

Abstract
Mast cells are critical in the pathogenesis of allergic disease due to the release of preformed and newly synthesized mediators, yet the mechanisms controlling mast cell activation are not well understood. Members of the tetraspanin family are recently emerging as modulators of FcεRI-mediated mast cell activation; however, mechanistic understanding of their function is currently lacking. The tetraspanin CD151 is a poorly understood member of this family and is specifically induced on mouse and human mast cells upon FcεRI aggregation but its functional effects are unknown. In this study, we show that CD151 deficiency significantly exacerbates the IgE-mediated late phase inflammation in a murine model of passive cutaneous anaphylaxis. Ex vivo, FcεRI stimulation of bone marrow–derived mast cells from CD151−/− mice resulted in significantly enhanced expression of proinflammatory cytokines IL-4, IL-13, and TNF-α compared with wild-type controls. However, FcεRI -induced mast cell degranulation was unaffected. At the molecular signaling level, CD151 selectively regulated IgE-induced activation of ERK1/2 and PI3K, associated with cytokine production, but had no effect on the phospholipase Cγ1 signaling, associated with degranulation. Collectively, our data indicate that CD151 exerts negative regulation over IgE-induced late phase responses and cytokine production in mast cells.
The high-affinity receptor for IgE (FcεRI) is a principal mast cell receptor mediating immune responses in allergic diseases (1). Crosslinking of IgE-bound FcεRI by Ags activates downstream signal transduction pathways, resulting in mast cell degranulation and de novo synthesis of cytokines (2–5). Proximal signaling through FcεRI involves phosphorylation of ITAMs within the FcεRI β and γ subunits by the Src-family protein tyrosine kinases Lyn, spleen tyrosine kinase (Syk), and Fyn (6, 7). In particular, activation of Syk is indispensable for FcεRI-mediated mast cell activation (5). This tyrosine kinase signaling induces two principal downstream signaling cascades: the phospholipase Cγ1 (PLCγ1)–protein kinase C (PKC)–Ca2+ cascade, which is required for degranulation and the release of preformed mediators stored in the mast cell’s cytoplasmic granules, and the Ras-Raf1-ERK1/2 cascade, which is critical for de novo synthesis of cytokines (7). Additionally, there are complementary pathways for amplification and maintenance of degranulation and cytokine production. The PI3K-dependent complementary pathway involved in degranulation is mediated via the recruitment of Btk kinase, as well as subsequent amplification and maintenance of PLCγ1-mediated latent calcium signals. Amplification of cytokine/chemokine production is regulated by PI3K via an independent pathway mediated by PDK1 and Akt signaling (8). The degranulation event is crucial for immediate-type allergic reactions, whereas mast cell–mediated late phase reactions and IgE-induced chronic allergic inflammatory processes are mainly dependent on the de novo production of inflammatory mediators (9, 10). At the same time, receptors bearing ITIM and ITAM motifs, protein tyrosine kinases, protein and lipid phosphatases, adaptors, and ubiquitin ligases provide a diverse regulatory network to achieve the desired response and limit a persistent or excessive “outside–in” signaling for mast cell activation (11–20).
Members of the tetraspanin family are classically recognized as “passive” facilitators that function as scaffolds in the assembly of signaling complexes at the cell membrane (21). Only recently, tetraspanins have started to emerge as “active” signaling molecules modulating outside–in signals for cellular activation. For example, tetraspanin CD9 negatively regulates LPS-induced macrophage activation and lung inflammation (22). It is also reported that macrophages from CD9 and CD81 null or CD9/CD81 double knockout mice show enhanced in vitro formation of multinucleated giant cells, which are known to contribute to inflammatory tissue damage through increased secretion of matrix metalloproteinases in vivo (23). In B cells, the tetraspanin CD37 has been shown to possess inhibitory functions upon ligation with an anti–CD37 small modular immunopharmaceutical (24). In fibroblasts, CD151 has been reported to negatively regulate the adhesion-dependent activation of Ras (25).
Mast cells constitutively express several members of the tetraspanin family, although the function of these molecules in mast cell FcεRI-mediated signaling is largely unknown (26). Interaction of tetraspanins with FcεRI in mast cells has been demonstrated in two previous studies using the RBL-2H3 mast cell line (27, 28). In both studies, Abs against the tetraspanins CD63 or CD81 inhibited in vitro and in vivo FcεRI-mediated mast cell degranulation, without affecting FcεRI-mediated Ca2+ mobilization or total tyrosine phosphorylation levels (27, 28). CD63 is a diagnostic marker in allergic diseases (29, 30), and the granular isoform of CD63 has also been reported as a molecular marker of degranulated human mast cells (31). Recently, it was demonstrated that the tetraspanin CD63 is required for IgE-mediated mast cell degranulation and anaphylactic response in mice, although the role of CD63 in the mechanisms that regulate degranulation was not defined (32). Tetraspanin CD9 has been recently reported as a regulator of mast cells chemotaxis, where aggregation of CD9 blocked Ag- and IL-13–induced chemotaxis of bone marrow–derived mast cells (BMMCs) (33). In contrast to other tetraspanins, CD151 was reported to be specifically induced upon IgE/Ag crosslinking of FcεRI receptors in umbilical cord–derived human cells (34). However, the functional significance of expression of this tetraspanin in mast cells is not known.
In this study, we used CD151−/− mice to investigate the role of CD151 in the regulation of IgE-dependent activation and effector functions of mast cells. Ex vivo, we demonstrated that deficiency of CD151 in BMMCs significantly enhanced FcεRI-mediated de novo synthesis of multiple cytokines without affecting mast cell degranulation or altering surface expression levels of FcεRI and c-Kit. In vivo, CD151 deficiency led to exaggerated IgE-mediated late phase responses in a model of passive cutaneous anaphylaxis (PCA). Mechanistically, we found CD151 to specifically target the late phase of mast cell activation via negative control over the canonical Ras/ERK1/2 and the complementary PI3K pathway for cytokine production. CD151 deficiency did not affect the upstream activation of PLCγ1, which regulates calcium flux and mast cell degranulation. Collectively, our data indicate that CD151 selectively exerts negative regulation over IgE-induced late phase allergic responses and de novo cytokine production by activated mast cells.
Materials and Methods
Reagents
Immunoblotting Ab against CD151 (H-80) was purchased from Santa Cruz Biotechnology (Dallas, TX). Alexa Fluor 568–conjugated anti-rat IgG secondary Ab was purchased from Invitrogen (Carlsbad, CA). FITC-conjugated anti-FcεRIα (MAR-1) Ab and cell stimulation mixture (PMA [40.5 μM] and ionomycin [670 μM] in ethanol [500×]) were from eBioscience (San Diego, CA). Allophycocyanin-conjugated anti-mouse CD117, purified anti-mouse CD16/CD32, and FITC-conjugated CD16/CD32 were from BD Biosciences (San Jose, CA). PE-conjugated anti-mouse CD9 Ab was from Santa Cruz Biotechnology (Dallas, TX). PE-conjugated anti-mouse CD63 and allophycocyanin/Cy7-conjugated anti-mouse CD117 flow cytometry Abs were from BioLegend (San Diego, CA). Allophycocyanin-conjugated anti-mouse CD151 flow cytometry Ab was from R&D Systems (Minneapolis, MN). ECL Prime Western blotting detection reagent was from GE Healthcare Biosciences (Pittsburgh, PA). Anti-phosphotyrosine 4G10 mAb was from Upstate Biotechnology (Lake Placid, NY). Abs against p-Akt (Ser473), Akt, p-Syk (Tyr317 and Tyr519/520), Syk, p-PLCγ1 (Tyr783), PLCγ1, p-ERK1/2 (Thr202/Tyr204), and ERK1/2 were purchased from Cell Signaling Technology (Danvers, MA). MEK/ERK inhibitor PD98056 and PI3K inhibitors LY294002 and wortmannin were obtained from Calbiochem (San Diego, CA). All other reagents were obtained from Sigma-Aldrich (St. Louis, MO) unless otherwise indicated.
Animals
CD151−/− mice on a C57BL/6 background (a gift from Dr. Jonathan Jones, Northwestern University) were previously described (35). C57BL/6J wild-type (WT) mice were purchased from The Jackson Laboratory (stock no. 000664). The mice were housed under barrier conditions and were specific pathogen-free as determined in sentinel mice. Female mice 6–8 wk of age were used in all experiments. All procedures were reviewed and approved by the Animal Care and Use Committee at Northwestern University.
Gene expression microarray data
For human mast cell CD151 expression, we consulted National Center for Biotechnology Information’s Gene Expression Omnibus (GEO) repository dataset GSE3982 (http://www.ncbi.nlm.nih.gov/genbank), which is a human immune cell transcriptomics study (36). In this study, nonstimulated human cord blood–derived mast cells (n = 2) were compared with mast cells stimulated with IgE for 2 h (n = 2). We performed statistical comparison of these groups by moderated t test with false discovery rate (FDR) p value (multiple testing) correction using the GEO2R analysis tool (National Center for Biotechnology Information). The 10% cut-off (p(corr) < 0.1) was used for determining statistical significance in differential expression. In this microarray, CD151 was represented by a single probe set (204306_s_at). CD151 gene expression data for mice were derived from our ongoing study of the transcriptomics of mast cell activation (P.J. Bryce, unpublished microarray data). Briefly, BMMCs from WT mice were plated on six-well plates at a density of 5 × 106 cells per well. Cells were either unstimulated or coated with 1 μg/ml DNP-IgE overnight, followed by 0.5 μg/ml DNP–human serum albumin (HSA) stimulation. Cells were collected 4 h after stimulation and RNA was extracted using an RNeasy kit (Qiagen). Samples were processed using Illumina bead array technology by the Genomics Core Facility at Northwestern University. Bioinformatics analysis was done using GeneSpring GX 12.6 software (Agilent Technologies). Data were normalized using a quantile normalization algorithm with no baseline transformation. Non-stimulated WT mast cells (n = 3) were compared with IgE-stimulated WT mast cells (n = 3) using a moderated t test with the Benjamini–Hochberg FDR multiple testing correction. The 10% cut-off (p(corr) < 0.1) was used for determining statistical significance in differential expression. CD151 was represented by three different probe sets (1400630, 6450390, 5720100).
