

Abstract
Fluorescence microscopy is one of the most important tools in cell biology research and it provides spatial and temporal information to investigate regulatory systems inside cells. This technique can generate data in the form of signal intensities at thousands of positions resolved inside individual live cells; however, given extensive cell-to-cell variation, methods do not currently exist to assemble these data into three- or four-dimensional maps of protein concentration that can be compared across different cells and conditions. Here, we have developed one such method and applied it to investigate actin dynamics in T cell activation. Antigen recognition in T cells by the T cell receptor (TCR) is amplified by engagement of the costimulatory receptor CD28 and we have determined how CD28 modulates actin dynamics. We imaged actin and eight core actin regulators under conditions where CD28 in the context of a strong TCR signal was engaged or blocked to yield over a thousand movies. Our computational analysis identified diminished recruitment of the activator of actin nucleation WAVE2 and the actin severing protein cofilin to F-actin as the dominant difference upon costimulation blockade. Reconstitution of WAVE2 and cofilin activity restored the defect in actin signaling dynamics upon costimulation blockade. Thus we have developed and validated an approach to quantify protein distributions in time and space for analysis of complex regulatory systems.
Introduction
As one of the great tools of cell biology, imaging enables the investigation of cellular processes as they occur in time and space inside live cells. Imaging generates a tremendous amount of data in the form of signal intensities at thousands of positions that are resolved inside each individual cell. Commonly, the scientific question to be answered enables the researcher to focus on specific reference elements within these data, for example, cytoskeletal structures or vesicular distributions, thus simplifying data analysis through the use of customized image quantification. However, as image acquisition becomes ever more efficient, the size and complexity of imaging data sets grow, thus bringing imaging into the realm of systems biology. With the growing size and complexity of data sets, the design and execution of customized analysis strategies becomes increasingly more difficult. As a generally applicable alternative, a strategy that uses the signal intensity at each resolved position within a cell would be highly advantageous. It would enable unbiased image analysis without the need for a previous focus on particular processes, enable efficient computational processing, and would do so using the entirety of the information contained in the images. We have developed such a computational image analysis routine and verified its usefulness by applying it to investigate the mechanism by which co-receptor usage regulates actin dynamics in T cells.
T cells become activated through direct interactions with antigen-presenting cells (APCs). The T cell receptor (TCR) recognizes antigen-derived peptide presented by the major histocompatibility complex (MHC) on the surface of the APC. Parallel engagement of costimulatory receptors by their APC ligands is required for efficient T cell activation. The most potent costimulatory receptor is CD28, which is activated by the B7 family ligands CD80 and CD86. T cell activation stimulates the rapid and transient accumulation of T cell actin at the interface between the T cell and the APC (a region known as the immunological synapse) (1), which is coregulated by the TCR and CD28 (2). Genetic and pharmacological interference with T cell actin dynamics suggests that they are critical for many aspects of T cell function including APC coupling, spatiotemporal organization of T cell signaling, and regulation of transcription (2–6); however, the molecular mechanisms by which costimulation contributes to the regulation of T cell actin dynamics are unresolved.
Understanding the regulation of actin dynamics by costimulation exemplifies two critical challenges caused by the increasing amounts of data generated by current cell biology: complexity and the need to determine alterations in protein function upon physiological perturbation rather than determining protein function per se. Actin regulation is a complex problem, because it occurs through the integrated interaction of numerous key actin regulators. The importance of individual actin regulators has been elegantly established by genetic means. The Arp2/3 complex is the dominant nucleator of actin in T cells (7). It is stimulated by the actin filament nucleation promoting factors Wiskott-Aldrich Syndrome protein (WASP), WASP family Verprolin-homologous protein (WAVE) 2, and cortactin homolog Hematopoietic-specific protein (HS) 1 (8–13). Lack of Arp2/3 activators impairs T cell coupling to APCs, calcium signaling, and cytokine secretion to a variable extent (14–20). Coronin 1A inhibits the generation of polymerized actin (F-actin). Coronin 1A‐deficient T cells display excessive accumulation of F-actin, yet reduced dynamics, and impaired calcium signaling (21, 22). Blocking the F-actin‐severing protein cofilin impairs immunological synapse organization, cytokine secretion, and T cell proliferation (23). The motor protein myosin II plays a context-dependent role in protein transport and signal amplification (24). The F-actin plus end-capping protein CP1α is likely to be important, because capping protein interactors play critical roles in the regulation of T cell activation (25, 26); however, how all of these proteins function as an integrated ensemble has remained largely inaccessible. With regard to the necessity to understand altered protein function, we need to investigate the contributions of costimulation to actin dynamics rather than just the regulation of overall actin turnover. Thus, the deletion or suppression of individual actin regulators should be replaced by approaches that monitor changes in the behavior of these regulators in the absence of costimulation. Again, individual proteins have been linked to actin regulation downstream of CD28, most prominently cofilin, L-plastin, and the capping protein interactor Rltpr, yet their functional integration remains unresolved (25, 27, 28).
Given the large number of interacting actin regulators and the need to determine changes in the behavior of each of them, a hypothesis-driven investigation of how costimulation regulates actin dynamics was inaccessible. As an unbiased alternative, we chose a large-scale imaging approach. Because actin turnover is local, we reasoned that recruitment of actin regulators to the local area of actin turnover was required for their efficient function. Thus, we could use diminished access of actin regulators to the local area of actin turnover in the context of costimulation blockade as a means to identify them as critically controlled by co-receptor stimulation. We generated time-lapse movies of T cell:APC conjugates in which we imaged actin and eight core actin regulators under full stimulus and costimulation-blocked T cell activation conditions, and then we developed an open source computational image analysis pipeline that provided maps of signal intensities at each resolved position within a cell such that they could be compared across all cells. In analyzing these maps, we used diminished recruitment to a core area of actin turnover to identify WAVE2 and cofilin as the principle targets of co-receptor signaling in actin regulation. We then showed that activation of either WAVE2 or cofilin alone in co-receptor-blocked T cells restored a critical element of costimulation-controlled actin dynamics. Restoration of T cell actin dynamics reconstituted spatiotemporal signaling defects in costimulation-blocked T cells, a set of experiments that also constitutes a second application of our computational methodology. These data support the physiological relevance of the mechanism of actin regulation that we described here and demonstrate the potential application of our computational methods to other scenarios.
Results
Microscopic imaging was used to investigate the regulation of actin dynamics by costimulation
In the physiological activation of primary T cells by APCs, actin distributions are precisely defined in time and space. Because actin turnover is regulated locally, the function of the various actin regulators themselves should also be spatiotemporally constrained. To determine how costimulation regulates actin dynamics, we therefore determined changes in the distributions of core actin regulators in cells for which the TCR was stimulated, but stimulation of the CD28 co-receptor was blocked (a condition referred to as costimulation blockade).
We determined the subcellular localization of actin and eight core actin regulators: Arp3 as a member of the Arp2/3 complex; WASP, WAVE2, and HS1 as activators of the Arp2/3 complex; and capping protein 1α, cofilin, coronin, and myosin II as regulators of actin filament stability. In vitro primed primary CD4+ T cells from 5C.C7 TCR transgenic mice were retrovirally transduced to express fluorescently tagged actin regulators. The concentrations of the endogenous actin regulators as determined by quantitative biochemistry ranged from 5 to 100 μM (Fig. 1). To maximize physiological relevance, the abundances of the green fluorescent protein (GFP)-tagged actin regulators (hereafter termed sensors) were limited such that the combined amounts of endogenous and GFP-tagged actin regulators remained within the 5 to 95% percentile of actin regulator abundance in non-transduced T cells for most sensors (Fig. 1). Time-lapsed spinning disk confocal microscopy was performed with the transduced T cells while they were activated by CH27 B cell lymphoma cells pulsed with 10μM MCC antigenic peptide (which acted as APCs) in the absence or presence of anti-CD80 and anti-CD86 antibodies to block stimulation of the CD28 co-receptor (Fig. 2). We generated 1672 movies of cell couples; on average, there were 93 movies per sensor and condition.
Fig. 1. Determination of the molar concentrations of actin regulators in 5C.C7 T cells.
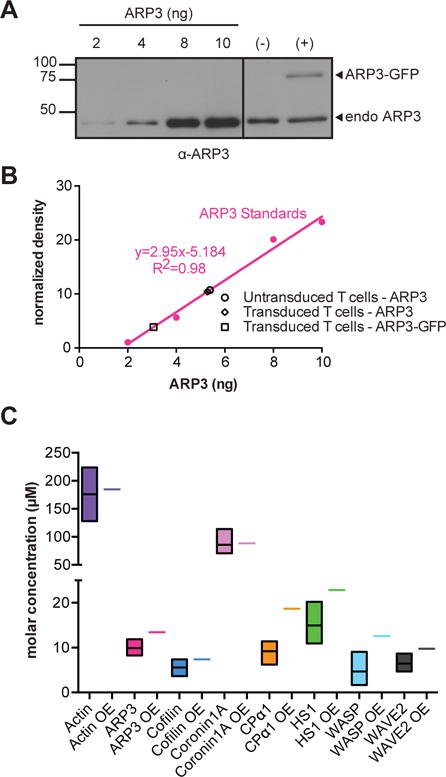
(A and B) 5C.C7 T cells that were left untransduced (−) or were retrovirally transduced to express Arp3-GFP (+) were lysed, resolved by SDS-PAGE together with the indicated quantities of purified Arp3 protein, and analyzed by Western blotting with antibody against Arp3 (A). Band intensities from the Arp3 Western blot were quantified and normalized to those corresponding to actin, which was a loading control for the cell extracts (B). Arp3 amounts in the cell extracts were determined from the calibration curve. Data are from a single experiment and are representative of three experiments. Raw data for other actin regulators are shown in fig. S6. (C) For each of the indicated actin regulators, protein abundance in 5C.C7 T cells was determined from three measurements, the average endogenous protein amount in non-transduced cells (black line in the middle of the colored box, as derived from experiments such as those shown in A, B), the 5 and 95 percentile of expression (colored bars, fig. S6), and the average combined amount of endogenous and GFP-tagged protein in retrovirally transduced cells (colored bar in the samples labelled OE (overexpression), as derived from experiments such as those shown in A, B). For actin regulators that were largely excluded from the nucleus (all but cofilin, Fig. 2), cytoplasmic volume was used in the conversion of the amounts of actin regulator in T cells to molar concentrations.
