

Abstract
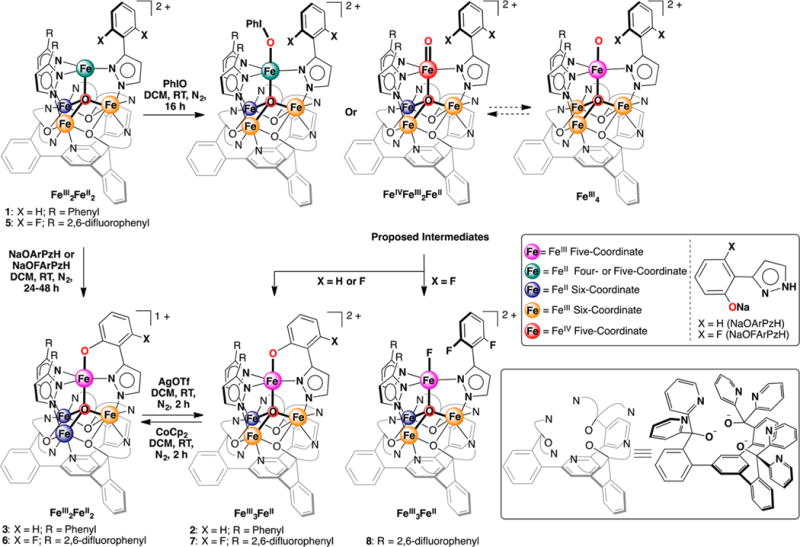
Herein we report the intramolecular arene C–H and C–F bond oxygenation by tetranuclear iron complexes. Treatment of [LFe3(PhPz)3OFe][OTf]2 (1) or its fluorinated analog [LFe3(F2ArPz)3OFe][OTf]2 (5) with iodosobenzene results in the regioselective hydroxylation of a bridging pyrazolate ligand, converting a C–H or C–F bond into a C–O bond. The observed reactivity suggests the formation of terminal and reactive Fe-oxo intermediates. With the possibility of intramolecular electron transfer within clusters in 1 and 5, different reaction pathways (FeIV-oxo vs FeIII-oxo) might be responsible for the observed arene hydroxylation.
Terminal metal-oxo species are proposed intermediates in a variety of biological transformations including water oxidation, dioxygen reduction, and C–H bond functionalization.1 While the synthesis and characterization of complexes modeling enzyme active sites that display terminal metal-oxo moieties have seen remarkable development,2 multinuclear analogs are significantly less studied.3 Except for synthetic dinuclear transition-metal complexes, accessing reactive terminal metal-oxo moieties on well-defined multinuclear iron complexes is rare.4,5 Nonetheless high-oxidation-state metal-oxo species on multimetallic scaffolds are desirable for systematic structure function studies in order to understand their reactivity.
We recently reported the rational synthesis of a family of tetranuclear iron clusters that are site-differentiated with an iron center in trigonal geometry and three metal centers six-coordinate.6 The site-differentiation allowed nitric oxide binding at the tripodal center and redox chemistry localized at the remaining sites, remotely tuning the degree of NO activation.6 Herein, we employ this platform to target terminal iron-oxo moieties on a metal cluster. Addition of oxygen atom-transfer reagents to the metal clusters leads to the regioselective conversion of ligand C–H and C–F bonds into C–O bonds, consistent with the formation of a terminal iron-oxo species as reactive intermediate. To the best of our knowledge, this is one of the very few examples suggesting the formation of highly reactive terminal oxo moieties on a multinuclear iron cluster.
High-oxidation-state iron–oxo complexes were targeted from the previously synthesized [LFe3(PhPz)3OFe][OTf]2 (1, Scheme 1; PhPz = 3-phenylpyrazolate).6 Treating 1 with iodosobenzene (PhIO; 1.1 equiv) resulted in significant changes in the 1H NMR spectrum and is consistent with the formation of an asymmetric species (2: Figure S19). Analysis of the reaction mixture by electrospray ionization mass spectrometry (ESI-MS) shows a shift of the peak for [LFe3(PhPz)3OFe]2+ (m/z = 762.1) to a peak consistent with [LFe3(PhPz)3OFe(O)]2+ (m/z = 769.6), indicating the incorporation of an oxygen atom together with the loss of an H atom (Figures S34–S35). Moreover, with tetrabutylammonium periodate (nBu4NIO4), similar results were obtained (Figure S22).