IgE-mediated PCA
PCA was performed as previously described (37). Briefly, mice were sensitized by intradermal injection of 100 ng anti-DNP IgE mAb (Sigma-Aldrich) into the left ears, whereas the right ears received saline as a control. After 24 h, mice were challenged by retro-orbital injection of 100 μg DNP-HSA (Sigma-Aldrich). The thickness of the ear was measured using a thickness micrometer at baseline and for the indicated time points. Changes in ear thickness were reported relative to baseline.
Histological quantification of mast cells in ear tissue
Mice were euthanized and ear tissue was fixed in formalin and embedded in paraffin. Four-micrometer tissue sections were stained for mast cells with toluidine blue. Twenty high-power fields were assessed per sample in a blinded fashion.
Mast cell cultures
BMMCs were obtained by flushing bone marrow from both femur bones using complete RPMI media (Invitrogen, Carlsbad, CA) with 10% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin, L-glutamine, 1 mM sodium pyruvate, and nonessential amino acids (1×). Cells were then cultured in complete mast cell growth media supplemented with 30 ng/ml recombinant mouse IL-3 (Shenandoah Biotechnology). For 6–8 wk, twice each week, non-adherent cells were removed, the media refreshed completely, and cells were replated in a new flask. Resulting mast cell culture purity was determined by flow cytometry for double staining of CD117+/FcεRI+ cells (>90% purity).
Toluidine blue staining of BMMCs
Cytospin slides were prepared by centrifugation of 105 cells at 400 rpm for 5 min in a CytoFuge. Cells were air dried overnight and then stained with toluidine blue solution with pH 2.0 (45 ml 1% sodium chloride plus 5 ml 1% toluidine blue O [Sigma-Aldrich] in 100 ml 70% EtOH) for 2–3 min. The slides were allowed to dry overnight before applying coverslips with Permount fixative (Fisher Scientific).
Flow cytometry
BMMCs or cell suspensions from lavage of the peritoneal cavity were washed once in PBS 1×, stained with Aqua Live/dead dye (Invitrogen) for 20 min at room temperature, blocked with anti-CD16/CD32 (mouse BD Biosciences Fc Block) for 10 min at 4°C in flow staining buffer, and consequently incubated with Abs in flow staining buffer for 30 min at 4°C. CD117 (c-Kit) and FcεRI double labeling was used to gate on mast cell populations. For surface staining of FcγRII/RIII, unblocked mast cells were labeled with FITC-conjugated CD16/CD32 Ab for 30 min at 4°C. Flow cytometry was performed on a BD LSR II flow cytometer (BD Biosciences) and analyzed using FlowJo v10 software.
β-Hexosaminidase assay
The degree of mast cell degranulation was measured by release of β-hexosaminidase. BMMCs (0.3 × 106 cells/ml) were preincubated overnight with anti-DNP IgE (100 ng/ml) in medium. Sensitized cells were stimulated for 20 min with indicated concentrations of DNP-HSA in Tyrode’s buffer, centrifuged, and the cell pellets were solubilized with 0.5% Triton X-100 in Tyrode’s buffer. The enzymatic activity of β-hexosaminidase in the supernatants and cell pellets was measured using p-nitrophenyl-N-acetyl-β-D-glucosaminide (Sigma-Aldrich) in 0.1 M sodium citrate (pH 4.5) as a substrate. The reaction was allowed to run for 30 min at 37°C and then stopped by adding 0.2 M glycine (pH 10.7). Release of the product, p-nitrophenol, was detected based on the absorbance at 405 nm. The percentage β-hexosaminidase release was determined by dividing the measurements detected in the supernatant by the total measurements detected in the supernatant plus those from the cell pellet.
Detection of gene expression by quantitative PCR
Gene expression of cytokines IL-4, IL-9, IL-10, IL-13, and TNF-α and chemokines CCL1 and CCL2 was determined by quantitative PCR (qPCR) from BMMCs sensitized overnight with 1 μg/ml anti-DNP IgE. Cells were pretreated or not for 1 h at 37°C with the following specific inhibitors: 10 μM PD98056, 10 μM LY204002, or 100 nM wortmannin. The cells were further nonstimulated or stimulated for 5 h with 0.5 μg/ml DNP-HSA. Total RNA was isolated from cells using the Qiagen RNeasy mini kit (Qiagen). cDNA was prepared from 500 ng mRNA/cDNA synthesis reaction using a qScript cDNA synthesis kit (Quanta BioSciences) and analyzed by real-time PCR on a 7500 real-time PCR system (Applied Biosystems) using primers/probes from Integrated DNA Technologies and Quanta BioSciences PerfeCTa qPCR FastMix. Amplification parameters were 50°C for 2 min, 95°C for 10 min, followed by 40 cycles of 95°C for 15 s alternating with 60°C for 1 min. Gene expression was determined based on the ΔCt values between gene of interest and housekeeping gene GAPDH and compared with the mean ΔCt values for the control group to determine a fold induction value, as previously described (38).
ELISA for cytokine secretion
BMMCs were sensitized overnight with 1 μg/ml anti-DNP IgE. Cells were pretreated (or not pretreated) for 1 h at 37°C with the following specific inhibitors: 10 μM PD98056, 10 μM LY204002, or 100 nM wortmannin. BMMCs were then stimulated with 100 ng/ml DNP-HSA for 3 or 24 h, or stem cell factor (SCF) 100 ng/ml or PMA/ionophore (0.081 nM/1.34 μM), after which the cell culture supernatants were assayed for cytokines TNF-α, IL-4, and IL-13 using Ready-SET-Go! ELISA kits purchased from eBioscience. ELISA assays were performed according to the manufacturer’s instructions.
Cell lysates and immunoblotting
BMMCs (2 × 106 cells/sample) were unprimed or primed with 1 μg/ml anti-DNP IgE for 24 h and stimulated with 0.5 μg/ml DNP-HSA for 5 and 10 min. Cell lysates were prepared in RIPA buffer (10 mM Tris [pH 7.4], 150 mM NaCl, 1% Triton X-100, 1% deoxycholic acid, 0.1% SDS, 5 mM EDTA) containing a protease inhibitor mixture (Sigma-Aldrich). Lysates were resolved by SDS-PAGE followed by semidry electroblotting transfer onto nitrocellulose membranes. Membranes were blocked with 5% nonfat milk in TBS (20 mM Tris [pH 7.5] and 0.15 M NaCl) containing 0.1% Tween 20 for 1 h at room temperature and incubated with Abs overnight at 4°C, followed by 1 h secondary incubation with HRP-conjugated goat IgG (Cell Signaling Technology). Proteins were detected by ECL Plus Western blotting detection reagent (GE Healthcare), and densitometry was performed on scanned Western blot images using ImageJ software (National Institutes of Health). The data were presented as the fold increase in the ratio of relative intensity of the band/the relative intensity of band for the loading control (β-actin or total protein for protein phosphorylation blots). Complete Western blot images are provided in Supplemental Fig. 4.
Calcium mobilization assay
Calcium flux was determined using the Fluo-4 NW calcium assay kit (F36206, Life Technologies, Grand Island, NY) according to the manufacturer’s instructions. Briefly, BMMCs were primed overnight with 1 μg/ml DNP-specific IgE, then cells were centrifuged and resuspended in assay buffer (13 HBSS, 20 mM HEPES) at a density of 2.5 × 106/ml. Cells were incubated at 37°C and 5% CO2 for 60 min to allow them to settle. The cell suspension was then mixed with an equal volume of dye loading solution (Fluo-4 dye mix plus assay buffer and 5 mM probenecid) and incubated at 37°C for 30 min followed by incubation at room temperature for 30 min. To measure calcium flux, the cells were analyzed by flow cytometry for 25 s to establish a baseline (prestimulation) FITC signal. Cells were treated with vehicle, DNP-HSA (5–500 ng/ml), or 0.25 μM ionomycin, the positive control. Cells were further analyzed for 30 s to determine a poststimulation FITC signal. The median pre- and poststimulation FITC signal values were determined using FlowJo v10 and subtracted from each other to determine the Δ median FITC or “relative Ca2+ flux.” A Student t test and two-way ANOVA were used to assess statistical differences for each individual stimulant concentration.
Statistical analysis
Data were analyzed by one-way ANOVA, followed by a Tukey multiple comparisons test. PCA and time course immunoblot data were analyzed using repeated measures ANOVA. All statistical tests were carried out in the Systat 13 statistical package (Systat Software). All data are presented as the mean ± SEM.