Fig. 2. Representative imaging data of actin regulators.
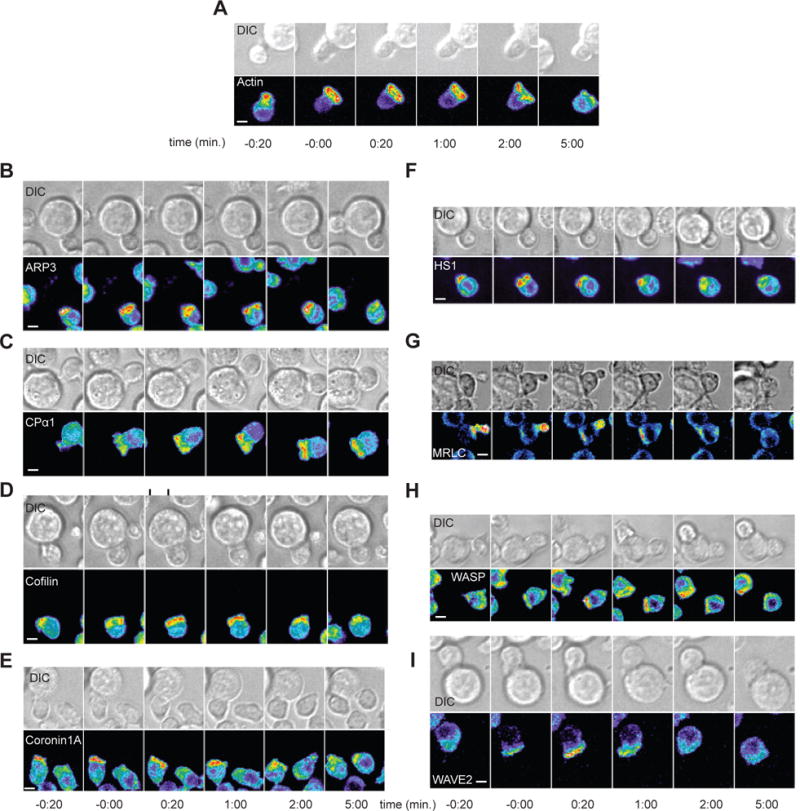
(A to I) Representative interactions of 5C.C7 T cells expressing the indicated GFP fusion proteins with CH27 cells loaded with 10 μM MCC at the indicated times relative to the formation of a tight cell-cell conjugate. Top: DIC images. Bottom: Top-down maximum projections of 3D fluorescence data are shown in a rainbow-like, false-color intensity scale (increasing from blue to red). Scale bars, 5 μm. Images are representative of 49 to 134 cells per protein from 4 to 7 experiments.
Analysis of fluorescence microscopy images generates maps of the three-dimensional subcellular concentrations of actin regulators
Imaging data contain three-dimensional (3D) maps of subcellular protein distributions as they change over time. Here, we computationally extracted these maps from the imaging data in three steps. First, individual T cells were computationally identified as separate objects (i.e., the image was segmented into regions containing single T cells). Second, T cells in different images were aligned with each other. Third, the slightly variable shapes of individual T cells were normalized such that a given position in one cell became directly comparable to the same position in another cell (Fig. 3).
Fig. 3. Illustration of the image analysis pipeline and resulting spatiotemporal models.
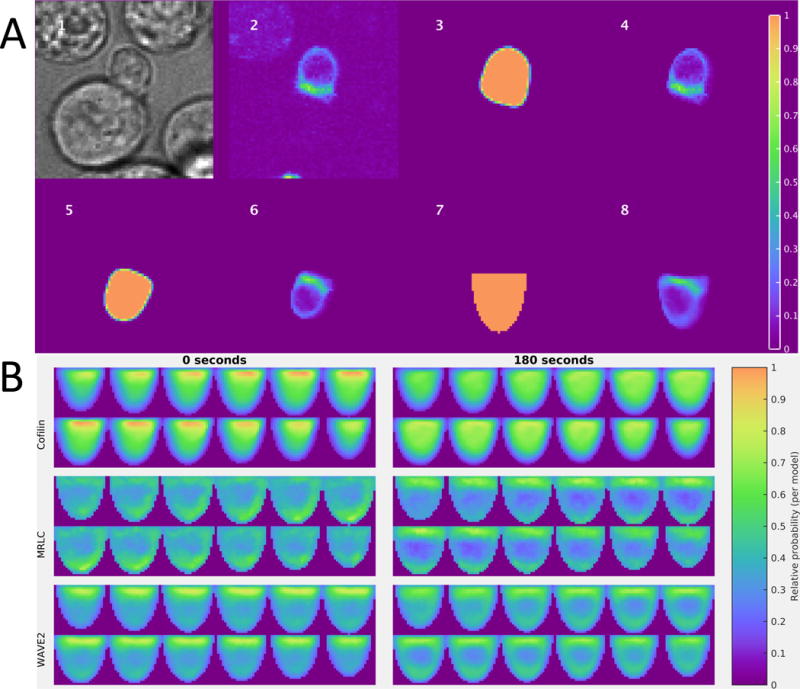
(A) Image 1: Brightfield image centered on a T cell:APC conjugate. Images 2 to 8: Single slices of 3D images that are approximately perpendicular to the immunological synapse, which is shown facing upwards in images 5 to 8. Image 2: False-colored raw coronin-1A–GFP fluorescence image. Image 3: Cell shape extracted by the segmentation algorithm. Image 4: Segmented intensity image. Image 5: Aligned segmentation (immunological synapse facing approximately upwards). Image 6: Aligned segmented intensity image. Image 7: Standard template shape. Image 8: Segmented intensity image deformed into the shape of a standardized cell. (B) Illustrations of spatiotemporal models for cofilin (top), MRLC (middle), and WAVE2 (bottom), which have distinct subcellular distributions at different times. Each panel contains slices perpendicular to the immunological synapse of the full model at 0 (left) or 180 s (right) after synapse formation for each sensor. Within a panel, slices are shown starting at the upper left corner and moving horizontally to the upper right, then wrapping to the lower left corner and continuing to move horizontally towards the lower right; these correspond to z positions from one side to the other relative to the midline of the cell. The models were constructed from 49 to 134 cells from 4 to 7 experiments (see Table S1).
The onset times of T cell:APC conjugate formation and the positions of the center of the T cell:APC interface at 12 time points relative to the time of onset were identified manually in overlays of differential interference contrast (DIC) bright field and maximum projection fluorescence images and were used as input into the computational analysis. The boundary of each T cell was found by first flattening the image histogram (assigning smaller intensity ranges to less frequent intensities) and then using an active contour method that initialized a spherical surface near the T cell:APC interface (immunological synapse) with the diameter of a typical cell and iteratively moved the surface towards the edges in the image. Next, cells were rotated so that the immunological synapse was facing approximately upwards (alignment). In a first iteration, we assumed that the orientation of the plane of the immunological synapse was perpendicular to a ray originating from the weighted centroid of the segmentation volume and directed through the center of the interface (whose coordinates were identified manually as described above). (Examples of the segmentations and alignments for individual cells are shown in fig. S1.) The shape of each cell was then morphed to that of a half-ellipsoid template by incrementally pushing and pulling the edges of the segmented cell (Fig. 3A5) until it matched the template (Fig. 3A7) using a nonrigid image registration algorithm. The fluorescence in each original voxel (3D pixel) (Fig. 3A6) was then adjusted by the same amount to yield the fluorescence in the standardized template (Fig. 3A8). This procedure and the analysis of the extent of cell shape variation at different times are detailed in Materials and Methods.
Thus, we generated > 15,000 shape-normalized maps of protein distributions in individual T cells at defined time points. Maps were then averaged to obtain a 3D model of the localization of actin and each of the eight actin regulators at each of the 12 time points under both conditions, thus resulting in the generation of 216 3D maps of population-averaged local concentrations. [Examples of the mean distributions are shown in Fig. 3B (and examples of the corresponding standard deviations are shown in fig. S2), and all maps are available both as images and numerical values at: http://murphylab.web.cmu.edu/data/TcellModels]. These maps were normalized to the fraction of total fluorescence for each sensor. Note that because the average concentrations of the sensors were determined earlier, these maps can be also expressed as absolute concentrations within each voxel (a file with the average concentrations and a Matlab function to convert the relative fluorescence values to concentrations are included in the dataset).
To evaluate the automated segmentation and alignment process, we visually examined hundreds of randomly selected frames. Only 5 ± 1% of frames showed poor segmentation. Furthermore, all poor segmentations could readily be explained by the close proximity of the analyzed T cell to either the edge of the field or a brighter neighbor. However, 13 ± 3% of frames showed misalignments at the immunological synapse, but these did not affect enrichment analysis. To improve alignment for precise analysis of the geometry at the immunological synapse, we used a two-point synapse annotation: we determined the coordinates of the end points of a line across the interface rather than the position of the interface center as input coordinates to align the T cells. In this case, we did not find any misalignments, likely because alignment became independent of T cell geometry (fig. S3). Even upon inclusion of T cells next to brighter neighbors and the edge of the field, 95% of the frames were accurately processed.
