

SUMMARY
Posttranscriptional regulation in eukaryotes requires cis- and trans-acting features and factors including RNA secondary structure and RNA-binding proteins (RBPs). However, a comprehensive view of the structural and RBP interaction landscape of nuclear RNAs has yet to be compiled for any organism. Here, we use our ribonuclease-mediated structure and RBP-binding site mapping approaches to globally profile these features in Arabidopsis seedling nuclei in vivo. We reveal anticorrelated patterns of secondary structure and RBP binding throughout nuclear mRNAs that demarcate sites of alternative splicing and polyadenylation. We also uncover a collection of protein-bound sequence motifs, and identify their structural contexts, co-occurrences in transcripts encoding functionally related proteins, and interactions with putative RBPs. Finally, using these motifs, we find that the chloroplast RBP CP29A also interacts with nuclear mRNAs. In total, we provide a simultaneous view of the RNA secondary structure and RBP interaction landscapes in a eukaryotic nucleus.
Graphical abstract
INTRODUCTION
RNA molecules are bound throughout their life cycle by dynamic complexes of proteins that regulate their splicing, polyadenylation, nuclear export, localization, translation, and degradation (Bailey et al., 2009). These RNA-binding proteins (RBPs) interact with their targets in a sequence- and secondary structure-specific manner (Cruz and Westhof, 2009). Therefore, both the bound RBPs and secondary structure are key regulatory features of these molecules (Ding et al., 2014; Li et al., 2012a, 2012b). For instance, recent studies have linked secondary structure of mRNA to translation efficiency, stability, splicing regulation, and polyadenylation (Ding et al., 2014; Li et al., 2012a, 2012b; Zheng et al., 2010).
Due to the importance of RNA secondary structure in eukaryotic posttranscriptional processing and regulation, several high-throughput approaches have been developed to globally profile single- and double-stranded RNAs (ssRNAs and dsRNAs, respectively) (Rouskin et al., 2014; Zheng et al., 2010). For example, ss- and dsRNA-seq employ single- and double-stranded RNases (ssRNases and dsRNases, respectively) to provide direct evidence for both single- and double-stranded regions of the transcriptome (Li et al., 2012a, 2012b; Zheng et al., 2010). Alternatively, dimethylsulfate sequencing (DMS-seq) is a technique where samples are treated with DMS, which specifically modifies unpaired adenines (As) and cytosines (Cs) resulting in the termination of reverse transcriptase products, providing evidence for unpaired As and Cs in RNAs (Ding et al., 2014; Rouskin et al., 2014). However, recent studies have demonstrated that DMS modification is obstructed at RBP-binding sites (Talkish et al., 2014), making protein-bound regions indistinguishable from truly structured regions of RNAs.
Most studies of RBP-RNA interactions identify the binding partners of a single protein of interest. This is often accomplished by crosslinking and immunoprecipitation (CLIP) (Ule et al., 2003), in which RNA-protein interactions are crosslinked via UV irradiation followed by immunoprecipitation of a protein of interest. Recently, two methods have reported development of unbiased approaches to study RNA-RBP binding (Baltz et al., 2012; Silverman et al., 2014). Protein interaction profile sequencing (PIP-seq) crosslinks RNA-protein interactions via formaldehyde and subsequently digests ssRNA and dsRNA using structure-specific RNases before high-throughput sequencing, providing a global view of both RNA secondary structure and RBP-bound RNA sequences across the transcriptome (Silverman et al., 2014). Additionally, global photoactivatable ribonucleoside CLIP (gPAR-CLIP) utilizes the incorporation of a synthetic nucleotide into RNAs to identify RNA-protein crosslinking events after exposure to long-wave UV radiation (Baltz et al., 2012). To date, there have been no global studies of either RBP binding or RNA secondary structure performed in the nucleus of any organism.
All aspects of posttranscriptional mRNA maturation are tightly controlled by RNA-protein interactions acting to positively or negatively regulate recruitment of catalytic molecular machines. For instance, splicing is performed by one of two large complexes, the U2- or U12-type spliceosomes, which identify and excise ~170,000 or ~1,800 introns in Arabidopsis, respectively (Marquez et al., 2012). In addition to being regulated by multiple spliceosomes, pre-mRNA transcripts can undergo alternative splicing (AS), resulting in mature mRNAs of different sequences (Wahl et al., 2009). In Arabidopsis, over 60% of introns are alternatively spliced, with failure to excise an intron (intron retention [IR]) or exclusion of an exon (exon skipping/cassette exon [CE]) in specific isoforms comprising > 64% of these events (Marquez et al., 2012). Additionally, more than 70% of Arabidopsis pre-mRNAs can undergo alternative polyadenylation (APA), resulting in transcript isoforms that differ in their 3′ termini (Hunt et al., 2012; Wu et al., 2011). Previous studies have shown that perturbing RNA secondary structure at alternatively spliced exons can result in decreased RBP recruitment and a shift in spliceoform abundance (Raker et al., 2009). Thus, both AS and APA are important regulatory processes driven by large collections of RBPs and their interactions with specific RNA sequences and structures.
The interplay between RBPs that bind functionally related genes has become a topic of great interest. Recent studies have attempted to identify posttranscriptional operons (Tenenbaum et al., 2011), transcripts with the same gene ontology that are bound by similar populations of RBPs. Thus, the binding of these RBPs would allow coregulation of genes encoding functionally related proteins. Evidence for posttranscriptional operons has been seen in human cells (Silverman et al., 2014); however, this analysis has yet to be performed in Arabidopsis.
Here, we simultaneously profile the global landscapes of RBP binding and RNA secondary structure in nuclei of 10-day-old Arabidopsis seedlings using our PIP-seq and structure-mapping approaches. In total, this study produces an unbiased view of RBP binding and RNA secondary structure for a nuclear transcriptome, providing a rich resource for future hypothesis generation and testing.
RESULTS AND DISCUSSION
PIP-seq on Purified Arabidopsis Seedling Nuclei
To probe the RNA-RBP interaction site and RNA secondary structure landscapes of the Arabidopsis nucleus, we performed PIP-seq (Silverman et al., 2014) on total nuclei from 10-day-old seedlings. The nuclei were crosslinked with formaldehyde prior to purification via the isolation of nuclei in tagged cell types (INTACT) approach (Deal and Henikoff, 2010). We confirmed nuclei purity by direct imaging (Figure S1A available online), revealing only DAPI-stained nuclei bound to the streptavidin-coated beads. Additionally, we found an enrichment of the nuclear histone H3 protein and undetectable levels of the mostly cytoplasmic ACT8 (Kandasamy et al., 1999), the ER-localized BIP1 and CNX1, and chloroplastic RUBISCO and PEPC proteins in our INTACT-purified nuclei preparations (Figure S1B), confirming that there is no chloroplastic, ER, or cytoplasmic contamination. We used ~2 million of these highly pure nuclei for each of two PIP-seq replicates, which were split into footprinting and structure-only samples (four total libraries per replicate) (Figure 1A). Our structure-only samples provide in vivo structure data, and additionally serve as a background to our footprinting samples accounting for regions that are insensitive to the structure-specific RNases.
Figure 1. Overview of PIP-seq in Arabidopsis Nuclei.
