

Abstract
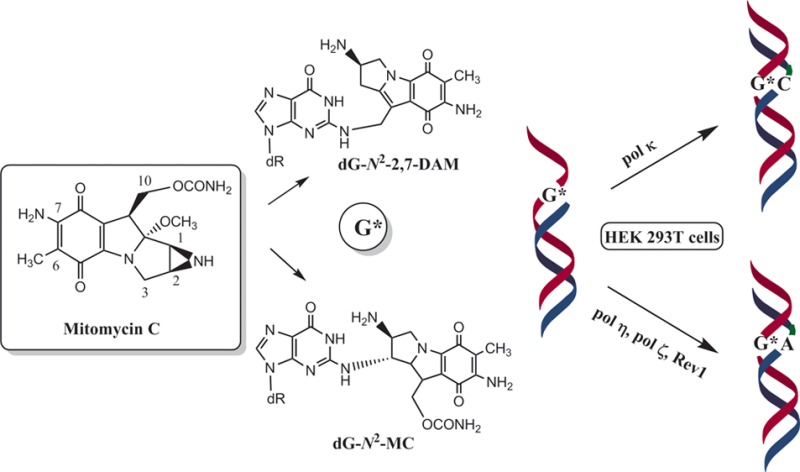
Mitomycin C (MC) is a cytotoxic and mutagenic antitumor agent that alkylates DNA upon reductive activation. 2,7-Diaminomitosene (2,7-DAM) is a major metabolite of MC in tumor cells, which also alkylates DNA. MC forms seven DNA adducts, including monoadducts and inter- and intrastrand cross-links, whereas 2,7-DAM forms two monoadducts. Herein, the biological effects of the dG-N2 adducts formed by MC and 2,7-DAM have been compared by constructing single-stranded plasmids containing these adducts and replicating them in human embryonic kidney 293T cells. Translesion synthesis (TLS) efficiencies of dG-N2-MC and dG-N2-2,7-DAM were 38 ± 3 and 27 ± 3%, respectively, compared to that of a control plasmid. This indicates that both adducts block DNA synthesis and that dG-N2-2,7-DAM is a stronger replication block than dG-N2-MC. TLS of each adducted construct was reduced upon siRNA knockdown of pol η, pol κ, or pol ζ. For both adducts, the most significant reduction occurred with knockdown of pol κ, which suggests that pol κ plays a major role in TLS of these dG-N2 adducts. Analysis of the progeny showed that both adducts were mutagenic, and the mutation frequencies (MF) of dG-N2-MC and dG-N2-2,7-DAM were 18 ± 3 and 10 ± 1%, respectively. For both adducts, the major type of mutation was G → T transversions. Knockdown of pol η and pol ζ reduced the MF of dG-N2-MC and dG-N2-2,7-DAM, whereas knockdown of pol κ increased the MF of these adducts. This suggests that pol κ predominantly carries out error-free TLS, whereas pol η and pol ζ are involved in error-prone TLS. The largest reduction in MF by 78 and 80%, respectively, for dG-N2-MC and dG-N2-2,7-DAM constructs occurred when pol η, pol ζ, and Rev1 were simultaneously knocked down. This result strongly suggests that, unlike pol κ, these three TLS polymerases cooperatively perform the error-prone TLS of these adducts.
Introduction
Mitomycin C (MC) is the most studied member of a group of potent antibiotics (reviewed recently in ref (1)), discovered in Japan in the 1950s, produced by the microorganism Streptomyces caespitosus.2,3 MC has been widely used in cancer chemotherapy.4 The anticancer activity of MC has been postulated to be due to its DNA alkylation activity and, notably, its ability to form interstrand DNA–DNA cross-links.5,6 MC (structure shown in Scheme 1) possesses two alkylating centers: the 1,2-aziridine and 10-carbamate groups. MC is quite unreactive in its native state, but reductive activation of the quinone moiety initiates its DNA alkylation and antitumor activity.7,8 Like MC, 10-decarbamoyl mitomycin C (DMC), the derivative obtained after chemical removal of the 10-carbamoyl group (Scheme 1), is highly toxic and a potent alkylating agent, despite lacking its carbamate group.9−11 Formation of DNA adducts by MC and DMC has been studied in EMT6 mouse mammary tumor cells, Fanconi anemia-A cells, normal human fibroblasts, and MCF-7 human breast cancer cells.11,12 Nine different covalent DNA adducts, including six monofunctional adducts and three cross-links, have been isolated from cancer cells treated with MC.12 Seven of these adducts are derived directly from MC (and DMC), in which the covalent linkage is formed at the exocyclic amino group of 2′-deoxyguanosine (dG) in DNA. These adducts include a set of two stereoisomeric monoadducts at the C1 position of MC and DMC, plus two stereoisomeric interstrand cross-links (ICL) and one intrastrand cross-link. The other two DNA adducts are formed at the N7 and 2-amino group of dG with 2,7-diaminomitosene (2,7-DAM), the major metabolite of MC. Isolation and characterization of these adducts were primarily carried out in Maria Tomasz’s laboratory over a period of 15 years (1986–2000) (reviewed in refs (13) and (14)). The first DNA adduct of MC to be isolated and identified was monoadduct 1 containing an α-linkage at the 1 position of MC with the exocyclic amino group of dG (Scheme 1).15,16 DMC forms the analogous monoadduct with a β-linkage, an epimer of 2.(11) Both MC and DMC form an ICL, 3, which contains an additional covalent bond between the C10 of MC and the exocyclic N2 position of dG in the complementary strand.11,17 Not shown in Scheme 1 are the three epimers of adducts 1–3, which contain a β-linkage to dG at the C1 position of MC (or DMC). Epimers of 2 and 3 are formed at a high level by DMC.11 An intrastrand cross-link, 4, has also been isolated.18 The structures of the two dG adducts of 2,7-DAM, 5 and 6, are shown in Scheme 1.19,20
Scheme 1. Chemical Structures of MC, DMC, and 2,7-DAM and Their Major DNA Adducts.

Not shown are the three epimers of adducts 1–3, which contain a β-linkage to dG at the C1 position of MC. Epimers of adducts 2 and 3 are formed at a high level by DMC.