As a first test of the maps of local concentrations, we assessed whether the computational analysis was consistent with features of the subcellular distributions of the actin regulators that were readily apparent. Through hierarchical clustering of the protein distributions in the computational models to determine spatiotemporal similarity, the distributions of the cofilin and myosin II regulatory light chain (MRLC) sensors were found to be distinct from those of all other actin regulators (Fig. 4). The distinct nature of the cofilin distribution was due to cofilin being the only sensor with a substantial nuclear presence. The accumulation of myosin II at the immunological synapse was delayed relative to that of all other actin regulators, and it seemed to be more concentrated at the center of the immunological synapse (Fig. 2 and fig. S4A). By plotting radial intensities at the immunological synapse as a function of time, we found that the more central accumulation of myosin II was readily apparent in comparison to that of actin (fig. S4B). Moreover, the well-established spreading of actin from an initial central contact area to the edge of the interface at the time of tight cell conjugate formation (2, 29, 30) can thus also be seen as the dissolution of the peripheral actin ring in favor of a more evenly spread distribution at late time points (fig. S4B).
Fig. 4. Hierarchical clustering of full models constructed for nine sensors under full stimulus and costimulation blockade conditions.
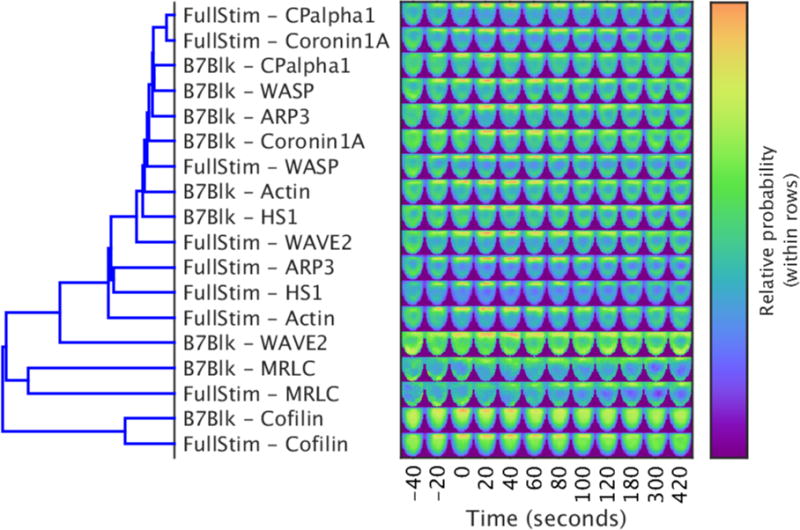
Left: The dendrogram shows xxx. Right: Each image shows a single slice from the 3D model of the distribution of the indicated sensor at the time shown on the horizontal axis. The slices are perpendicular to the synapse and through the middle of the model (the synapse is facing upwards). The cophenetic coefficient of the dendrogram is 0.842. Images are representative of xxx experiments.
Quantitative analysis of sensor enrichment at the immunological synapse identifies WAVE2 and cofilin as central targets of the regulation of actin dynamics by costimulation
Because actin dynamics are restricted in time and space, the reduced recruitment of actin regulators to core areas of actin turnover in the context of costimulation blockade was expected to efficiently identify actin regulators that were dependent on CD28 signaling. Using the shape-normalized computational models of the local concentrations of all actin regulators under control and costimulation-blocked conditions, we determined the tenth of the model volume with the highest average sensor concentration (Fig. 5A). We then analyzed each model separately for the recruitment of actin regulators to this core volume of actin turnover. Four proteins showed the greatest reduction in recruitment to the immunological synapse in the context of CD28 blockade: WAVE2, HS1, Arp3, and cofilin (Fig. 5, B to G). The reduced accumulation of HS1 was restricted to the first minute of tight cell coupling, whereas that of the other three proteins was more sustained, which was suggestive of a more substantial defect. Because WAVE2 and HS1 are both activators of the Arp2/3 complex, all three proteins together constitute the joint first regulatory target of costimulation: F-actin generation. The severing protein cofilin then constitutes the second independent target. The joint regulation of actin dynamics by WAVE2, HS1, Arp3, and cofilin is an attractive scenario, because parallel activation of F-actin generation and severing would be expected to accelerate actin turnover without substantially altering the overall amount of F-actin. Blocking co-receptor signaling markedly affected the geometry of MRLC (Fig. 4), but did not affect its enrichment; therefore we did not pursue it further.
Fig. 5. Kinetics of protein enrichment in the immunological synapse region.
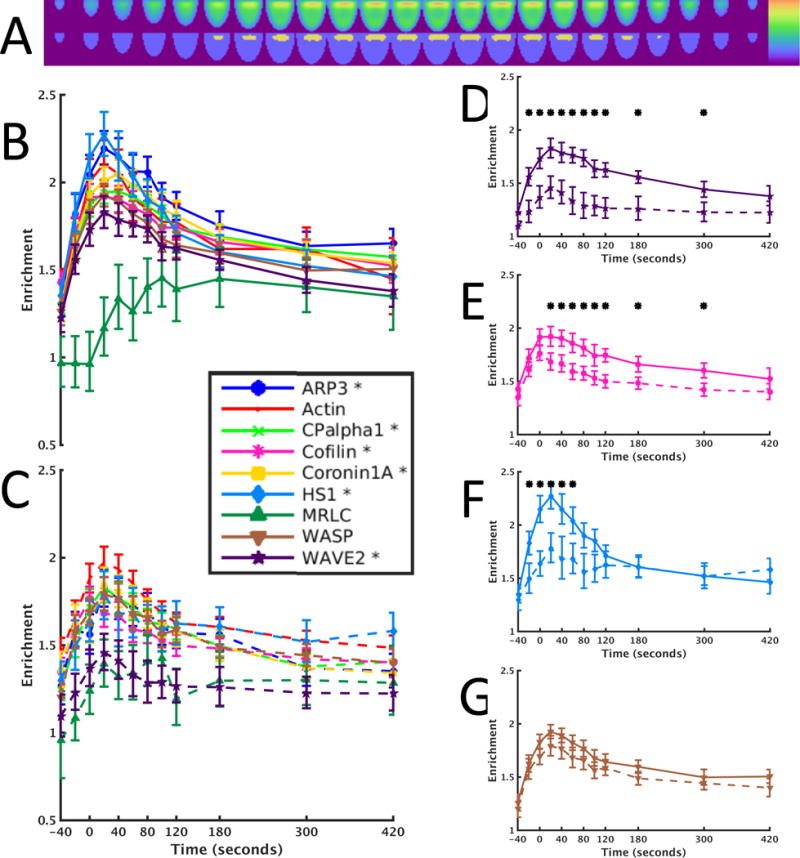
(A to C) To compute the enrichment of a sensor in the core volume of actin turnover, we defined the core region (A) that approximates the immunological synapse as described in Materials and Methods and calculated the ratio of the amount of the sensor in the region to the average amount across the whole cell. This was done for all time points either under conditions of full stimulus (B) or costimulation blockade (C). An asterisk in the legend indicates a sensor that had at least one time point with a statistically significant difference between the two different conditions. (D to G) Detailed comparisons of enrichment for full stimulus (solid lines) and costimulation blockade (dashed lines) for WAVE2 (D), cofilin (E), HS1 (F), and WASP (G). Asterisks indicate time points with statistically different values between the two conditions. Detailed statistics are provided in table S4.
Selective activation of WAVE2 and cofilin under costimulation-blocked conditions restores a critical element of costimulation-dependent actin dynamics
To test the importance of WAVE2-HS1-Arp3 and cofilin for actin regulation by co-receptor signaling, we sought to selectively restore the activity of WAVE2 as the dominant T cell actin nucleation-promoting factor (18, 19) and cofilin under costimulation-blocked conditions. This required an experimental strategy to activate WAVE2 and cofilin and to identify an element of actin dynamics under almost complete control by costimulation. To activate WAVE2 and cofilin, we used the S3A constitutively active mutant of cofilin (cofilinca) together with the constitutively active L61 mutant of Rac1 (Rac1ca) to activate the WAVE2 complex. Both proteins were delivered into primary T cells by protein transduction to ensure parallel, short-term, and quantitative delivery. As an initial control, 1 μM Rac1ca together with 250 nM Cofilinca restored the recruitment of WAVE2 to the immunological synapse in the activation of 5C.C7 T cells by CH27 APCs and peptide, as intended (Fig. 6A). Nevertheless, roles for additional targets of active Rac1 cannot be excluded. As a largely static measure of actin, the amounts of actin at the immunological synapse were only moderately reduced upon costimulation blockade (Fig. 6B), and thus were not suitable to assess how WAVE2 and Cofilin restore costimulation-dependent elements of actin dynamics.
Fig. 6. Active Rac and cofilin restore the recruitment of WAVE2 to the immunological synapse during costimulation blockade.