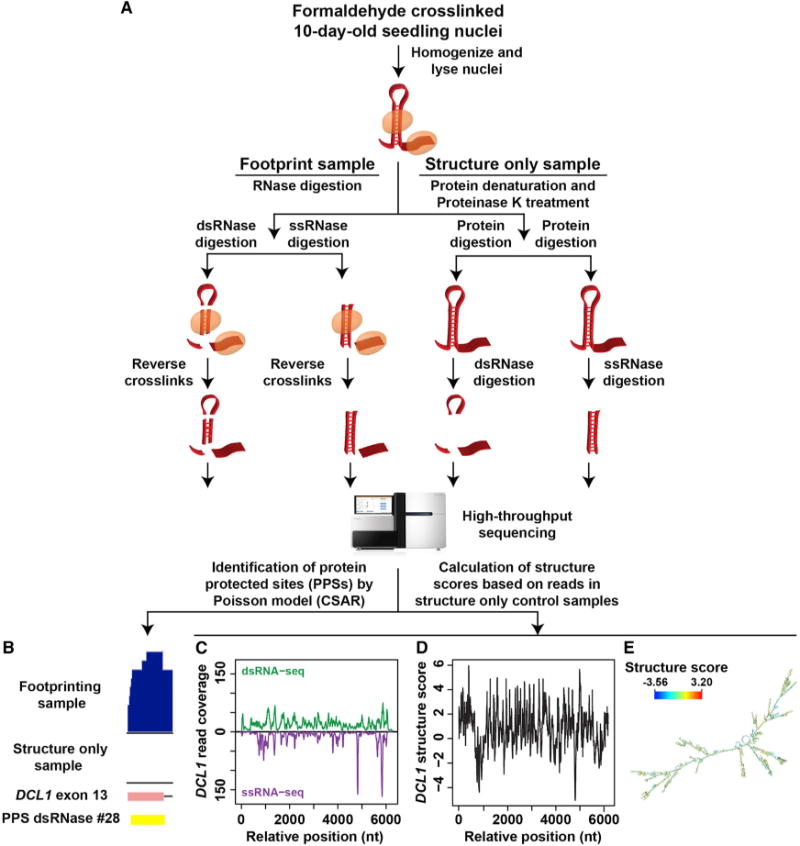
(A) The PIP-seq approach in the Arabidopsis nucleus. Nuclei were purified from 10-day-old Arabidopsis seedlings that were crosslinked using a 1% formaldehyde solution. Nuclei were lysed and separated into footprinting and structure-only samples. Four total sequencing libraries were then prepared for each replicate experiment as previously described (Silverman et al., 2014).
(B) An example of PPS identification (dsRNase #28) in exon 13 of DCL1.
(C) Read coverage across the DCL1 transcript for the ds- (top, green line) and ssRNA-seq (bottom, purple line) structure-only samples.
(D) Structure scores for the DCL1 transcript based on read coverage seen in (C).
(E) mRNA secondary structure model for DCL1 determined using our methodology. See also Figures S1–S3.
Footprint samples were directly treated with either an ss- or dsRNase (see Experimental Procedures). In contrast, the structure-only samples first had proteins denatured in SDS and degraded with Proteinase K prior to RNase digestion. Denaturation of RBPs before RNase treatment will make protein-bound sequences in the footprinting sample accessible to RNases in these reactions. Thus, RBP-bound sequences were enriched in footprinting relative to structure-only samples (Figure 1B). Additionally, analysis of the structure-only samples as previously described (Li et al., 2012a) allowed us to determine the native (protein-bound) RNA base-pairing probabilities for the Arabidopsis nuclear transcriptome (example shown in Figures 1C–1E).
The resulting high-quality PIP-seq libraries (Figures S2A and S2B) were sequenced and provided ~24–38 million raw reads per library. To determine reproducibility, we used a 50 nucleotide (nt) sliding window to define the correlation of nonredundant sequence read abundance between biological replicates of footprinting and structure-only libraries. We observed a high correlation in read counts between all footprinting and structure-only libraries (Pearson correlation > 0.81) (Figures S3A–S3D). Similarly, principle component analysis of read coverage in 500 nt bins revealed that replicates of each library type clustered together (Figure S3E), further indicating the high quality and reproducibility of our PIP-seq libraries.
The RNA-Protein Interaction Landscape of the Arabidopsis Nucleus
To identify protein-protected sites (PPSs), we used a Poisson distribution model to identify enriched regions in the footprinting compared to the structure-only libraries at a false-discovery rate of 5% as previously described (Silverman et al., 2014) (Figure 1B). We identified 61,632 total PPSs in our experiments, 64.7% of which overlap between the two replicates (Figure 2A). Consolidation of all PPSs yields 40,131 distinct sites (Table S1) with an average size of 68 nt (Figure S4A). This reproducibility is much higher than many CLIP-seq experiments, which often produce < 35% overlap between replicates (Lebedeva et al., 2011). The majority of PPSs were identified by the dsRNase (~30,000 PPSs) as compared to the ssRNase (~10,000 PPSs) (Figures S4B and S4C) treatment, with ~50% of the sites uncovered by the ssRNase overlapping those from the dsRNase libraries (Figures S4D and S4E).
Figure 2. Characterization of Arabidopsis Nuclear PPSs.
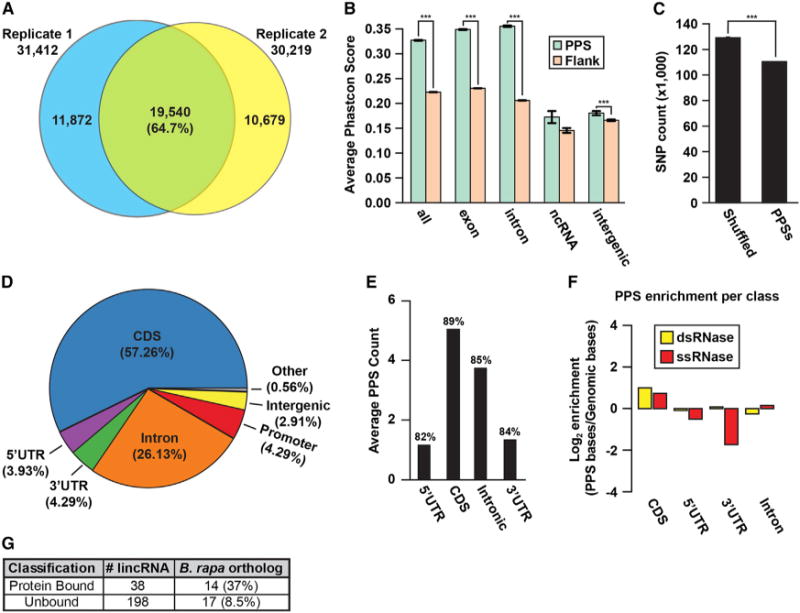
(A) Overlap between PPSs identified from two replicate nuclear PIP-seq experiments.
(B) Comparison of average PhastCons scores between PPSs (green bars) and equal-sized flanking regions (orange bars) for various genomic regions. *** denotes p value < 1×10−10, Kolmogorov-Smirnov test. Error bars, ± SEM.
(C) Analysis of the total number of SNPs identified by the 1001 Genomes Project (Cao et al., 2011) in PPSs compared to a shuffled background control. *** denotes p value < 1×10−10, χ2 test. Error bars, ± SD.
(D) Absolute distribution of PPSs throughout various RNA species and transcript regions.
(E) Average PPS count per pre-mRNA transcript region. Percentages indicate the fraction of annotated RNAs that contain sequencing information for that region.
(F) Genomic enrichment of PPS density, measured as log2 enrichment of the fraction of PPS base coverage normalized to the fraction of genomic bases covered by indicated nuclear mRNA regions for the dsRNase (yellow bars) and ssRNase (red bars) libraries.
(G) Breakdown of bound compared to unbound nuclear lincRNAs that are conserved between Arabidopsis thaliana and Brassica rapa. See also Figure S4 and Table S1.
Given the high reproducibility between our PIP-seq replicates (Figures 2A, S3, and S4), we focused on the complete set of 40,131 distinct PPSs for all subsequent analyses. To estimate the functional relevance of these nuclear PPSs, we compared flowering plant PhastCons conservation scores (Li et al., 2012b) for PPSs versus same-sized flanking regions. We found that PPS sequences were significantly (p values < 1×10−200, Kolmogorov-Smirnov test) more evolutionarily conserved than flanking regions (Figures 2B and S4F). Importantly, this was true for PPS sequences in both exonic and intronic portions of the nuclear collection of mature and pre-mRNA transcripts (nuclear mRNAs), but not for ncRNAs (Figure 2B). These results support the notion that nuclear mRNA sequences are constrained by their ability to interact with RBPs, while decreased PPS conservation within ncRNAs is consistent with their low conservation rates across plant species (Liu et al., 2012).
We also reasoned that functional RBP-interacting sequences would contain less nucleotide diversity across closely related strains when compared to an equal number of same-sized regions randomly selected from detected transcripts. To address this, we used data from the 1001 Genomes Project, which has cataloged naturally occurring single-nucleotide polymorphisms (SNPs) between 80 strains of Arabidopsis thaliana (Cao et al., 2011). We found a significant (p value < 2.2×10−16, χ2 test) decrease in nucleotide diversity within PPSs compared to shuffled regions (Figure 2C). Therefore, Arabidopsis PPSs resist the effects of random genetic drift occurring in the numerous populations across the globe, indicating their functional significance.