In various tumor cell lines, 2,7-DAM is either nontoxic or barely cytotoxic, whereas both MC and DMC are highly cytotoxic (reviewed in ref (21)). Surprisingly, DMC is somewhat more cytotoxic than MC in cell cultures, but the level of ICL formation seems to correlate with their cytotoxicity.11 Interestingly, DMC forms 20–30-fold more monoadducts than MC, and its covalent bond formation to dG-N2 exhibits opposite stereochemical preference to MC (i.e., MC forms monoadduct 1, whereas DMC forms the epimer of 2).11,22,23
Although it is generally accepted that ICLs are largely responsible for the cytotoxicity of MC and DMC, the repair and replication of the various monoadducts are also of significant interest to determine if they might be responsible for generating secondary tumors. MC is mutagenic in bacterial and mammalian cells.24,25 However, a convincing link between specific DNA adducts formed by MC and its mutagenesis has not been established. Monoadduct 1 is cytotoxic but not mutagenic in Escherichia coli.26 In vitro studies of monoadducts 1 and 2 by several prokaryotic DNA polymerases showed that they are strong blocks of replication.27,28 In each case, replication stopped at the base 3′ of the adduct site. Even with the bypass polymerase human DNA polymerase η (hpol η), no further extension was observed. In contrast, for 2,7-DAM adduct 5, full-length extension of a primer by Klenow (exo-) and T7 (exo-) DNA polymerases and partial extension by hpol η were observed.28 Adducts 1 and 5 were also compared in vivo in E. coli cells, which showed that MC adduct 1 is highly toxic and reduces viability of the construct by nearly 90% even with SOS, whereas 2,7-DAM adduct 5 decreased the viability by ∼50%.28 However, neither adduct was found to be mutagenic in E. coli.28 Adduct 5, in addition, failed to induce mutations in simian kidney cells.28
In the current work, we have compared the mutagenicity of MC-derived monoadduct 1 and 2,7-DAM-derived adduct 6, both at the N2 position of dG, in human embryonic kidney (HEK) 293T cells. Both adducts block replication, but adduct 6 is a stronger block of replication than adduct 1. For the first time, we also show that both adducts are mutagenic, inducing primarily G → T mutations, and that adduct 1 is more mutagenic than adduct 6. Employing an siRNA knockdown approach, we also established that pol κ carries out predominantly error-free translesion synthesis (TLS) of both lesions, whereas pol η, pol ζ, and Rev1 are involved in error-prone TLS.
Experimental Procedures
Materials
dNTP solutions (100 mM) were purchased from New England Biolabs (Ipswich, MA) or GE Healthcare (formerly Amersham Biosciences, Piscataway, NJ). [γ-32P]ATP was purchased from PerkinElmer (Waltham, MA). Unmodified oligonucleotides were purchased from Midland Certified Reagents (Midland, TX).
siRNAs
Synthetic siRNA duplexes against PolH (SI02663619), PolK (SI04930884), and Rev1 (SI00115311) and negative control siRNA (1027280) were purchased from Qiagen (Valencia, CA), whereas the same for Rev3 was purchased from Integrated DNA Technologies (Coralville, IA). Sequences of all siRNAs have been reported.29
Methods
12-mer Containing a dG-N2-2,7-DAM Adduct
The synthetic approach followed a postoligomerization method, as reported in ref (30). Briefly, diethylamine (20 μL), DMSO (150 μL), and the N2-(trimethylsilyl)ethoxycarbonyl-protected triaminomitosene (6 mg, 0.015 mmol) were added to oligonucleotide 5′-CTAGTG(X)TATCC-3′ (with X = 2-fluoro-O6-(2-trimethylsilylethyl)-deoxyinosine; 204 nmol, 30 A260 units), and the mixture was stirred at room temperature. The reaction was monitored by HPLC, which showed that no starting material remained after 72 h, when the reaction was stopped. The mixture was diluted with 750 μL of water and 100 μL of 3 M NaOAc (pH 5). Ethanol (3 mL) was added, and it was left at −20 °C for 20 min. The suspension was centrifuged (13 000 rpm for 15 min), and the supernatant was discarded. The brown residue was washed twice with ethanol (750 μL), and the crude modified oligonucleotide was dried in air. It was dissolved in a ZnBr2 solution (300 μL, made by dissolving 250 mg of ZnBr2 in 150 μL of nitromethane and 150 μL of isopropanol) and incubated at room temperature for 48 h. It was quenched with EDTA (12 mL of a 0.05 M solution), and the volume was reduced by one-half. The mixture was subjected to Sephadex G-25 gel filtration, and the void fraction containing the desired oligonucleotide was lyophilized and purified by HPLC as described below.
12-mer Containing a dG-N2-MC Adduct
Oligonucleotide 5′-CTAGTCGTATCC-3′ (676 nmol, 100 A260 units), the partially complementary 7-mer 5′-TA(5meC)GACT-3′ (752 nmol, 63 A260 units, where 5meC represents the 5 methylated cytosine), and 48.9 μmole of mitomycin C were dissolved in phosphate buffer (5.8 mL of 0.1 mM buffer, pH 7.5). The solution was warmed to 55 °C for 5 min with shaking. It was then slowly cooled to room temperature and left at 4 °C for 30 min. A deaerated solution of Na2SO4 (22.3 μmole, 554 μL of a 40.2 mM solution in 0.1 M potassium phosphate buffer) was added to the oligonucleotide mixture with continuous bubbling of argon. The solution was stirred in air for 1.5 h, and then the solvents were lyophilized. The mixture was diluted with 2.25 mL of water and 300 μL of 3 M NaOAc (pH 5). Ethanol (9 mL) was added, and the mixture was left at −20 °C for 20 min. The suspension was centrifuged (13 000 rpm for 15 min), and the supernatant was discarded. The brown residue was washed twice with ethanol (2.25 mL), and the desired adducted oligonucleotide was purified by HPLC as described below.
