

Abstract
Eukaryotic DNA is packaged into chromatin where nucleosomes form the basic building unit. Knowing the precise positions of nucleosomes is important because they determine the accessibility of underlying regulatory DNA sequences. Here we describe a detailed method to map on a genomic scale the locations of nucleosomes with very high resolution. Micrococcal nuclease (MNase) digestion followed by chromatin immunoprecipitation and facilitated library construction for deep sequencing provides a simple and accurate map of nucleosome positions.
1. Introduction
Nucleosomes form the building blocks of chromatin, consisting of ∼ 147 base-pair (bp) DNA that is tightly wrapped around a histone octamer core (Richmond & Davey, 2003). This wrapping requires substantial bending of DNA. At some nucleosome positions, DNA sequences such as the AA/TT dinucleotides may be enriched every 10 bp and along with regions of higher GC content near the nucleosome dyad (midpoint) collaborate to favor nucleosome formation (Albert et al., 2007; Satchwell, Drew, & Travers, 1986; Segal & Widom, 2009; Tillo & Hughes, 2009). In contrast, Poly dA:dT tracts tend to be intrinsically rigid and hence disfavor nucleosome formation (Anderson & Widom, 2001; Bao, White, & Luger, 2006; Iyer & Struhl, 1995).
In addition to the role of DNA sequence, adenosine triphosphate (ATP)-dependent chromatin remodeling enzymes use the energy of ATP hydrolysis to alter histone–DNA contacts and control nucleosome positions. In vivo, the action of these enzymes may lead to diverse outcomes, ranging from shifting nucleosome positions to partial and/or complete eviction of nucleosomes and histone replacement (Hartley & Madhani, 2009; Langst & Becker, 2001; Whitehouse, Rando, Delrow, & Tsukiyama, 2007). Recent work has suggested specific roles of distinct chromatin remodelers. Isw2 functions adjacent to promoters, where it repositions nucleosomes at the interface between genic and intergenic regions (Whitehouse et al., 2007). The RSC complex is responsible for maintaining nucleosome-free promoter regions and its flanking nucleosomes (Hartley & Madhani, 2009). Isw1, Isw2, and Chd1 remodelers might work together in maintaining genome-wide nucleosome organization (Gkikopoulos et al., 2011). In vitro studies have also indicated the role of ATP-dependent activities in reconstituting proper nucleosome positioning across the genome (Zhang et al., 2011).
Nucleosome locations affect every cellular process requiring access to DNA, from influencing evolution to regulating gene expression, development, aging, and human health (Zhang & Pugh, 2011). Thus, determining precise nucleosome positions is of importance and an area of active research. Changes in nucleosome positions, even by a few base pairs, can have significant outcomes.
In this chapter, we describe the experimental details of mapping nucleosome positions across the yeast genome with high precision. In brief, standard chromatin immunoprecipitation (ChIP) methods are initially employed, whereby cell cultures are first treated with formaldehyde, which cross-links proteins to DNA (Fragoso & Hager, 1997). Cells are then harvested, and the chromatin was isolated and fragmented by micrococcal nuclease (MNase) digestion. Histone H3 is immunopurified under semidenaturing conditions to ensure that isolated nucleosomal DNA is cross-linked to histone H3 in vivo, as cross-linking can be quite inefficient. Importantly, if such a requirement is not imposed, then there remains a possibility that at least some nucleosomes, particularly mutants, may become repositioned during the chromatin isolation and MNase digestion. A key novel feature of the protocol described here is the construction of the nucleosomal sequence library while it is present within the immunoprecipitate. This facilitates subsequent sample clean up.
2. Methodology
2.1. Harvesting
We use ∼107−108 yeast cells as the starting material for mapping nucleosomes. Yeast cells are grown in YPD growth medium and utilize dextrose as the carbon source. We autoclave dextrose for 20 min and have not encountered any issues with crystallization of the media.
Reagents
Prepare ahead of time.
-
1 × YPD
10 g Yeast extract
20 g Bacto-Peptone
20 g Dextrose
In 1 l water.
Autoclave at 121 °C for 20 min.
-
ST buffer
mM Tris–Cl, pH 7.5
mM NaCl
Filter 0.22 μm, store at 4 °C.