A classification of all distinct PPSs revealed the majority of these sites were located in nuclear mRNAs, with the largest fractions occupying the coding sequence (CDS) (57.3%) and introns (26.1%) (Figure 2D). Closer examination of PPSs broken down by genic features (e.g., 5′ and 3′ UTR, CDS, and intron) revealed that detected Arabidopsis nuclear mRNAs contained multiple binding events in both the CDS (~5 total/gene) and introns (~4 total/gene), while the 5′ and 3′ UTRs averaged only a single interaction per expressed transcript (Figure 2E).
We then tested the enrichment of PPSs in specific nuclear mRNA regions (e.g., 3′ and 5′ UTRs) normalized to the number of bases annotated as these features in the TAIR10 Arabidopsis genome. We found that PPSs identified by both RNases were enriched in CDSs, while being underrepresented in 5′ UTRs (Figure 2F). Interestingly, both introns and 3′ UTRs show opposite enrichment trends for ds- and ssRNases, suggesting that PPSs preferentially occur in more highly or lowly structured regions, respectively. When interrogating the enrichment of PPSs in the CDSs of nuclear mRNAs we found that the intron flanking ends of exons tend to be more protein bound than their middle segments (Figure S5A). This binding suggests that we can detect a high level of splicing factor/machinery binding through nuclear PIP-seq as described below. In total, our results reveal that the CDSs of mRNAs are enriched for RBP binding in the Arabidopsis nucleus.
Although PPSs in ncRNAs were not conserved, this category consists of many RNA subgroups, thus conserved classes might be obscured. Long intergenic noncoding RNAs (lincRNAs) are a recently discovered class of ncRNAs that are necessary for vertebrate development (Cech and Steitz, 2014; Sauvageau et al., 2013), but are not well characterized in plants (Hacisuleyman et al., 2014; Liu et al., 2012). We examined the relationship between our PIP-seq data and a set of ~2,700 curated lincRNAs in Arabidopsis (Liu et al., 2012) to identify nuclear protein-bound RNAs. We detected 236 lincRNAs in our nuclear sequencing data, 38 of which contained one to four PPSs (Figure 2G). We found that these protein-bound lincRNAs were significantly (p value < 4.5×10−30, χ2 test) more conserved within the related crop species Brassica rapa (37%, 14 total) as compared to unbound nuclear lincRNAs (8.5%, 17 total) (Figure 2G). The combination of nuclear protein binding and conservation in B. rapa suggests that RBP-bound nuclear lincRNAs have important functions in plant systems.
Patterns of RNA Secondary Structure and RBP Binding Are Anticorrelated
To interrogate the landscape of RBP binding and RNA secondary structure in specific regions of nuclear mRNAs, we calculated the structure scores and PPS densities and examined the average profiles for all detectable transcripts. The structure score is a generalized log ratio of dsRNA-seq to ssRNA-seq reads at each nucleotide position, with positive and negative scores indicating ds- and ssRNA, respectively (see Supplemental Experimental Procedures). To examine the relationship between PPS density and structure score, we focused on the boundaries between the UTRs and CDS of nuclear mRNAs. We observed the highest PPS density in the CDS with decreased occupancy within the 5′ and 3′ UTRs (Figures 3A and 3B), consistent with the gross PPS localization and enrichment analysis (Figures 2D–2F). Interestingly, we observed significantly (p value < 8.2×10−32, Wilcoxon test) higher levels of protein binding directly over the start codon (Figure 3A) relative to flanking regions. Similarly, we examined the start codons at high-confidence upstream open reading frames (uORFs) (von Arnim et al., 2014) and found a significant (p value < 0.01, Wilcoxon test) increase in PPS density over uORF start codons relative to the upstream flanking region (Figure S5B). Similar increases in PPS density over the start and stop codon were speculated to be due to ribosome binding (Baltz et al., 2012; Silverman et al., 2014). However, the nuclear preparations used in this study are free of the cellular compartments containing functional ribosomes (cytoplasm and ER) (Figure S1B), and RBP-binding profiles for transcripts that are not translated in the rough ER (Figure S5C) or are unspliced and likely localized in the nucleus (Figure S5D) demonstrate very similar protein-binding profiles. Taken together, these results suggest that one or more nuclear RBPs occupy this region.
Figure 3. Patterns of Protein Occupancy and Secondary Structure in Specific Nuclear mRNA Regions.
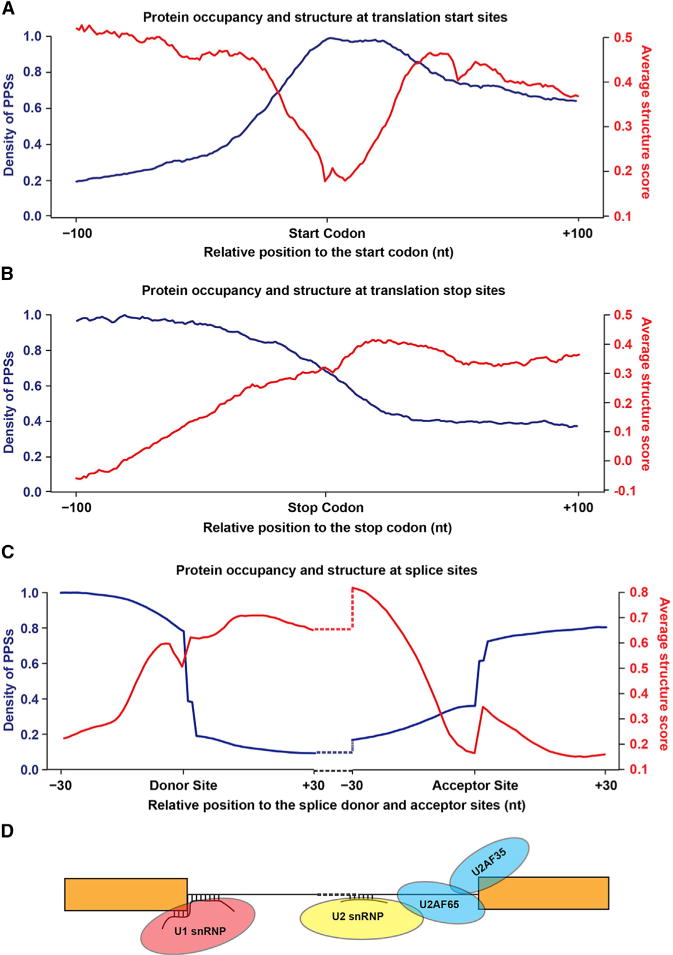
(A and B) PPS density and structure score profiles for nuclear mRNAs based on our PIP-seq experiments. Average PPS density (blue lines) and structure scores (red lines) at each position ± 100 nt from canonical (A) start and (B) stop codons for Arabidopsis nuclear mRNAs.
(C) PPS density and structure score profiles for exon/intron boundaries of nuclear mRNAs. Average PPS density (blue lines) and structure scores (red lines) at each position ± 30 nt from splice donor and acceptor sites.
(D) Model depicting the canonical protein and RNA interactions of the U2-type spliceosome at the splice donor and acceptor sites depicted in (C). See also Figure S5.
In contrast to RBP occupancy, we found that secondary structure was higher in both UTRs compared to the CDS at the regions analyzed, with a significant (p values < 0.05, Wilcoxon test) dip directly over uORF and canonical start codons, as well as upstream of the stop codon, as observed previously (Ding et al., 2014; Li et al., 2012b) (Figures 3A, 3B, and S5B). Thus, these structural characteristics at the start and stop codons seem to be a consistent feature of both Arabidopsis nuclear and mature mRNAs. Interestingly, our analyses revealed that secondary structure and PPS density are anticorrelated to one another. Specifically, we looked at both PPS density and structure score simultaneously, and found a significant (p value < 2.2×10−16, asymptotic t approximation) anticorrelation (Spearman’s rho < −0.82) between these metrics at both canonical start and stop codons. Although the correlation is milder (likely due to fewer instances), there is a significant (p value < 3.6×10−9, asymptotic t approximation) negative correlation (Spearman’s rho < −0.55) for uORF start codons as well.
