

Abstract
One to two percent of all children are born with a developmental disorder requiring pediatric hospital admissions. For many such syndromes, the molecular pathogenesis remains poorly characterized. Parallel developmental disorders in other species could provide complementary models for human rare diseases by uncovering new candidate genes, improving the understanding of the molecular mechanisms and opening possibilities for therapeutic trials. We performed various experiments, e.g. combined genome-wide association and next generation sequencing, to investigate the clinico-pathological features and genetic causes of three developmental syndromes in dogs, including craniomandibular osteopathy (CMO), a previously undescribed skeletal syndrome, and dental hypomineralization, for which we identified pathogenic variants in the canine SLC37A2 (truncating splicing enhancer variant), SCARF2 (truncating 2-bp deletion) and FAM20C (missense variant) genes, respectively. CMO is a clinical equivalent to an infantile cortical hyperostosis (Caffey disease), for which SLC37A2 is a new candidate gene. SLC37A2 is a poorly characterized member of a glucose-phosphate transporter family without previous disease associations. It is expressed in many tissues, including cells of the macrophage lineage, e.g. osteoclasts, and suggests a disease mechanism, in which an impaired glucose homeostasis in osteoclasts compromises their function in the developing bone, leading to hyperostosis. Mutations in SCARF2 and FAM20C have been associated with the human van den Ende-Gupta and Raine syndromes that include numerous features similar to the affected dogs. Given the growing interest in the molecular characterization and treatment of human rare diseases, our study presents three novel physiologically relevant models for further research and therapy approaches, while providing the molecular identity for the canine conditions.
Author Summary
Rare developmental disorders make a major contribution to pediatric hospital admissions and mortality. There is a growing interest in the development of therapeutics for these conditions, but that requires understanding of the genetic cause and pathology. Research can be facilitated by physiologically relevant models, such as dogs with corresponding disorders. We have characterized the clinical features and genetic causes of three developmental syndromes in dogs, including craniomandibular osteopathy (CMO), a previously undescribed skeletal syndrome, and dental hypomineralization, for which we identified mutations in the canine SLC37A2, SCARF2 and FAM20C genes, respectively. CMO is a clinical equivalent to an infantile cortical hyperostosis (Caffey disease) for which SLC37A2 is a new candidate gene. SLC37A2 is a glucose-phosphate transporter in osteoclasts, and its defect suggests an impaired glucose homeostasis in developing bone, leading to hyperostosis. Mutations in the SCARF2 and FAM20C genes have been associated with the human van den Ende-Gupta and Raine syndromes. Our study provides molecular identity for the canine conditions and presents three novel physiologically relevant models of human rare diseases.
Introduction
One to two percent of all children are born with a developmental disorder, such as a heart defect, skeletal abnormality, or mental retardation as a result of errors in embryogenesis and early neurodevelopment. These disorders make a major contribution to pediatric hospital admissions and mortality [1]. Rare pediatric disorders are typically with homozygous, compound heterozygous, or de novo pathogenic variants. The advent of cost-efficient next generation sequencing (NGS) technologies drives gene discovery in many disorders without a cognate gene [2] and thousands of rare variants have been described (www.orpha.net). There is also a growing interest in the development of therapeutics for rare diseases, which requires the identification of the genetic defects, comprehensive understanding of the molecular pathology and access to physiologically relevant animal models.
Developmental disorders are frequent also in other species, including dogs, which as large animals bear very close physiologic and genetic resemblance with us. Dogs give birth to litters with multiple puppies. However, often some of the littermates are affected by developmental or other abnormalities, and perinatal mortality (stillbirth, fetal and neonatal death) is common with prevalence ranging from 5 to 35% [3]. The causes include respiratory distress syndrome/hypoxia, infectious diseases, severe malformations and suspected hereditary diseases. Culling is a common practice among dog breeders and the deceased or abnormal puppies are not always presented to the veterinarian. Therefore, numerous syndromes with likely genetic origin remain unknown. It would be important to investigate the extent of this common phenomenon at the clinical and molecular level to better understand the diverse causes of the morbidity and to better manage it through advised breeding programs. At the same time, the identification of the causative gene could inform gene functions, disease etiology, molecular pathology and phenotypic overlap across species. Importantly, physiological similarity of dogs with human would establish relevant therapeutic models to human rare disorders. Online Mendelian Inheritance in Animals (OMIA), a catalogue of inherited disorders and associated genes in animals, reports more than 350 inherited diseases in dogs as potential models for human disease (http://omia.angis.org.au/).