-
Aliquot stocks
Complete, Mini, EDTA-free Protease Inhibitor Cocktail (CPI) tablets—Roche, catalog #04693159001
2.5 M Glycine
37% Formaldehyde
-
Other
Liquid nitrogen
Day 1
Make a primary inoculum by inoculating 5–10 ml of YPD with cells of interest.
Grow in shaker at 25 °C, 250–300 rpm until cultures have grown sufficiently to start large cultures at desired OD600 nm. This will take usually at least 6 h or overnight. The doubling time of wild-type yeast when grown at 25 °C is around 2 h. Alternatively, you can also grow yeast at 30 °C with a doubling time of 90 min.
Day 2
-
3
Check and record the OD600 nm of starter cultures.
-
4
Inoculate 500 ml YPD with enough of starter culture to bring the culture to the desired starting OD600 nm.
Calculation: (desired large culture OD)/(starter culture OD) × volume of large culture = ml's to inoculate.
-
5
Grow in shaker at 25 °C, 250–300 rpm.
Day 3
-
6
Monitor growth carefully until OD600 nm reaches ∼0.8. Do not go higher than OD of 1.0.
-
7
Add 37% formaldehyde to a final concentration of ∼ 1.0%.
-
8
Return flasks to a room-temperature shaker for 15 min to cross-link.
-
9
Quench cross-linked culture by adding 2.5 M glycine to a final concentration of 125 mM. Incubate for 5 min at room temperature with constant shaking.
-
10
Transfer the contents of the flask to GS3 tubes or equivalent (maximum of 500 ml per tube).
-
11
Centrifuge in a Sorvall RC6 + centrifuge at 4 °C for 3 min at 4000 rpm or ∼2800 ×g.
-
12
Resuspend one cell pellet with 1 ml ice-cold ST buffer to which fresh protease inhibitor cocktail is added.
-
13
Resuspend the second pellet with this cell lysate. Continue to repeat until all pellets are combined and resuspended. All the above steps are done on ice to keep the cells cold.
-
14
Divide the resuspended cells into microcentrifuge tubes (USA Scientific, catalog #1420-8700) to get the equivalent of ∼ 100 ml of cells per tube.
-
15
Spin the tubes in bench-top microcentrifuge at 4 °C for 2 min at ∼9300×g.
-
16
Resuspend each cell pellet in 0.5 ml of ST buffer.
-
17
Spin the tubes at 4 °C for 2 min at ∼9300×g. Aspirate off the supernatant and freeze the cells in liquid nitrogen.
2.2. Cell lysis, MNase digestion
We use mechanical force (by agitating ceramic zirconia beads at high speed) to break yeast cell walls, when isolating chromatin. Alternatively, one can use glass beads coupled to a vortexer, or enzymatic treatment (zymolase or other enzymes) for cell wall digestion. Efficiency of cell disruption can be monitored by observing the cells under a microscope. The isolated chromatin is subsequently fragmented using MNase treatment. MNase is a unique endo–exonuclease, which results in cleavage at nucleosome linker regions. We include an additional step of brief sonication to completely solubilize the fragmented chromatin from the pellet into the supernatant. This sonication does not further fragment the nucleosomal DNA. A portion of the digestion chromatin is then extracted and visualized by agarose gel electrophoresis to obtain an MNase ladder corresponding to multiples of the nucleosomes core with the linker DNA.
Reagents
Prepare ahead of time.
-
FA lysis buffer
50 mM HEPES/KOH, pH 8.0
150 mM NaCl
2.0 mM EDTA
1.0% (v/v) Triton X-100
0.1% (w/v) Sodium deoxycholate
Filter 0.22 μm, store at 4 °C.
-
NP-S buffer
0.5 mM Spermidine
0.075% (v/v) IGEPAL
50 mM NaCl
10 mM Tris–Cl, pH 7.5
5 mM MgCl2
1 mM CaCl2
Filter 0.22 μm, store at 4 °C
-
2 × Proteinase K buffer
40 mM of 1 M Tris–Cl, pH 7.5
40 mM EDTA
2% (w/v) SDS
Filter 0.22 μm, store at 4 °C
-
TE buffer
10 mM Tris–Cl, pH 8
1 mM EDTA
Filter 0.22 μm, store at 4 °C
-
Enzyme stocks
-
40 U/μl MNase—Worthington Biochemical.
MNase is supplied as a lyophilized powder, which is resuspended in NP-S buffer containing 30% glycerol to a final concentration of 40 U/μl and stored at —20 °C.