It is worth noting that although the majority of PPSs were identified in the dsRNase-treated samples, this does not necessitate that the interacting RBPs are binding dsRNA. In support of this hypothesis, we found that more highly structured regions generally surrounded PPSs, with a lower average structure score directly over the RBP-bound sequence (Figure S6A). Although the dsRNase-identified PPSs have a significantly (p value < 2.2×10−16, Wilcoxon test) higher average structure score than those uncovered by the ssRNase (Figure S6A), the dip in structure score directly over these regions suggests that they can be ds- and/or ssRNAs. Taken together, these results suggest that many Arabidopsis RBPs bind ssRNA flanked by structured regions.
It should also be noted that the higher overall structure of the UTRs compared to the CDS is opposite to what has been observed previously both in vivo and in vitro when profiling total (mostly mature cytoplasmic) RNA in Arabidopsis (Ding et al., 2014; Li et al., 2012b). Together, these results suggest that the structural landscape of the nucleus is distinct from that of the cytoplasm. These differences in secondary structure in specific cellular locales will need to be further investigated.
As we were probing the nuclear transcriptome, we next examined the PPS density and structure scores across all TAIR10 annotated splice donor and acceptor sites (Figure 3C). We first determined that the RNA population consisted of a high percentage of unspliced pre-mRNA. Specifically, we found that ~40% of reads mapping to the first and last constitutively spliced intron junctions cross the exon-intron boundary in total RNA sequencing data sets from congruently purified nuclei (see Supplemental Experimental Procedures), suggesting comparable levels of spliced and unspliced transcripts in our data sets (Figure S6B). Despite the large percentage of detectable unspliced transcripts (pre-mRNAs), exonic and intronic regions cannot be directly compared due to slightly lower read coverage in introns. Therefore, we first compared 30 nt regions up- or downstream of acceptor and donor intron sites, respectively, and found that the 3′ end of introns had significantly (p value < 1×10−30, Wilcoxon test) higher protein binding relative to the 5′ end. These results are consistent with the U2 auxiliary factors (U2AFs) occupying the acceptor splice site (Wahl et al., 2009). Intriguingly, there were distinct patterns of secondary structure at both the splice donor and acceptor sites (Figure 3C). Upstream of the donor site, we observed a dramatic decrease in secondary structure from nt −3 to −1, corresponding to the U1 snRNA binding site (−3 to +8) (Chiou et al., 2013). This dip in secondary structure mirrors what we have seen over the translation start codon (Figure 3A), revealing that this region is more accessible to intermolecular RNA pairing than flanking sequences, perhaps facilitating binding of the U1 snRNA. Additionally, we found a drop in secondary structure immediately upstream of the splice acceptor site, suggesting an increased accessibility to U2AFs and other splicing factors in this region (Wahl et al., 2009) (Figure 3D).
We again observed opposing patterns of secondary structure and PPS density at all regions examined in these analyses (Figure 3C). Specifically, we found that this anticorrelation (Spearman’s rho < −0.93) between PPS density and RNA secondary structure was significant (p value < 2.2×10−16, asymptotic t approximation) at regions flanking the acceptor sites, as well as the upstream exonic sequence at donor sites. The proximal intronic region at donor sites had a milder (Spearman’s rho < −0.38), but still significant (p value < 0.05, asymptotic t approximation) anticorrelation between structure score and PPS density, which may be due to the intermolecular base pairing between the U1 snRNA and the intron (Figure 3D) that occurs at 8 of the 30 nt probed. In total, our findings reveal that RBP binding and RNA secondary structure are anticorrelated features in the Arabidopsis nuclear transcriptome.
Distinct RNA Secondary Structure and RBP-Binding Profiles Demarcate AS and Polyadenylation Sites
The specific patterns of RBP binding and RNA secondary structure at exon/intron boundaries suggest that these features may also have distinct distributions at sites of AS. Therefore, we compared the profiles for these two features at several types of alternatively spliced exons. To do this, we used ASTALAVISTA (Foissac and Sammeth, 2007) to annotate AS events in the TAIR10 transcript assembly, and isolated all examples of CE and IR. We also focused on TAIR10 introns that have been previously described as U12-type splice sites (Marquez et al., 2012). We compared average PPS density and structure score for 50 nt in the exonic region and 30 nt in the intronic sequence at both the splice donor and acceptor sites for these splicing events (Figure 4A). We found that IR events have significantly (p values < 4.3×10−7, Wilcoxon test) higher PPS density in the 40 nt upstream (−40 to −1) of the splice donor, while CE and U12-type introns do not significantly (p value > 0.05, Wilcoxon test) differ from constitutive introns. This trend for increased PPS density continues in IR events 30 nt into the intron at splice donor sites, with these events showing ~4.5-fold higher protein binding than constitutive introns (p value < 1.9×10−44, Wilcoxon test) (Figure 4B). The increased binding within these introns is consistent with the presence of intronic splicing silencers, cis elements that recruit proteins to inhibit spliceosome assembly (Chen and Manley, 2009). We observed increased PPS density at the splicing acceptor for both CE and IR sites in the downstream exon (p values < 6.7×10−6, Wilcoxon test) and in the 30 nt of intron directly upstream of this splice site (p values < 0.001, Wilcoxon test) (Figure 4B). This can likely be explained by recruitment of RBPs through a combination of both positive and negative cis regulatory elements, such as exonic splicing silencers to induce exon skipping, and intronic splicing enhancers to increase inclusion, working additively to regulate each exon in a cell type-specific manner (Chen and Manley, 2009). These same trends are observed when specifically examining CE and IR events with adjacent constitutive exons (Figures S6C and S6D). In total, these results reveal that IR and CE events can be differentiated from one another based on the patterns of protein binding density just up- and downstream of both splice sites.
Figure 4. Protein Occupancy and Secondary Structure Landscapes at Alternative Splicing and Polyadenylation Sites.
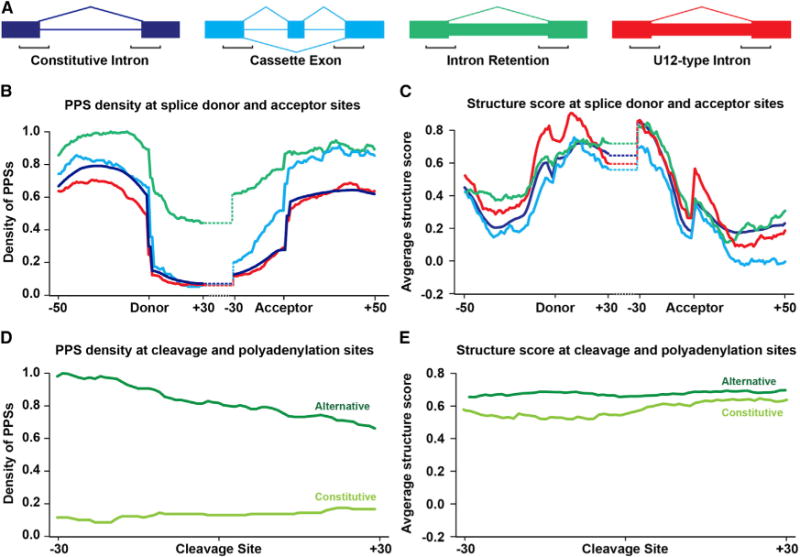
(A) Diagram of constitutive introns (blue), cassette exons (turquoise), intron retention events (green), and U12-type introns (red). Large boxes represent exons, lines represent constitutive introns, and small boxes represent alternatively spliced sequences, with the black brackets indicating the regions graphed in (B) and (C) for reference.
(B) PPS density profiles for constitutive and alternative splicing events in Arabidopsis. Average PPS density at each position −50 to +30 nt at the donor splice site, and −30 to +50 nt at the acceptor splice site. Line colors correspond to examples shown in (A).
(C) Structure score profiles for constitutive and alternative splicing events in Arabidopsis covering the same regions as (B). Line colors correspond to examples shown in (A).
(D) PPS density profiles for constitutive and alternative poly(A) sites of nuclear mRNAs. Average PPS density at each position ± 30 nt from constitutive (light-green line) and alternative (dark-green line) cleavage and polyadenylation sites.
(E) Average structure score profiles for constitutive and APA sites covering the same regions as (D). See also Figure S6.