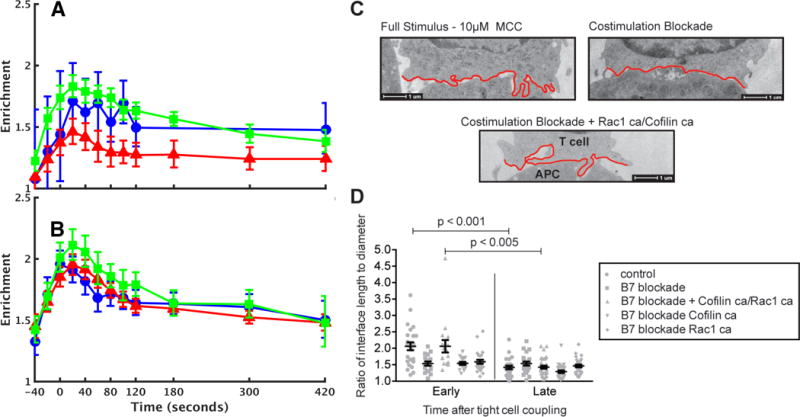
(A and B) Enrichment of WAVE2 (A) and actin (B) in the immunological synapse region (calculated as described in Fig. 5) is shown for full stimulus conditions (green), costimulation blockade (red), and costimulation blockade in cells transduced with 250 nM cofilinca and 1 μM Rac1ca (blue). Note that the recruitment of WAVE2 was restored by active cofilin and Rac1. (C) Representative EM micrographs of cell-cell conjugates formed between a 5C.C7 T cell and a CH27 cell (APC) in the presence of 10 μM MCC peptide under the indicated conditions. The T cell membranes at the interface are highlighted in red. Micrographs are representative of 41 to 51 cells from 4 to 5 experiments. (D) Ratios of interface length (that is, the red outline in C) to diameter (that is, the straight line across) are given at an early, undulation-rich time point (< 2 min after tight cell coupling) and a late time point (3 to 5 min). T cells were treated with buffer only (control), 250 nM active cofilinca, 1 μM Rac1ca, or both. APCs were treated with buffer only (control) or with anti-CD80 (10 μg/ml) and anti-CD86 (10 μg/ml) antibodies (B7 blockade). On average 24 cell couples were analyzed per condition, for a total of 244.
As an actin-driven dynamic structure at the early peak time of accumulation of actin at the immunological synapse, we next investigated undulations of the T cell:APC interface (31). A greater number and size of such undulations during the first 2 min of tight cell coupling relative to that of later time points was entirely costimulation-dependent (Fig. 6C). We therefore determined whether the activation of WAVE2 and cofilin could restore such undulations in cells in which CD28 stimulation was blocked (Fig. 6D). When used independently, 1 μM Rac1ca and 250 nM cofilinca had no effect on the undulations; however, when they were used together, undulations in the costimulation-blocked cells were fully restored. These data support our imaging-derived hypothesis that WAVE2 and cofilin are key downstream effectors of costimulation in the regulation of T cell actin dynamics.
Selective activation of WAVE2 and cofilin under costimulation-blocked conditions restores LAT localization as a costimulation-dependent element of T cell signaling
Costimulation is the principal amplifier of T cell signaling, which is evident from the reduced recruitment, in the absence of co-receptor signaling, of numerous signaling intermediates to two principal cellular structures that mediate T cell activation: a large, highly cross-linked protein complex at the center of the immunological synapse and a large actin-based lamellum that selectively forms during the first five minutes of cell coupling and traps smaller signaling complexes (6, 31–33). To determine the importance of costimulation-controlled actin dynamics in driving such signaling organization, we asked whether the reduced localization of a prototypical element of T cell signaling upon costimulation blockade could be restored through the activation of WAVE2 and cofilin. We generated 149 movies in which we imaged the localization of linker of activated T cells (LAT) as a critical proximal signaling intermediate in T cell activation (34) in conjugates formed between 5C.C7 T cells and CH27 cells (as APCs) upon control, costimulation blockade, and actin reconstitution conditions. Under control, full stimulus, conditions, LAT was rapidly recruited to the center of the immunological synapse and was retained there (Fig. 7A); however, upon costimulation blockade, early LAT accumulation was rapidly moved into a large invagination with accompanying loss of sustained accumulation at the immunological synapse.
Fig. 7. Active Rac and cofilin restore defective LAT localization upon costimulation-blockade.
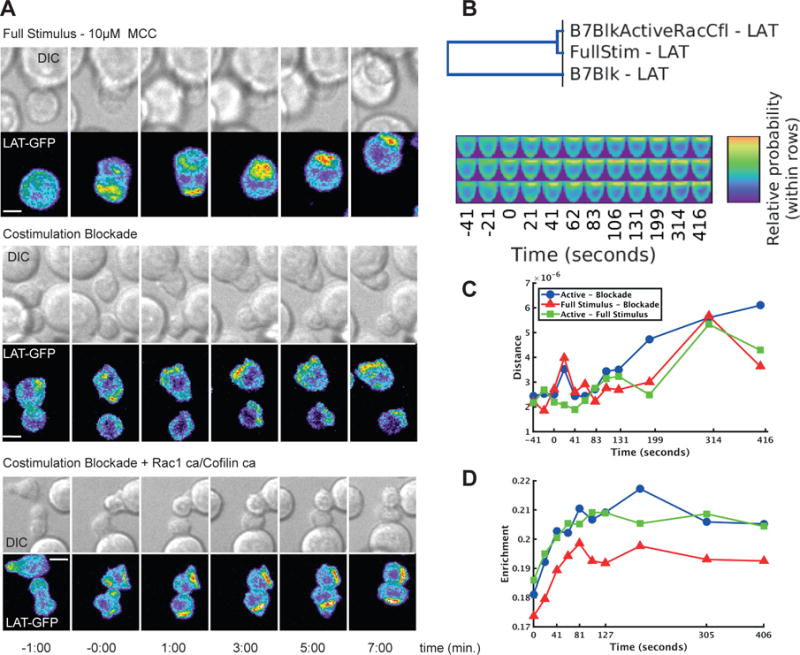
(A) Representative images of interactions between 5C.C7 T cells expressing LAT-GFP and CH27 cells loaded with 10μM MCC at the indicated times relative to the formation of a tight cell conjugate are given for cells subjected to a full stimulus, cells subjected to costimulation blockade with anti-CD80 antibody (10 μg/ml) and anti-CD86 antibody (10 μg/ml) (B7 blockade), and cells subjected to costimulation blockade and in which actin dynamics were restored with 250 nM cofilinca and 1 μM Rac1ca, as indicated. Top: DIC images. Bottom: Top-down maximum projections of 3D fluorescence data are shown in a rainbow-like, false-color intensity scale (increasing from blue to red). Scale bars, 5 μm. (B) Comparison of the 4D distributions of LAT between the three conditions. The dendrogram measures the overall similarity of the distributions across all time points, indicating that the costimulation blockade condition is different from the other two conditions (the cophenetic coefficient of the dendrogram is 0.987). The maps shown are in the same order as the dendrogram. (C) Comparison of the 3D distributions of LAT at each time point for different pairs of conditions. The Euclidean distance is shown between full stimulus and costimulation blockade (red triangles), full stimulus and reconstitution (green squares), and costimulation blockade and reconstitution (blue circles). (D) Enrichment of LAT in a cylindrical region at the center of the immunological synapse as a function of time for full stimulus (green squares), costimulation blockade (red triangles), and reconstitution (blue circles).
For an unbiased quantitative analysis of these observations, and of the effects of actin restoration, we generated shape-normalized maps of LAT distributions at 12 time points with “two-point synapse annotation,” that is, coordinates of a line across the interface, to align the cells (fig. S5). In hierarchical clustering of LAT concentrations in all maps at all time points, the subcellular LAT distribution was very similar under full stimulus and actin-reconstituted conditions, but was distinct upon costimulation blockade, which was suggestive of the full restoration of LAT localization upon reconstitution of actin dynamics (Fig. 7B). Substantial differences occurred 20 s after cell coupling, consistent with cell surface as opposed to invagination accumulation under full stimulus and reconstituted conditions (Fig. 7C). At the early time points, LAT accumulation at the central quarter of the immunological synapse was less intense upon costimulation blockade (Fig. 7D) and was shifted to the interior of the T cell (fig. S5). This is consistent with the shift in the localization of LAT from the center into the invagination upon costimulation blockade (Fig. 7D). This invagination is thought to be a site of protein internalization that leads to degradation (35). The computational analysis thus readily identified a diverse set of spatial and temporal defects upon costimulation blockade that could be confirmed by intuitive image inspection. Biologically, the LAT data suggest that costimulation uses actin regulation by WAVE2 and cofilin to control spatiotemporal distributions in T cell signaling. For LAT, this may constitute a feedback loop in which LAT-stimulated proximal signaling regulates the actin-dependent inclusion of LAT in the central signaling complex, as amplified by costimulation.
Discussion
Imaging and image analysis can play a major role in systems biology by allowing the analysis of complex processes that require the integrated action of large numbers of proteins and providing objective analysis and quantitative models (36–39). The chance for serendipitous, unbiased discovery also substantially increases with the amount of data. To take advantage of the potential of large-scale imaging to systems understanding and discovery, image quantification approaches are required that access the data in an unbiased fashion. Ideally, the entirety of the information present in images, the signal intensity at each resolved position within a cell, should be used for analysis.
Here, we developed a general image analysis strategy to access signal intensities voxel by voxel. Two key features enable this analysis. First, in large data sets, segmentation becomes more challenging, because signal distributions in cells can vary substantially between sensors and cellular activation conditions. By combining the flattening of the image intensity histogram with an active contour method, we took advantage of two invariable features of virtually all fluorescence distributions. Despite all of its variability, the signal intensity within a cell is on average higher than the intensity outside of the cell, and it decreases at the cell edge. Our segmentation method thus promises to be widely applicable. Second, we shape-transformed or “morphed” cell shape to a standardized template to account for variation in cell shape. Through the process of morphing, cells can be compared voxel by voxel, thus opening the entire data set to comparative analysis. We thus determined a core area of actin turnover and the enrichment of actin regulators in this region. Demonstrating the flexibility of this method, we also analyzed a diverse set of spatial and temporal distinctions in LAT distributions. For future mathematical modeling of complex signaling processes, we can combine normalized protein distributions with the quantification of average molar protein concentrations (Fig. 1). This would generate 3D maps of local protein concentrations as the most comprehensive input possible for spatiotemporally resolved computational models.