Proteinase K—Roche
DNase-free RNase—Roche
-
-
Aliquot stocks
20% (w/v) SDS
14.3 M β-Mercaptoethanol (BME)
0.1 M Phenylmethylsulfonyl fluoride (PMSF)
0.5 M EDTA
-
Phenol:chloroform:iso-amyl alcohol (PCIA).
PCIA aliquots should be kept at 4 °C, stored in aluminum foil wrapped tubes as it is light sensitive. PCIA should be handled with care as it can cause skin burns.
20 mg/ml Glycogen
Isopropanol, stored at room temperature
70% (v/v) Ethanol, stored at room temperature
-
6 × Xylene cyanol DNA-loading dye.
30% (v/v) Glycerol.
0.02% (w/v) Xylene cyanol
CPI stock
-
Other
Zirconia/silica beads—BioSpec Products, catalog #11079105z
Agarose—OmniPur, EMD Chemicals, catalog #2125
2.2.1 Day1: Bead-beating lysis
Briefly (around 2 min) thaw 100-ml cell pellet aliquots of cross-linked cells. Vortex and spin the cells before use.
Resuspend the cell pellet in 1.0 ml of FA lysis buffer to which fresh protease inhibitor cocktail and PMSF (to a final concentration of 0.2 mM) are added.
Add 1.0 ml of zirconia beads.
To lyse the cells, use the Mini-Beadbeater-96 machine (BioSpec Products, catalog #1001) for three cycles of 3 min each with a gap of 5 min in between the cycles. (Keep the Eppendorf holder at — 20 °C for 10 min before lysis and on an iced block for the 5-min intervals.) Alternatively, one can use glass beads and vortex at maximum speed for 2 h at 4 °C.
Pierce the top and bottom of the tube with a hot 22-gauge needle and spin the extracts into a collection tube using a bench-top clinical centrifuge for 5 s at ∼3500 ×g. Mix and transfer into 1.5-ml tube.
Spin the tubes in bench-top centrifuge at 4 °C for 10 min at ∼ 16000 ×g. Discard the supernatant.
2.2.2 MNase digestion of lysed cells
-
7
Resuspend the chromatin pellet in 600 μl NP-S buffer by pipetting (or vortex 30 s at maximum speed). Spin down the chromatin at 4 °C for 5 min at 16,000 ×g and discard the supernatant.
-
8
Add BME to NP-S buffer to a final concentration of 1 mM. BME is not stable in solution, so it must be added to the buffer on the day of experiment.
-
9
Resuspend the chromatin pellet in 300 μl NP-S buffer to which BME is added. Mix thoroughly.
-
10
Remove 5 ml cell equivalent of the sample as “input” to MNase digestion for visualizing on the gel.
-
11
For each sample, titrate the amount of MNase added to determine the optimum MNase concentration where ∼80% of the material is in mononucleosomal form.
-
12
Incubate for 20 min at 37 °C in thermomixer with shaking. Alternatively, one can use a heat-block or thermocycler set at a particular temperature and mix by pipetting every 5 min.
-
13
Halt digestion by transferring samples to 4 °C and adding 0.5 MEDTA to a final concentration of 10 mM. Incubate the samples on ice for 10 min.
-
14
Spin at 4 °C for 10 min at 16,000 ×g. Collect the supernatant in a fresh tube and label as supe1.
-
15
To completely solubilize chromatin from the pellet, wash the pellet with 300 μl of NP-S buffer containing 0.2% SDS and then sonicate for four cycles (30-s pulse) at medium strength.
-
16
Spin at 4 °C for 15 min at 16,000 ×g and add this second supernatant to supe1.
-
17
Remove 5 ml cell equivalent of the pellet and supernatant sample for visualization on gel.
-
18
Store the remaining supernatant and the pellet sample at — 80 °C. The pellet sample can be discarded after DNA extraction once you establish that it does not have any significant amount of insoluble DNA.
2.2.3 Reversing cross-links for sample checks
Exposure to high temperature reverses the protein–DNA cross-links. Proteinase K treatment digests all DNA-bound proteins.
-
19
Take 5 ml cell equivalent of the input, pellet and supernatant sample.
-
20
Make up the volume of all samples to 200 μl by adding NP-S buffer.
-
21
Add equal volume of 2× proteinase K buffer.
-
22
Add 3 μl of 20 mg/ml proteinase K enzyme to each sample.