NGS approaches are rapidly changing the diagnostic landscape in veterinary medicine in companion animals and enable now a feasible approach to tackle the molecular background of developmental conditions in small pedigrees with translational potential to human rare disease. For example, we have previously discovered a new gene (ATG4D) responsible for a neurodegenerative vacuolar storage disease in Lagotto Romagnolos [4] and a missense variant in canine FAM83G causing palmoplantar hyperkeratosis and demonstrating its role in maintaining the integrity of the palmoplantar epidermis [5]. A CNGB1 frameshift variant has been identified to cause a progressive retinal atrophy in dogs [6] and the same gene has been associated with retinal degeneration in human as well [7]. Similarly, we have found that a congenital skeletal disease in Brazilian Terriers is caused by a pathogenic variant in the GUSB orthologue responsible for human pediatric disorder, mucopolysaccharidosis type VII [8].
This study addressed the clinical and genetic background of three developmental disorders in dogs; craniomandibular osteopathy (CMO; OMIA: 000236–9615) in West Highland White Terriers (WHWT), Cairn Terriers and Scottish Terriers, a previously undescribed developmental syndrome in Wire Fox Terriers, and dental hypomineralization in Border Collies. CMO is a self-limiting proliferative bone disease seen in young dogs [9]. It manifests between 4 to 8 months of age with typical signs including swelling of the jaw, periodical fever, lack of appetite, pain, difficulty opening the mouth and dysphagia. The excessive proliferation causes bony lesions primarily on the skull bones, especially on the mandible and tympanic bulla, but occasionally also on the metaphyses of long bones. Signs of the disease usually resolve with time, when the growth period is finished. CMO exists in several breeds with the highest frequency in WHWT and has been suggested to be an autosomal recessive trait [10–12]. Canine CMO corresponds to human Caffey disease [MIM: 114000] and its genetic characterization might reveal insights into similar painful human swelling disorders [10]. We identified a novel CMO gene that represents a candidate gene for human Caffey disease. The two other syndromes have not previously been reported in dogs. We describe the detailed clinical features for them in our study. Moreover, we demonstrate their shared genetic etiology with the corresponding human syndromes, van den Ende-Gupta (VDEGS [MIM: 600920]) and Raine syndromes [MIM: 259775].
Results
A splicing variant in SLC37A2 causes craniomandibular osteopathy in three terrier breeds
We performed a genome-wide association study (GWAS) to map the CMO locus with Illumina’s 22K canine SNP chips in a cohort of 51 WHWTs, including 10 cases (diagnosed by radiography; Fig 1A) and 41 controls. A case-control association test revealed significant association on CFA5 with the best SNP (BICF2S23544899) at 8,953,507 Mb (praw = 1.2 x 10−7, pgenome = 0.02) (genomic inflation factor λ = 1.17) (Fig 1B). Manual assessment of genotypes at the CFA5 locus revealed a shared 1.9-Mb homozygosity block in all affected dogs spanning from 7,764,955 bp to 9,707,794 bp. The same homozygosity block was seen in five controls as well. The associated region was replicated and fine-mapped using 105 additional SNPs in 88 samples from three related breeds, WHWT, Cairn Terriers and Scottish Terriers. Fine mapping confirmed the association in all three breeds with the best SNP BICF2S23134295 at 8,183,669 (p = 2.09x10-15).