Purification of Adducted Oligonucleotides
Adducted oligonucleotides were purified by HPLC on a semiprep C-18 reverse-phase column; the system was equipped with a diode array detector and monitored at 260 and 320 nm (flow rate: 5 mL/min). The following gradient was used for the oligonucleotide containing the dG-N2-2,7-DAM adduct: initial conditions 99% NH4OAc (0.1 M in water)–1% acetonitrile, then 60 min linear gradient to 80% acetonitrile, and 5 min at 80% acetonitrile, followed by 5 min linear gradient to initial conditions. The gradient used for the oligonucleotide containing the dG-N2-MC adduct was as follows: buffer A: 0.1 M TEAA, 0.1 mM EDTA; buffer B: 70% buffer A and 30% acetonitrile. Initial conditions: 25% buffer B, then 96 min linear gradient to 60% buffer B%, and 5 min at 60% buffer B, followed by 5 min linear gradient to initial conditions.
Characterization of Adducted Oligonucleotides
A part of the adducted oligonucleotides (containing dG-N2-2,7-DAM or dG-N2-2,7-MC) was each dissolved (8.3 nmol, 1.3 A260 units) in the following mixture: 700 μL of water, 200 μL of 0.5 M Tris-HCl, pH 8.6, and 350 μL of 0.025 M MgCl2 and incubated with snake venom phosphodiesterase (4 units) and alkaline phosphatase (4 units) at room temperature for 8 h. The mixtures were analyzed by reverse-phase HPLC using the following gradient: 5–60% buffer B in buffer A in 75 min (buffer A: 0.3 M potassium phosphate, pH 5.8; buffer B: 70% buffer A, 30% acetonitrile). The modified nucleosides (dG-N2-2,7-DAM and dG-N2-MC) were identified by comparison with an authentic sample on the basis of their retention times and UV spectra. The adducted oligonucleotides also were subjected to ESI-MS analysis. The m/z of dG-N2-MC-12-mer gave 3898.4 Da (negative electrospray; deconvolution), which is 302 Da higher than the calculated mass of the unmodified 12-mer (3596.4 Da), indicating the presence of MC (334.3 Da) (minus OCH3 and H) in the 12-mer template. The dG-N2-2,7-DAM-12-mer gave m/z 3877.7 Da, which is 241.3 Da higher than the unmodified 12-mer (3636.4 Da), indicating the presence of 2,7-DAM moiety in the 12-mer. The purity of the adducted oligonucleotides was examined by electrophoresis on a 16% polyacrylamide gel containing 8 M urea (Figure S1), which showed that the adducted 12-mers ran slightly slower than the unmodified 12-mers and that they were >98% pure.
Construction of a pMS2 Vector Containing a Single dG-N2-2,7-DAM or dG-N2-MC and Their Replication in HEK 293T Cells
We have constructed a single adduct-modified single-stranded vector, pMS2, with neomycin and ampicillin resistance genes, similarly as reported elsewhere.31,32 The ligation efficiency of the adducted oligonucleotides to both sides of the gapped pMS2 plasmid was in excess of 60%. The HEK 293T cells were grown to ∼90% confluence and transfected with 50 ng of construct in 6 μL of Lipofectamine cationic lipid reagent (Invitrogen, Carlsbad, CA). Following transfection with modified or unmodified pMS2, the cells were allowed to grow at 37 °C in 5% CO2 for 24 h and the plasmid DNA was collected and purified.33 It was used to transform E. coli DH10B, and transformants were analyzed by oligonucleotide hybridization followed by DNA sequence analysis.32,34
TLS Assay in Human Cells
The adduct-containing or control pMS2 construct was mixed with an equal amount of a single-stranded pMS2 DNA containing a different 12-mer sequence (i.e., 5′-GTGCGTGTTTGT-3′ in place of 5′-CTAGTGGTATCC-3′ or 5′-CTAGTCGTATCC-3′) constructed in a manner similar to the construction of the dG-N2-2,7-DAM or dG-N2-MC (or control) construct. The mixed DNA was used to transfect HEK 293T cells and processed as described above. Oligonucleotide probes for the complementary sequences for both the wild-type and mutant (i.e., containing a different 12-mer sequence) plasmids were used to analyze the progeny. The mutant DNA was used as an internal control, and it gave equal number of progeny as the control construct.
Mutational Analyses of TLS Products from Human Cells with Polymerase Knockdowns
Prior to transfection of the control and dG-N2-2,7-DAM- or dG-N2-MC-containing vectors, synthetic siRNA duplexes were transfected into HEK 293T cells using Lipofectamine. HEK 293T cells were plated in 6-well plates at 50% confluence. After a 24 h incubation, they were transfected with 100 pmole of siRNA duplex mixed with Lipofectamine, diluted in Opti-MEM (Gibco), per well. One day before transfection of the plasmid, cells were seeded in 24-well plates at 70% confluence. Cells were then cotransfected with another aliquot of siRNA and either control plasmid or lesion-containing plasmid at a ratio of 2:1. After a 24 h incubation, progeny plasmids were isolated as described.
RT-PCR Analysis and Western blotting
Total RNA was extracted from the cells 72 h after the first transfection of siRNA duplexes, and 100 ng of total RNA was used for RT-PCR analysis. Using primers specific to TLS DNA polymerases and GAPDH as the control gene, siRNA knockdown efficiency was determined as previously described.29,35 Reverse transcription and the initial activation step of the PCR were performed for 30 min at 50 °C and 15 min at 95 °C, respectively. Details of the amplification of PolH, PolK, PolI, and Rev1 as well as GAPDH are described in detail in ref (29). RT-PCR products were analyzed on a 2% agarose gel run at 100 V for 3 h in 1× TBE buffer.
The specifics of the western blotting procedure have been reported in ref (29). Briefly, cells were washed with cold phosphate buffered saline and lysed in ice-cold RIPA buffer containing protease inhibitor cocktail. After a 1 h incubation on ice, the mixture was centrifuged at 10 000 rpm for 15 min at 4 °C, the protein concentration was determined, and western blotting was performed on the supernatant. The protein extracts were boiled in sample loading buffer. Proteins were separated on either a 5 or 7% SDS-PAGE gel by electrophoresis for 2 h and transferred onto PVDF membranes. The membranes were blocked with 5% milk and incubated with antibodies that specifically recognize human PolH, PolK, PolI, Rev3, or Rev1. Human β-actin antibody was used to confirm equal gel loading. Horseradish peroxidase-conjugated goat anti-rabbit and goat anti-mouse secondary antibodies were used at 1:5000 dilutions. The signals were developed using Pierce ECL western blotting substrate, and images were taken using a PhosphorImager.