-
23
Incubate the samples at 65 °C overnight for proteinase K treatment.
2.2.4 Day 2: DNA extraction for sample checks
-
24
Extract the samples with 450 μl of PCIA. Vortex the samples for 20 s.
-
25
Spin in a bench-top microcentrifuge at 16,000 ×g for 6 min at room temperature.
-
26
Carefully remove the upper aqueous layer and transfer to fresh Eppendorf tube.
-
27
Repeat “PCIA” extraction once more.
-
28
Add 1 μl of 20 mg/ml glycogen.
-
29
Precipitate DNA from aqueous layer with 0.6 volume of isopropanol (room temperature). Mix well and then incubate for 30 min at room temperature.
-
30
Pellet by microcentrifugation for 30 min, 16,000 ×g at room temperature. Check for the presence of pellet and then decant the supernatant into the sink.
-
31
Wash the pellet with 500 μl of 70% ethanol (room temperature). Mix by pipetting. Spin for 10 min, 16000 ×g at room temperature.
-
32
Repeat 70% ethanol wash once more.
-
33
Decant the supernatant. Dry the pellet for ∼ 15 min (or until dry) in a speed vac (medium temperature setting).
-
34
Add 30 μl TE buffer containing 50 μg/ml RNase to each sample.
-
35
Incubate the samples at 37 °C for 2 h (minimum of 30 min).
-
36
Dissolve the samples in 6 × xylene cyanol loading dye and run on 2% agarose gel at 135 V for 35 min.
-
37
Visualizing DNA from the supernatant sample provides a determination of whether optimum MNase digestion had occurred—see Fig. 10.1A. Any DNA present in the pellet is an indication of unsolubilized DNA— see Fig. 10.1B.
-
38
Proceed with the best titration sample for immunoprecipitation.
Figure 10.1.
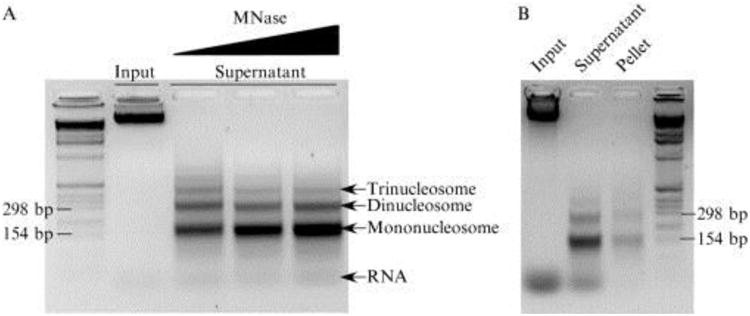
MNase digestion results. The input lane indicates undigested chromatin prior to MNase treatment. (A) An MNase ladder is visible in all three MNase-treated supernatant lanes. As indicated in the gel, with increasing MNase concentration, there is an increase in mononucleosomal DNA and decrease in higher molecular weight bands corresponding to “polynucleosomes.” Left-most lane indicates a 1-kb ladder. (B) The pellet lane indicates a small amount of unsolubilized DNA trapped in the pellet prior to sonication.
2.3. ChIP and library preparation
We enrich for nucleosomal DNA by immunoprecipitation (ChIP) with anti-H3 antibody and select using magnetic Protein A beads. We use 0.05% SDS during IP to remove uncross-linked DNA fragments. Enriched ChIP DNA is then processed for sequencing library preparation while on the magnetic beads. Here we describe library preparation when using Illumina as the sequencing platform, but this procedure can be modified according to the sequencing platform used.
Reagents
Prepare ahead of time.
FA lysis buffer, see above
-
FA high-salt wash buffer
50 mM HEPES-KOH, pH 7.5
1 M NaCl
1.0% (v/v) Triton X-100
1.0% (w/v) Sodium deoxycholate
2 mM EDTA, pH 8.0
Filter 0.22 μm. Store at 4 °C.
-
FA wash buffer 3
10 mM Tris–Cl, pH 8.0
25 mM LiCl
1.0% (v/v) IGEPAL
1.0% (w/v) Sodium deoxycholate
2 mM EDTA, pH 8.0
Filter 0.22 μm. Store at 4 °C.