Fig 1. Genetic analyses in canine craniomandibular osteopathy identify a leaky splicing variant in exon 15 of SLC37A2, resulting in a frameshift that truncates the protein.

A. Laterolateral (left) and ventrodorsal (right) radiograph of the skull of two different WHWT affected with CMO. The typical palisade-like periosteal new bone formation is commonly located along the corpus mandibulae (left, white arrowheads) or around the Bulla tympanica (right, black arrowheads). B. Manhattan plot from GWAS indicates the associated region on chromosome 5 (pgenome = 0.02). C. A synonymous variant, c. 1332C>T, in exon 15 of SLC37A2. D. ESEfinder suggests that the synonymous variant affects a splicing enhancer site. The binding scores for the different splicing factors (colored blocks) for 79 nucleotides of exon 15 are shown for the wild-type and mutant allele. The c.1332C>T mutation eliminates a potential binding site for the SF2/ASF splicing factor (shown in purple). Binding scores for different splicing proteins (shown on the y-axis) are based on different weight matrices and cannot be compared directly to each other. E. A semi-quantitative RT-PCR in the wild-type (CC), carrier (CT) and affected (TT) WHWTs confirms the splicing defect (79 bp deletion) caused by the variant, and reveals a splicing leakage in the carrier and affected dogs. B2M was used as a loading control. F. A schematic 12-transmembrane secondary structure of SLC37A2 predicted by TMHMM. The truncated region in the mutated protein is indicated by a dashed line.
To identify candidate variants, the associated and fine mapped region (1.8 Mb) was captured from two affected and two healthy WHWT with opposite haplotypes followed by a paired-end NGS by HiSeq2000. The shared variants identified in the two affected WHWT dogs were filtered against the two WHWT controls and 32 additional healthy dogs from four different breeds. We found altogether three homozygous variants shared in cases (S1 Table); two intergenic indels and a synonymous variant in exon 15 of solute carrier family 37 member 2 gene (SLC37A2 c.1332 C>T) (Fig 1C). As an additional independent verification to avoid potential targeted capture biases, we performed whole genome sequencing in one CMO-affected WHWT and 188 control dogs from other breeds (S2 Table) to compare the variants in the associating region. This analysis yielded a single case-specific variant (chr5 g.9,387,327G>A) that was the same as the one identified in the capture experiment.
Although the variant, c.1332C>T in exon 15 of SLC37A2 is synonymous, it was predicted to affect a splicing enhancer element based on ESEfinder analysis (Fig 1D). The mutant T allele eliminates a potential binding site for the splicing factor ASF/SF-2. To confirm the predicted effect on splicing, we amplified the region between exons 7 and 18 in lymphocyte mRNAs from three cases, six carriers and seven controls. RT-PCR experiments showed that two alternatively spliced SLC37A2 transcripts were expressed in all dogs that carried one or two copies of the mutant T allele at the SLC37A2 SNP, regardless of disease status (Fig 1E). Sequencing of the RT-PCR products indicated that the smaller band corresponded to a mutant SLC37A2 transcript lacking 79 bp from exon 15. The splice variant resulted in a frameshift and premature stop codon at the beginning of exon 16. This altered splicing was predicted to lead to a C-terminally truncated protein lacking 75 amino acids compared to the wild-type SLC37A2 protein (Fig 1F). The RT-PCR indicated that both wild-type and mutant transcripts were expressed in even the CMO affected dogs, although the expression of the wild-type transcript was significantly reduced in the affected homozygous dogs (Fig 1E). The reduction of the wild type transcript level was more moderate in healthy heterozygous carrier dogs. None of the examined wild-type dogs expressed the mutant transcript. Overall, these results demonstrate the leaky nature of the splice site mutation.