Results
dG-N2-MC and dG-N2-2,7-DAM Are Replication-Blocking Lesions in HEK 293T Cells
Each dG-N2 adduct construct and an unmodified plasmid (as an internal control) were cotransfected into HEK 293T cells. Following a 24 h incubation to allow for the replication of the plasmids, the progeny DNA was isolated and used to transform E. coli DH10B cells. The fractions of the colonies originating from the adduct-containing plasmid relative to the unmodified plasmid, which indicates the TLS efficiency, were determined by oligonucleotide hybridization. As shown in Figure 1, compared to the unmodified plasmid, TLS efficiencies of dG-N2-MC and dG-N2-2,7-DAM were 38 ± 3 and 27 ± 3%, respectively. This indicates that both adducts block DNA synthesis and that dG-N2-2,7-DAM is a stronger replication block compared to that of dG-N2-MC.
Figure 1.
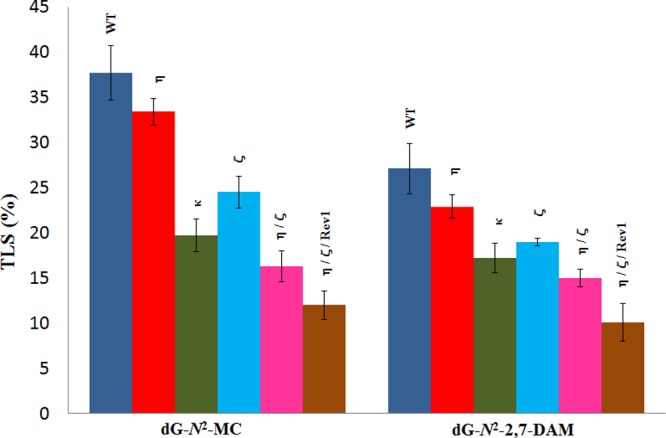
Effect of siRNA knockdown of TLS polymerases on the extent of replicative bypass of dG-N2-MC (in 5′-CTAGTCG*TATCC-3′) and dG-N2-2,7-DAM (in 5′-CTAGTGG*TATCC-3′). Percent TLS in various polymerase knockdowns was measured using an internal control of unmodified plasmid in which a different 12-mer was ligated. The data represent the mean and standard deviation of results from two independent experiments. HEK 293T cells were treated with negative control (NC) siRNA (WT), whereas the other single, double, or triple polymerase knockdowns are indicated above the bar. TLS result from each knockdown experiment was considered statistically significant (p < 0.02) (except in pol η-knockdown cells) compared to that from HEK 293T cells treated with NC siRNA (WT). The p value of %TLS for each knockdown was calculated using a two-tailed, unpaired Student’s t test.
Roles of pol η, pol κ, pol ζ, and Rev1 in TLS Efficiency of dG-N2-MC and dG-N2-2,7-DAM
In order to define the replication-blocking characteristics of these dG-N2 adducts and identify the polymerases involved in their bypass, we knocked down several TLS polymerases prior to replicating the adduct-containing plasmid in HEK 293T cells. Upon knockdown of pol η, pol κ, or pol ζ, TLS of each construct containing the MC or 2,7-DAM adduct was reduced. For dG-N2-MC, knockdown of pol η, pol κ, or pol ζ, respectively, resulted in 12, 48, or 35% reduction in TLS. For dG-N2-2,7-DAM, the trend was similar, and knockdown of pol η, pol κ, or pol ζ, respectively, resulted in 15, 37, or 30% reduction in TLS. Evidently, in each case, the most significant reduction occurred with knockdown of pol κ, which suggests that pol κ plays a major role in TLS of these adducts. Further reduction in TLS was observed upon simultaneous knockdown of two or three TLS polymerases. Simultaneous knockdown of pol η, pol ζ, and Rev1, for example, resulted in 68 and 63% reductions in TLS for the dG-N2-MC and dG-N2-2,7-DAM constructs, respectively. Generally, the trend of reduced TLS with knockdowns followed an analogous trend in dG-N2-MC and dG-N2-2,7-DAM constructs, although the extent of reduction in TLS was more pronounced in the case of the former.
Mutagenicity of dG-N2-MC and dG-N2-2,7-DAM in HEK 293T Cells and the Roles of TLS Polymerases
While dG-N2-2,7-DAM was a stronger block to replication than dG-N2-MC, the latter was found to be more mutagenic than the former in HEK 293T cells (Figure 2). The mutation frequencies of dG-N2-MC (adduct 1) and dG-N2-2,7-DAM (adduct 6) were 18 ± 3 and 10 ± 1%, respectively. For both adducts, the major type of mutation was G → T, which occurred in 12 and 6% frequencies in dG-N2-MC and dG-N2-2,7-DAM, respectively. They also induced G → A (∼3% for each adduct) and low levels of G → C and semitargeted mutations (Table S2A). Knockdown of pol η and pol ζ reduced the MF of dG-N2-MC and dG-N2-2,7-DAM, whereas knockdown of pol κ increased the MF of these adducts. MFs of dG-N2-MC and dG-N2-2,7-DAM constructs were reduced by 44 and 20%, respectively, upon knockdown of pol η, whereas reductions by 33 and 11% of the same was observed when pol ζ was knocked down (Figure 2 and Table S2B,D). In contrast, MF of dG-N2-MC and dG-N2-2,7-DAM constructs was increased by 39 and 50%, respectively, when pol κ was knocked down (Figure 2 and Table S2C). This suggests that pol κ predominantly carries out error-free TLS, whereas pol η and pol ζ are involved in error-prone TLS of these two dG-N2 adducts. We also investigated the MF when pol η and pol ζ were simultaneously knocked down, which exhibited a synergy in reducing the MF (by 56 and 60%, respectively, for dG-N2-MC and dG-N2-2,7-DAM constructs) (Figure 2 and Table S2E). The largest reduction in MF, 78 and 80% for dG-N2-MC and dG-N2-2,7-DAM constructs, respectively, was observed when pol η, pol ζ, and Rev1 were simultaneously knocked down (Figure 2 and Table S2F). This result suggests that these three TLS polymerases cooperatively perform error-prone TLS of these adducts.