We next probed the structural profiles for each of these subsets of introns across splice sites (Figure 4C). The most striking feature we observed was the dramatic difference in overall profile shape between U12-type introns and constitutive introns upstream of the donor splice site (−16 to −1). We found a significantly (p value < 0.01, Wilcoxon test) higher structure score for these introns in this region, which have a PPS profile that is indistinguishable from constitutive introns. This structural profile likely influences the identity of the proteins binding this region (Cech and Steitz, 2014), resulting in distinct RBP populations at each type of intron. Additionally, IR events are also significantly (p value < 4.5×10−3, Wilcoxon test) more structured 40 nt upstream of the donor splice site (−40 to −1). Specifically, these profiles reveal highly structured regions that are associated with increased binding levels of regulatory proteins. Thus, in both U12-type and IR events, the increased structure in specific regions likely limits the accessibility of binding sites to specific RBPs allowing for a tighter control over the splicing machinery. Interestingly, CEs are the only subset of events that are consistently less structured than constitutive introns. This trend is only statistically significant (p value < 0.05, Wilcoxon test) upstream of the acceptor site (−30 to −1), but the analysis is limited by a low number of annotated events (< 700) (Figure 4C). Constitutive exon-flanked CE and IR events exhibit similar patterns (Figure S6E). In total, these results reveal that each of these three subtypes of AS has a distinct combination of PPS and structural profiles, supporting the idea that both structure and protein occupancy are required for their proper regulation.
Addition of the poly(A) tail (polyadenylation) during eukaryotic mRNA maturation is also highly regulated. Therefore, we calculated average PPS density and structure score 30 nt up- and downstream of expressed transcripts with constitutive or APA sites (Sherstnev et al., 2012). We found that APA events were on average 3.7-fold (p value < 4.8×10−16, Wilcoxon test) more protein bound up- and downstream of the cleavage site as compared to constitutive events (Figure 4D). Interestingly, there is no significant (p value > 0.05, Wilcoxon test) difference in structure scores between the alternative and constitutive sites (Figure 4E), revealing that this differential protein binding is independent of secondary structure. These results indicate that APA sites do not exhibit altered secondary structure compared to constitutive sites; however, the increased protein binding could be used to differentiate these two types of events from one another.
The Structural Landscape of Protein-Bound RNA Motifs
To identify RBP-bound motifs, we employed the motif finding algorithms MEME (Bailey et al., 2009) and HOMER (Heinz et al., 2010) on PPSs partitioned by specific region (e.g., CDS) or on the entire collection, respectively. We identified one GAN repeat motif by MEME that was common to both the CDS and 5′ UTR (Figure 5A), while HOMER identified 40 octamers that were significantly (p values < 10−7) enriched in our PPSs (Table S2), of which we further characterized four of the most significantly enriched (p values < 1.0×10−67) (Figures 5B–5E).
Figure 5. The Landscape of Protein-Bound RNA Motifs.
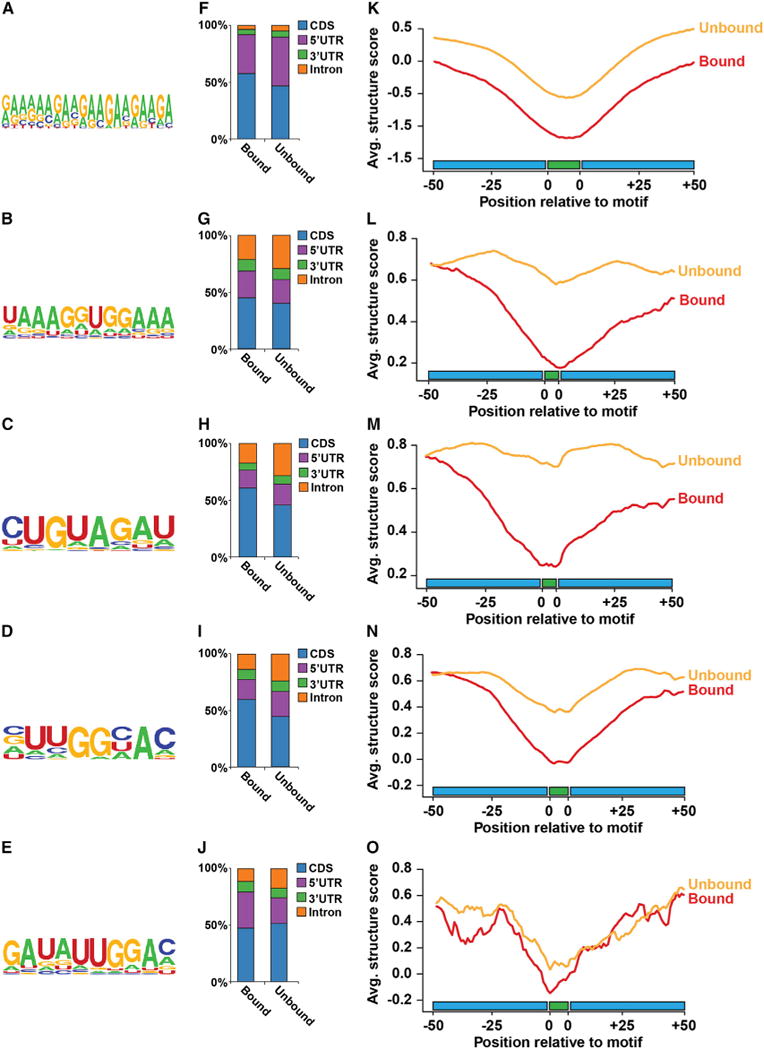
(A–E) Overrepresented sequence motifs identified by MEME (A) or HOMER (B–E) analysis of PPS sequences.
(F–J) The relative distribution of protein-bound and unbound motifs from (A) to (E) throughout specific pre-mRNA regions, including the CDS (blue), 5′ UTR (purple), 3′ UTR (green), and intron (orange).
(K–O) Structure score profiles at RBP-binding motifs from (A) to (E). Average structure score at each position ± 50 nt up- and downstream of bound (red lines) and unbound (orange lines) motif occurrences from (A) to (E).
We identified the percentage of PPS-bound and -unbound motif occurrences in specific regions of nuclear mRNAs normalized by their overall length in the genome (Figures 5F–5J). Comparing the localization of bound and unbound motif instances revealed stark differences. We saw an overall enrichment of bound sites within the CDS and 5′ UTR. Conversely, the unbound HOMER motif instances were generally more prevalent in introns (Figures 5G–5J), while the 5′ UTR is overrepresented in the unbound GAN repeat occurrences (Figure 5F). In total, these results indicate that within the nucleus RBP binding is enriched within 5′ UTR and CDS instances of specific sequence motifs.
To define the structural context at these five sequence motifs, we calculated average structure scores at the core motif and 50 nt flanking regions for bound and unbound instances. We observed that the five motifs have low structure scores, but are flanked by more structured regions (Figures 5K–5O). As mentioned above, the high levels of this conformation within the nuclear transcriptome may explain increased PPS identification by the dsRNase (Figures S4B, S4C, and S6A). Interestingly, protein-bound instances of all five motifs and their flanking sequences are significantly (p values < 7.3×10−12, Wilcoxon test) less structured relative to unbound instances of these sequences (Figures 5K–5O). In total, these findings support the observations that PPSs occur preferentially at less-structured regions of transcripts. Whether this is a cause or consequence of protein binding to these sequence elements will need to be further investigated.
Evidence of Posttranscriptional Operons in the Arabidopsis Nuclear Transcriptome
RBP-interacting motifs often co-occur in functionally related genes in human cells (Silverman et al., 2014), but it is not known if this happens in the Arabidopsis nuclear transcriptome. To address this, we interrogated the interactions between protein-bound motifs discovered by our PIP-seq approach. Thus, we identified all bound instances of each identified motif (Table S2) in target RNAs using the HOMER suite (Bailey et al., 2009) on the total set of nuclear PPSs. We then quantified co-occurrences of each pair of these protein-bound motifs within all nuclear mRNAs. We used k-means clustering of the resultant weighted adjacency matrix and identified three clusters of motifs that co-occur on highly similar sets of target transcripts (Figure 6A). Interestingly, Clusters 1 and 2 have only five and four motifs, respectively, while Cluster 3 consisted of the remaining 32 motifs, although no transcripts contained more than four of these co-occurring PPS-bound motifs. The number of transcripts containing at least three bound motifs within each cluster varied greatly, with Clusters 2 and 3 having 188 and 204 transcripts, respectively, while Cluster 1 had the most co-occurring bound motifs with 5,887. These findings indicate that many Arabidopsis transcripts contain numerous RBP-interacting motifs.