To investigate the segregation and frequency of the variant across the CMO affected breeds, we performed a large variant screening by genotyping the c.1332C>T variant altogether in 1052 dogs, including 695 WHWT, 249 Scottish Terriers and 108 Cairn Terriers (S3 Table). We found 123 homozygous dogs in the WHWT breed, of which 48% had been reported with CMO. About 40% of WHWT (275 dogs) carried the pathogenic variant, of which 10 dogs (3,6%) were reported with CMO. In Scottish Terriers, 10 dogs (4%) were homozygous and all were reported with CMO, and 43 dogs (17%) were carriers, of which 3 dogs (7%) were reported with CMO. In Cairn Terriers, 9 dogs (8%) were homozygous and reported with CMO, and 15 dogs (14%) carried the pathogenic variant, from which 3 dogs (20%) were reported with CMO. We found one wild-type dog in both Scottish and Cairn Terriers with CMO, and screened the coding regions of the entire SLC37A2 gene in these two dogs for possible other pathogenic variants, but did not find any. This suggests phenocopies, misdiagnoses or genetic heterogeneity. The analysis of the pathogenic variant in the three main breeds with 96 cases resulted in a highly significant association (p = 6.62x10-303) with CMO.
In addition to the above three breeds, we screened the c.1332C>T variant in 458 dogs in 124 breeds, but found only a single heterozygous carrier dog in Jack Russell Terrier breed (S3 Table). The phenotype information for this dog was not available. The variant was also screened in the known CMO cases from seven breeds (two Bull Terriers, one Curly Coated Retriever, two Border Collies, one Australian Terrier, one Basset, one German Wirehaired Pointer, one Old English Sheepdog), but they did not have it.
Collectively, our results suggest that CMO is inherited as dominant disease with incomplete penetrance. Canine CMO is equivalent to Caffey disease and our data reveals a novel candidate gene, SLC37A2, for the syndrome.
Truncated SCARF2 causes a skeletal syndrome in Wire Fox Terriers
Wire Fox Terrier breeders contacted us for help in the characterization of an unknown congenital syndrome with severe mandibular prognathia and other skeletal features, mainly severe patellar luxation, in the breed. Two affected 7-week-old puppies from different litters, two unaffected littermates and two affected adult Wire Fox Terriers were examined by radiography. Additionally, a computed tomography (CT) study of the skull was made on two affected dogs (one adult and one puppy), and three dogs were studied for general clinical characteristics and neurological examination.
A prominent underbite with short maxilla (brachygnathia superior) was evident in all affected Wire Fox Terriers except one adult dog (Fig 2A and 2B). The caudodorsal border of the maxilla was slightly convex in all affected animals. In CT images, the nasal septum deviated prominently to the left at the level of the dorsocaudal frontal bone in both examined dogs (Fig 2C). The number and position of the vertebrae were normal, but the mid-thoracic spinous processes were thinner, longer and more horizontally aligned in the affected than in the normal dogs. One adult dog had an abnormally wide second rib. The adult dog had marked spondylosis of the spine. One affected puppy had unilateral congenital elbow luxation (Fig 2D), and in the other the secondary ossification centers of the olecranon were non-mineralized. The secondary ossification centers of the tibial tuberosities were small in both affected puppies, when compared to a healthy puppy (Fig 2E and 2F). The proximal epiphyses of the fibulae were not mineralized in the affected puppies and the patellae were medially luxated. The femurs of the affected dogs had medial bowing of mid-shafts of the bone. In an eye examination of a puppy and two affected adult dogs, the eyes appeared small and the sclera thinner than normal.
Fig 2. Radiographic and CT findings in a novel canine developmental syndrome.

A. Lateral radiograph of a 7-week-old affected Wire Fox Terrier puppy, where prominent underbite (long arrow) and dorsally convex maxilla (short arrow) are clearly visible. B. Healthy littermate with normal scissor bite and flat dorsal border of the maxilla. C. Transverse CT image of the nasal cavities of an affected puppy. Marked lateral deviation of the nasal septum is evident (arrow). D. Lateral radiograph of stifle joints of an affected puppy. The ossification centers are poorly mineralized (long arrows) and the proximal fibular epiphyses are non-mineralized (short arrows). E. Lateral image of an unaffected puppy indicates that the ossification centers are normal for the age (long arrows) and proximal epiphysis of the fibula is clearly seen (short arrow). F. Ventrodorsal radiograph of an affected puppy. Lateral luxation of the left radial head is evident.