Results
IgE stimulation induces upregulation of tetraspanin CD151 expression by both mouse and human mast cells
We consulted both mouse and human gene expression microarray studies to determine whether tetraspanin CD151 was expressed basally in mast cells and whether its transcription was induced by stimulation with IgE. In mouse BMMCs (P.J. Bryce, unpublished microarray study of transcriptomics of mast cell activation), CD151 was found to be significantly upregulated at 4 h poststimulation (the only time point considered in the experiment) (Fig. 1A). All three probe sets for CD151 showed statistically significant upregulation of CD151 upon activation of mast cells by IgE. In human cord blood–derived mast cells (GEO dataset GSE3982), CD151 was significantly upregulated at 2 h after IgE stimulation (also the only time point considered) (Fig. 1A). Additionally, in another study of human mast cell IgE-dependent activation, CD151 was reported to be significantly upregulated at 6 h (34). Out of 34 tetraspanin family members measured in these microarray experiments, only CD151 was consistently upregulated by IgE stimulation in both mouse and human arrays (data for expression of other tetraspanins not shown). Additionally, we assessed protein level expression of CD151 by flow cytometry in mouse WT BMMCs stimulated with IgE/Ag for 4 h. CD151 expression was induced by IgE stimulation of WT BMMCs (Fig. 1B).
FIGURE 1.
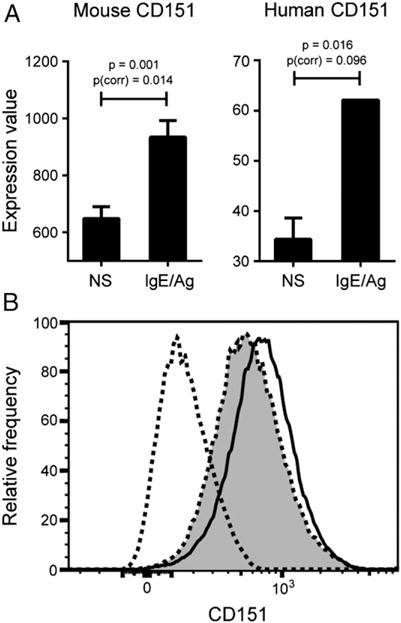
IgE stimulation induces upregulation of tetraspanin CD151 expression by both mouse and human mast cells. (A) BMMCs from WT mice were either unstimulated or primed with 1 μg/ml DNP-IgE overnight, followed by 0.5 μg/ml DNP-HSA stimulation. Cells were collected 4 h after stimulation and subjected to gene expression microarray analysis using Illumina bead array technology. Nonstimulated mast cells (n = 3) were compared with IgE-stimulated mast cells (n = 3) using a t test with the Benjamini–Hochberg FDR multiple testing correction. This graph represents the average of three probe sets used to measure CD151 expression in the arrays (each individual probe showed statistical significance). Both raw t test p value and p value after FDR correction for multiple testing [p(corr)] are shown. Data for human mast cell CD151 expression are derived from GEO2R analysis of human immune cell transcriptomics study GSE3982 (National Center for Biotechnology Information’s GEO database). In this study, human cord blood–derived mast cells were either left unstimulated (n = 2) or stimulated with IgE for 2 h (n = 2). The p values shown are both unadjusted and adjusted by the Benjamini–Hochberg FDR correction [p(corr)]. p < 0.05 and p(corr) < 0.1 are deemed to be significant. (B) CD151 is upregulated in WT BMMCs at 4 h as measured by flow cytometry.
CD151 deficiency significantly enhances the late phase of FcεRI-mediated PCA
We used a murine model of PCA to examine the role of CD151 in regulation of mast cell–dependent responses in vivo (Fig. 2A). CD151−/− and WT mice were sensitized by intradermal injection of anti-DNP IgE or saline 24 h prior to Ag challenge by retro-orbital injection of 100 μg DNP-HSA or saline control solution. The early phase response to anti-DNP IgE, measured within 2 h by localized tissue swelling at the site of injection, was not significantly different between CD151−/− and WT controls (Fig. 2A). However, CD151−/− mice exhibited a significant increase in magnitude of the late phase response compared with the WT mice, as assessed by a significant increase in thickness of ear tissue between 24 and 36 h after Ag challenge (Fig. 2A). To exclude the possibility that the increased cutaneous allergic response in CD151−/− mice was caused by a basal level increase in CD151−/− mast cell numbers, the ears of CD151−/− and WT mice were sectioned in paraffin followed by histological staining and quantification of mast cells. There were no differences in basal numbers of tissue mast cells between WT and CD151−/− mice (Fig. 2B). These data suggest that CD151 may exert negative influences over the development of the IgE-dependent late-phase cutaneous response. This prompted us to extend our findings to mast cells derived from the bone marrow of CD151−/− and WT mice and determine a mechanism by which CD151 negatively regulates FcεRI-mediated mast cell activation.
FIGURE 2.
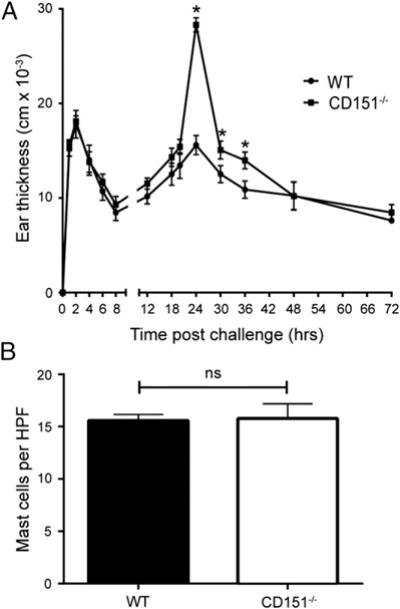
CD151 deficiency leads to enhanced late phase of FcεRI-mediated PCA. (A) WT and CD151−/− mice were sensitized passively by an intradermal injection with 100 ng IgE into the right ear and saline into the left ear as a control. After 24 h, mice were challenged retro-orbitally with 100 μg DNP-HSA. Ear swelling was measured at each time point shown (mean ± SEM, n = 5 mice/group). The DNP-HSA–induced increase in ear thickness was measured as the Δ difference from thickness of the same mouse control ears injected with saline. At 24–36 h, a significant increase in IgE-induced ear thickness was observed in CD151−/− mice compared with WT mice (*p < 0.05). This experiment was repeated three times. (B) Mast cells were quantified by histology in 4-μm paraffin sections of ear tissue stained by toluidine blue. HPF, high-powered field.
Mast cells develop normally in absence of CD151
Mast cells were cultured from the bone marrow of CD151−/− or WT mice for 4–6 wk in the presence of murine recombinant IL-3. The IL-3–stimulated growth and proliferation of mast cells cultured from the bone marrow of CD151−/− mice was not significantly different from that of BMMCs from WT mice (Fig. 3A). We next determined whether CD151 is constitutively expressed at mRNA and protein levels by nonstimulated BMMCs ex vivo and peritoneal mast cells in vivo. Constitutive expression of CD151 was detected by qPCR and Western blot analysis of total cell lysates of WT BMMCs with an anti-CD151 Ab that recognizes the extracellular domain of this protein. CD151 was constitutively expressed by BMMCs derived from WT mice. As expected, there was no CD151 present in mast cells from CD151−/− mice (Fig. 3A). In vivo, CD151 was constitutively expressed by FcεRI+c-Kit+ peritoneal mast cells in WT mice (Fig. 3A). The morphology of resulting mast cells was assessed by toluidine blue staining. The appearance and presence of toluidine blue–positive, granule-containing cells was similar in the cultures of BMMCs derived from WT and CD151−/− mice (Fig. 3B). No significant difference in the expression levels of FcεRI and c-Kit or resulting culture purity was observed between BMMCs from CD151−/− and WT mice, as assessed by flow cytometry (Fig. 3B). Because decreased levels of the inhibitory Fcγ receptors may be associated with an exaggerated IgE response (39–41), we determined whether the BMMCs derived from CD151−/− mice had FcγRII/III expression comparable to WT controls. No significant changes in the expression of Fcγ receptors were observed in BMMCs derived from CD151−/− mice (Supplemental Fig. 1). Basal surface expression of two other constitutively expressed mast cell tetraspanins, CD9 and CD63, was comparable between WT and CD151−/− BMMCs, as measured by flow cytometry (Supplemental Fig. 1). Taken together, these findings suggest that the basal development of BMMCs was not affected by CD151 deficiency, and that the resulting cells exhibit normal expression levels of several key cell surface receptors.
FIGURE 3.
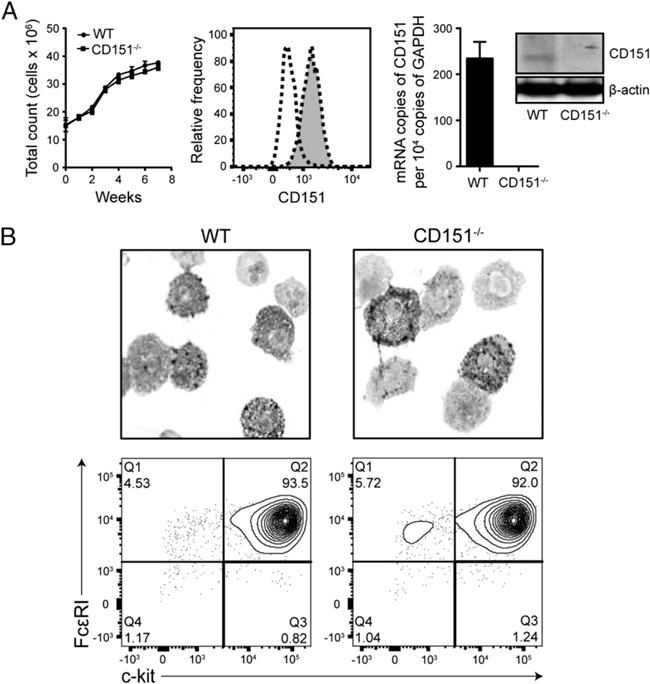
CD151 deficiency does not alter basal phenotype of cultured BMMCs. (A) CD151 deficiency does not affect growth dynamics of mast cell culture. WT and CD151−/− BMMCs were cultured in IL-3–conditioned media for 8 wk and the total numbers of live cells in culture were counted at each time point indicated (left). Flow cytometry for CD151 expression on peritoneal mast cells (flow cytometry chart) (middle), qPCR detection of CD151 mRNA in WT and CD151−/− BMMCs (bar graph, right), and Western blot analysis of total-protein lysates for CD151 expression (Western blot, right) all confirm constitutive CD151 expression in WT mast cells. WT peritoneal mast cells are shown as gray-filled histogram with dotted line and negative control as transparent histogram with dotted line. In immunoblotting, actin was used as a loading control. (B) Five-week-old BMMCs from WT and CD151−/− mice were stained with toluidine blue and images were obtained with an original magnification of ×100. Flow cytometry analysis of FcεRI and c-Kit surface expression and purity of WT and CD151−/− BMMC cultures. All data are representative of three independent experiments. Data are represented as mean ± SEM. *p < 0.05.