We have demonstrated the usefulness of large imaging data sets in conjunction with their computational quantification by resolving how T cell costimulation regulates actin dynamics. This is a prototypical problem in complex cellular regulation. First, the number of system components involved is so large that combinatorial regulation becomes experimentally inaccessible. Although we have investigated each of the eight actin regulators one-by-one, it would have been exceedingly difficult to image the 28 pair-wise combinations thereof, and impossible for any higher order combinations. However, by normalizing the large image data set covering all of the actin regulators to a common frame of reference, any combination can be probed computationally as a powerful initial screen to guide more directed experimentation. This type of approach has been referred to as “computational multiplexing” (39); a key difference is that we have analyzed the distribution of each sensor in the entire cell rather than just in regions in which events occur. We found the reduced recruitment of four actin regulators upon costimulation blockade, such that the joint activation of two actin regulators, WAVE2 and cofilin, restored actin dynamics in the activation of 5C.C7 T cells by CH27 APCs and peptide. Second, we sought to elucidate how protein function changed with cellular activation conditions rather than define what the roles of the proteins were per se. When comparing cellular activation conditions, RNAi-treated or gene-deficient organisms may not give an accurate picture. Unbiased, large-scale imaging proved to be a powerful alternative. Based on the assumption that subcellular localization is related to protein function, we used unbiased computational analysis of shape-normalized models of subcellular distributions of an entire small system of core actin regulation to discover that actin-generating and -severing proteins together regulated actin dynamics downstream of costimulation; this was proven correct with conventional cell biological restoration approaches. Because investigations of changes in complex regulatory systems upon altered cellular stimulation are of increasing interest as cell biology moves toward system approaches, unbiased large-scale imaging in conjunction with quantitative image analysis as we developed here should become ever more important.
Materials and Methods
Antibodies, purified proteins, and reagents
Antibodies used for quantitative Western blotting were as follows: rabbit anti-β-actin (Cell Signaling #4967), mouse anti-ARP3 clone FM5338 (Sigma-Aldrich #A5979), chicken anti-Capping Protein α1 (Genway Biotech #15-28821950F), mouse anti-cofilin (Cell Signaling #3312), mouse anti-Coronin 1A (Santa Cruz Biotechnology #SC-100925), rabbit anti-HS1 (Cell Signaling #4557), rabbit anti-WASP (Cell Signaling #4860), rabbit anti-WAVE2 (D2C8) XP (Cell Signaling #3659), and mouse anti-GFP living colors JL-8 (Clontech #632380). Antibodies used for the blockade of B7-1- and B7-22-dependent CD28 costimulation were anti-mouse CD80 Clone 16-10-A1 (BD Pharmingen #553736) and anti-CD86 Clone GL1 (BD Pharmingen #553689). Purified proteins used for quantitative Western blotting standards were as follows: β-actin (abcam #40624), ARP2/3 protein complex (Cytoskeleton #RP01), capping protein α1 (Genway Biotech #10-288-21950F), cofilin (Cytoskeleton #CF01-A), and EGFP (BioVision #4999-100).
Mice and cells
T cells extracted from the lymph nodes of B10.BR 5C.C7 TCR transgenic mice were used for all experiments unless otherwise noted in the figure legends. 5C.C7 mice harbor only CD4+ T cells, which express a TCR that recognizes the moth cytochrome c peptide fragment (amino acid residues 88 to 103, ANERADLIAYLKQATK) in the context of I-ek (40). Single-cell suspensions were made from the lymph nodes of 6- to 8-week-old mice of either gender. The cells were adjusted to 4 × 106 cells/ml and MCC peptide was added to a final concentration of 3 μM. All animals were maintained in the pathogen-free animal care facility at the University of Texas Southwestern Medical Center, and the care and use of the mice were approved by the University of Texas Southwestern Animal Care and Use Committee. The CH27 B cell lymphoma cell line was used in all experiments as APCs. To load the APCs, the cells were incubated in the presence of 10 μM MCC peptide for at least 4 hours. All cells were maintained in T cell medium composed of RPMI with L-glutamine, 10% fetal bovine serum (FBS, Hyclone), penicillin (100 IU/mL), streptomycin (100 μg/ml), and 0.5μM β-mercaptoethanol. Interleukin-2 (IL-2) was added at a final concentration of 0.05 U/ml during parts of the retroviral transduction procedure. These T cells were transduced with MMLV-derived retroviruses (33) encoding a fusion protein composed of GFP and one of nine proteins (actin, ARP3, capping protein α, cofilin, coronin, HS1, myosin II regulatory light chain, WASP, and WAVE2). Each batch of T cells was imaged every 20 s for 15 min and subjected to one of two experimental conditions: full stimulus and B7 blockade as further described below.
Quantitative Western blotting analysis
These procedures were used to quantify the amount of the regulatory proteins in 5C.C7 T cells (fig. S6). Briefly, precise numbers of T cells expressing each of the actin regulatory proteins were sorted into phosphate-buffered saline (PBS) and lysed in 1% Triton X-100 lysis buffer (pH 7.6) containing 20 mM Tris-HCl, 150 mM NaCl, 2 mM EDTA, and supplemented with protease and phosphatase inhibitors (1 mM PMSF, 50 μM leupeptin, 1 μM pepstatin A, 10 mM NaF, and 200 μM sodium Orthovanadate). For quantitative Western blotting, the lysate from a known number of T cells was loaded onto a 10% SDS-PAGE gel alongside purified protein standards. A standard curve was generated from the densitometry measurements for each purified protein standard, and the nanogram amount of endogenous protein and retrovirally expressed GFP-tagged protein was simultaneously calculated per T cell based on the standard curve. This nanogram amount of protein was converted into an average single T cell molar concentration based on the average size of a 5C.C7 T cell and the mass of the protein (in Kd). In the case of proteins for which the purified protein was not available, the GFP-tagged protein band was used to estimate the endogenous amount. Based on previous work (33), we determined that the average single-cell molar concentration of GFP-tagged protein in our sort gate (1 to 1.5 logs from the negative population) was 2.6 μM. Thus, the ratio of the densitometry measurements of the endogenous protein band and the retrovirally expressed GFP-tagged protein band was used to estimate the amount of endogenous protein.
T cell volumetric measurements
T cells were labelled with SNARF-1 (Invitrogen #S22801) as a whole-cell stain and with Hoechst 34580 (Invitrogen #H21486) to delineate the nucleus. T cells were imaged with a Perkin Elmer spinning-disk confocal microscope. Whole-cell volume measurements and nuclear volume measurements of T cells were calculated by first measuring the full-width half-maximum of the Gaussian profile from line scans across the center plane of each T cell (SNARF, red channel) and nucleus (Hoechst, blue channel). Once the radii of the cell and nucleus were calculated, the upper and lower intensity values were set for each channel, and object surfaces and volumes were measured for the whole T cell and the nucleus in Imaris (Bitplane) (fig. S7).
Expression and purification of cell-permeable, tat-tagged proteins
Using protein transduction methodology previously described and validated for use in 5C.C7 primary T cells (41), the cDNAs encoding a constitutively active mutant cofilin (S3A) and a constitutively active mutant Rac1 (Q61L) were cloned into a ptat-nde vector, which provides an N-terminal tat-tag for protein transduction and a 6× His tag for purification with Ni-NTA. The proteins were expressed in BL21(DE3) Escherichia coli at room temperature overnight after induction of expression with 1 mM isopropyl-beta-D-thiogalactopyranoside (IPTG). The bacteria were lysed under native conditions by sonication in lysis buffer containing 4× PBS, 0.01% IGEPAL, 10% glycerol, 10 mM imidazole, 1 mM MgCl2 (only added for Rho family GTPases), 1% lysoszyme, 0.14% β-mercaptoethanol, and protease inhibitors at pH 7.5. The proteins were purified over Ni-NTA resin. After protein binding, the resin was washed twice with wash buffer containing 4× PBS, 20 mM imidazole, 1 mM MgCl2, and 0.14% β-mercaptoethanol. The protein was then released from the column with elution buffer containing 4× PBS, 250 mM imidazole, 1 mM MgCl2, and 0.14% β-mercaptoethanol. After elution, the protein was dialyzed into PBS and quantified by SDS-PAGE.
Time-lapsed imaging of T cell:APC interactions
Time-lapse fluorescence microscopy was performed with retrovirally transduced T cells and CH27 cells loaded with 10μM MCC unless otherwise noted in the figure legends. The T cells and CH27 cells were imaged in imaging buffer (PBS, 10% FBS, 1 mM CaCl2, 0.5 mM MgCl2) on 384-well glass-bottom plates. All imaging of actin regulatory proteins and actin was performed on a Perkin Elmer spinning disk confocal systems fitted onto a Zeiss Axiovert 200M microscope body equipped with full environmental control, a Hamamatsu C9100-50 EMCCD, and 6 laser lines from 405 to 647nm. A Zeiss PlanFluoro 40× oil objective (NA=1.3) was used for all imaging. Automated control of the microscope was performed with Volocity software (Perkin Elmer). For experiments in which the B7-1- and B7-2-dependent activation of CD28 was blocked, peptide-loaded CH27 cells were incubated on ice for 30 min in the presence of anti- CD80 Clone 16-10-A1 (10 μg/ml) and anti- CD86 Clone GL1 (10 μg/ml)(BD Pharmingen) antibody before the CH27 cells were transferred to the imaging plate with the T cells. For experiments in which cells were reconstituted with tat-tagged Rac and cofilin proteins, T cells were incubated for 30 min at 37°C with the protein transduction reagents at the indicated concentrations in the imaging plate before the addition of the peptide-loaded CH27 cells. Image acquisition then proceeded as described earlier. Each time-lapse image was generated by taking a two-dimensional (2D) brightfield image and a 3D image of the GFP channel every 20 s for an average of 45 (range: 26 to 46) frames. Voxels in these 3D images were of size 0.406 μm in the horizontal plane and 0.4 μm along the optical axis. The number of movies used for each condition-sensor combination is listed in table S1.