-
ChIP buffers
FA lysis buffer+0.2% (w/v) SDS
10 mM Tris–HCl, pH 7.5
-
Antibody
Anti-histone H3 antibody—Abcam, catalog #ab1791
-
Enzymes
T4 DNA polymerase (NEB)
T4 DNA ligase (NEB)
Phi29 DNA polymerase (NEB)
Klenow fragment exo- (NEB)
Taq DNA polymerase (NEB)
-
Enzyme buffers
10 × NEBuffer 2 (NEB)
10 × T4 DNA ligase buffer (NEB)
10 × Phi29 polymerase buffer (NEB)
10 × Taq polymerase buffer (NEB)
-
Aliquot stocks
20% (w/v) SDS
3 mM dNTPs
25 mM dNTPs
3 mM dATP
1 × BSA (1 mg/ml)
CPI stock
-
Adaptors and primers
Sequencing adaptors (15 μM)
PCR primers (20 μM)
Index (20 μM)
-
Other
Magna ChIP Protein A Magnetic Beads (Millipore)
DynaMag—15 magnet—Invitrogen, catalog #123-01D
DynaMag—2 magnet—Invitrogen, catalog #123-21D
Agarose, see above
Qiagen QIAquick Gel Extraction Kit
2.3.1 Day 1: Antibody-histone attachment
Thaw the MNase-digested supernatant from — 80 °C storage. Make sure samples go on ice as soon as thawing is complete.
Adjust the sample with NP-S buffer to have a minimal volume of 500 μl. Then add 20% SDS for a final concentration of 0.05%. Add 1.25 μl of 20% SDS to a 500-μl sample without SDS. Dilute the sample with NP-S buffer if it contains an SDS concentration higher than 0.05%.
Thaw one aliquot of anti-H3 antibody for each sample. Each antibody aliquot should contain 10 μg.
Add a 10-μg aliquot of antibody to each sample and incubate the samples on inverting rototorque, 4 °C overnight (12–16h). Check after ∼ 10 min to make sure samples are not leaking.
2.3.2 Day 2: ChIP
Notes
Do not let the beads remain “dry” for longer than about 30 s. If necessary, aspirate a subset of tubes, then add buffer.
Only spin the samples down before and during enzymatic reactions.
Follow these instructions for all washes (not just the ChIP washes).
Wash buffers should be kept on ice, do not set buffers out at room temperature.
Briefly resuspend Protein A Magna beads, then immediately add 50 μl Magna beads to each sample in a 15-ml Falcon tube.
Mix the sample by inverting the tube.
Incubate the reactions for 1.5 h at 4 °C on rototorque (slowly rotating).
After incubation, spin down the samples briefly.
Place in a DynaMag—15 magnet (which can hold 15-ml Falcon tubes) for 1 min.
Using P1000 pipette, carefully transfer the supernatant (flow-through) to new tubes labeled “FT.”
Store flow-through at —80 °C until the resulting data set is validated, then it may be discarded.
2.3.3 ChIP wash series
-
Wash the beads with FA lysis buffer+0.025% SDS.
Add 0.8 ml FA lysis buffer+0.025% SDS+CPI (4 °C) to each sample.
Mix by pipetting up and down and transfer to a new 1.5-ml tube (including beads).
Place in a DynaMag—2 magnet (magnetic rack) for ∼ 1 min.
Aspirate off the supernatant (Wash 1).
Add 1.4 ml FA lysis buffer+0.025% SDS+CPI.
Mix gently by repeated inversions.
Spin down the sample briefly, place on magnetic rack for ∼ 1 min.
Aspirate off the supernatant (Wash 2).
Add 1.4 ml FA lysis buffer+0.025% SDS+CPI.
Mix gently by repeated inversions, rotate on rototorque for 15 min.
k. Spin down the sample briefly, place on magnetic rack for ∼ 1 min.
Aspirate off the supernatant (Wash 3).
-
Wash the beads with FA lysis buffer
Add 1.4 ml FA lysis buffer+CPI.
Mix gently by repeated inversions.
Spin down the sample briefly, place on magnetic rack for ∼ 1 min
Aspirate off the supernatant (Wash 4).
-
Wash the beads with FA high-salt buffer.
Add 1.4 ml FA high-salt buffer+CPI.
Mix gently by repeated inversions.
Spin down the sample briefly, place on magnetic rack for ∼ 1 min.
Aspirate off the supernatant (Wash 5).
Add 1.4 ml FA high-salt buffer+CPI.
Mix gently by repeated inversions, rotate on rototorque for 15 min.
Spin down the sample briefly, place on magnetic rack for ∼ 1 min.
