microglia (mediated by TLR4 antagonism) can be strategized to gain optimum benefits through generation of tolerogenic microenvironment. LPS-Rs, through inhibition of cognate and non cognate interactions, resulted in generation of tolerogenic T regulatory cells with subsequent decrease in the Th1/Th17 cytokines. Additionally, we reported that the Treg cytokines, IL-10 transfers the tolerogenic ability to microglia as evidenced by decreased production of TNF-α and nitric oxide.
Conclusively, our study demonstrated microglial TLR4 and downstream MAPK as a key mediators of neuroinflammation. LPS-Rs through TLR4 antagonism prevents the neuroinflammatory circuit and the downstream kinases were proven to be essential in mediating the activity. TLR4 antagonism leads to decreased neuronal apoptosis and phagocytosis. The antagonism transfers microglial tolerogenicity to render generation of IL-10 producing Treg cells. The immunosuppressive IL-10 confers bidirectional gain through inhibition of microglia activation to re-instigate neurosupportive microenvironment Figure 1. Cautious manipulations of microglia activation and signaling pathways, through selective manipulation, might promote neurosupportive/regenerative microenvironment.
The work was funded by Department of Science and Technology, Government of India (Grant No. SR/CSI/59/2011(G)) and Gujarat State Biotechnology Mission (Grant No. GSBTM/MD/PROJECTS/SSA/3385/2012/2013).
References
- Barochia A, Solomon S, Cui X, Natanson C, Eichacker PQ. Eritoran tetrasodium (E5564) treatment for sepsis: review of preclinical and clinical studies. Expert Opin Drug Metab Toxicol. 2011;7:479–494. doi: 10.1517/17425255.2011.558190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Paola M, Mariani A, Bigini P, Peviani M, Ferrara G, Molteni M, Gemma S, Veglianese P, Castellaneta V, Boldrin V, Rossetti C, Chiabrando C, Forloni G, Mennini T, Fanelli R. Neuroprotective effects of toll-like receptor 4 antagonism in spinal cord cultures and in a mouse model of motor neuron degeneration. Mol Med. 2012;18:971–981. doi: 10.2119/molmed.2012.00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaikwad S, Agrawal-Rajput R. Lipopolysaccharide from rhodobacter sphaeroides attenuates microglia-mediated inflammation and phagocytosis and directs regulatory T cell response. Int J Inflam 2015. 2015 doi: 10.1155/2015/361326. 361326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez H, Pacheco R. T-cell-mediated regulation of neuroinflammation involved in neurodegenerative diseases. J Neuroinflammation. 2014;11:201. doi: 10.1186/s12974-014-0201-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnardt S, Massillon L, Follett P, Jensen FE, Ratan R, Rosenberg PA, Volpe JJ, Vartanian T. Activation of innate immunity in the CNS triggers neurodegeneration through a Toll-like receptor 4-dependent pathway. Proc Natl Acad Sci U S A. 2003;100:8514–8519. doi: 10.1073/pnas.1432609100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher JJ, Neniskyte U, Zhao JW, Bal-Price A, Tolkovsky AM, Brown GC. Inhibition of microglial phagocytosis is sufficient to prevent inflammatory neuronal death. J Immunol. 2011;186:4973–4983. doi: 10.4049/jimmunol.1003600. [DOI] [PubMed] [Google Scholar]
- Opal SM, et al. Effect of eritoran, an antagonist of MD2-TLR4, on mortality in patients with severe sepsis: the ACCESS randomized trial. JAMA. 2013;309:1154–1162. doi: 10.1001/jama.2013.2194. [DOI] [PubMed] [Google Scholar]
- Varnum MM, Ikezu T. The classification of microglial activation phenotypes on neurodegeneration and regeneration in Alzheimer's disease brain. Arch Immunol Ther Exp (Warsz) 2012;60:251–266. doi: 10.1007/s00005-012-0181-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker CL, Liu NK, Xu XM. PTEN/PI3K and MAPK signaling in protection and pathology following CNS injuries. Front Biol (Beijing) 2013;8 doi: 10.1007/s11515-013-1255-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Lapointe BM, Clark SR, Zbytnuik L, Kubes P. A requirement for microglial TLR4 in leukocyte recruitment into brain in response to lipopolysaccharide. J Immunol. 2006;177:8103–8110. doi: 10.4049/jimmunol.177.11.8103. [DOI] [PubMed] [Google Scholar]