CD151 deficiency does not alter the level of degranulation by BMMCs
We next examined whether CD151 played a role in FcεRI-mediated mast cell degranulation ex vivo. It was previously established that Abs against the tetraspanins CD63 or CD81 inhibit in vitro and in vivo FcεRI-mediated mast cell degranulation (27, 28). Moreover, CD63−/− mast cells exhibit reduced mast cell degranulation compared with WT cells (32). To investigate whether CD151 participates in regulating mast cell degranulation, WT and CD151−/− BMMCs were primed with anti-DNP IgE overnight. Degranulation was induced by incubating mast cells with different concentrations of DNP-HSA. There was no difference in degranulation levels between BMMCs from CD151−/− and WT mice, as assessed by release of β-hexosaminidase (Fig. 4A). This finding was consistent with the lack of effect on the mast cell degranulation-dependent early phase PCA reaction in vivo (Fig. 2A). β-Hexosaminidase release initiated by the positive degranulation control compound 48/80 was also comparable in BMMCs from CD151−/− and WT mice (Fig. 4A). We also found no significant differences in calcium flux responses with mast cell CD151 deficiency compared with WT BMMCs (Fig. 4B, Supplemental Fig. 2). For this experiment, sensitized BMMCs were loaded with 1.5 μM Fluo-4 and stimulated with DNP-HSA. The kinetics of calcium flux was monitored by the reduction in green fluorescence (FITC) intensity. Both CD151−/− and WT mast cells generated the same level of intracellular calcium rise either upon IgE plus DNP-HSA stimulation or in the presence of ionophore ionomycin, which bypasses the need for FcεRI stimulation.
FIGURE 4.
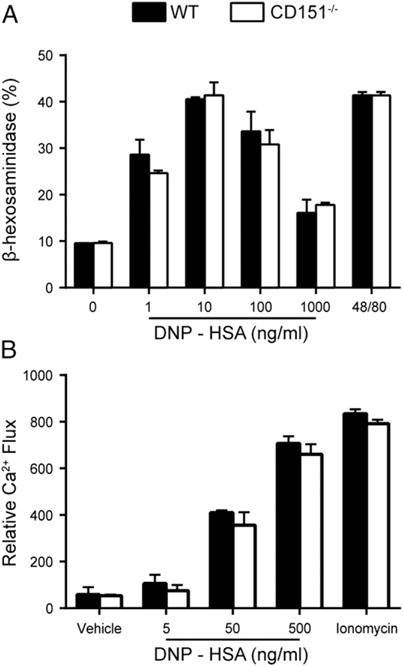
CD151 deficiency does not affect BMMC degranulation or calcium mobilization. (A) β-Hexosaminidase release by stimulated CD151−/− and WT BMMCs shows no difference in mast cell degranulation ex vivo. Data are represented as the mean ± SEM from three individual assay wells/group over two independent experiments. (B) BMMCs were precoated with DNP-IgE overnight and incubated with a dye loading solution (Fluo-4 dye mix, assay buffer, and 5 mM probenecid) and incubated for 60 min prior to 0.5 μg/ml DNP-HSA stimulation. Cells were also stimulated with 0.25 μM ionomycin to confirm the capability of cells to induce calcium influx. Filled columns indicate WT; open columns indicate CD151−/−.
CD151 deficiency significantly increases FcεRI-mediated cytokine production by BMMCs
Activated mast cells play a significant role in the activation of other cell types and in the development of the late-phase allergic response through the production and release of cytokines and chemokines (42). For this reason, we investigated whether FcεRI-mediated cytokine production was altered in CD151−/− mast cells. WT and CD151−/− BMMCs were sensitized with DNP-specific IgE and then stimulated with DNP-HSA for 3 or 24 h. After 3 h, FcεRI crosslinking of CD151-deficient BMMCs did not result in any changes in protein levels of released cytokines measured in culture supernatants (Fig. 5A). However, IgE/Ag stimulation for 24 h did induce a significant increase in cytokine production as measured by ELISA, suggesting an effect on de novo synthesis of cytokines. Specifically, protein levels of the cytokines IL-4, IL-13, and TNF-α were significantly greater in CD151−/− BMMCs upon DNP-HSA stimulation (Fig. 5B). Gene expression of IL-4, IL-13, and TNF-α measured by qPCR was also significantly increased in CD151−/− BMMCs compared with WT BMMC controls at 24 h (Supplemental Fig. 3). This effect of CD151 on cytokine production was specific to IgE/Ag stimulation, because no increase in cytokine levels was observed between WT and CD151−/− BMMCs stimulated with SCF or PMA (Fig. 5C). FcεRI-mediated expression of cytokines IL-9 and IL-10, as well as chemokines CCL1 and CCL2, was unaffected by the absence of CD151 (Supplemental Fig. 3). In summary, CD151 appeared to selectively negatively regulate FcεRI-mediated de novo production of cytokines IL-4, IL-13, and TNF-α.
FIGURE 5.
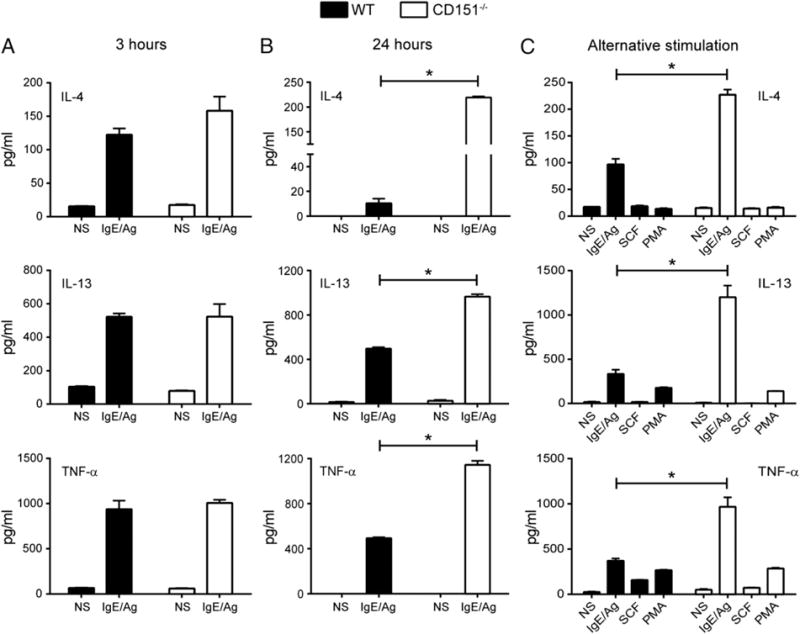
Increased cytokine release by CD151−/− BMMCs after IgE/Ag stimulation. Cytokine release by BMMCs was measured in culture supernatants by ELISA. BMMCs were unprimed or primed with IgE-DNP, then exposed to 100 ng/ml DNP-HSA for 3 h (A) or 24 h (B) and culture supernatants were tested for levels (pg/ml) of IL-4, IL-13, and TNF-α. (C) Comparison of stimulation effects of IgE, SCF, and PMA on cytokine release by WT and CD151 BMMCs. ELISA data represent the mean ± SEM from four individual assay wells/group over two independent experiments. Filled columns indicate WT; open columns indicate CD151−/−. *p < 0.05.
Enhanced and sustained ERK1/2 and Akt phosphorylation in FcεRI-stimulated mast cells with CD151 deficiency
To understand the mechanism by which CD151 deficiency enhances mast cell cytokine production, we next examined signaling events downstream of FcεRI receptor activation. WT and CD151−/− BMMCs were sensitized with anti–DNP-IgE and activated with DNP-HSA. At indicated time points, the cells were lysed and analyzed by immunoblotting using the specified Abs. The phosphorylation of Syk at Tyr519/520 within its activation loop is essential for Syk’s activity and is the most proximal signal in the FcεRI signaling cascade (43–45). There was no difference in Syk Tyr519/520 phosphorylation between WT and CD151−/− BMMCs, both at 5 and 10 min poststimulation (Fig. 6). We also quantified Syk phosphorylation at residue Tyr317, which is an inhibitory site limiting Syk activation (46). In CD151−/− BMMCs, Syk Tyr317 phosphorylation was similar to WT cells at 5 min poststimulation; however, the signal persisted for 10 min, although it returned to basal levels in WT controls (Fig. 6). PLCγ1 phosphorylation is crucial for FcεRI-induced diacylglycerol production and mast cell degranulation (2, 5). PLCγ1 phosphorylation at Tyr783 was not affected by CD151 deficiency in mast cells (Fig. 6). A complementary pathway for cytokine production is mediated by PI3K signaling and activation of Akt (8, 47). We detected an increase in phosphorylation of Akt at both 5 and 10 min poststimulation (Fig. 6). Downstream signaling through MAPKs is critical for cytokine release by mast cells (7). Immunoblotting experiments revealed that ERK1/2 phosphorylation at Thr203/Tyr205 reached its peak at 5 min following FcεRI aggregation, and it was significantly increased in CD151−/− BMMCs compared with WT controls (Fig. 6). Moreover, in CD151−/− BMMCs, the ERK1/2 phosphorylation signal was sustained at 10 min, whereas it decreased 2-fold in WT cells. No differences in total tyrosine phosphorylation were detected between WT and CD151−/− mast cells (Fig. 6). In summary, deletion of CD151 resulted in sustained ERK1/2 and Akt phosphorylation, but no changes in activation of Syk, PLCγ1, or total tyrosine phosphorylation levels in activated BMMCs. This is consistent with the lack of change in calcium release and the increase in cytokine production by CD151−/− BMMCs.