Aspirate off the supernatant (Wash 6).
-
Wash the beads with FA wash buffer 3
Add 1.4 ml FA wash buffer 3+CPI.
Mix gently by repeated inversions.
Place on magnetic rack for ∼ 1 min.
Aspirate off the supernatant (Wash 7).
Add 1.4 ml FA wash buffer 3+CPI.
Mix gently by repeated inversions.
Spin down the sample briefly, place on magnetic rack for ∼ 1 min.
Aspirate off the supernatant (Wash 8).
-
Wash the beads with 10 mM Tris–HCl, pH 8.0
Add 1 ml of 10 mM Tris–HCl, pH 8.0+CPI.
Transfer the sample to fresh 1.7-ml LoBind tube.
Place on magnetic rack for ∼ 1 min.
Carefully remove the supernatant with P1000 pipette (Wash 9).
-
Wash the beads with double-distilled water+CPI.
Add 0.5 ml double-distilled water+CPI.
Mix gently by repeated inversions, place on magnetic rack for ∼ 1 min.
Carefully remove the supernatant with P1000 pipette.
2.4. Sequencing library preparation
2.4.1 Kinase reaction
This step is essential as MNase leaves a 3′ phosphate, whereas a 5′ phosphate is needed. T4 polynucleotide kinase (PNK) removes 3′ phosphates and adds 5′ phosphates in the presence of ATP.
Preheat thermomixer to 37 °C.
-
Prepare Master Mix with 1 × T4 DNA ligase buffer and 1.5 U T4 PNK to a total volume of 18 μl per reaction tube.
Ligase buffer contains DTT, which can form a white precipitate when cold. Ensure all DTT is resuspended before use.
Add 18 ml Kinase Mix to 2 ml bead-bound DNA for total of 20 μl. Pipette up/down ∼10 times gently.
Incubate the samples on thermomixer at 37 °C for 30 min.
-
Wash the beads with FA high-salt buffer.
Add 0.5 ml FA high-salt buffer.
Mix gently by repeated inversions, place on magnetic rack for ∼ 1 min.
Aspirate off the supernatant.
-
Wash the beads with 10 mM Tris–HCl, pH 8.0.
Add 1 ml of 10 mM Tris–HCl, pH 8.0.
Mix gently by repeated inversions, place on magnetic rack for ∼ 1 min.
Carefully remove the supernatant with P1000 pipette.
2.4.2 A-tailing
This step adds a single adenosine nucleotide to the 3′ end of the DNA, which will increase the efficiency of ligation to the Illumina adapters (which have a T overhang). Other sequencing platforms may not use this strategy, in which case the A-tailing step can be skipped.
Prepare Master Mix with 1 × NEBuffer 2, 100 μM dATP, and 10 U Klenow fragment, exo- to a total volume of 28 μl per reaction tube.
Add 28 μl A-Tail Mix to 2 ml bead-bound DNA for total of 30 μl. Pipette up/down ∼10 times gently.
Incubate the samples on shaking thermocycler at 37 °C for 30 min.
-
Wash the beads with FA high-salt buffer.
Add 0.5 ml FA high-salt buffer,
Mix gently by repeated inversions, place on magnetic rack for ∼ 1 min.
Aspirate off the supernatant.
-
Wash the beads with 10 mM Tris–HCl, pH 8.0.
Add 1 ml of 10 mM Tris–HCl, pH 8.0.
Transfer the sample to fresh 1.7-ml LoBind tube.
Mix gently by repeated inversions, place on magnetic rack for ∼ 1 min.
Carefully remove the supernatant with P1000 pipette.
2.4.3 Adaptor ligation
Here, it is necessary to ligate the sequencing adaptor that is appropriate for the sequencing instrument. If planning to multiplex, then each sample gets assigned a unique identity by ligating an individual index sequence for each sample.
Preheat thermomixer to 15 °C.
-
Prepare Master Mix with 15pmol of each adaptor, 30 pmol index (if multiplexing), 1 × T4 DNA ligase buffer, and 1800 U T4 ligase to a total volume of 48 μl.
We use nonphosphorylated adapters to minimize primer dimer. This necessitates a later phi29 fill-in step.
Add 48 μl ligation mix to bead-bound DNA for a total of 50 μl. Pipette up/down ∼10 times gently.
Incubate the samples on thermomixer at 15 °C overnight.