Figure 2.
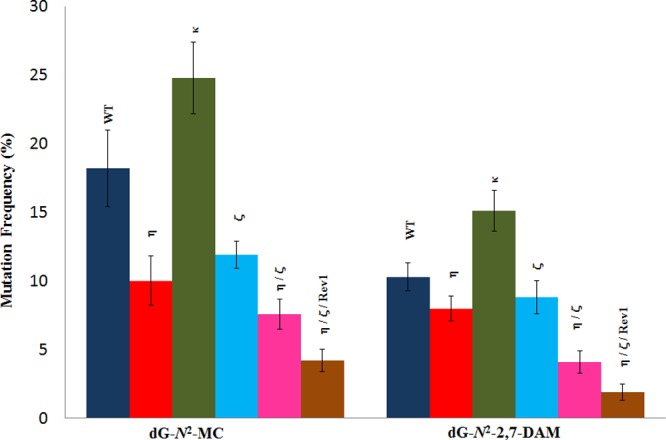
Mutational frequency of dG-N2-MC (in 5′-CTAGTCG*TATCC-3′) and dG-N2-2,7-DAM (in 5′-CTAGTGG*TATCC-3′) in HEK 293T cells cotransfected with NC siRNA (WT) or siRNA for single, double, or triple polymerase knockdowns (shown above the bar). The data represent the average of two independent experiments (shown in Table S2A–F).
Discussion
Our result that dG-N2-2,7-DAM is a stronger replication block than dG-N2-MC is interesting since it was reported that 2,7-DAM is not cytotoxic and does not activate the p53 pathway, whereas MC and DMC are cytotoxic and activate the p53 pathway.36,37 However, the lack of substantial cytotoxicity of 2,7-DAM was attributed to its inability to form cross-links in DNA, which is considered to be the hallmark of MC’s ability to kill malignant cells.6,13,21 Of the various monoadducts of MC and 2,7-DAM, in an earlier work in E. coli, we found that dG-N2-MC is more toxic than dG-N7-2,7-DAM, with the latter being the other monoadduct (structure 5 in Scheme 1) formed by 2,7-DAM.28 In addition, dG-N2-MC was found to be a complete block to pol η in vitro, and replication stopped one nucleotide before the adduct, whereas pol η carried out primer extension to full-length product past dG-N7-2,7-DAM.28 However, TLS of dG-N2-MC and dG-N2-2,7-DAM was not compared in any prior study, and the current investigation is the first to show that dG-N2-2,7-DAM is a stronger block to replication than dG-N2-MC in HEK 293T cells. It should be noted, however, that the 5′ base of the two adducts is not identical. The 5′ base of dG-N2-MC is C, whereas dG-N2-2,7-DAM has a 5′ G. It is unclear if this difference in the 5′ base has any effect on the TLS and MF of the two adducts.
A high-resolution NMR solution structure of dG-N7-2,7-DAM in duplex DNA indicated that the drug moiety is not intercalated and lies in the major groove of a relatively unperturbed B-DNA structure.38 In contrast, the MC moiety of the dG-N2-MC lies tightly in the slightly widened minor groove of duplex B-DNA,39 but there are extensive noncovalent contacts between the drug and the minor groove of the DNA duplex. Structural information on the dG-N2-2,7-DAM is lacking, but it is conceivable that it occupies the minor groove in a manner similar to that of the dG-N2-MC adduct. It was proposed that DNA adducts localized in the solvent-exposed major groove (as in the case of dG-N7-2,7-DAM) are better tolerated than adducts located in the minor groove (such as the two adducts used in this study) that interact with the polymerase surface.40 This suggestion is consistent with the results that show efficient replication past dG-N7-2,7-DAM, whereas both dG-N2-2,7-DAM and dG-N2-MC are strong blocks of DNA synthesis.
In view of the result that dG-N2-MC is significantly mutagenic in HEK 293T cells, another intriguing question is why it is not mutagenic in E. coli, even though in a uvrA strain ∼0.2% semitargeted mutants were detected.26 We believe that the rationalization for this apparent discrepancy lies in the abundance of TLS polymerases, which allows for a much higher level of TLS in human cells compared to that in E. coli, and the identity of the polymerases that bypass it. TLS of the dG-N2-MC construct in E. coli was reported to be ∼7%, which increased 2-fold with SOS,26 in comparison to 38% TLS in HEK 293T cells, as determined in the current work. Structural studies of a replication-blocking benzo[a]pyrene-dG-N2 adduct indicate that the steric constraints of the active site of a high-fidelity DNA polymerase result in structural disruptions that are incompatible with DNA replication of this type of bulky dG-N2 adduct.40 Accordingly, we hypothesize that replicative polymerases, such as DNA polymerase III in E. coli and pol δ and ε in human cells, cannot bypass this adduct and that only the TLS polymerases are able to carry out TLS. It also appears that the TLS polymerases in human cells, with the exception of pol κ, though more efficient in TLS of this adduct, are also more error-prone. However, of the three TLS polymerases in E. coli, the identity of the polymerase responsible for TLS of dG-N2-MC (and dG-N2-2,7-DAM) is yet to be determined, but this polymerase is less error-prone in comparison to the coordinated TLS carried out by pol η, pol ζ, and Rev1.