Scheme 1.
Synthesis of Tetranuclear Iron Complexes
Single crystal X-ray diffraction (XRD) analysis of 2 revealed the regioselective hydroxylation of the bridging pyrazolate ligand (Figure 1). The coordination environment around the apical iron center (Fe4) is distorted trigonal bipyramidal (τ5=0.72),7 with the μ4-oxido (O1) and phenoxido (O40) in the axial positions and N14|N24|N34 spanning the trigonal plane. The apical iron center is connected to the Fecore (Fecore = Fe1–Fe3) through the μ4-oxido O1, resulting in a tetrahedral [Fe4(μ4-O)] motif. In previously reported complexes, the Fecore–O1 bond distances are characteristic of the presence of FeII or FeIII centers.6,8 However, due to crystallographic disorder, we could not assign the iron oxidation states with certainty. Nevertheless, for Fe4, the Fe4–O1 distance of 1.931(4) Å is shorter compared to those in previously reported complexes 1 (1.971(2) Å) and [LFe3(PhPz)3OFe][OTf]3 (2.031(2) Å), suggesting a more oxidized iron center (FeIII) at the apical position (Fe4). The iron oxidation states in 2 were assigned by using zero-field 57Fe Mössbauer spectroscopy and by independent synthesis of 2 (vide infra). The Mössbauer spectrum of 2 (Figures 2 and S38) features four quadrupole doublets in a 1:1:1:1 ratio. The quadrupole doublet at δ = 1.17 mm/s (|ΔEQ| = 2.96 mm/s) is consistent with a high-spin ferrous center in the triiron core (Figure 2; blue trace).9 The quadrupole doublets with identical isomer shifts of δ = 0.47 mm/s and quadrupole splittings of |ΔEQ| = 0.56 and 0.99 mm/s, are indicative of the presence of two high-spin ferric ions (Figure 2; orange traces).9 The observed Mössbauer parameters (Table S4) are similar to our previously reported triiron-oxo/hydroxo clusters8 and to our, and other reported, complexes featuring a discrete [Fe4(μ4-O)] core.6,8,10 Based on these parameters the oxidation state of the triiron core in 2 is thus assigned as [FeIIFeIII2].
Figure 1.
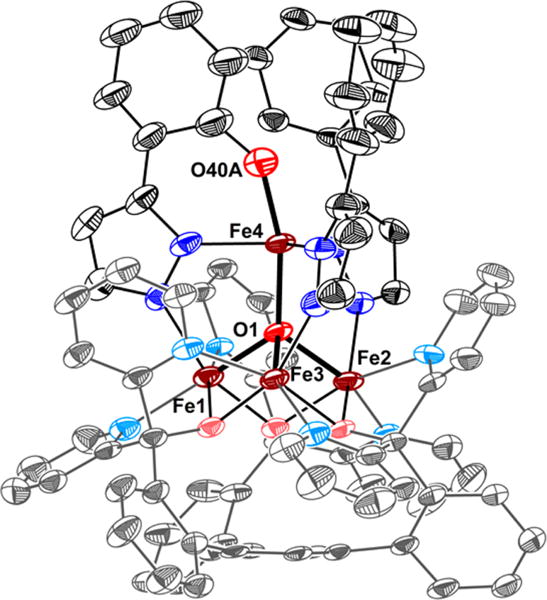
Crystal structure of [LFe3(PhPz)2(OArPz)OFe][OTf]2 (2). Thermal ellipsoids are shown at the 50% probability level. Hydrogen atoms, outer sphere counter ions, and co-crystallized solvents molecules are not shown for clarity.
Figure 2.