Day 3: Continuation of Section 2.4.3
-
5
Wash the beads with FA high-salt buffer.
Add 0.5 ml FA high-salt buffer.
Mix gently by repeated inversions, place on magnetic rack for ∼ 1 min.
Aspirate off the supernatant.
-
6
Wash the beads with 10 mM Tris–HCl, pH 8.0.
Add 1 ml of 10 mM Tris–HCl, pH 8.0.
Transfer the sample to a fresh 1.7-ml LoBind tube.
Mix gently by repeated inversions, place on magnetic rack for ∼ 1 min.
Carefully remove the supernatant with P1000 pipette.
2.4.4 Phi29 fill in
Phi29 DNA polymerase activity will start at a nick and continue to the end of the DNA. It has a strand displacement activity.
Preheat thermomixer to 30 °C.
Prepare Master Mix with 200 μg/ml BSA, 1 × phi29 Buffer, 200 μM dNTPs, and 10 U phi29 polymerase to a total volume of 48 μl.
Add 48 μl Fill in Mix to 2 μl bead-bound DNA for total of 50 μl. Pipette up/down ∼ 10 times gently.
Incubate the samples on thermomixer at 30 °C for 20 min.
-
Wash the beads with FA high-salt buffer.
Add 0.5 ml FA high-salt buffer.
Mix gently by repeated inversions, place on magnetic rack for ∼ 1 min.
Aspirate off the supernatant.
-
Wash the beads with 10 mM Tris–HCl, pH 8.0.
Add 1 ml of 10 mM Tris–HCl, pH 8.0.
Mix gently by repeated inversions, place on magnetic rack for ∼ 1 min.
Carefully remove the supernatant with P1000 pipette.
Resuspend the 2-ml bead-bound DNA in 48 μl double-distilled water+CPI.
2.4.5 Ligation-mediated PCR
Prepare Master Mix with 1 × Taq buffer, 0.25 mM each dNTP, and 0.3 μM PCR primers to a total volume of 49 μl.
Add 49 μl PCR Mix to 50 μl bead-bound DNA for a total of 99 μl. Pipette up/down ∼10 times gently.
Run the samples in thermocycler with the following program: “Hot start” the samples by adding 1 μl of Taq DNA polymerase to the reaction mix at the “forever” 95 °C step and then continue. Alternatively, a latent heat-activated DNA polymerase may be used.
| Time | Temperature (°C) | Cycles |
|
| ||
| Forever | 72 | 1 |
|
| ||
| 20 min | 72 | 1 |
|
| ||
| Forever | 95 | 1 |
|
| ||
| 5 min | 95 | 1 |
|
| ||
| 15 s | 95 | 18 |
|
| ||
| 15 s | 52 | |
|
| ||
| 1 min | 72 | |
|
| ||
| 5 min | 72 | 1 |
|
| ||
| Forever | 4 | Hold |
|
| ||
It is not recommended to go above 18 cycles of PCR amplification, rather < 18 cycles is preferable if sufficient sample is present.
2.4.6 Gel purification
It is important to size select for appropriate size DNA and remove small molecular weight DNAs that preferentially get amplified.
Thoroughly clean and rinse an appropriate size gel box.
Pour 2% agarose gel with thick combs.
Mix 1/6th volume of 6× sequencing-grade xylene cyanol dye with each sample.
Load 7 μl of 1-kb ladder. Load the samples (region B) on agarose gel between 1-kb ladders (regions A and C) as shown in Fig. 10.2.
Excise Lanes A and C (dashed black lines) from the gel. Leave “region B” of the gel at your bench.
Visualize Lanes A and C on a short-wavelength UV transilluminator and mark the desired DNA fragment size on the DNA ladders encompassing the 200 and 400 bp markers (blue boxes) by cutting the gel in that area.
Reassemble the gel at your bench; using Lanes A and C as markers, excise the sections of agarose-containing DNA fragments of the desired size (red box).
Place each gel excision into a 1.7-ml LoBind tube
Record the weight of each excised piece. Write this weight directly onto the LoBind tube.
Take short-wavelength UV image of excised gel.
Figure 10.2.

Agarose gel purification. Regions A and C indicate 1-kb ladder. Region B: red box indicates the excised gel portion.
2.4.7 Qiagen cleanup
Notes
All buffers and centrifuge spins should be at room temperature.