Manual annotation of immunological synapse positions
We manually tracked the 2D locations of immunological synapses in the brightfield movies. First, the location and frame number of each immunological synapse formation event were identified as when either the T cell:APC interface had reached its full width or the cells had been in contact for 40 s, whichever came first. Thus, negative time points may contain small nascent interfaces. Second, the position of the center of the immunological synapse for each cell-to-cell conjugate was marked for 12 time points before and after synapse formation. We tracked an average of 15 synapses per movie for a total of 1401 tracked cell conjugates. The number of manual annotations used to construct the final model of each condition-sensor combination is listed in table S1 and is less than or equal to the number of annotations because of occasional failures of image segmentation in the automated pipeline. In some cases, two-point annotation was used, in which points on the left and right edges of the immunological synapse were marked.
Image segmentation
For each manually marked immunological synapse, we extracted a 71×71 voxel window from all Z slices of the corresponding video frame to make downstream operations faster. We designed our segmentation method to produce segmentations of small objects with high internal contrast and low contrast with background, and to produce smooth segmentation surfaces near the edges of objects with widely varying shapes. These two design goals are addressed by the following edge detection and segmentation steps. Window images were transformed into edge magnitude images before being passed to the segmentation algorithm as follows. First, we subtracted background intensity, which we estimated from the mode of intensity values below the mean intensity value. Second, we normalized intensities by dividing by the 99.9th percentile of intensity, an approximation to the maximum intended to provide some robustness. Third, we used global histogram equalization to suppress intensity variation within cells after observing that some of the sensors showed high contrast patterns of subcellular location. Such patterns resulted in strong edges in the image, and these edges were often larger than those at the boundaries of cells. The output from this step showed much more uniform intensity inside cells. Fourth, we used anisotropic diffusion (42) to reduce noise and enhance edges. Fifth, we produced an edge potential image using the gradient magnitude at each voxel, where the gradient was computed with Scharr’s 5×5 filter (43) instead of centered finite differences. Although Scharr’s design was for precision in gradient direction estimation, we primarily used this filter to compute gradients with larger neighborhoods to combat noise sensitivity and weak corner gradients. The segmentation for a cell was initialized to be a sphere of approximately the same radius as a typical T cell (20 voxels) and centered near the cell’s manually specified synapse point. The center of the sphere was found by initializing it to have the same XY coordinates as the manual point; setting the Z coordinate to place the point in the middle Z slice; and finding the nearest local maximum of intensity in a smoothed (Gaussian filter, standard deviation of 4.6 voxels; that is, one fifth of the aforementioned 23 voxel maximum cell diameter) version of the histogram-equalized preprocessed image by hill climbing, starting from that point and moving up the gradient until convergence. We used the active contour method (44) as extended to 3D segmentations represented as triangle meshes (45) to segment individual cells within the windows of the fluorescence images of the sensors (we used a simplified implementation of this method that did not use hierarchical approximation). The snake method was run on the edge images produced as described earlier, and the method’s parameters were manually tuned to these edge images. The snake method was run in two stages with distinct sets of parameters, a coarse stage and a fine stage, in which the coarse stage was initialized with the aforementioned sphere and produced a triangle mesh as output, whereas the fine stage was initialized with the output mesh from the coarse stage. The coarse stage’s parameters were selected to find a rough shape for each cell with more severe smoothness constraints in an attempt to prevent over- and under-segmentation, whereas the fine stage’s parameters were chosen to enable the surface of the segmentation more flexibility in order to gain precision. See table S2 for a list of the parameters used. Note that autofluorescence within the T cell combined with the smoothness constraint in the active contour were sufficient to distinguish the cell boundary from the extracellular region even for sensors that exhibited highly localized distributions.
Rigid alignment with respect to the immunological synapse
To represent the probability distribution of the relative concentration of a protein in various parts of the cell as measured from multiple imaged cells, each image of a cell must be assigned a coordinate system in which anatomically similar positions in each cell are assigned similar coordinates. In general, cells of the same tissue or cell line can vary widely in shape in general. The helper T cells imaged in this study were no exception, but they had certain anatomical markers that helped in determining the coordinates for each voxel in an image of the cell. When the immunological synapse has formed, there is usually a relatively flat interface between the T cell and the APC that establishes a polarization axis that can be viewed as one of the coordinates. The rest of the cell will be amorphous, but largely rounded or elongated. For these reasons, our previous work (33) used half-ellipsoidal diagrams to illustrate the spatial patterning of sensors in T cells. We used the same idealized shape to establish a coordinate system for each cell. The shapes of these cells as determined by the segmentation method are triangle meshes. To process the segmentations as images, the meshes were rasterized into 3D images by testing if each voxel were within the mesh, and setting the voxel to one if so and to zero if otherwise. For a more precise representation of the segmentation, we anti-aliased this image by rasterizing the mesh at twice the size in each dimension and then down-sampling this rasterization by a factor of two.
One-point immunological synapse annotation
We approximately extracted the orientation and position of each cell as follows. First, we assumed that the manual annotation for the synapse point for a cell was approximately at the center of the synapse, so the location in the horizontal plane was set to the synapse point and the vertical coordinate to the weighted centroid was computed by weighting each voxel of the segmentation by the negative exponential of distance in the horizontal plane from the synapse point. Second, we assumed that the orientation of the synapse plane was perpendicular to a ray originating from the weighted centroid of the segmentation volume and directed through the synapse point. Third, we assumed that the cells should have approximately the same distribution regardless of volume, so we uniformly scaled the images so that the segmentations had the same volume.
Two-point immunological synapse annotation
For some sensors (actin and LAT), an alternative method was used to estimate the synapse position. In this case, the coordinates of the left and right edges of the immunological synapse were determined in transparent overlays of the fluorescence onto the bright field DIC data and automatically written into a data file. A vertical coordinate was assigned to these points by the weighting method used for one-point annotation. The orientation of the synapse plane was assumed to be perpendicular to a ray from the weighted centroid of the segmentation and directed through the midpoint of the line connecting the left and right edges of the immunological synapse. The volume was scaled as described for the one-point annotation.
Non-rigid standardization of cell shape
The next step was to standardize the coordinate system within each cell to enable comparisons to be made. At this point in the pipeline, cells were aligned, but coordinates in one cell were not matched to coordinate in other cells due to variation in cell shape. We addressed this by finding a shape transformation such that each cell was shaped like a common template shape (in our case, a half-ellipsoid) after applying the transformation. A nonrigid image registration method like large distance diffeomorphic mapping (LDDMM) (46) can determine for each voxel of the segmentation image the corresponding position in the space of a given template shape. We used a method that is an approximation to LDDMM, specifically an extension to the Christensen algorithm (46–48). The amount of morphing required on average for each sensor and time point is shown in table S3. As expected, the amount of morphing required was somewhat larger for those cells before immunological synapse formation (because the cell shapes were more variable before that point), but was similar for all sensors and time points after immunological synapse formation. To enable assessment of how much cell shape changed over time and whether shape change was consistent between conditions, we noted the variation in cell shape for each time point of the full stimulus condition, as well as the average magnitude of the morphing field (fig. S8). This also revealed more variation before the immunological synapse was formed, and that variation after formation was modest. Analysis of the morphing fields for the full stimulus condition (fig. S8B) showed that very minor morphing was required to bring the edges of individual cells in towards the edge of the template (peripheral regions colored in magenta) and that there were frequently slight adjustments right at the edge (regions shown in warmer colors). The morphing fields did not change substantially for the B7 blockade condition (fig. S8C). After morphing to the standard shape, the fluorescence distribution in each cell at a given time point was represented as a standardized vector (of length 6628) formed from the intensity values of each of the voxels within the template shape, where the intensities for each time point were normalized so that the values of the vector were probabilities (that is, fractions of total intensity). Our model for each sensor for a given condition at each time point was generated by calculating the mean vector (and its standard deviation vector). The overall model for each sensor for a given condition was generated by concatenating the models for all time points.
Statistical testing of enrichment
To compare sensor distributions and to test for sensors with statistically significant changes between conditions, we first generated a region that approximated the immunological synapse. This region was defined as the 10% most fluorescent voxels of the average probability distribution across all cells, for all time points, and for all sensors. This definition was used to enable averaging over more cells and improve the signal-to-noise ratio for finding the edges of the region. We defined enrichment to be the ratio of the mean probability in the distribution of that sensor for that cell at that time point within a region corresponding to the immunological synapse and the mean probability in the entire cell. We compared enrichment between conditions with Welch’s t test. We assumed that the enrichment was lognormal distributed because it was always greater than zero and, for most of the imaged proteins, greater than 1.0 and so obviously not normally distributed. This resulted in a total of 108 tests, and we applied Bonferroni–Holm correction to keep the false positive rate at most 0.05 (see table S4 for the corrected P values for all comparisons). For the analysis of LAT enrichment, an enrichment region was defined as a cylinder centered on the immunological synapse with half of its diameter and extending eight slices into the template (this corresponds to the central 25% of the immunological synapse area and approximately 12% of the total volume). The fraction of sensor contained in this region was calculated for each time point for the three different conditions.