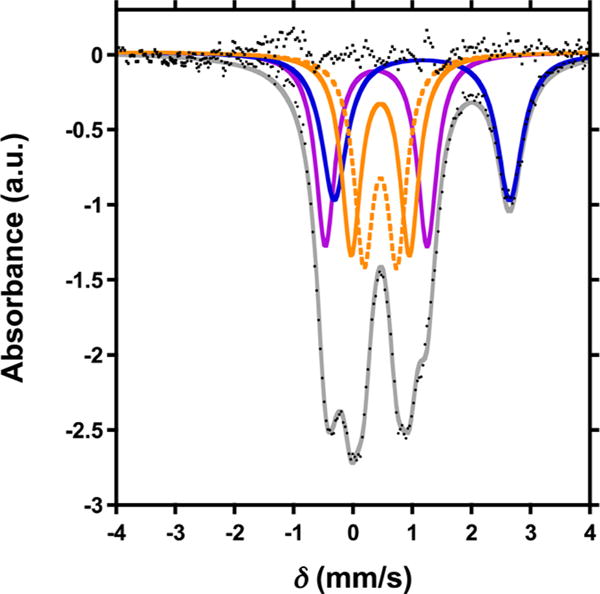
Zero-field 57Fe Mössbauer spectra (80 K) of [LFe3(PhPz)2(OArPz)OFe][OTf]2 (2).
The remaining quadrupole doublet at δ = 0.40 mm/s (|ΔEQ| = 1.72 mm/s) is assigned to the apical iron Fe4, as a FeIII metal center (Figure 2; purple trace).9 Such values are consistent with high-spin FeIII.9 A detailed discussion on the assignment of the quadrupole doublets in the Mössbauer spectra is presented in the Supporting Information. Further studies are required to determine the exact spin-states in complexes 2–8.
The assignment of the apical iron as a FeIII metal center suggests an overall oxidation state in 2 as [LFeIIFeIII2 (PhPz)2 (OArPz)OFeIII ][OTf]2. However, an assignment of [LFeIIFeIII2(PhPz)2(HOArPz)OFeII ][OTf]2 is also plausible, where the apical iron is FeII and the oxygenated ligand is a phenol rather than a phenoxide. In order to rule out any ambiguity, we independently synthesized 2 from 1. Treatment of 1 with NaOArPzH leads to complex 3 by protonolysis of a PhPz ligand. Oxidation of 3 with AgOTf provides 2 based on 1H NMR spectroscopy (Scheme 1; Figure S21). XRD analysis of 3 shows that Fecore–O1 bond distances of 2.154(3) Å (Fe1–O1), 2.119(3) Å (Fe2–O1), and 1.938(3) Å (Fe3–O1) are consistent with an assignment of a [FeII2FeIII] triiron core (Table S1; Figure S45). Based on charge balance with 1, from which 3 was synthesized, the apical iron Fe4 is a FeIII metal center. The isomer shift and quadrupole splitting (δ = 0.46 mm/s; |ΔEQ| = 1.84 mm/s) for the apical iron Fe4 (Figure S39, Table S4) is similar to 2 and supports the assignment of the apical iron center as FeIII. Furthermore, the short Fe4–O1 (1.867(3) Å) and Fe4–O40 (1.911(4) Å) distances are in agreement to those found in complex 2 (Table S1) and are in line with other reported Fe(III)–phenoxide complexes.11 Taking these data into account, the formal iron oxidation states are assigned as [LFeII2FeIII(PhPz)2(OArPz)-OFeIII][OTf] (3) and [LFeIIFeIII2(PhPz)2(OArPz)OFeIII]-[OTf]2 (2). Thus, the isolated product 2 shows overall C–H bond oxygenation with formal loss of a hydrogen atom.
Similar hydroxylation pathways have been observed in other monometallic nonheme iron complexes, where FeIV-oxo moieties are the (proposed) reactive intermediates.11,12 The observed intramolecular hydroxylation in 1 thus hints at the formation of a high-valent FeIV-oxo as intermediate, although other reaction pathways cannot be excluded, including hydroxylation from a PhIO adduct (Scheme 1).13 Further studies might reveal more mechanistic details of this process. Nonetheless, the Fe-oxo species implicated here is unusual in that the tetrairon cluster, a multinuclear motif with four redox-active metal centers, supports it.
In order to prolong the lifetime of the putative FeIV-oxo species, by avoiding intramolecular C–H bond hydroxylation, we synthesized the fluorinated analog of 1; [LFe3(F2ArPz)3OFe][OTf]2 (5; F2ArPz = 3-(2,6-difluorophenyl)pyrazolate, Figure S46). Treatment of 5 with PhIO (1.1 equiv) results in a mixture of at least two species: (i) [LFe3(F2ArPz)2(FArPz)OFe(O)]2+ (7; m/z = 814.6) and (ii) [LFe3(F2ArPz)3OFe(F)]2+ (8; m/z = 825.6), as judged by ESI-MS and 1H NMR (Figures S26 and S36–S37). Using nBu4NIO4 as an oxygen atom-transfer reagent resulted only in the formation of 7 (Figure S30). These data are consistent with incorporation of an oxygen atom with loss of a fluorine atom, suggesting C–F to C–O bond conversion (Scheme 1).