Figure 6. Clusters of Motifs Are Present in Functionally Related Genes.
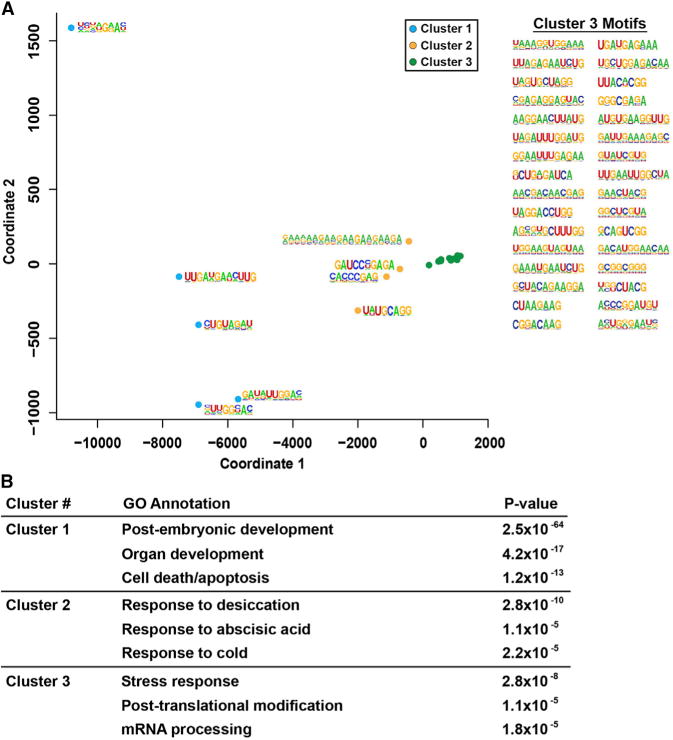
(A) Multidimensional scaling analysis of RBP-bound motif co-occurrence in Arabidopsis transcripts. The motifs used for this analysis were identified by HOMER- and MEME-based analyses of PPS sequences. Sequences for all of the motifs used in this analysis can be seen in the figure and found in Table S2. Colored dots indicate cluster membership as defined by k-means clustering (k = 3).
(B) The most significantly enriched biological processes (and corresponding p value) for target transcripts of the specified clusters of motifs identified in (A) where three or more of the motifs are protein-bound and co-occurring. See also Table S2.
We used agriGO (Du et al., 2010) to interrogate overrepresented biological processes for these collections of RNAs with co-occurring RBP-bound motifs (Figure 6A). We found that the most highly overrepresented functional terms were related to distinct processes, including cell death/apoptosis and postembryonic and organ development (Cluster 1); response to desiccation, abscisic acid, and cold (Cluster 2); as well as stress response, posttranslational modification, and mRNA processing (Cluster 3) (Figure 6B). The identification of groups of functionally related transcripts bound by the same collection of RBPs during their nuclear life cycle supports the idea of posttranscriptional operons (Keene and Tenenbaum, 2002; Tenenbaum et al., 2011) functioning in the Arabidopsis nucleus.
CP29A Localizes to the Arabidopsis Nucleus
After identifying enriched motifs within our PPS list we used these motifs to identify putative Arabidopsis RBPs. To begin, we confirmed that these sequences interact in vitro with specific RBPs using a UV crosslinking assay with radiolabeled RNA probes (from Figures 5A–5E; Table S3) or a scrambled control sequence. We found that each sequence motif interacted with one or more distinct RBPs (Figure S7A). We then used these same probes in RNA-affinity chromatography followed by mass spectrometry analysis. Using this approach with four significant HOMER motifs (Figures 5B–5E), we identified 25 proteins with peptides that were enriched over our negative controls, with four proteins that passed a threshold of > 6-fold enrichment for interaction with at least one sequence (Figures S7B and 7A; Table S4). Interestingly, CVP2 as well as the LRR family and DUF544-containing proteins do not have canonical RNA-binding domains (RBDs). This is similar to recent findings in human RBP identification (Baltz et al., 2012; Castello et al., 2012), suggesting that these proteins interact with their target motifs via noncanonical RBDs or an RBP partner.
The GAN repeat motif is of particular interest because it has been linked to splicing regulation in Physcomitrella patens (Wu et al., 2014). The UV crosslinking assay indicated that numerous proteins were capable of binding this motif, with several 25–40 kDa proteins significantly (p value < 0.05, Fisher’s t test) enriched over the negative control (Figure S7A). However, from mass spectrometry analysis of interacting proteins only four passed a threshold of 6-fold enrichment over negative controls, with the strongest candidate RBPs being CP29A (> 18-fold enrichment) (Figure 7B). This protein has previously been identified as an RBP that functions in the chloroplast (Ye et al., 1991), but nuclear localization had not been demonstrated. We used an Arabidopsis CP29A monoclonal antibody (Kupsch et al., 2012) to perform western blots on lysates from INTACT-purified nuclei and 10-day-old seedlings. Although at low levels, we could reproducibly detect CP29A in the Arabidopsis nucleus (Figure 7C), in contrast to other chloroplastic proteins (Figure S1B), showing that a subset of CP29A is localized in the nucleus.
Figure 7. Identification of Arabidopsis RNA-Interacting Proteins.
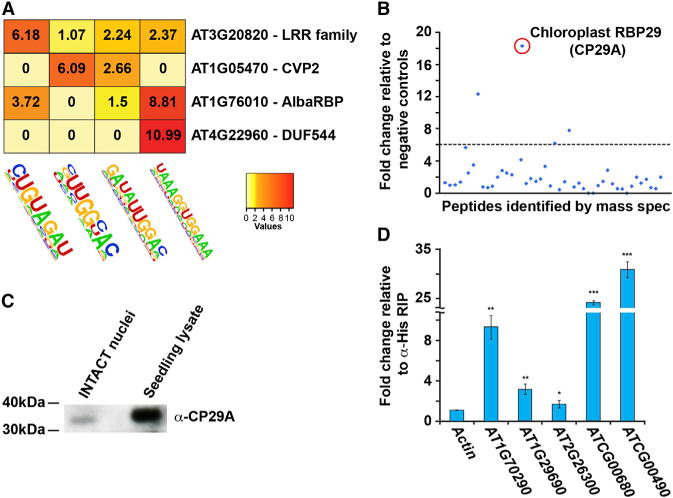
(A and B) Identification of proteins that interact with specific overrepresented sequence motifs.
(A) The fold enrichment over negative control of peptides from each designated protein identified by mass spectrometry analysis of eluates after RNA-affinity chromatography with each specified motif.
(B) The fold enrichment of peptides from proteins identified by mass spectrometry analysis after RNA-affinity chromatography with the GAN repeat motif (Figure 5A). The top candidate identified by this analysis, CP29A, is annotated and denoted with a red circle. Dotted line indicates cutoff of 6-fold enrichment.
(C) Western blot analysis of INTACT-purified nuclei and Arabidopsis 10-day-old seedling lysates using an antibody to CP29A.
(D) RT-qPCR analysis of three nuclear GAN motif-containing genes (AT1G70290, AT1G29690, and AT2G26300), two positive control chloroplast transcripts (ATCG00680 and ATCG00490 [also with motif]), and an ACTIN negative control following RIP with an α-CP29A or α-His antibody. The data is presented as the fold change in the α-CP29A relative to α-His RIP samples. Error bars, ± SD. *, **, and *** indicate p value < 0.05, < 0.001, and < 1×10−10, respectively, Fisher’s t test. See also Figure S7 and Table S4.
To confirm that CP29A could interact with both nuclear and chloroplast transcripts containing the predicted GAN repeat motif in vivo we performed RNA immunoprecipitation (RIP). We took lysates from formaldehyde-treated leaves and incubated them with either a monoclonal α-CP29A or α-His antibody (negative control) (Figure S7C) followed by RT-qPCR for three nuclear transcripts and two chloroplast RNAs as positive controls. All three nuclear and one chloroplast (ATCG00490) transcript contain the GAN repeat motif. We found that all five transcripts were significantly (all p values < 0.05) enriched > 1.5-fold in the α-CP29A compared to the α-His control RIP samples, as opposed to the ACTIN negative control (Figure 7D). Taken together, these results indicate that CP29A localizes to both the chloroplast and nucleus, and interacts with a subset of GAN repeat motif-containing transcripts in Arabidopsis, suggesting a new functionality for this plant RBP.