Clinical examination of three affected dogs indicated swollen knee joints and patellar luxation. Neurologically, all the examined dogs were alert and exhibited no remarkable neurological deficits. Postmortem examination of two puppies (one newborn and one 7 weeks old) did not reveal any additional gross abnormalities.
To identify the genetic cause of the syndrome, we performed GWAS with Illumina’s CanineHD array in a cohort of 12 Wire Fox Terriers including 4 cases and 8 controls. A case-control association test revealed association on chromosome 26 with seven nearby SNPs at 29,607,333 to 31,863,083 Mb (praw = 7.74x10-6, pgenome = 0.05) (genomic inflation factor λ = 1.10) (Fig 3A). Manual assessment of genotypes at the CFA5 locus revealed a shared homozygosity segment of ~3 Mb in the affected dogs spanning from 29,176,909 to 32,226,403 bp (Fig 3B). The associated region was captured and resequenced from five samples including two affected, two healthy and one obligate carrier Wire Fox Terrier (S4 Table). We identified a 2-bp homozygous deletion in exon 6 of the SCARF2 gene in the affected dogs after filtering the data according to an autosomal recessive model and against additional 169 unaffected control dogs from different breeds (S2 Table). The identified SCARF2 c.865_866delTC variant results in a frameshift and a premature stop codon, (p.S289Gfs*15), leading to a truncated protein in the first half of the coding region (Fig 3C).
Fig 3. Genetic analyses in a developmental syndrome in Wire Fox Terriers reveal a deletion in exon 6 of SCARF2, resulting in an early truncation of the predicted protein.

A. A Manhattan plot from a GWAS indicates a locus on chromosome 26 (pgenome = 0.05). B. A 3.3-Mb disease-associated haplotype in the affected dogs. C. Targeted resequencing revealed a 2-bp deletion in exon 6 (c.865-866delTC), resulting in a severe truncation at Ser289 that removes the transmembrane and cytoplasmic domains of the predicted protein as indicated by a dashed line in the schematic structure. D. RT-PCR in the affected and control Wire Fox Terrier indicate expression of SCARF2 mRNA and suggest that the mutated SCARF2 transcript is not directed to nonsense-mediated RNA decay.
Screening of the variant within the Wire Fox Terrier breed (57 dogs) confirmed full segregation with the disease (S5 Table). The four cases from the GWA study and an additional case were homozygous for the variant, while obligate carriers were heterozygous. The larger screening for the mutation revealed one homozygous dog that was a littermate of one of the genotyped affected dogs. This dog was then clinically examined, including neurological examination, radiography and CT scanning. The clinical findings confirmed the disease, including mandibular prognathia and patellar luxation. The carrier frequency among the population controls (n = 45) was 22%.
The effect of the 2-bp deletion on the stability of the SCARF2 mRNA was investigated by RT-PCR in postmortem skin samples from one affected and one unaffected dog (Fig 3D). The SCARF2 mRNA was detected both in affected and unaffected samples suggesting that the mutated transcript is not affected by nonsense-mediated RNA decay in the studied tissue.
SCARF2 defects have been reported in the rare human bone disease van den Ende-Gupta syndrome. Our results thus established an orthologous canine model with clinical similarity.
A missense variant in FAM20C causes dental hypomineralization in Border Collies
We were approached by a Border Collie breeder with a family of several affected dogs that suffered from severe tooth wear resulting in pulpitis and requiring extraction of those teeth. Further inspection of the tooth problem in the breed identified additional related cases, suggesting an autosomal recessive mode of inheritance (S1 Fig). Two affected dogs were subjected to a clinical study, including dental examination and radiography, as well as to histology of the extracted teeth and were regularly followed up in the next years. In addition, dental radiographs were available from two other cases.