In order to establish the identity of species 7 and 8, they were synthesized independently (Scheme 1). Treating 5 with NaOFArPzH results in the formation of a new species, [LFe3(F2ArPz)2(OFArPz)OFe][OTf] (6; Figure S28). Subsequent oxidation of 6 with silver triflate (AgOTf; 1.0 equiv) produced a species with 1H NMR characteristics identical to species 7 (Figures S27 and S29). The crystal structure of 7 (Figure 3A) is nearly identical to 2. The coordination environment around the apical iron center is a distorted trigonal bipyramid (τ5 = 0.89).7 The Fecore–O1 bond distances of 2.198(4) Å (Fe1–O1), 1.978(4) Å (Fe2–O1), and 1.986(4) Å (Fe3–O1) are consistent with an oxidation state of [FeIIFeIII2] for the triiron core. Similar to 2, the apical iron is assigned as FeIII based on the short Fe4–O1 bond distance (1.914(3) Å, Table S1) and the zero-field 57Fe Mössbauer parameters (Figure S42 and Table S4).6,8,10 The byproduct of C–F bond oxygenation (8) was independently synthesized from complex 5 and xenon difluoride (XeF2; 1.0 equiv). Species 8 was identified as [LFeIIFeIII2(F2ArPz)3FeIII(F)][OTf]2 by XRD analysis (Figure 3B). The Mössbauer spectra and parameters resemble those reported for 2 and 7 (Figure S43 and Table S4). The independent synthesis of 7 and 8 demonstrates the regioselective conversion of the pyrazolate C–F bond into a C–O bond, by a putative Fe-oxo species. The overall reaction is balanced, with the oxygenated product losing fluoride and formally oxidizing by one electron the cluster that accepts the fluoride ligand.
Figure 3.
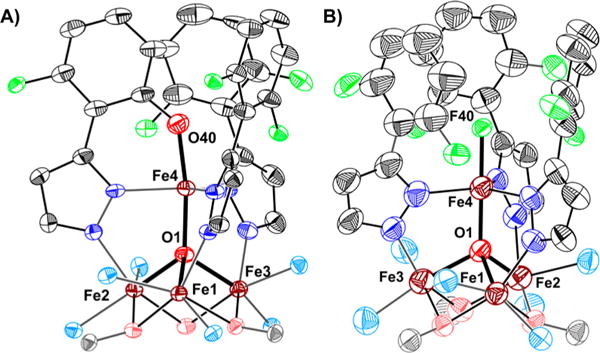
Crystal structure of (A) [LFe3(F2ArPz)2(OFArPz)OFe]-[OTf]2 (7) and (B) [LFe3(F2ArPz)3OFe(F)][OTf]2 (8). Thermal ellipsoids are shown at the 50% probability level. Hydrogen atoms, parts of the ligand L, outer sphere counter ions, and co-crystallized solvents molecules are not shown for clarity.
While C–H bond hydroxylation has ample precedent with Fe-oxo species, C–F hydroxylation is very rare. This reactivity has been recently observed with a nonheme iron complex where treatment of with oxygen atom-transfer reagents resulted in intramolecular C–F bond hydroxylation.14 That reactivity resulted from a detectable FeIV-oxo intermediate. Recently, a copper facilitated C–F bond oxygenation was reported, proposed to involve a high-oxidation-state CuIII2(O)2 species.15 In both examples, the orientations of the C–F bond and the aromatic π-system were deemed important for the observed reactivity. Although we have not observed the proposed terminal Fe-oxo intermediate, such species are in line with precedent for both C–H and C–F bond functionalization.11,12,14 The present multinuclear clusters also present the distinct mechanistic possibility of internal electron transfer, leading to a FeIII-oxo species, expected to be very basic.16 Recent studies have shown that the reactivity of such intermediates (FeIV-oxo vs FeIII-oxo) can be very different.17 A FeIII-oxo could undergo C–F activation via a nucleophilic attack, although typically high-oxidation-state metal-oxo species are proposed to perform arene hydroxylation via electrophilic mechanisms.11c,12f,14,18 Additionally, the spin state of the Fe-oxo species, shown to influence reactivity, can be affected in the reported clusters by metal–metal interactions. Current efforts are directed toward elucidating the mechanism of C–H and C–F bond activation from 1 and 5.