Conclusion
Here, we characterized the global landscapes of RNA secondary structure and RBP occupancy of the Arabidopsis nuclear transcriptome (Figure 1). We demonstrated that these data are highly reproducible, and that the identified protein-binding sites are significantly more conserved than their flanking sequences (Figure 2). Additionally, we calculated the structure score for nuclear RNAs that passed filtering criteria (see Supplemental Experimental Procedures), creating a comprehensive database of in vivo RNA secondary structure for the Arabidopsis nucleus (Figures 1C–1E). Together, these data sets provide a vast resource of RBP binding and secondary structure information for the Arabidopsis nuclear transcriptome that can inform future experiments focused on understanding posttranscriptional regulation.
Using the data generated here, we searched for patterns of global RBP binding and RNA secondary structure. The most striking association that we identified was a distinct anticorrelation between RNA secondary structure and RBP occupancy within the RNA regions that were examined (Figures 3, S5B, 4, and 5). This pattern was present when focusing on uORF and canonical translation start codons (Figures 3A and S5B), stop codons (Figure 3B), exon/intron junctions (Figure 3C), and specific RBP-binding motifs (Figures 5K–5O). Furthermore, we found that the RBP-interacting motifs identified by our study tend to be less structured when protein bound (Figures 5K–5O). Although we cannot discern causality, our findings reveal that in general RBPs bind to unstructured sequence elements in target transcripts resulting in the overall opposing patterns of these features in the Arabidopsis nucleus.
When initially examining these data we questioned whether the structure score was artificially lowered in regions of high PPS density by occlusion of the RNase through the incomplete digestion of bound RBPs. However, if this were true these regions would not be called PPSs in our initial analyses because their read levels would be artificially raised in the structure-only libraries. Furthermore, we find that the presence of PPSs is actually associated with more negative structure scores (Figures 5K and S6A). Thus, our results are likely true biological observations of decreased structure at RBP-binding sites, not an artifact of the PIP-seq methodology.
We also examined subsets of annotated alternative exons and identified unique profiles of PPS density and secondary structure in constitutive, CE, IR, and U12-type introns (Figures 4B and 4C). These profiles suggest that gross protein binding can regulate AS, while secondary structure can influence the population of proteins that occupies each region. Although it is known that RBP binding in the exon or intron can regulate AS (Chen and Manley, 2009; Simpson et al., 2010), our observations demonstrate that protein occupancy levels in regions near the splice site can differentiate subsets of alternative exons. Our observations have provided the resources for identifying these populations of proteins and specific structural features in these alternative events.
Finally, we uncovered motifs that were enriched within our PPSs and identified co-occurrences of RBP-bound instances of these sequences in functionally related transcripts (Figure 6). These findings are similar to previous observations in human cells (Silverman et al., 2014), and support a model in which RNA transcripts encoding proteins with related functions also share a set of interacting RBPs through underlying sequence motifs allowing their coregulation. Taken together, our findings suggest that both plants and humans use different groups of RBPs to allow specific sets of proteins, especially those functioning in development, stress responses, and apoptosis, to be precisely coregulated in an operon-like fashion.
EXPERIMENTAL PROCEDURES
Supplemental Experimental Procedures
Further details on the experimental procedures, high-throughput sequencing, and processing, mapping, and analysis of PIP-seq data are provided in the Supplemental Experimental Procedures.
INTACT-Purified Nuclei
Seedlings of UBQ10:NTF/ACT2p:BirA Arabidopsis thaliana ecotype Col-0 were grown for 10 days (20°C, 16 hr light/8 hr dark) before RNA-protein interactions were crosslinked in a 1% formaldehyde solution under a vacuum and subsequently quenched with 125 mM glycine. INTACT purification was then performed as previously described (Deal and Henikoff, 2010). This same ecotype of Arabidopsis was used for all analyses in this study.
PIP-seq and PPS Analysis
We used 2 million purified nuclei for each PIP-seq replicate, which was performed as previously described (Silverman et al., 2014). Read processing and alignment, PPS identification, and all other PPS analyses were done as previously described (Silverman et al., 2014).
Supplementary Material
Highlights.
Patterns of RNA secondary structure and RBP binding are anticorrelated
Alternative splice sites have distinct RBP binding and secondary structure profiles
Groups of Arabidopsis RBP-bound motifs co-occur on functionally related mRNAs
The chloroplast RBP CP29A also interacts with nuclear mRNAs
Acknowledgments
The authors thank Dr. Christian Schmitz-Linneweber for providing the α-CP29A antibody, Alsu Ibragimova for optimizing the RNA-affinity chromatography, Jennifer Nemhauser for providing the UBQ10p:NTF/ACT2p:BirA transgenic line, and members of the B.D.G. lab for helpful discussions. This work was funded by NSF grant MCB-1243947 to B.D.G. and NIGMS 5T32GM008216-26 to I.M.S.
Footnotes
ACCESSION NUMBERS
The raw and processed data for PIP-seq and total RNA sequencing from our analyses have been deposited into the NCBI Gene Expression Omnibus (GEO) database under the accession number GSE58974. All of our data (i.e., files of all identified PPSs, complete list of nuclear mRNA structure scores, etc.) can also be accessed at http://gregorylab.bio.upenn.edu/PIPseq_AtTotalNuc.
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures, four tables, and Supplemental Experimental Procedures and can be found with this article online at http://dx.doi.org/10.1016/j.molcel.2014.12.004.
AUTHOR CONTRIBUTIONS
S.J.G., S.W.F., R.B.D., and B.D.G. conceived the study and designed the experiments. S.J.G., S.W.F., D.W., N.S., A.D.L.N., M.A.B., F.D., and B.D.G. performed the experiments. S.J.G., S.W.F., I.M.S., and B.D.G. analyzed the data. S.W.F., S.J.G., I.M.S., R.B.D., and B.D.G. wrote the paper with assistance from all authors. The authors have read and approved the manuscript for publication.