Cluster analysis
To visualize the relative similarity between the spatiotemporal distributions of a reduced number of sensors (eight) under the two conditions, we applied single-linkage hierarchical clustering to the overall models, that is, vectors formed by concatenating probability models of a condition-sensor combination for all time points. This clustering method starts with each model as being in a distinct cluster and iteratively merges the closest two clusters until only one cluster remains. This produces a binary tree structure (which can be displayed as a dendrogram) where, at each branch point, models in one branch are closer to each other than they are to the other branch. We measured the faithfulness of a dendrogram’s representation of the distances between clustered models by computing the cophenetic coefficient (the correlation between the dendrogram’s dissimilarity measure between each pair of models and the Euclidean distance between those two models). Because this is a measure of correlation, values closer to 1.0 are better.
Electron microscopy
Sample preparation, data acquisition, and analysis of membrane undulation were performed as described previously (31).
Supplementary Material
Fig. S1. Example images from the segmentation, alignment and morphing pipeline.
Fig. S2. Illustrations of the standard deviations of the spatiotemporal models.
Fig. S3. Spatiotemporal models for actin distribution using two-point annotation.
Fig. S4. Analysis of radial distributions confirms differences between myosin and actin.
Fig. S5. Comparison of LAT spatiotemporal dynamics reveals restoration of costimulation by active Rac and cofilin.
Fig. S6. Quantification of actin regulatory proteins in primary mouse T cells.
Fig. S7. Volume measurements of primary mouse T cells.
Fig. S8. Variation in cell shape and morphing across time points and conditions.
Table S1. The number of imaging fields and manually marked immunological synapses used to construct the spatiotemporal model of each condition-sensor combination.
Table S2. The manually tuned parameters used for both stages of segmentation by the active contour method.
Table S3. Average amount of morphing required for each sensor at each time point.
Table S4. Statistical testing of differences in sensor enrichment in the immunological synapse region between the full stimulus condition and costimulation blockade.
Movie S1. 4D Maps of three representative sensors.
Acknowledgments
We thank Kate-Luby Phelps and Mark Jepson for imaging support, Angela Mobley and Andrew Herman for cell sorting, and Gustavo Rohde for helpful discussions.
Funding: This work was supported in part by National Institutes of Health grant P41 GM103712, by National Science Foundation grants MCB1121793 and MCB1121919, by European Research Council grant PCIG11-GA-2012-321554, by a National Research Foundation of Korea Grant (MOEHRD, Basic Research Promotion Fund, KRF-2008-D00316) (to B.H.C.), and by a scholarship from the China Scholarship Council (to J.Z.).
Footnotes
Author contributions: C.W. and R.F.M. conceived the study, K.T.R. and C.W. designed experiments, K.T.R., D.J.C, R.A., H.M.T., P.V. and C.W. executed experiments and analyzed accumulation data, T.B., X.R. and R.F.M. designed the image analysis and constructed the maps, P.V. performed electron microscopy experiments, and K.T.R., T.B., X.R. C.W. and R.F.M. wrote the manuscript.
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: A zip file with maps for all of the sensors at all time points may be found at: http://murphylab.web.cmu.edu/data/TcellModels Folders are included for both the average values and the standard deviations across all cells for each sensor, time point, and condition. Included within each folder are images (scaled to the maximum average value for a given map) and comma-separated value (csv) files containing the relative fluorescence intensity values (scaled to a fraction of the total fluorescence). A csv file containing the average cellular concentration of each probe (except MRLC) and a Matlab function that can read the csv files and return 3D images either as relative fluorescence or concentration values are also included. All source code and intermediate results are available as a reproducible research archive from http://murphylab.web.cmu.edu/software.
References and Notes
- 1.Burkhardt JK, Carrizosa E, Shaffer MH. The actin cytoskeleton in T cell activation. Annu Rev Immunol. 2008;26:233–259. doi: 10.1146/annurev.immunol.26.021607.090347. [DOI] [PubMed] [Google Scholar]
- 2.Tskvitaria-Fuller I, Rozelle AL, Yin HL, Wülfing C. Regulation of sustained actin dynamics by the TCR and costimulation as a mechanism of receptor localization. J Immunol. 2003;171:2287–2295. doi: 10.4049/jimmunol.171.5.2287. [DOI] [PubMed] [Google Scholar]
- 3.Nolz JC, Fernandez-Zapico ME, Billadeau DD. TCR/CD28-stimulated actin dynamics are required for NFAT1-mediated transcription of c-rel leading to CD28 response element activation. J Immunol. 2007;179:1104–1112. doi: 10.4049/jimmunol.179.2.1104. [DOI] [PubMed] [Google Scholar]
- 4.Valitutti S, Dessing M, Aktories K, Gallati H, Lanzavecchia A. Sustained signaling leading to T cell activation results from prolonged T cell receptor occupancy. Role of T cell actin cytoskeleton. J Exp Med. 1995;181:577–584. doi: 10.1084/jem.181.2.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kumari S, Depoil D, Martinelli R, Judokusumo E, Carmona G, Gertler FB, Kam LC, Carman CV, Burkhardt JK, Irvine DJ, Dustin ML. Actin foci facilitate activation of the phospholipase C-gamma in primary T lymphocytes via the WASP pathway. eLife. 2015;4 doi: 10.7554/eLife.04953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roybal KT, Mace EM, Clark DJ, Leard AD, Herman A, Verkade P, Orange JS, Wulfing C. Modest Interference with Actin Dynamics in Primary T Cell Activation by Antigen Presenting Cells Preferentially Affects Lamellal Signaling. PloS one. 2015;10:e0133231. doi: 10.1371/journal.pone.0133231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Billadeau DD, Nolz JC, Gomez TS. Regulation of T-cell activation by the cytoskeleton. Nat Rev Immunol. 2007;7:131–143. doi: 10.1038/nri2021. [DOI] [PubMed] [Google Scholar]
- 8.Higgs HN, Pollard TD. Regulation of actin filament network formation through Arp2/3 complex: Activation by a diverse array of proteins. Ann Rev Biochem. 2001;70:649–676. doi: 10.1146/annurev.biochem.70.1.649. [DOI] [PubMed] [Google Scholar]
- 9.Pollard TD, Borisy GG. Cellular motility driven by assembly and disassembly of actin filaments: Integration of signals to the Arp2/3 complex. Cell. 2003;112:453–465. doi: 10.1016/s0092-8674(03)00120-x. [DOI] [PubMed] [Google Scholar]
- 10.Ibarra N, Pollitt A, Insall RH. Regulation of actin assembly by SCAR/WAVE proteins. Biochem Soc Trans. 2005;33:1243–1246. doi: 10.1042/BST0331243. [DOI] [PubMed] [Google Scholar]
- 11.Soderling SH, Scott JD. WAVE signalling: from biochemistry to biology. Biochem Soc Trans. 2006;34:73–76. doi: 10.1042/BST0340073. [DOI] [PubMed] [Google Scholar]
- 12.Stradal TE, Scita G. Protein complexes regulating Arp2/3-mediated actin assembly. Curr Opin Cell Biol. 2006;18:4–10. doi: 10.1016/j.ceb.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 13.Badour K, Zhang J, Siminovitch KA. The Wiskott-Aldrich syndrome protein: forging the link between actin and cell activation. Immunol Rev. 2003;192:98–112. doi: 10.1034/j.1600-065x.2003.00031.x. [DOI] [PubMed] [Google Scholar]
- 14.Snapper SB, Rosen FS, Mizoguchi E, Cohen P, Khan W, Liu CH, Hagemann TL, Kwan SP, Ferrini R, Davidson L, Bhan AK, Alt FW. Wiskott-Aldrich syndrome protein-deficient mice reveal a role for WASP in T but not B cell activation. Immunity. 1998;9:81–91. doi: 10.1016/s1074-7613(00)80590-7. [DOI] [PubMed] [Google Scholar]
- 15.Cannon JL, Labno CM, Bosco G, Seth A, McGavin MH, Siminovitch KA, Rosen MK, Burkhardt JK. Wasp recruitment to the T cell:APC contact site occurs independently of Cdc42 activation. Immunity. 2001;15:249–259. doi: 10.1016/s1074-7613(01)00178-9. [DOI] [PubMed] [Google Scholar]
- 16.Labno CM, Lewis CM, You D, Leung DW, Takesono A, Kamberos N, Seth A, Finkelstein LD, Rosen MK, Schwartzberg PL, Burkhardt JK. Itk functions to control actin polymerization at the immune synapse through localized activation of Cdc42 and WASP. Curr Biol. 2003;13:1619–1624. doi: 10.1016/j.cub.2003.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zeng R, Cannon JL, Abraham RT, Way M, Billadeau DD, Bubeck-Wardenberg J, Burkhardt JK. SLP-76 coordinates Nck-dependent Wiskott-Aldrich syndrome protein recruitment with Vav-1/Cdc42-dependent Wiskott-Aldrich syndrome protein activation at the T cell-APC contact site. J Immunol. 2003;171:1360–1368. doi: 10.4049/jimmunol.171.3.1360. [DOI] [PubMed] [Google Scholar]