In summary, we have demonstrated the intramolecular oxygenation of C–H and C–F bonds upon treatment of tetranuclear iron complexes 1 and 5 with oxygen atom-transfer reagents. These processes suggest the involvement of a high-oxidation-state Fe-oxo, which is rare on well-defined multinuclear scaffolds. The possibility of intramolecular electron transfer offers several potential mechanistic pathways for bond activation engendered by the presence of proximal spin- and redox-active metals.
Supplementary Material
Acknowledgments
This research was supported by the NIH (R01-GM102687A). T.A. is grateful for Sloan, Dreyfus, and Cottrell fellowships. T.A. and G.de R. are grateful for a Camille & Henry Dreyfus Environmental Chemistry Fellowship. We thank Lawrence M. Henling for assistance with crystallography.
Footnotes
Supporting Information
- Crystallographic data (CIF)
- General considerations, physical methods, and synthetic procedures. Figures S1–S48, Tables S1–S4 (PDF)
Notes
The authors declare no competing financial interest.
References
- 1.(a) Solomon EI, Heppner DE, Johnston EM, Ginsbach JW, Cirera J, Qayyum M, Kieber-Emmons MT, Kjaergaard CH, Hadt RG, Tian L. Chem Rev. 2014;114:3659. doi: 10.1021/cr400327t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yano J, Yachandra V. Chem Rev. 2014;114:4175. doi: 10.1021/cr4004874. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Poulos TL. Chem Rev. 2014;114:3919. doi: 10.1021/cr400415k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Tinberg CE, Lippard SJ. Acc Chem Res. 2011;44:280. doi: 10.1021/ar1001473. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Costas M, Mehn MP, Jensen MP, Que L. Chem Rev. 2004;104:939. doi: 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]; (f) Feig AL, Lippard SJ. Chem Rev. 1994;94:759. [Google Scholar]
- 2.(a) Boaz NC, Bell SR, Groves JT. J Am Chem Soc. 2015;137:2875. doi: 10.1021/ja508759t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Baglia RA, Prokop-Prigge KA, Neu HM, Siegler MA, Goldberg DP. J Am Chem Soc. 2015;137:10874. doi: 10.1021/jacs.5b05142. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Taguchi T, Gupta R, Lassalle-Kaiser B, Boyce DW, Yachandra VK, Tolman WB, Yano J, Hendrich MP, Borovik AS. J Am Chem Soc. 2012;134:1996. doi: 10.1021/ja210957u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Park YJ, Ziller JW, Borovik AS. J Am Chem Soc. 2011;133:9258. doi: 10.1021/ja203458d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Lacy DC, Gupta R, Stone KL, Greaves J, Ziller JW, Hendrich MP, Borovik AS. J Am Chem Soc. 2010;132:12188. doi: 10.1021/ja1047818. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) McGown AJ, Kerber WD, Fujii H, Goldberg DP. J Am Chem Soc. 2009;131:8040. doi: 10.1021/ja809183z. [DOI] [PubMed] [Google Scholar]; (g) Rohde J-U, In J-H, Lim MH, Brennessel WW, Bukowski MR, Stubna A, Münck E, Nam W, Que L. Science. 2003;299:1037. doi: 10.1126/science.299.5609.1037. [DOI] [PubMed] [Google Scholar]; (h) MacBeth CE, Golombek AP, Young VG, Yang C, Kuczera K, Hendrich MP, Borovik AS. Science. 2000;289:938. doi: 10.1126/science.289.5481.938. [DOI] [PubMed] [Google Scholar]; (i) Groves JT, Haushalter RC, Nakamura M, Nemo TE, Evans BJ. J Am Chem Soc. 1981;103:2884. [Google Scholar]; (j) Puri M, Que L. Acc Chem Res. 2015;48:2443. doi: 10.1021/acs.accounts.5b00244. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Nam W. Acc Chem Res. 2015;48:2415. doi: 10.1021/acs.accounts.5b00218. [DOI] [PubMed] [Google Scholar]; (l) Cook SA, Borovik AS. Acc Chem Res. 2015;48:2407. doi: 10.1021/acs.accounts.5b00212. [DOI] [PMC free article] [PubMed] [Google Scholar]; (m) McDonald AR, Que L., Jr Coord Chem Rev. 2013;257:414. [Google Scholar]; (n) Hohenberger J, Ray K, Meyer K. Nat Commun. 2012;3:720. doi: 10.1038/ncomms1718. [DOI] [PubMed] [Google Scholar]; (o) Borovik AS. Chem Soc Rev. 2011;40:1870. doi: 10.1039/c0cs00165a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (p) Que L. Acc Chem Res. 2007;40:493. doi: 10.1021/ar700024g. [DOI] [PubMed] [Google Scholar]; (q) Nam W. Acc Chem Res. 2007;40:522. doi: 10.1021/ar700027f. [DOI] [PubMed] [Google Scholar]
- 3.(a) Khenkin AM, Kumar D, Shaik S, Neumann R. J Am Chem Soc. 2006;128:15451. doi: 10.1021/ja0638455. [DOI] [PubMed] [Google Scholar]; (b) de Visser SP, Kumar D, Neumann R, Shaik S. Angew Chem Int Ed. 2004;43:5661. doi: 10.1002/anie.200453867. [DOI] [PubMed] [Google Scholar]
- 4.(a) Stoian SA, Xue G, Bominaar EL, Que L, Münck E. J Am Chem Soc. 2014;136:1545. doi: 10.1021/ja411376u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kodera M, Kawahara Y, Hitomi Y, Nomura T, Ogura T, Kobayashi Y. J Am Chem Soc. 2012;134:13236. doi: 10.1021/ja306089q. [DOI] [PubMed] [Google Scholar]; (c) Do LH, Xue G, Que L, Lippard SJ. Inorg Chem. 2012;51:2393. doi: 10.1021/ic202379b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Kudrik EV, Afanasiev P, Alvarez LX, Dubourdeaux P, Clémancey M, Latour J-M, Blondin G, Bouchu D, Albrieux F, Nefedov SE, Sorokin AB. Nat Chem. 2012;4:1024. doi: 10.1038/nchem.1471. [DOI] [PubMed] [Google Scholar]; (e) Xue G, De Hont R, Münck E, Que L. Nat Chem. 2010;2:400. doi: 10.1038/nchem.586. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Xue G, Fiedler AT, Martinho M, Münck E, Que L. Proc Natl Acad Sci U S A. 2008;105:20615. [Google Scholar]
- 5.For some recent iron clusters studied for other applications, including their redox properties, catalysis, and small molecule reactivity, see:; (a) Hernández Sánchez R, Zheng S-L, Betley TA. J Am Chem Soc. 2015;137:11126. doi: 10.1021/jacs.5b06453. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nguyen AD, Rail MD, Shanmugam M, Fettinger JC, Berben LA. Inorg Chem. 2013;52:12847. doi: 10.1021/ic4023882. [DOI] [PubMed] [Google Scholar]; (c) Lee Y, Sloane FT, Blondin G, Abboud KA, García-Serres R, Murray LJ. Angew Chem, Int Ed. 2015;54:1499. doi: 10.1002/anie.201409676. [DOI] [PubMed] [Google Scholar]
- 6.de Ruiter G, Thompson NB, Lionetti D, Agapie T. J Am Chem Soc. 2015;137:14094. doi: 10.1021/jacs.5b07397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Addison AW, Rao TN, Reedijk J, van Rijn J, Verschoor G. C J Chem Soc, Dalton Trans. 1984:1349. [Google Scholar]
- 8.Herbert DE, Lionetti D, Rittle J, Agapie T. J Am Chem Soc. 2013;135:19075. doi: 10.1021/ja4104974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gutlich P, Eckhard B, Trautwein AX. Mössbauer Spectroscopy and Transition Metal Chemistry. Springer; Berlin: 2011. [Google Scholar]