FIGURE 6.
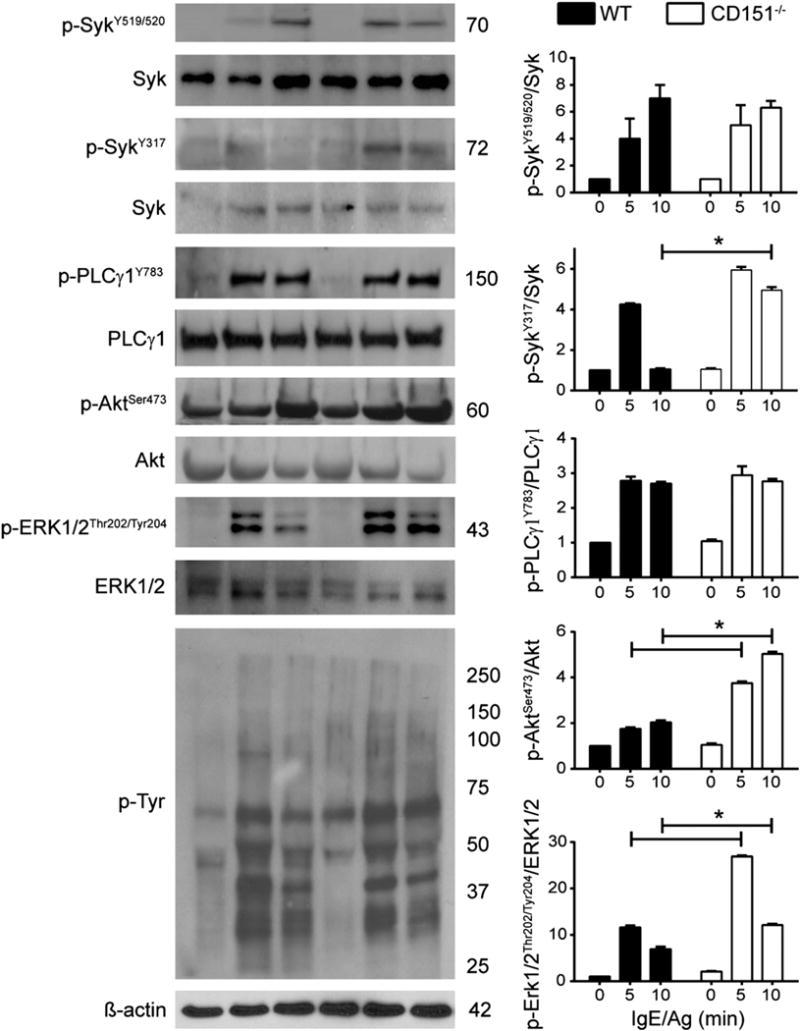
CD151 deficiency leads to enhanced and sustained ERK1/2 and Akt activation in IgE-stimulated mast cells. BMMCs from WT and CD151−/− mice were sensitized with 1 μg/ml IgE for 24 h and then stimulated with 0.5 μg/ml DNP-HSA for the time points indicated. Cell lysates were subjected to immunoblotting analysis of the following phosphorylation events: Syk phosphorylation at Tyr519/520 and Tyr317, PLCγ1 at Tyr783, Akt phosphorylation at Ser473, ERK1/2 phosphorylation at Thr202/Tyr204, and total phosphotyrosine detection, which represents total phosphorylation events following IgE stimulation. Representative Western blots are shown on the left, as phosphorylation bands followed by total protein loading controls. Bar graphs on the right show intensities of Western blot bands quantified by densitometry analysis. Fold increase in phosphorylation intensity was measured relative to total levels of detected proteins of interest. Densitometry values are mean ± SEM of three independent Western blots. Filled columns indicate WT; open columns indicate CD151−/−. *p < 0.05.
CD151 negatively regulates cytokine production in activated BMMCs by inhibiting ERK pathway activation
In the “principal” FcεRI signaling cascade in activated mast cells, activation of the MAPK family members is central to downstream activity of numerous transcription factors important for cytokine production (9, 48). As demonstrated in Fig. 6, ERK1/2 phosphorylation was significantly enhanced and sustained in CD151−/− BMMCs compared with WT mast cells. Thus, to determine whether the ERK pathway contributed to elevated cytokine production by CD151−/− BMMCs, we treated mast cells with PD98056, a specific inhibitor of MEK, which is a crucial component of the ERK1/2 MAPK pathway. The addition of 10 μM PD98056 decreased mRNA levels of the cytokines IL-4, IL-13, and TNF-α in stimulated WT and CD151−/− cells (Fig. 7). Alternatively, the “complementary” or amplification signaling cascade in activated mast cells may involve the PI3K pathway as another positive regulator of IgE-induced cytokine production (48–50). Moreover, the ERK signaling pathway itself can be also activated in a PI3K-dependent manner upon FcεRI stimulation (51). To test possible involvement of PI3K kinase in cytokine production by WT and CD151−/− mast cells, we employed two different PI3K inhibitors: LY294002 and wortmannin. We used two inhibitors rather than one because all PI3K inhibitors possess some off-target effects, which tend to be different from one inhibitor to another (52–54). Similar biological responses by the two different inhibitors suggested that their effect was through the inhibition of PI3K. LY294002 and wortmannin both suppressed IL-4, IL-13, and TNF-α gene expression in response to IgE/Ag activation (Fig. 7). The degree of cytokine suppression was similar to the reduction in cytokine signaling caused by ERK inhibitor PD98056 (Fig. 7). In summary, both ERK and PI3K pathways were responsible for IgE/Ag-induced cytokine production in WT and CD151−/− BMMCs. Moreover, these results suggest that CD151 functions as a negative regulator of both principal and complementary pathways for cytokine production by activated mast cells.
FIGURE 7.
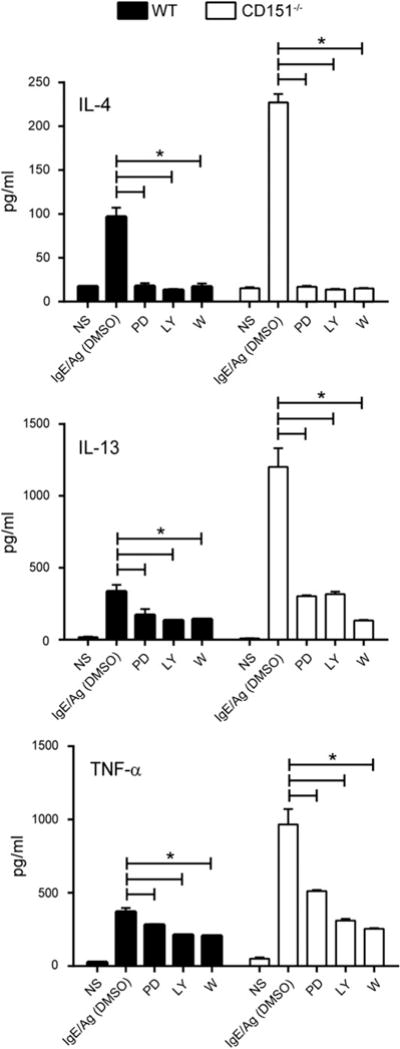
CD151 negatively regulates cytokine production in activated BMMCs by inhibiting ERK and PI3K pathway activation. BMMCs were unprimed or primed with IgE, then exposed to 100 ng/ml DNP-HSA for 24 h in the absence or presence of 10 μM LY294002 (LY), 100 nM Wortmannin (W), 10 μM PD98056 (PD) or DMSO vehicle control. NS, not stimulated controls. IL-4, IL-13, and TNF-α cytokine release by BMMCs was measured in culture supernatants by ELISA. Results shown are the mean ± SEM of three determinations. Filled columns indicate WT; open columns indicate CD151−/−. *p < 0.05.
Discussion
To our knowledge, this study is the first to report a regulatory function of tetraspanin CD151 in mast cells. Moreover, it is one of the first reports, to our knowledge, addressing the signaling mechanism of modulation of mast cell activation by any member of the tetraspanin family. In the present study, we demonstrated that CD151 deficiency exacerbated late-phase allergic inflammation in mice in vivo and enhanced proinflammatory cytokine production by cultured BMMCs ex vivo. Moreover, BMMCs deficient in CD151 showed enhanced and sustained FcεRI-induced ERK1/2 and Akt phosphorylation compared with WT cells. Conversely, CD151 deficiency had no effect on mast cell degranulation or the acute phase of PCA. Thus, our data demonstrate that the tetraspanin CD151 functions to selectively inhibit late-phase anaphylaxis responses and the de novo synthesis of cytokines by activated mast cells.