Despite the difference in magnitude, the nearly identical pattern of TLS and mutagenesis data for replication past dG-N2-MC and dG-N2-2,7-DAM in this work strongly suggests that the mechanism of error-free and error-prone bypass of these two adducts are the same. Of the four human Y family DNA polymerases, pol κ is the only enzyme that has homologues in prokaryotes and archaea, such as DinB (pol IV) in E. coli and Dbh and Dpo4 in Sulfolobus solfataricus. However, pol κ is set apart from pol IV, Dpo4, and the others by an extension at its N-terminus, which is conserved only in eukaryotes and is critical for pol κ’s activity in lesion bypass and mispair extension.41 Even when pol κ is inefficient at inserting a base opposite certain DNA lesions, it efficiently carries out extension after another polymerase incorporates a base opposite the DNA adduct.42 Specifically, for dG-N2 adducts, however, pol κ promotes both error-free insertion and extension efficiently.43 The finding that pol κ performs error-free TLS of the MC and 2,7-DAM adducts is indeed consistent with a substantial body of work that established pol κ’s role in accurate and efficient replication past many dG-N2 adducts, including the major dG-N2 adduct of benzo[a]pyrene.43−45 The active site of pol κ is more open at the minor groove side of the DNA template compared to that of other Y family polymerases, which allows it to accommodate a bulky dG-N2 adduct in its anti conformation to base-pair with C and continue error-free replication.46 The extension at the N-terminus, known as the N-clasp of human pol κ, plays a crucial role in stabilizing the single-stranded template and interacting with all three (i.e., finger/thumb/little finger) DNA binding domains.45,46 As shown in Figure 1, pol κ appears to be responsible for a major fraction of TLS of the two MC and 2,7-DAM dG-N2 adducts. A large increase in the mutation frequency in the absence of pol κ underscores its ability to perform accurate TLS (Figure 2). In contrast, pol η, pol ζ, and Rev1 together are responsible for the major fraction of mutagenic TLS (Figure 2). On the basis of the current state of knowledge regarding TLS,47−49 we postulate that pol η incorporates the wrong base (predominantly A) opposite the dG-N2 adduct of MC or 2,7-DAM, which is extended by pol ζ, whereas Rev1 performs a noncatalytic role by physically interacting with the other two polymerases. Additional in vitro and cellular experiments may be able to test this hypothesis in the future.
Glossary
Abbreviations
- MC
mitomycin C
- 2,7-DAM
2,7-diaminomitosene
- DMC
10-decarbamoyl mitomycin C
- HEK
human embryonic kidney
- TLS
translesion synthesis
- MF
mutation frequency
- dG-N2-MC and dG-N2-2,7-DAM
2′-deoxyguanosine-N2 monoadducts of MC and 2,7-DAM, respectively
- dG-N7-2,7-DAM
2′-deoxyguanosine-N7 monoadduct of 2,7-DAM
- NC
negative control
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.chemrestox.6b00087.
Autoradiogram of lesion-containing and control oligonucleotides and tables detailing TLS and mutation data (PDF)
This work was supported by NIH grants ES09127 and ES023350 to A.K.B. and 5SC3GM105460 to E.C.
The authors declare no competing financial interest.
Supplementary Material
References
- Bass P. D.; Gubler D. A.; Judd T. C.; Williams R. M. (2013) Mitomycinoid alkaloids: mechanism of action, biosynthesis, total syntheses, and synthetic approaches. Chem. Rev. 113, 6816–6863. 10.1021/cr3001059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata T.; Hoshi T.; Kanamori K.; Matsumae A.; Sano Y.; Shima T.; Sugawara R. (1956) Mitomycin, a new antibiotic from Streptomyces. I. J. Antibiot. 9, 141–146. [PubMed] [Google Scholar]
- Wakaki S.; Marumo H.; Tomioka K.; Shimizu G.; Kato E.; Kamada H.; Kudo S.; Fujimoto Y. (1958) Isolation of new fractions of antitumor mitomycins. Antibiot. Chemother. 8, 228–240. [PubMed] [Google Scholar]
- Bradner W. T. (2001) Mitomycin C: a clinical update. Cancer Treat. Rev. 27, 35–50. 10.1053/ctrv.2000.0202. [DOI] [PubMed] [Google Scholar]
- Iyer V. N.; Szybalski W. (1963) A molecular mechanism of mitomycin action: Linking of complementary DNA strands. Proc. Natl. Acad. Sci. U. S. A. 50, 355–362. 10.1073/pnas.50.2.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer V. N.; Szybalski W. (1964) Mitomycins and porfiromycin: Chemical mechanism of activation and cross-linking of DNA. Science 145, 55–58. 10.1126/science.145.3627.55. [DOI] [PubMed] [Google Scholar]
- Tomasz M.; Chawla A. K.; Lipman R. (1988) Mechanism of monofunctional and bifunctional alkylation of DNA by mitomycin C. Biochemistry 27, 3182–3187. 10.1021/bi00409a009. [DOI] [PubMed] [Google Scholar]
- Suresh Kumar G.; Lipman R.; Cummings J.; Tomasz M. (1997) Mitomycin C-DNA adducts generated by DT-diaphorase. Revised mechanism of the enzymatic reductive activation of mitomycin C. Biochemistry 36, 14128–14136. 10.1021/bi971394i. [DOI] [PubMed] [Google Scholar]
- Kinoshita S.; Uzu K.; Nakano K.; Takahashi T. (1971) Mitomycin derivatives. 2. Derivatives of decarbamoylmitosane and decarbamoylmitosene. J. Med. Chem. 14, 109–112. 10.1021/jm00284a006. [DOI] [PubMed] [Google Scholar]
- Rockwell S.; Kim S. Y. (1995) Cytotoxic potential of monoalkylation products between mitomycins and DNA: studies of decarbamoyl mitomycin C in wild-type and repair-deficient cell lines. Oncol. Res. 7, 39–47. [PubMed] [Google Scholar]