Dental examination of a neutered 9-year-old female Border Collie revealed that all remaining teeth had significant wear. The previous dental treatment was performed 2 years earlier and multiple teeth were extracted. The length of the crowns was reduced. Lower incisor teeth were worn close to gingival margin. The enamel appeared dull and had light brown discoloration. The worn occlusal surfaces were discolored dark brown and there was reparative dentin formation. There were five teeth that had pulp exposure and pulpitis as a result of the wear (Fig 4A). The dog’s occlusion was normal and, therefore, the dental wear was not caused by abnormal tooth-to-tooth contact (attrition). Calcitriol (1,25(OH)2D3), phosphate and alkaline phosphatase levels in blood were normal. The other dental examination was performed for a neutered 10-year-old male Border Collie, revealing similar findings (Fig 4B and 4C). Other external causes such as abrasive hard chews were excluded as a cause of dental wear in both affected dogs.
Fig 4. The dental phenotype in the Border Collies with a homozygous FAM20C variant.

A. X-ray of the anterior mandibular teeth of a 9-year-old female Border Collie. Arrows indicate apical inflammations. B. X-ray of the anterior mandibular molars and premolars of a 10 year old male Border Collie indicating dental wear and apical inflammations (arrows). C. Anterior mandibular molars and premolars of the same dog indicating dental wear causing pulpal exposure. D. Ground section of a deciduous incisor from an unaffected Border Collie showing intact enamel and normal dentin, the crack is artefactual. E. Ground section of a permanent incisor of an affected 9-year-old female Border Collie. The enamel layer (e) preserved cervically is thin as compared to the control tooth (D). Wear at the edge (arrow) of tooth extends to the peripheral regular dentin (rd) and deeper to the abnormal globular dentin (gd), not present in the control (D). F. Ground section from deeper position of the same tooth as in (E) showing the layerwise alteration in the dentin structure. Peripheral dentin (rd) has a normal tubular pattern with no globules, the middlemost zone is pronouncedly globular (gd), while the central zone (cd) next to the diminished or almost completely obliterated pulp chamber (p) shows a slightly irregular tubular pattern but no globules. G. Haematoxylin-eosin stained paraffin section of a premolar from a 9-year old female. Contiguous interglobular spaces (arrows) in the middlemost dentin zone stain lightly with eosin. Tubules run uninterruptedly in stained areas. Bars, 50 um (D,E,F) and 200 um (G).
Extracted teeth from two affected dogs, a 9-year-old female Border Collie described above and its female littermate, were submitted to histopathological examinations. The analysis of ground sections did not reveal structural aberrations but the enamel of the incisor was smooth and slightly hypoplastic as compared to the unaffected control dog (Fig 4D and 4E). The enamel of the premolar had largely worn and cracked, but the cervical enamel, which was preserved, showed no structural defects. Coronal dentin of both teeth comprised three distinct, circumferential zones. The tubular pattern and the structure of the matrix in the thick peripheral zone subjacent to the enamel were regular. The middlemost zone was pronouncedly globular (Fig 4E and 4F). The neighboring globules largely adapted to each other and the tubules ran uninterrupted. Wider interglobular spaces were filled with air. The globules diminished and disappeared in the central direction. The central dentin zone next to the reduced pulp showed no matrix defects, but the tubular pattern was slightly irregular (Fig 4F). The proportion of globular dentin gradually reduced in the apical direction.
The analysis of paraffin sections demonstrated significant wear of the dentin. The structure of the peripheral zone was regular, whereas the middlemost zone was globular. Between the globules there were wide, contiguous defects with an angular contour, void of matrix and tubules and filled with amorphous, barely detectable material (Fig 4G). A pulp chamber did not become visible at the level of the sections. Tubules in the central dentin zone were slightly irregular. Pulp tissue in the root canal was necrotic. A patchy, chronic inflammatory cell infiltrate with plasma cells predominating was present apically. The structure of the acellular cementum was regular. Lacunae in the cellular cementum and at the periphery of the alveolar bone trabeculae were in places obliterated. Demarcated areas in the periodontal ligament facing the alveolar bone showed an amorphous, fibrotic texture. Overall, clinical and histopathological analyses indicate severe hypomineralization of teeth in the affected dogs.