Mast cells possess mechanisms for fine tuning cellular activation that allow initial FcεRI-mediated signaling to proceed in a controlled fashion and further permit the termination of the activated signals in a timely manner. Tetraspanins are classically known as passive membrane “organizer” proteins that form complexes with integrins and regulate cellular events involving cytoskeletal reorganization such as cell fusion and motility (55). Only recently have they begun to emerge as molecules with a specific regulatory capacity for signal transduction in various cell types (22–25). In mast cells specifically, tetraspanins CD63 and CD81 have been shown to be negative regulators of mast cell degranulation, both in vivo and in vitro (27, 28, 32). Moreover, tetraspanin CD63 has long been known as an activation marker of human basophils (29, 30). In contrast to other mast cell tetraspanins, CD151 is specifically induced at 6 h following FcεRI aggregation in human mast cells (34) at a time when degranulation is largely complete, but prior to FcεRI-mediated cytokine production. To build on information from this study, we revisited raw data from additional mast cell microarray studies. In agreement with Jayapal et al. (34), we found that, following IgE stimulation, CD151 transcription was significantly enhanced at 4 h poststimulation in mouse mast cells and at 2 h in human mast cells. This timing coincides with onset of the late-phase reaction, which is usually associated with cytokine production by activated mast cells. Notably, tetraspanin CD63, implicated in control of mast cell degranulation and early phase anaphylactic reactions, was not upregulated with IgE treatment in either mouse or human microarray studies that we examined. Curiously, out of 34 tetraspanins considered, only tetraspanin CD151 showed consistent IgE-induced upregulation of transcription by both mouse and human mast cells (data for other tetraspanins not shown). To further support CD151 as a potential negative regulator of cytokine production by activated mast cells, we demonstrated the enhanced production of cytokines IL-4, IL-13, and TNF-α by Ag-activated CD151−/− mast cells ex vivo. This function of CD151 as an inhibitor of cytokine production in mast cells is consistent with our finding that CD151−/− mice had an exaggerated FcεRI-mediated late-phase allergic response in a mast cell–dependent model of PCA in vivo, which requires production of cytokines and recruitment of inflammatory cells.
CD151 is a membrane scaffolding molecule that is known to regulate signaling molecules, such as phosphatidylinositol-4-kinase and PKC in epithelial cells (1, 56). Therefore, it could have complex roles in multiple signaling pathways affecting homeostasis and activation of mast cells. Basally, CD151 deficiency did not alter development of BMMCs ex vivo or numbers of cutaneous mast cells in vivo. CD151−/− BMMCs were not deficient in surface levels of receptors FcεRI and c-Kit, or expression of inhibitory receptor FcγRII/III and other tetraspanins known to be expressed on mast cells. In general, the basal phenotype of CD151−/− mast cells that we are reporting is consistent with reports of deficiency of tetraspanin CD63 not affecting basal development of human mast cells (32).
However, interactions of CD151 with signaling pathways directly or indirectly associated with mast cell activation and exocytosis are not known. To gain further insight into the molecular mechanism behind the inhibitory role of CD151 in mast cell activation, we interrogated FcεRI-mediated signaling transduction in CD151−/− BMMCs. In a “classical” mast cell activation pathway, propagation of FcεRI signaling depends on phosphorylation of protein tyrosine kinases Syk and Lyn, which results in phosphorylation of PLCγ1 (48, 57). Degranulation follows the activation of PLCγ1 and calcium mobilization (Ca2+)/PKC pathway, whereas cytokine synthesis depends primarily on downstream activation of the Ras–Raf1–MEK (MEK1 and 2)–MAPK (ERK1/2)–transcription factor pathway (9, 10, 43). Phosphoproteomics studies show that ERK1 phosphorylation at the autophosphorylation sites Thr203/Tyr205 and ERK2 autophosphorylation at Thr183/Tyr185 are the most prominent phosphorylation events that follow IgE receptor aggregation in mouse mast cells (44). Similarly, we observed a 5-min peak in ERK1/2 phosphorylation at the residues Thr203/Tyr205 and Thr183/Tyr185. However, ERK1/2 phosphorylation was significantly enhanced in CD151−/− BMMCs compared with WT mast cells, and this increase was sustained at 10 min following IgE receptor stimulation. Moreover, we found that Akt phosphorylation was also increased at 5 and 10 min poststimulation. In contrast to activation of pathways leading to cytokine production, phosphorylation status of PLCγ1 and Ag-induced calcium flux were not affected by CD151 deficiency. There was no difference in Syk phosphorylation at tyrosine residue Tyr519/520 in the activation loop of the kinase. Interestingly, at 10 min, but not 5 min, CD151−/− BMMCs exhibited significantly greater increase in Syk phosphorylation at tyrosine residue Tyr317, which is an inhibitory site in the linker region of Syk, and binds the negative regulator Cbl, which negatively regulates the signals leading to degranulation (46). This would further prevent hyperactivation of pathways associated with degranulation in CD151−/− mast cells. Additionally, we showed that a selective inhibitor of MEK/ERK pathway inhibited Ag-induced expression of IL-4, IL-13, and TNF-α in WT and CD151−/− BMMCs. Our results showing that MEK/ERK inhibitors inhibited cytokine induction are consistent with those reported previously (58–60). Moreover, inhibitors of PI3K significantly attenuated cytokine production by activated WT and CD151−/− BMMCs to a degree comparable with ERK inhibition, thus also implicating a complementary PI3K-dependent signaling pathway for cytokine production. The aforementioned signaling events were specific to IgE/Ag-induced stimulation, because we observed no signaling differences between WT and CD151−/− mast cells upon stimulation with SCF, which activates mast cells via its binding to the c-Kit receptor, or PMA, which bypasses outside–in receptor signaling. Collectively, these findings reinforce the conclusion that the tetraspanin CD151 has a focused inhibitory function in FcεRI-dependent mast cell activation, specifically targeting components of the ERK1/2 and PI3K/Akt signaling cascade downstream of Syk, leading to de novo cytokine production.
Selective inhibition of ERK1/2-PI3K-Akt signaling by CD151 may be explained either by 1) the tetraspanin’s direct involvement in the ERK1/2 activation pathway or 2) CD151 functioning as an inhibitory receptor via recruitment of phosphatases inhibitory to the MAPK activity. In fibroblasts, CD151 was reported to directly interact with and inhibit Ras (Raf-1) activity (25). Furthermore, activation of PKB/c-Akt and ERK1/2, downstream targets in the Ras signaling pathway, was also diminished in fibroblasts overexpressing CD151 (25). As a scaffold, CD151 has potential to recruit inhibitory adaptor proteins that negatively regulate the Ras/Raf1/ERK1/2 pathway, although this possibility remains to be tested. For instance, recruitment of inhibitory adaptor protein Dok-1 negatively regulates Ras by recruiting Ras GTPase or SHIP-1, which inhibits the entire Ras/Raf1/ERK pathway in Ag-stimulated RBL-2H3 mast cells (16). Another potential mechanism for the inhibitory action of tetraspanins is the recruitment of inhibitory phosphatases to ITIM-like motifs found in cytoplasmic domains of several members of this protein family. ITIM motifs are known to recruit phosphatases such as SHIP-1/2, SHP-1/2, and the dual specificity protein phosphatase-1, which all have capacity to dephosphorylate and inactivate MAPKs (17, 18). Upon ligation with anti–CD37 small modular immunopharmaceutical, the ITIM-like motif of tetraspanin CD37 recruits the phosphatase SHP-1, which exerts negative regulation of survival of B cells by inhibiting phosphorylation of MAPK/ERK and AKT (24). Moreover, tetraspanins CD53 and CD63 were reported to immunoprecipitate an unidentified phosphatase with inhibitory activity in lymphoid and RBL-2H3 mast cells (61). Interestingly, this hyperactivation phenotype of CD151−/− mast cells matches the phenotype of mast cells derived from mice with specific knockouts of phosphatases PTPα (62), SHIP-1 (63), and SHIP-2 (64). Although CD151 does not possess a confirmed ITIM or ITIM-like motif, it is possible that through its scaffolding interactions or yet unknown tyrosine-based motifs, CD151 recruits a phosphatase for the specific inhibition of MAPK activity. This remains a subject of future investigation by our group.
Although all evidence points to a direct modulation of classical IgE receptor signaling responses by CD151, we cannot exclude the possibility that CD151 may also be involved in the regulation of other mast cell receptors, some of which could be modulatory for IgE downstream signaling pathways. Notably, downstream of ERK1/2 activation, CD151 deficiency resulted in overproduction of only selected cytokines, whereas expression of other cytokines or chemokines was not altered. This implies existence of a fine-tuning mechanism of ERK1/2-driven transcriptional regulation of cytokine production, which may be time- or context-dependent and may involve additional targets of CD151 at the level of ERK1/2 signaling.
In conclusion, we demonstrated that CD151 deficiency exacerbated inflammation during the late phase of IgE-induced PCA and that, mechanistically, in the FcεRI signaling pathway, CD151 is inhibitory for the activation of the ERK1/2 and PI3K/Akt signal transduction cascades, which further control downstream transcription of selected proinflammatory cytokines. These results reveal a significant role for CD151 in mast cell activation and provide further evidence that members of the tetraspanin family have specific and nonredundant functions in IgE-mediated signaling.
Supplementary Material
Acknowledgments
We thank Holly Ann Schroeder (the Bryce Lab, Northwestern University) for sharing technical expertise in conducting mast cell activation experiments.