- 18.Nolz JC, Gomez TS, Zhu P, Li S, Medeiros RB, Shimizu Y, Burkhardt JK, Freedman BD, Billadeau DD. The WAVE2 complex regulates actin cytoskeletal reorganization and CRAC-mediated calcium entry during T cell activation. Curr Biol. 2006;16:24–34. doi: 10.1016/j.cub.2005.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zipfel PA, Bunnell SC, Witherow DS, Gu JJ, Chislock EM, Ring C, Pendergast AM. Role for the Abi/wave protein complex in T cell receptor-mediated proliferation and cytoskeletal remodeling. Curr Biol. 2006;16:35–46. doi: 10.1016/j.cub.2005.12.024. [DOI] [PubMed] [Google Scholar]
- 20.Gomez TS, McCarney SD, Carrizosa E, Labno CM, Comiskey EO, Nolz JC, Zhu P, Freedman BD, Clark MR, Rawlings DJ, Billadeau DD, Burkhardt JK. HS1 functions as an essential actin-regulatory adaptor protein at the immune synapse. Immunity. 2006;24:741–752. doi: 10.1016/j.immuni.2006.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Foger N, Rangell L, Danilenko DM, Chan AC. Requirement for coronin 1 in T lymphocyte trafficking and cellular homeostasis. Science. 2006;313:839–842. doi: 10.1126/science.1130563. [DOI] [PubMed] [Google Scholar]
- 22.Mugnier B, Nal B, Verthuy C, Boyer C, Lam D, Chasson L, Nieoullon V, Chazal G, Guo XJ, He HT, Rueff-Juy D, Alcover A, Ferrier P. Coronin-1A links cytoskeleton dynamics to TCR alpha beta-induced cell signaling. PLoS One. 2008;3:e3467. doi: 10.1371/journal.pone.0003467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eibert SM, Lee KH, Pipkorn R, Sester U, Wabnitz GH, Giese T, Meuer SC, Samstag Y. Cofilin peptide homologs interfere with immunological synapse formation and T cell activation. Proc Natl Acad Sci U S A. 2004;101:1957–1962. doi: 10.1073/pnas.0308282100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hammer JA, 3rd, Burkhardt JK. Controversy and consensus regarding myosin II function at the immunological synapse. Curr Opin Immunol. 2013 doi: 10.1016/j.coi.2013.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liang Y, Cucchetti M, Roncagalli R, Yokosuka T, Malzac A, Bertosio E, Imbert J, Nijman IJ, Suchanek M, Saito T, Wulfing C, Malissen B, Malissen M. The lymphoid lineage-specific actin-uncapping protein Rltpr is essential for costimulation via CD28 and the development of regulatory T cells. Nat Immunol. 2013;14:858–866. doi: 10.1038/ni.2634. [DOI] [PubMed] [Google Scholar]
- 26.Zhao J, Bruck S, Cemerski S, Zhang L, Butler B, Dani A, Cooper JA, Shaw AS. CD2AP links cortactin and capping protein at the cell periphery to facilitate formation of lamellipodia. Mol Cell Biol. 2013;33:38–47. doi: 10.1128/MCB.00734-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee KH, Meuer SC, Samstag Y. Cofilin: a missing link between T cell co-stimulation and rearrangement of the actin cytoskeleton. Eur J Immunol. 2000;30:892–899. doi: 10.1002/1521-4141(200003)30:3<892::AID-IMMU892>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 28.Wabnitz GH, Kocher T, Lohneis P, Stober C, Konstandin MH, Funk B, Sester U, Wilm M, Klemke M, Samstag Y. Costimulation induced phosphorylation of L-plastin facilitates surface transport of the T cell activation molecules CD69 and CD25. Eur J Immunol. 2007;37:649–662. doi: 10.1002/eji.200636320. [DOI] [PubMed] [Google Scholar]
- 29.Babich A, Li S, O’Connor RS, Milone MC, Freedman BD, Burkhardt JK. F-actin polymerization and retrograde flow drive sustained PLCgamma1 signaling during T cell activation. J Cell Biol. 2012;197:775–787. doi: 10.1083/jcb.201201018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yi J, Wu XS, Crites T, Hammer JA., 3rd Actin retrograde flow and actomyosin II arc contraction drive receptor cluster dynamics at the immunological synapse in Jurkat T cells. Mol Biol Cell. 2012;23:834–852. doi: 10.1091/mbc.E11-08-0731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roybal KT, Mace EM, Mantell JM, Verkade P, Orange JS, Wulfing C. Early Signaling in Primary T Cells Activated by Antigen Presenting Cells Is Associated with a Deep and Transient Lamellal Actin Network. PloS one. 2015;10:e0133299. doi: 10.1371/journal.pone.0133299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roybal KT, Sinai P, Verkade P, Murphy RF, Wulfing C. The actin-driven spatiotemporal organization of T-cell signaling at the system scale. Immunological reviews. 2013;256:133–147. doi: 10.1111/imr.12103. [DOI] [PubMed] [Google Scholar]
- 33.Singleton KL, Roybal KT, Sun Y, Fu G, Gascoigne NR, van Oers NS, Wulfing C. Spatiotemporal patterning during T cell activation is highly diverse. Sci Signal. 2009;2:ra15. doi: 10.1126/scisignal.2000199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Balagopalan L, Barr VA, Kortum RL, Park AK, Samelson LE. Cutting edge: cell surface linker for activation of T cells is recruited to microclusters and is active in signaling. J Immunol. 2013;190:3849–3853. doi: 10.4049/jimmunol.1202760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Singleton K, Parvaze N, Dama KR, Chen KS, Jennings P, Purtic B, Sjaastad MD, Gilpin C, Davis MM, Wulfing C. A large T cell invagination with CD2 enrichment resets receptor engagement in the immunological synapse. J Immunol. 2006;177:4402–4413. doi: 10.4049/jimmunol.177.7.4402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Glory E, Murphy RF. Automated Subcellular Location Determination and High Throughput Microscopy. Developmental Cell. 2007;12:7–16. doi: 10.1016/j.devcel.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 37.Zhao T, Murphy RF. Automated learning of generative models for subcellular location: building blocks for systems biology. Cytometry Part A. 2007;71A:978–990. doi: 10.1002/cyto.a.20487. [DOI] [PubMed] [Google Scholar]
- 38.Bakal C, Aach J, Church G, Perrimon N. Quantitative morphological signatures define local signaling networks regulating cell morphology. Science. 2007;316:1753–1756. doi: 10.1126/science.1140324. [DOI] [PubMed] [Google Scholar]
- 39.Machacek M, Hodgson L, Welch C, Elliott H, Pertz O, Nalbant P, Abell A, Johnson GL, Hahn KM, Danuser G. Coordination of Rho GTPase activities during cell protrusion. Nature. 2009;461:99–103. doi: 10.1038/nature08242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kaye J, Hsu ML, Sauron ME, Jameson SC, Gascoigne NR, Hedrick SM. Selective development of CD4+ T cells in transgenic mice expressing a class II MHC-restricted antigen receptor. Nature. 1989;341:746–749. doi: 10.1038/341746a0. [DOI] [PubMed] [Google Scholar]
- 41.Tskvitaria-Fuller I, Mistry N, Sun S, Wulfing C. Protein transduction as a means of effective manipulation of Cdc42 activity in primary T cells. J Immunol Methods. 2007;319:64–78. doi: 10.1016/j.jim.2006.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kroon DJ, Slump CH, Maal TJ. Medical Image Computing and Computer-Assisted Intervention–MICCAI 2010. Springer; 2010. pp. 221–228. [DOI] [PubMed] [Google Scholar]
- 43.Scharr H. Complex Motion. Springer; 2007. pp. 14–29. [Google Scholar]
- 44.Kass M, Witkin A, Terzopoulos D. Snakes: Active contour models. International journal of computer vision. 1988;1:321–331. [Google Scholar]
- 45.Lürig C, Kobbelt L, Ertl T. Hierarchical solutions for the deformable surface problem in visualization. Graphical Models. 2000;62:2–18. [Google Scholar]
- 46.Beg MF, Miller MI, Trouvé A, Younes L. Computing large deformation metric mappings via geodesic flows of diffeomorphisms. International journal of computer vision. 2005;61:139–157. [Google Scholar]
- 47.Christensen GE, Rabbitt RD, Miller MI. Deformable templates using large deformation kinematics. Image Processing, IEEE Transactions on. 1996;5:1435–1447. doi: 10.1109/83.536892. [DOI] [PubMed] [Google Scholar]
- 48.Johnson GR, Buck TE, Sullivan DP, Rohde GK, Murphy RF. Joint Modeling of Cell and Nuclear Shape Variation. Mol Biol Cell. 2015;26:4046–4056. doi: 10.1091/mbc.E15-06-0370. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Example images from the segmentation, alignment and morphing pipeline.
Fig. S2. Illustrations of the standard deviations of the spatiotemporal models.
Fig. S3. Spatiotemporal models for actin distribution using two-point annotation.
Fig. S4. Analysis of radial distributions confirms differences between myosin and actin.
Fig. S5. Comparison of LAT spatiotemporal dynamics reveals restoration of costimulation by active Rac and cofilin.
Fig. S6. Quantification of actin regulatory proteins in primary mouse T cells.
Fig. S7. Volume measurements of primary mouse T cells.
Fig. S8. Variation in cell shape and morphing across time points and conditions.
Table S1. The number of imaging fields and manually marked immunological synapses used to construct the spatiotemporal model of each condition-sensor combination.
Table S2. The manually tuned parameters used for both stages of segmentation by the active contour method.
Table S3. Average amount of morphing required for each sensor at each time point.
Table S4. Statistical testing of differences in sensor enrichment in the immunological synapse region between the full stimulus condition and costimulation blockade.
Movie S1. 4D Maps of three representative sensors.