Do not heat the gel to dissolve the gel slice as it may result in denaturation of the library. Instead, dissolve the gel slice by vortexing at room temperature until completely dissolved.
Use QIAquick Gel Extraction Kit to purify the DNA from the gel. Quantify DNA using bioanalyzer and/or qPCR and then send for deep sequencing. It is recommended that you make clear to the sequencing facility that your library has already been constructed, so that they do not attempt to construct a library with your sample. The sequencing facility should be able to map your DNA sequences to the reference genome that you specify. Those sequencing reads can then be clustered into peak calls using publicly available peak calling algorithms, and nucleosome positions and occupancy levels defined, as discussed elsewhere (Zhang & Pugh, 2011).
Acknowledgments
We thank previous lab member Elissa Ward for initial standardization of the protocol. We also thank Matt Rossi and Vinesh Vinayachandran for their suggestions.
References
- Albert I, Mavrich TN, Tomsho LP, Qi J, Zanton SJ, Schuster SC, et al. Translational and rotational settings of H2A.Z nucleosomes across the Saccharomyces cerevisiae genome. Nature. 2007;446:572–576. doi: 10.1038/nature05632. [DOI] [PubMed] [Google Scholar]
- Anderson JD, Widom J. Poly(dA-dT) promoter elements increase the equilibrium accessibility of nucleosomal DNA target sites. Molecular and Cellular Biology. 2001;21:3830–3839. doi: 10.1128/MCB.21.11.3830-3839.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao Y, White CL, Luger K. Nucleosome core particles containing a poly(dA.dT) sequence element exhibit a locally distorted DNA structure. Journal of Molecular Biology. 2006;361:617–624. doi: 10.1016/j.jmb.2006.06.051. [DOI] [PubMed] [Google Scholar]
- Fragoso G, Hager GL. Analysis of in vivo nucleosome positions by determination of nucleosome-linker boundaries in crosslinked chromatin. Methods. 1997;11:246–252. doi: 10.1006/meth.1996.0411. [DOI] [PubMed] [Google Scholar]
- Gkikopoulos T, Schofield P, Singh V, Pinskaya M, Mellor J, Smolle M, et al. A role for Snf2-related nucleosome-spacing enzymes in genome-wide nucleosome organization. Science. 2011;333:1758–1760. doi: 10.1126/science.1206097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley PD, Madhani HD. Mechanisms that specify promoter nucleosome location and identity. Cell. 2009;137:445–458. doi: 10.1016/j.cell.2009.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer V, Struhl K. Poly(dA:dT), a ubiquitous promoter element that stimulates transcription via its intrinsic DNA structure. The EMBO Journal. 1995;14:2570–2579. doi: 10.1002/j.1460-2075.1995.tb07255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langst G, Becker PB. Nucleosome mobilization and positioning by ISWI-containing chromatin-remodeling factors. Journal of Cell Science. 2001;114:2561–2568. doi: 10.1242/jcs.114.14.2561. [DOI] [PubMed] [Google Scholar]
- Richmond TJ, Davey CA. The structure of DNA in the nucleosome core. Nature. 2003;423:145–150. doi: 10.1038/nature01595. [DOI] [PubMed] [Google Scholar]
- Satchwell SC, Drew HR, Travers AA. Sequence periodicities in chicken nucleosome core DNA. Journal of Molecular Biology. 1986;191:659–675. doi: 10.1016/0022-2836(86)90452-3. [DOI] [PubMed] [Google Scholar]
- Segal E, Widom J. What controls nucleosome positions? Trends in Genetics. 2009;25:335–343. doi: 10.1016/j.tig.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tillo D, Hughes TR. G+C content dominates intrinsic nucleosome occupancy. BMC Bioinformatics. 2009;10:442. doi: 10.1186/1471-2105-10-442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehouse I, Rando OJ, Delrow J, Tsukiyama T. Chromatin remodelling at promoters suppresses antisense transcription. Nature. 2007;450:1031–1035. doi: 10.1038/nature06391. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Pugh BF. High-resolution genome-wide mapping of the primary structure of chromatin. Cell. 2011;144:175–186. doi: 10.1016/j.cell.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Wippo CJ, Wal M, Ward E, Korber P, Pugh BF. A packing mechanism for nucleosome organization reconstituted across a eukaryotic genome. Science. 2011;332:977–980. doi: 10.1126/science.1200508. [DOI] [PMC free article] [PubMed] [Google Scholar]