Abbreviations
- BMMC
bone marrow–derived mast cell
- FDR
false discovery rate
- GEO
Gene Expression Omnibus
- HSA
human serum albumin
- PCA
passive cutaneous anaphylaxis
- PKC
protein kinase C
- PLCγ1
phospholipase Cγ1
- qPCR
quantitative PCR
- SCF
stem cell factor
- Syk
spleen tyrosine kinase
- WT
wild-type
Footnotes
The online version of this article contains supplemental material.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Levy S, Shoham T. Protein-protein interactions in the tetraspanin web. Physiology (Bethesda) 2005;20:218–224. doi: 10.1152/physiol.00015.2005. [DOI] [PubMed] [Google Scholar]
- 2.Rivera J, Gilfillan AM. Molecular regulation of mast cell activation. J Allergy Clin Immunol. 2006;117:1214–1225. doi: 10.1016/j.jaci.2006.04.015. quiz 1226. [DOI] [PubMed] [Google Scholar]
- 3.Metcalfe DD, Baram D, Mekori YA. Mast cells. Physiol Rev. 1997;77:1033–1079. doi: 10.1152/physrev.1997.77.4.1033. [DOI] [PubMed] [Google Scholar]
- 4.Galli SJ, Nakae S, Tsai M. Mast cells in the development of adaptive immune responses. Nat Immunol. 2005;6:135–142. doi: 10.1038/ni1158. [DOI] [PubMed] [Google Scholar]
- 5.Kambayashi T, Koretzky GA. Proximal signaling events in FcεRI-mediated mast cell activation. J Allergy Clin Immunol. 2007;119:544–552. doi: 10.1016/j.jaci.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 6.Kraft S, Kinet JP. New developments in FcεRI regulation, function and inhibition. Nat Rev Immunol. 2007;7:365–378. doi: 10.1038/nri2072. [DOI] [PubMed] [Google Scholar]
- 7.Siraganian RP. Mast cell signal transduction from the high-affinity IgE receptor. Curr Opin Immunol. 2003;15:639–646. doi: 10.1016/j.coi.2003.09.010. [DOI] [PubMed] [Google Scholar]
- 8.Kim MS, Rådinger M, Gilfillan AM. The multiple roles of phosphoinositide 3-kinase in mast cell biology. Trends Immunol. 2008;29:493–501. doi: 10.1016/j.it.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Galli SJ, Kalesnikoff J, Grimbaldeston MA, Piliponsky AM, Williams CM, Tsai M. Mast cells as “tunable” effector and immunoregulatory cells: recent advances. Annu Rev Immunol. 2005;23:749–786. doi: 10.1146/annurev.immunol.21.120601.141025. [DOI] [PubMed] [Google Scholar]
- 10.Williams CM, Galli SJ. The diverse potential effector and immunoregulatory roles of mast cells in allergic disease. J Allergy Clin Immunol. 2000;105:847–859. doi: 10.1067/mai.2000.106485. [DOI] [PubMed] [Google Scholar]
- 11.Katz HR. Inhibitory receptors and allergy. Curr Opin Immunol. 2002;14:698–704. doi: 10.1016/s0952-7915(02)00400-4. [DOI] [PubMed] [Google Scholar]
- 12.Odom S, Gomez G, Kovarova M, Furumoto Y, Ryan JJ, Wright HV, Gonzalez-Espinosa C, Hibbs ML, Harder KW, Rivera J. Negative regulation of immunoglobulin E-dependent allergic responses by Lyn kinase. J Exp Med. 2004;199:1491–1502. doi: 10.1084/jem.20040382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Furumoto Y, Brooks S, Olivera A, Takagi Y, Miyagishi M, Taira K, Casellas R, Beaven MA, Gilfillan AM, Rivera J. Cutting edge: lentiviral short hairpin RNA silencing of PTEN in human mast cells reveals constitutive signals that promote cytokine secretion and cell survival. J Immunol. 2006;176:5167–5171. doi: 10.4049/jimmunol.176.9.5167. [DOI] [PubMed] [Google Scholar]
- 14.Ota Y, Samelson LE. The product of the proto-oncogene c-cbl: a negative regulator of the Syk tyrosine kinase. Science. 1997;276:418–420. doi: 10.1126/science.276.5311.418. [DOI] [PubMed] [Google Scholar]
- 15.Paolini R, Molfetta R, Beitz LO, Zhang J, Scharenberg AM, Piccoli M, Frati L, Siraganian R, Santoni A. Activation of Syk tyrosine kinase is required for c-Cbl-mediated ubiquitination of FcεRI and Syk in RBL cells. J Biol Chem. 2002;277:36940–36947. doi: 10.1074/jbc.M204948200. [DOI] [PubMed] [Google Scholar]
- 16.Hiragun T, Peng Z, Beaven MA. Dexamethasone up-regulates the inhibitory adaptor protein Dok-1 and suppresses downstream activation of the mitogen-activated protein kinase pathway in antigen-stimulated RBL-2H3 mast cells. Mol Pharmacol. 2005;67:598–603. doi: 10.1124/mol.104.008607. [DOI] [PubMed] [Google Scholar]
- 17.Lang R, Hammer M, Mages J. DUSP meet immunology: dual specificity MAPK phosphatases in control of the inflammatory response. J Immunol. 2006;177:7497–7504. doi: 10.4049/jimmunol.177.11.7497. [DOI] [PubMed] [Google Scholar]
- 18.Owens DM, Keyse SM. Differential regulation of MAP kinase signalling by dual-specificity protein phosphatases. Oncogene. 2007;26:3203–3213. doi: 10.1038/sj.onc.1210412. [DOI] [PubMed] [Google Scholar]
- 19.Kimura T, Sakamoto H, Appella E, Siraganian RP. The negative signaling molecule SH2 domain-containing inositol-polyphosphate 5-phosphatase (SHIP) binds to the tyrosine-phosphorylated β subunit of the high affinity IgE receptor. J Biol Chem. 1997;272:13991–13996. doi: 10.1074/jbc.272.21.13991. [DOI] [PubMed] [Google Scholar]
- 20.Karra L, Levi-Schaffer F. Down-regulation of mast cell responses through ITIM containing inhibitory receptors. Adv Exp Med Biol. 2011;716:143–159. doi: 10.1007/978-1-4419-9533-9_9. [DOI] [PubMed] [Google Scholar]
- 21.Hemler ME. Tetraspanin functions and associated microdomains. Nat Rev Mol Cell Biol. 2005;6:801–811. doi: 10.1038/nrm1736. [DOI] [PubMed] [Google Scholar]
- 22.Suzuki M, Tachibana I, Takeda Y, He P, Minami S, Iwasaki T, Kida H, Goya S, Kijima T, Yoshida M, et al. Tetraspanin CD9 negatively regulates lipopolysaccharide-induced macrophage activation and lung inflammation. J Immunol. 2009;182:6485–6493. doi: 10.4049/jimmunol.0802797. [DOI] [PubMed] [Google Scholar]
- 23.Zhu XW, Price NM, Gilman RH, Recarvarren S, Friedland JS. Multinucleate giant cells release functionally unopposed matrix metalloproteinase-9 in vitro and in vivo. J Infect Dis. 2007;196:1076–1079. doi: 10.1086/521030. [DOI] [PubMed] [Google Scholar]
- 24.Lapalombella R, Yeh YY, Wang L, Ramanunni A, Rafiq S, Jha S, Staubli J, Lucas DM, Mani R, Herman SE, et al. Tetraspanin CD37 directly mediates transduction of survival and apoptotic signals. Cancer Cell. 2012;21:694–708. doi: 10.1016/j.ccr.2012.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sawada S, Yoshimoto M, Odintsova E, Hotchin NA, Berditchevski F. The tetraspanin CD151 functions as a negative regulator in the adhesion-dependent activation of Ras. J Biol Chem. 2003;278:26323–26326. doi: 10.1074/jbc.C300210200. [DOI] [PubMed] [Google Scholar]
- 26.Köberle M, Kaesler S, Kempf W, Wölbing F, Biedermann T. Tetraspanins in mast cells. Front Immunol. 2012;3:106. doi: 10.3389/fimmu.2012.00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fleming TJ, Donnadieu E, Song CH, Laethem FV, Galli SJ, Kinet JP. Negative regulation of FcεRI-mediated degranulation by CD81. J Exp Med. 1997;186:1307–1314. doi: 10.1084/jem.186.8.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kraft S, Fleming T, Billingsley JM, Lin SY, Jouvin MH, Storz P, Kinet JP. Anti-CD63 antibodies suppress IgE-dependent allergic reactions in vitro and in vivo. J Exp Med. 2005;201:385–396. doi: 10.1084/jem.20042085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eberlein-König B, Varga R, Mempel M, Darsow U, Behrendt H, Ring J. Comparison of basophil activation tests using CD63 or CD203c expression in patients with insect venom allergy. Allergy. 2006;61:1084–1085. doi: 10.1111/j.1398-9995.2006.01122.x. [DOI] [PubMed] [Google Scholar]
- 30.Ebo DG, Lechkar B, Schuerwegh AJ, Bridts CH, Clerck LSDe, Stevens WJ. Comments regarding “Marked improvement of the basophil activation test by detecting CD203c instead of CD63” by Boumiza et al. Clin Exp Allergy. 2003;33:849. author reply 852–853. [PubMed] [Google Scholar]
- 31.Schäfer T, Starkl P, Allard C, Wolf RM, Schweighoffer T. A granular variant of CD63 is a regulator of repeated human mast cell degranulation. Allergy. 2010;65:1242–1255. doi: 10.1111/j.1398-9995.2010.02350.x. [DOI] [PubMed] [Google Scholar]