References
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37:W202–W208. doi: 10.1093/nar/gkp335. Web Server issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltz AG, Munschauer M, Schwanhäusser B, Vasile A, Murakawa Y, Schueler M, Youngs N, Penfold-Brown D, Drew K, Milek M, et al. The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol Cell. 2012;46:674–690. doi: 10.1016/j.molcel.2012.05.021. [DOI] [PubMed] [Google Scholar]
- Cao J, Schneeberger K, Ossowski S, Günther T, Bender S, Fitz J, Koenig D, Lanz C, Stegle O, Lippert C, et al. Whole-genome sequencing of multiple Arabidopsis thaliana populations. Nat Genet. 2011;43:956–963. doi: 10.1038/ng.911. [DOI] [PubMed] [Google Scholar]
- Castello A, Fischer B, Eichelbaum K, Horos R, Beckmann BM, Strein C, Davey NE, Humphreys DT, Preiss T, Steinmetz LM, et al. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell. 2012;149:1393–1406. doi: 10.1016/j.cell.2012.04.031. [DOI] [PubMed] [Google Scholar]
- Cech TR, Steitz JA. The noncoding RNA revolution—trashing old rules to forge new ones. Cell. 2014;157:77–94. doi: 10.1016/j.cell.2014.03.008. [DOI] [PubMed] [Google Scholar]
- Chen M, Manley JL. Mechanisms of alternative splicing regulation: insights from molecular and genomics approaches. Nat Rev Mol Cell Biol. 2009;10:741–754. doi: 10.1038/nrm2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiou NT, Shankarling G, Lynch KW. hnRNP L and hnRNP A1 induce extended U1 snRNA interactions with an exon to repress spliceosome assembly. Mol Cell. 2013;49:972–982. doi: 10.1016/j.molcel.2012.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz JA, Westhof E. The dynamic landscapes of RNA architecture. Cell. 2009;136:604–609. doi: 10.1016/j.cell.2009.02.003. [DOI] [PubMed] [Google Scholar]
- Deal RB, Henikoff S. A simple method for gene expression and chromatin profiling of individual cell types within a tissue. Dev Cell. 2010;18:1030–1040. doi: 10.1016/j.devcel.2010.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y, Tang Y, Kwok CK, Zhang Y, Bevilacqua PC, Assmann SM. In vivo genome-wide profiling of RNA secondary structure reveals novel regulatory features. Nature. 2014;505:696–700. doi: 10.1038/nature12756. [DOI] [PubMed] [Google Scholar]
- Du Z, Zhou X, Ling Y, Zhang Z, Su Z. agriGO: a GO analysis toolkit for the agricultural community. Nucleic Acid Res. 2010;38:W64–W70. doi: 10.1093/nar/gkq310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foissac S, Sammeth M. ASTALAVISTA: dynamic and flexible analysis of alternative splicing events in custom gene datasets. Nucleic Acids Res. 2007;35:W297–W299. doi: 10.1093/nar/gkm311. Web Server issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacisuleyman E, Goff LA, Trapnell C, Williams A, Henao-Mejia J, Sun L, McClanahan P, Hendrickson DG, Sauvageau M, Kelley DR, et al. Topological organization of multichromosomal regions by the long intergenic noncoding RNA Firre. Nat Struct Mol Biol. 2014;21:198–206. doi: 10.1038/nsmb.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt AG, Xing D, Li QQ. Plant polyadenylation factors: conservation and variety in the polyadenylation complex in plants. BMC Genomics. 2012;13:641. doi: 10.1186/1471-2164-13-641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandasamy MK, McKinney EC, Meagher RB. The late pollen-specific actins in angiosperms. Plant J. 1999;18:681–691. doi: 10.1046/j.1365-313x.1999.00487.x. [DOI] [PubMed] [Google Scholar]
- Keene JD, Tenenbaum SA. Eukaryotic mRNPs may represent posttranscriptional operons. Mol Cell. 2002;9:1161–1167. doi: 10.1016/s1097-2765(02)00559-2. [DOI] [PubMed] [Google Scholar]
- Kupsch C, Ruwe H, Gusewski S, Tillich M, Small I, Schmitz-Linneweber C. Arabidopsis chloroplast RNA binding proteins CP31A and CP29A associate with large transcript pools and confer cold stress tolerance by influencing multiple chloroplast RNA processing steps. Plant Cell. 2012;24:4266–4280. doi: 10.1105/tpc.112.103002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebedeva S, Jens M, Theil K, Schwanhäusser B, Selbach M, Landthaler M, Rajewsky N. Transcriptome-wide analysis of regulatory interactions of the RNA-binding protein HuR. Mol Cell. 2011;43:340–352. doi: 10.1016/j.molcel.2011.06.008. [DOI] [PubMed] [Google Scholar]
- Li F, Zheng Q, Ryvkin P, Dragomir I, Desai Y, Aiyer S, Valladares O, Yang J, Bambina S, Sabin LR, et al. Global analysis of RNA secondary structure in two metazoans. Cell Rep. 2012a;1:69–82. doi: 10.1016/j.celrep.2011.10.002. [DOI] [PubMed] [Google Scholar]
- Li F, Zheng Q, Vandivier LE, Willmann MR, Chen Y, Gregory BD. Regulatory impact of RNA secondary structure across the Arabidopsis transcriptome. Plant Cell. 2012b;24:4346–4359. doi: 10.1105/tpc.112.104232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Jung C, Xu J, Wang H, Deng S, Bernad L, Arenas-Huertero C, Chua NH. Genome-wide analysis uncovers regulation of long intergenic noncoding RNAs in Arabidopsis. Plant Cell. 2012;24:4333–4345. doi: 10.1105/tpc.112.102855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquez Y, Brown JWS, Simpson C, Barta A, Kalyna M. Transcriptome survey reveals increased complexity of the alternative splicing landscape in Arabidopsis. Genome Res. 2012;22:1184–1195. doi: 10.1101/gr.134106.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raker VA, Mironov AA, Gelfand MS, Pervouchine DD. Modulation of alternative splicing by long-range RNA structures in Drosophila. Nucleic Acids Res. 2009;37:4533–4544. doi: 10.1093/nar/gkp407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouskin S, Zubradt M, Washietl S, Kellis M, Weissman JS. Genome-wide probing of RNA structure reveals active unfolding of mRNA structures in vivo. Nature. 2014;505:701–705. doi: 10.1038/nature12894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauvageau M, Goff LA, Lodato S, Bonev B, Groff AF, Gerhardinger C, Sanchez-Gomez DB, Hacisuleyman E, Li E, Spence M, et al. Multiple knockout mouse models reveal lincRNAs are required for life and brain development. eLife. 2013;2:e01749. doi: 10.7554/eLife.01749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherstnev A, Duc C, Cole C, Zacharaki V, Hornyik C, Ozsolak F, Milos PM, Barton GJ, Simpson GG. Direct sequencing of Arabidopsis thaliana RNA reveals patterns of cleavage and polyadenylation. Nat Struct Mol Biol. 2012;19:845–852. doi: 10.1038/nsmb.2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman IM, Li F, Alexander A, Goff L, Trapnell C, Rinn JL, Gregory BD. RNase-mediated protein footprint sequencing reveals protein-binding sites throughout the human transcriptome. Genome Biol. 2014;15:R3. doi: 10.1186/gb-2014-15-1-r3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson CG, Manthri S, Raczynska KD, Kalyna M, Lewandowska D, Kusenda B, Maronova M, Szweykowska-Kulinska Z, Jarmolowski A, Barta A, Brown JW. Regulation of plant gene expression by alternative splicing. Biochem Soc Trans. 2010;38:667–671. doi: 10.1042/BST0380667. [DOI] [PubMed] [Google Scholar]
- Talkish J, May G, Lin Y, Woolford JL, Jr, McManus CJ. Modseq: high-throughput sequencing for chemical probing of RNA structure. RNA. 2014;20:713–720. doi: 10.1261/rna.042218.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenenbaum SA, Christiansen J, Nielsen H. The post-transcriptional operon. Methods Mol Biol. 2011;703:237–245. doi: 10.1007/978-1-59745-248-9_16. [DOI] [PubMed] [Google Scholar]
- Ule J, Jensen KB, Ruggiu M, Mele A, Ule A, Darnell RB. CLIP identifies Nova-regulated RNA networks in the brain. Science. 2003;302:1212–1215. doi: 10.1126/science.1090095. [DOI] [PubMed] [Google Scholar]
- von Arnim AG, Jia Q, Vaughn JN. Regulation of plant translation by upstream open reading frames. Plant Sci. 2014;214:1–12. doi: 10.1016/j.plantsci.2013.09.006. [DOI] [PubMed] [Google Scholar]
- Wahl MC, Will CL, Lührmann R. The spliceosome: design principles of a dynamic RNP machine. Cell. 2009;136:701–718. doi: 10.1016/j.cell.2009.02.009. [DOI] [PubMed] [Google Scholar]
- Wu X, Liu M, Downie B, Liang C, Ji G, Li QQ, Hunt AG. Genome-wide landscape of polyadenylation in Arabidopsis provides evidence for extensive alternative polyadenylation. Proc Natl Acad Sci USA. 2011;108:12533–12538. doi: 10.1073/pnas.1019732108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HP, Su YS, Chen HC, Chen YR, Wu CC, Lin WD, Tu SL. Genome-wide analysis of light-regulated alternative splicing mediated by photoreceptors in Physcomitrella patens. Genome Biol. 2014;15:R10. doi: 10.1186/gb-2014-15-1-r10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye LH, Li YQ, Fukami-Kobayashi K, Go M, Konishi T, Watanabe A, Sugiura M. Diversity of a ribonucleoprotein family in tobacco chloroplasts: two new chloroplast ribonucleoproteins and a phylogenetic tree of ten chloroplast RNA-binding domains. Nucleic Acids Res. 1991;19:6485–6490. doi: 10.1093/nar/19.23.6485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Q, Ryvkin P, Li F, Dragomir I, Valladares O, Yang J, Cao K, Wang LS, Gregory BD. Genome-wide double-stranded RNA sequencing reveals the functional significance of base-paired RNAs in Arabidopsis. PLoS Genet. 2010;6:e1001141. doi: 10.1371/journal.pgen.1001141. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.