- Palom Y.; Suresh Kumar G.; Tang L. Q.; Paz M. M.; Musser S. M.; Rockwell S.; Tomasz M. (2002) Relative toxicities of DNA cross-links and monoadducts: new insights from studies of decarbamoyl mitomycin C and mitomycin C. Chem. Res. Toxicol. 15, 1398–1406. 10.1021/tx020044g. [DOI] [PubMed] [Google Scholar]
- Paz M. M.; Ladwa S.; Champeil E.; Liu Y.; Rockwell S.; Boamah E. K.; Bargonetti J.; Callahan J.; Roach J.; Tomasz M. (2008) Mapping DNA adducts of mitomycin C and decarbamoyl mitomycin C in cell lines using liquid chromatography/ electrospray tandem mass spectrometry. Chem. Res. Toxicol. 21, 2370–2378. 10.1021/tx8002615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasz M. (1995) Mitomycin C: small, fast and deadly (but very selective). Chem. Biol. 2, 575–579. 10.1016/1074-5521(95)90120-5. [DOI] [PubMed] [Google Scholar]
- Paz M. M.; Pritsos C. A. (2012) The molecular toxicology of mitomycin C. Adv. Mol. Toxicol. 6, 243–299. 10.1016/B978-0-444-59389-4.00007-0. [DOI] [Google Scholar]
- Tomasz M.; Lipman R.; Verdine G. L.; Nakanishi K. (1986) Reassignment of the guanine-binding mode of reduced mitomycin C. Biochemistry 25, 4337–4344. 10.1021/bi00363a024. [DOI] [PubMed] [Google Scholar]
- Tomasz M.; Chowdary D.; Lipman R.; Shimotakahara S.; Veiro D.; Walker V.; Verdine G. L. (1986) Reaction of DNA with chemically or enzymatically activated mitomycin C: isolation and structure of the major covalent adduct. Proc. Natl. Acad. Sci. U. S. A. 83, 6702–6706. 10.1073/pnas.83.18.6702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasz M.; Lipman R.; Chowdary D.; Pawlak J.; Verdine G. L.; Nakanishi K. (1987) Isolation and structure of a covalent cross-link adduct between mitomycin C and DNA. Science 235, 1204–1208. 10.1126/science.3103215. [DOI] [PubMed] [Google Scholar]
- Bizanek R.; McGuinness B. F.; Nakanishi K.; Tomasz M. (1992) Isolation and structure of an intrastrand cross-link adduct of mitomycin C and DNA. Biochemistry 31, 3084–3091. 10.1021/bi00127a008. [DOI] [PubMed] [Google Scholar]
- Palom Y.; Belcourt M. F.; Kumar G. S.; Arai H.; Kasai M.; Sartorelli A. C.; Rockwell S.; Tomasz M. (1998) Formation of a major DNA adduct of the mitomycin metabolite 2,7-diaminomitosene in EMT6 mouse mammary tumor cells treated with mitomycin C. Oncol. Res. 10, 509–521. [PubMed] [Google Scholar]
- Palom Y.; Belcourt M. F.; Musser S. M.; Sartorelli A. C.; Rockwell S.; Tomasz M. (2000) Structure of adduct X, the last unknown of the six major DNA adducts of mitomycin C formed in EMT6 mouse mammary tumor cells. Chem. Res. Toxicol. 13, 479–488. 10.1021/tx000024j. [DOI] [PubMed] [Google Scholar]
- Bargonetti J.; Champeil E.; Tomasz M. (2010) Differential toxicity of DNA adducts of mitomycin C. J. Nucleic Acids 2010, 1–6. 10.4061/2010/698960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz M. M. (2010) Cross-linking of dithiols by mitomycin C. Chem. Res. Toxicol. 23, 1384–1392. 10.1021/tx100134h. [DOI] [PubMed] [Google Scholar]
- Bueren-Calabuig J. A.; Negri A.; Morreale A.; Gago F. (2012) Rationale for the opposite stereochemistry of the major monoadducts and interstrand crosslinks formed by mitomycin C and its decarbamoylated analogue at CpG steps in DNA and the effect of cytosine modification on reactivity. Org. Biomol. Chem. 10, 1543–1552. 10.1039/c1ob06675g. [DOI] [PubMed] [Google Scholar]
- Maier P.; Feldman D. B.; Ficsor G. (1978) Host-mediated assay in rhesus monkey (Macaca mulatta): mutagenicity of Mitomycin C. Mutat. Res., Fundam. Mol. Mech. Mutagen. 57, 91–95. 10.1016/0027-5107(78)90238-5. [DOI] [PubMed] [Google Scholar]
- Ferguson L. R.; Palmer B. D.; Denny W. A. (1988) Microbial mutagenicity of chlorambucil, its half-mustard and mitomycin C: a modified screening strategy for genetic toxicology of bis-alkylating anti-tumour drugs. Anti-Cancer Drug Des. 3, 67–76. [PubMed] [Google Scholar]
- Ramos L. A.; Lipman R.; Tomasz M.; Basu A. K. (1998) The major mitomycin C-DNA monoadduct is cytotoxic but not mutagenic in Escherichia coli. Chem. Res. Toxicol. 11, 64–69. 10.1021/tx970163+. [DOI] [PubMed] [Google Scholar]
- Basu A. K.; Hanrahan C. J.; Malia S. A.; Kumar S.; Bizanek R.; Tomasz M. (1993) Effect of site-specifically located mitomycin C-DNA monoadducts on in vitro DNA synthesis by DNA polymerases. Biochemistry 32, 4708–4718. 10.1021/bi00069a004. [DOI] [PubMed] [Google Scholar]
- Utzat C. D.; Clement C. C.; Ramos L. A.; Das A.; Tomasz M.; Basu A. K. (2005) DNA adduct of the mitomycin C metabolite 2,7-diaminomitosene is a nontoxic and nonmutagenic DNA lesion in vitro and in vivo. Chem. Res. Toxicol. 18, 213–223. 10.1021/tx049813h. [DOI] [PubMed] [Google Scholar]