- 32.Kraft S, Jouvin MH, Kulkarni N, Kissing S, Morgan ES, Dvorak AM, Schröder B, Saftig P, Kinet JP. The tetraspanin CD63 is required for efficient IgE-mediated mast cell degranulation and anaphylaxis. J Immunol. 2013;191:2871–2878. doi: 10.4049/jimmunol.1202323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hálová I, Dráberová L, Bambousková M, Machyna M, Stegurová L, Smrz D, Dráber P. Cross-talk between tetraspanin CD9 and transmembrane adaptor protein non-T cell activation linker (NTAL) in mast cell activation and chemotaxis. J Biol Chem. 2013;288:9801–9814. doi: 10.1074/jbc.M112.449231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jayapal M, Tay HK, Reghunathan R, Zhi L, Chow KK, Rauff M, Melendez AJ. Genome-wide gene expression profiling of human mast cells stimulated by IgE or FcεRI-aggregation reveals a complex network of genes involved in inflammatory responses. BMC Genomics. 2006;7:210. doi: 10.1186/1471-2164-7-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wright MD, Geary SM, Fitter S, Moseley GW, Lau LM, Sheng KC, Apostolopoulos V, Stanley EG, Jackson DE, Ashman LK. Characterization of mice lacking the tetraspanin superfamily member CD151. Mol Cell Biol. 2004;24:5978–5988. doi: 10.1128/MCB.24.13.5978-5988.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jeffrey KL, Brummer T, Rolph MS, Liu SM, Callejas NA, Grumont RJ, Gillieron C, Mackay F, Grey S, Camps M, et al. Positive regulation of immune cell function and inflammatory responses by phosphatase PAC-1. Nat Immunol. 2006;7:274–283. doi: 10.1038/ni1310. [DOI] [PubMed] [Google Scholar]
- 37.Hsu CL, Neilsen CV, Bryce PJ. IL-33 is produced by mast cells and regulates IgE-dependent inflammation. PLoS ONE. 2010;5:e11944. doi: 10.1371/journal.pone.0011944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ujike A, Ishikawa Y, Ono M, Yuasa T, Yoshino T, Fukumoto M, Ravetch JV, Takai T. Modulation of immunoglobulin (Ig)E-mediated systemic anaphylaxis by low-affinity Fc receptors for IgG. J Exp Med. 1999;189:1573–1579. doi: 10.1084/jem.189.10.1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Robbie-Ryan M, Tanzola MB, Secor VH, Brown MA. Cutting edge: both activating and inhibitory Fc receptors expressed on mast cells regulate experimental allergic encephalomyelitis disease severity. J Immunol. 2003;170:1630–1634. doi: 10.4049/jimmunol.170.4.1630. [DOI] [PubMed] [Google Scholar]
- 41.Takai T. Roles of Fc receptors in autoimmunity. Nat Rev Immunol. 2002;2:580–592. doi: 10.1038/nri856. [DOI] [PubMed] [Google Scholar]
- 42.Brown JM, Wilson TM, Metcalfe DD. The mast cell and allergic diseases: role in pathogenesis and implications for therapy. Clin Exp Allergy. 2008;38:4–18. doi: 10.1111/j.1365-2222.2007.02886.x. [DOI] [PubMed] [Google Scholar]
- 43.de Castro RO. Regulation and function of Syk tyrosine kinase in mast cell signaling and beyond. J Signal Transduct. 2011;2011:507291. doi: 10.1155/2011/507291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cao L, Yu K, Banh C, Nguyen V, Ritz A, Raphael BJ, Kawakami Y, Kawakami T, Salomon AR. Quantitative time-resolved phosphoproteomic analysis of mast cell signaling. J Immunol. 2007;179:5864–5876. doi: 10.4049/jimmunol.179.9.5864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang J, Billingsley ML, Kincaid RL, Siraganian RP. Phosphorylation of Syk activation loop tyrosines is essential for Syk function. An in vivo study using a specific anti-Syk activation loop phosphotyrosine antibody. J Biol Chem. 2000;275:35442–35447. doi: 10.1074/jbc.M004549200. [DOI] [PubMed] [Google Scholar]
- 46.Sada K, Zhang J, Siraganian RP. Point mutation of a tyrosine in the linker region of Syk results in a gain of function. J Immunol. 2000;164:338–344. doi: 10.4049/jimmunol.164.1.338. [DOI] [PubMed] [Google Scholar]
- 47.Kitaura J, Asai K, Maeda-Yamamoto M, Kawakami Y, Kikkawa U, Kawakami T. Akt-dependent cytokine production in mast cells. J Exp Med. 2000;192:729–740. doi: 10.1084/jem.192.5.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gilfillan AM, Tkaczyk C. Integrated signalling pathways for mast-cell activation. Nat Rev Immunol. 2006;6:218–230. doi: 10.1038/nri1782. [DOI] [PubMed] [Google Scholar]
- 49.Gilfillan AM, Rivera J. The tyrosine kinase network regulating mast cell activation. Immunol Rev. 2009;228:149–169. doi: 10.1111/j.1600-065X.2008.00742.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Poderycki M, Tomimori Y, Ando T, Xiao W, Maeda-Yamamoto M, Sauer K, Kawakami Y, Kawakami T. A minor catalytic activity of Src family kinases is sufficient for maximal activation of mast cells via the high-affinity IgE receptor. J Immunol. 2010;184:84–93. doi: 10.4049/jimmunol.0901590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Takayama G, Ohtani M, Minowa A, Matsuda S, Koyasu S. Class I PI3K-mediated Akt and ERK signals play a critical role in FcεRI-induced degranulation in mast cells. Int Immunol. 2013;25:215–220. doi: 10.1093/intimm/dxs105. [DOI] [PubMed] [Google Scholar]
- 52.Knight ZA, Gonzalez B, Feldman ME, Zunder ER, Goldenberg DD, Williams O, Loewith R, Stokoe D, Balla A, Toth B, et al. A pharmacological map of the PI3-K family defines a role for p110α in insulin signaling. Cell. 2006;125:733–747. doi: 10.1016/j.cell.2006.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hazeki K, Nigorikawa K, Hazeki O. Role of phosphoinositide 3-kinase in innate immunity. Biol Pharm Bull. 2007;30:1617–1623. doi: 10.1248/bpb.30.1617. [DOI] [PubMed] [Google Scholar]
- 54.Hayakawa M, Kawaguchi K, Kaizawa H, Koizumi T, Ohishi T, Yamano M, Okada M, Ohta M, Tsukamoto S, Raynaud FI, et al. Synthesis and biological evaluation of sulfonylhydrazone-substituted imidazo[1,2-a]pyridines as novel PI3 kinase p110α inhibitors. Bioorg Med Chem. 2007;15:5837–5844. doi: 10.1016/j.bmc.2007.05.070. [DOI] [PubMed] [Google Scholar]
- 55.Hemler ME. Tetraspanin proteins mediate cellular penetration, invasion, and fusion events and define a novel type of membrane microdomain. Annu Rev Cell Dev Biol. 2003;19:397–422. doi: 10.1146/annurev.cellbio.19.111301.153609. [DOI] [PubMed] [Google Scholar]
- 56.Yauch RL, Berditchevski F, Harler MB, Reichner J, Hemler ME. Highly stoichiometric, stable, and specific association of integrin α3β1 with CD151 provides a major link to phosphatidylinositol 4-kinase, and may regulate cell migration. Mol Biol Cell. 1998;9:2751–2765. doi: 10.1091/mbc.9.10.2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jabril-Cuenod B, Zhang C, Scharenberg AM, Paolini R, Numerof R, Beaven MA, Kinet JP. Syk-dependent phosphorylation of Shc. A potential link between FcεRI and the Ras/mitogen-activated protein kinase signaling pathway through SOS and Grb2. J Biol Chem. 1996;271:16268–16272. doi: 10.1074/jbc.271.27.16268. [DOI] [PubMed] [Google Scholar]
- 58.Koranteng RD, Swindle EJ, Davis BJ, Dearman RJ, Kimber I, Flanagan BF, Coleman JW. Differential regulation of mast cell cytokines by both dexamethasone and the p38 mitogen-activated protein kinase (MAPK) inhibitor SB203580. Clin Exp Immunol. 2004;137:81–87. doi: 10.1111/j.1365-2249.2004.02510.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang C, Baumgartner RA, Yamada K, Beaven MA. Mitogen-activated protein (MAP) kinase regulates production of tumor necrosis factor-α and release of arachidonic acid in mast cells. Indications of communication between p38 and p42 MAP kinases. J Biol Chem. 1997;272:13397–13402. doi: 10.1074/jbc.272.20.13397. [DOI] [PubMed] [Google Scholar]
- 60.Masuda A, Yoshikai Y, Aiba K, Matsuguchi T. Th2 cytokine production from mast cells is directly induced by lipopolysaccharide and distinctly regulated by c-Jun N-terminal kinase and p38 pathways. J Immunol. 2002;169:3801–3810. doi: 10.4049/jimmunol.169.7.3801. [DOI] [PubMed] [Google Scholar]
- 61.Carmo AM, Wright MD. Association of the transmembrane 4 superfamily molecule CD53 with a tyrosine phosphatase activity. Eur J Immunol. 1995;25:2090–2095. doi: 10.1002/eji.1830250743. [DOI] [PubMed] [Google Scholar]
- 62.Samayawardhena LA, Pallen CJ. PTPα activates Lyn and Fyn and suppresses Hck to negatively regulate FcεRI-dependent mast cell activation and allergic responses. J Immunol. 2010;185:5993–6002. doi: 10.4049/jimmunol.1001261. [DOI] [PubMed] [Google Scholar]
- 63.Haddon DJ, Antignano F, Hughes MR, Blanchet MR, Zbytnuik L, Krystal G, McNagny KM. SHIP1 is a repressor of mast cell hyperplasia, cytokine production, and allergic inflammation in vivo. J Immunol. 2009;183:228–236. doi: 10.4049/jimmunol.0900427. [DOI] [PubMed] [Google Scholar]
- 64.Leung WH, Bolland S. The inositol 59-phosphatase SHIP-2 negatively regulates IgE-induced mast cell degranulation and cytokine production. J Immunol. 2007;179:95–102. doi: 10.4049/jimmunol.179.1.95. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.