- 10.(a) Sutradhar M, Carrella LM, Rentschler E. Eur J Inorg Chem. 2012;2012:4273. [Google Scholar]; (b) Murali M, Nayak S, Costa JS, Ribas J, Mutikainen I, Turpeinen U, Clémancey M, Garcia-Serres R, Latour J-M, Gamez P, Blondin G, Reedijk J. Inorg Chem. 2010;49:2427. doi: 10.1021/ic902360x. [DOI] [PubMed] [Google Scholar]
- 11.(a) Mehn MP, Fujisawa K, Hegg EL, Que L. J Am Chem Soc. 2003;125:7828. doi: 10.1021/ja028867f. [DOI] [PubMed] [Google Scholar]; (b) Harman WH, Chang CJ. J Am Chem Soc. 2007;129:15128. doi: 10.1021/ja076842g. [DOI] [PubMed] [Google Scholar]; (c) Sahu S, Widger LR, Quesne MG, de Visser SP, Matsumura H, Moënne-Loccoz P, Siegler MA, Goldberg DP. J Am Chem Soc. 2013;135:10590. doi: 10.1021/ja402688t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Cook SA, Ziller JW, Borovik AS. Inorg Chem. 2014;53:11029. doi: 10.1021/ic501531g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bigi JP, Harman WH, Lassalle-Kaiser B, Robles DM, Stich TA, Yano J, Britt RD, Chang CJ. J Am Chem Soc. 2012;134:1536. doi: 10.1021/ja207048h. [DOI] [PubMed] [Google Scholar]; (c) England J, Guo Y, Farquhar ER, Young VG, Jr, Münck E, Que L., Jr J Am Chem Soc. 2010;132:8635. doi: 10.1021/ja100366c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) England J, Martinho M, Farquhar ER, Frisch JR, Bominaar EL, Münck E, Que L. Angew Chem, Int Ed. 2009;48:3622. doi: 10.1002/anie.200900863. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Nielsen A, Larsen FB, Bond AD, McKenzie CJ. Angew Chem, Int Ed. 2006;45:1602. doi: 10.1002/anie.200502656. [DOI] [PubMed] [Google Scholar]; (f) Jensen MP, Lange SJ, Mehn MP, Que EL, Que L. J Am Chem Soc. 2003;125:2113. doi: 10.1021/ja028478l. [DOI] [PubMed] [Google Scholar]; (g) Mekmouche Y, Ménage S, Toia-Duboc C, Fontecave M, Galey J-B, Lebrun C, Pécaut J. Angew Chem, Int Ed. 2001;40:949. [PubMed] [Google Scholar]; (h) Hegg EL, Ho RYN, Que L. J Am Chem Soc. 1999;121:1972. [Google Scholar]
- 13.(a) Collman JP, Chien AS, Eberspacher TA, Brauman JI. J Am Chem Soc. 2000;122:11098. [Google Scholar]; (b) Hong S, Wang B, Seo MS, Lee Y-M, Kim MJ, Kim HR, Ogura T, Garcia-Serres R, Clémancey M, Latour J-M, Nam W. Angew Chem, Int Ed. 2014;53:6388. doi: 10.1002/anie.201402537. [DOI] [PubMed] [Google Scholar]
- 14.Sahu S, Quesne MG, Davies CG, Dürr M, Ivanović-Burmazović I, Siegler MA, Jameson GNL, de Visser SP, Goldberg DP. J Am Chem Soc. 2014;136:13542. doi: 10.1021/ja507346t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Serrano-Plana J, Garcia-Bosch I, Miyake R, Costas M, Company A. Angew Chem, Int Ed. 2014;53:9608. doi: 10.1002/anie.201405060. [DOI] [PubMed] [Google Scholar]
- 16.Gupta R, Borovik AS. J Am Chem Soc. 2003;125:13234. doi: 10.1021/ja030149l. [DOI] [PubMed] [Google Scholar]
- 17.Usharani D, Lacy DC, Borovik AS, Shaik S. J Am Chem Soc. 2013;135:17090. doi: 10.1021/ja408073m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.(a) Ansari A, Kaushik A, Rajaraman G. J Am Chem Soc. 2013;135:4235. doi: 10.1021/ja307077f. [DOI] [PubMed] [Google Scholar]; (b) de Visser SP, Oh K, Han A-R, Nam W. Inorg Chem. 2007;46:4632. doi: 10.1021/ic700462h. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.