- Pande P.; Malik C. K.; Bose A.; Jasti V. P.; Basu A. K. (2014) Mutational analysis of the C8-guanine adduct of the environmental carcinogen 3-nitrobenzanthrone in human cells: critical roles of DNA polymerases eta and kappa and Rev1 in error-prone translesion synthesis. Biochemistry 53, 5323–5331. 10.1021/bi5007805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Champeil E.; Paz M. M.; Ladwa S.; Clement C. C.; Zatorski A.; Tomasz M. (2008) Synthesis of an oligodeoxyribonucleotide adduct of mitomycin C by the postoligomerization method via a triamino mitosene. J. Am. Chem. Soc. 130, 9556–9565. 10.1021/ja802118p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalam M. A.; Haraguchi K.; Chandani S.; Loechler E. L.; Moriya M.; Greenberg M. M.; Basu A. K. (2006) Genetic effects of oxidative DNA damages: comparative mutagenesis of the imidazole ring-opened formamidopyrimidines (Fapy lesions) and 8-oxo-purines in simian kidney cells. Nucleic Acids Res. 34, 2305–2315. 10.1093/nar/gkl099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt D. L.; Utzat C. D.; Hilario P.; Basu A. K. (2007) Mutagenicity of the 1-nitropyrene-DNA adduct N-(deoxyguanosin-8-yl)-1-aminopyrene in mammalian cells. Chem. Res. Toxicol. 20, 1658–1664. 10.1021/tx700131e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirt B. (1967) Selective extraction of polyoma DNA from infected mouse cell cultures. J. Mol. Biol. 26, 365–369. 10.1016/0022-2836(67)90307-5. [DOI] [PubMed] [Google Scholar]
- Kalam M. A.; Basu A. K. (2005) Mutagenesis of 8-oxoguanine adjacent to an abasic site in simian kidney cells: tandem mutations and enhancement of G-- > T transversions. Chem. Res. Toxicol. 18, 1187–1192. 10.1021/tx050119r. [DOI] [PubMed] [Google Scholar]
- Yoon J. H.; Prakash L.; Prakash S. (2009) Highly error-free role of DNA polymerase eta in the replicative bypass of UV-induced pyrimidine dimers in mouse and human cells. Proc. Natl. Acad. Sci. U. S. A. 106, 18219–18224. 10.1073/pnas.0910121106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbas T.; Olivier M.; Lopez J.; Houser S.; Xiao G.; Kumar G. S.; Tomasz M.; Bargonetti J. (2002) Differential activation of p53 by the various adducts of mitomycin C. J. Biol. Chem. 277, 40513–40519. 10.1074/jbc.M205495200. [DOI] [PubMed] [Google Scholar]
- Boamah E. K.; White D. E.; Talbott K. E.; Arva N. C.; Berman D.; Tomasz M.; Bargonetti J. (2007) Mitomycin-DNA adducts induce p53-dependent and p53-independent cell death pathways. ACS Chem. Biol. 2, 399–407. 10.1021/cb700060t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam G.; Paz M. M.; Suresh Kumar G.; Das A.; Palom Y.; Clement C. C.; Patel D. J.; Tomasz M. (2001) Solution structure of a guanine-N7-linked complex of the mitomycin C metabolite 2,7-diaminomitosene and DNA. Basis of sequence selectivity. Biochemistry 40, 10473–10484. 10.1021/bi010965a. [DOI] [PubMed] [Google Scholar]
- Sastry M.; Fiala R.; Lipman R.; Tomasz M.; Patel D. J. (1995) Solution structure of the monoalkylated mitomycin C-DNA complex. J. Mol. Biol. 247, 338–359. 10.1006/jmbi.1994.0143. [DOI] [PubMed] [Google Scholar]
- Hsu G. W.; Huang X.; Luneva N. P.; Geacintov N. E.; Beese L. S. (2005) Structure of a high fidelity DNA polymerase bound to a benzo[a]pyrene adduct that blocks replication. J. Biol. Chem. 280, 3764–3770. 10.1074/jbc.M411276200. [DOI] [PubMed] [Google Scholar]
- Uljon S. N.; Johnson R. E.; Edwards T. A.; Prakash S.; Prakash L.; Aggarwal A. K. (2004) Crystal structure of the catalytic core of human DNA polymerase kappa. Structure 12, 1395–1404. 10.1016/j.str.2004.05.011. [DOI] [PubMed] [Google Scholar]
- Lone S.; Townson S. A.; Uljon S. N.; Johnson R. E.; Brahma A.; Nair D. T.; Prakash S.; Prakash L.; Aggarwal A. K. (2007) Human DNA polymerase kappa encircles DNA: implications for mismatch extension and lesion bypass. Mol. Cell 25, 601–614. 10.1016/j.molcel.2007.01.018. [DOI] [PubMed] [Google Scholar]
- Choi J. Y.; Angel K. C.; Guengerich F. P. (2006) Translesion synthesis across bulky N2-alkyl guanine DNA adducts by human DNA polymerase kappa. J. Biol. Chem. 281, 21062–21072. 10.1074/jbc.M602246200. [DOI] [PubMed] [Google Scholar]
- Avkin S.; Goldsmith M.; Velasco-Miguel S.; Geacintov N.; Friedberg E. C.; Livneh Z. (2004) Quantitative analysis of translesion DNA synthesis across a benzo[a]pyrene-guanine adduct in mammalian cells: the role of DNA polymerase kappa. J. Biol. Chem. 279, 53298–53305. 10.1074/jbc.M409155200. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Yang Y.; Tang T. S.; Zhang H.; Wang Z.; Friedberg E.; Yang W.; Guo C. (2014) Variants of mouse DNA polymerase kappa reveal a mechanism of efficient and accurate translesion synthesis past a benzo[a]pyrene dG adduct. Proc. Natl. Acad. Sci. U. S. A. 111, 1789–1794. 10.1073/pnas.1324168111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha V.; Bian C.; Xing G.; Ling H. (2016) Structure and mechanism of error-free replication past the major benzo[a]pyrene adduct by human DNA polymerase kappa. Nucleic Acids Res. gkw204. 10.1093/nar/gkw204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson R. E.; Washington M. T.; Haracska L.; Prakash S.; Prakash L. (2000) Eukaryotic polymerases iota and zeta act sequentially to bypass DNA lesions. Nature 406, 1015–1019. 10.1038/35023030. [DOI] [PubMed] [Google Scholar]
- Haracska L.; Unk I.; Johnson R. E.; Johansson E.; Burgers P. M.; Prakash S.; Prakash L. (2001) Roles of yeast DNA polymerases delta and zeta and of Rev1 in the bypass of abasic sites. Genes Dev. 15, 945–954. 10.1101/gad.882301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shachar S.; Ziv O.; Avkin S.; Adar S.; Wittschieben J.; Reissner T.; Chaney S.; Friedberg E. C.; Wang Z.; Carell T.; Geacintov N.; Livneh Z. (2009) Two-polymerase mechanisms dictate error-free and error-prone translesion DNA synthesis in mammals. EMBO J. 28, 383–393. 10.1038/emboj.2008.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.