To identify the cause of the mineralization defect we sequenced the whole genomes of three affected dogs. The variants of the affected dogs were filtered against two unaffected obligate carriers and fourteen other unaffected Border Collie genomes assuming recessive transmission, resulting in the identification of a case-specific non-synonymous homozygous variant, c.899C>T, in the FAM20C gene. This leads to a missense change, p.A300V, in a highly conserved position in the kinase domain of the FAM20C protein (Fig 5). Bioinformatic predictions by SIFT (with a score of 0.00) and Polyphen2 (with a HumVar score of 0.992) suggested pathogenicity. Genotyping the pedigree and additional Border Collies (191 dogs) demonstrated complete segregation of the variant with the disease phenotype (S1 Fig and S6 Table) and showed 11% carrier frequency in the breed. We also genotyped 186 dogs from 20 additional breeds (S6 Table), but did not find any carriers in the other breeds suggesting that this pathogenic variant is specific for Border Collies.
Fig 5. Whole genome sequencing of three Border Collies affected with tooth attrition reveals a missense variant, FAM20C c.899C>T, in the conserved kinase domain of the protein (p.A300V).
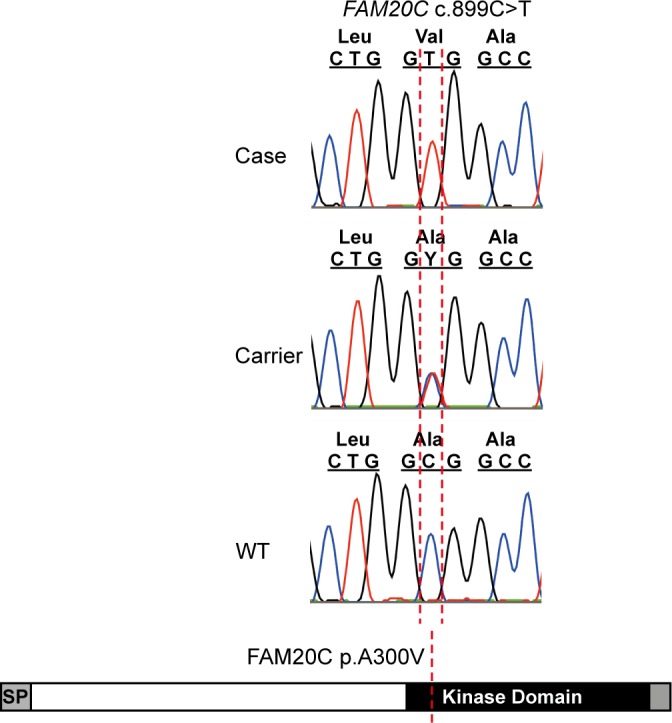
Defects in FAM20C have been associated with a rare human disease, Raine syndrome/FGF23-related hypophosphatemia characterized by dental and bone hypomineralization. Our results indicated a causative variant in the kinase domain of FAM20C and established a canine model for human Raine syndrome.
Discussion
This study demonstrates the power of NGS approaches to establish molecular diagnosis and categorization for unknown congenital canine disorders with high relevance to rare human diseases. We identified a novel candidate gene, SLC37A2, for the corresponding human disease, infantile cortical hyperostosis, also known as Caffey disease, and implicated SCARF2 and FAM20C variants in the canine forms of van den Ende-Gupta and Raine syndromes, respectively. All three spontaneous canine models closely resemble the human syndromes and provide physiologically relevant models to better understand poorly characterized gene functions in each condition and the entire molecular pathologies. Given the growing interest in the development of new therapies for rare human diseases, our study highlights the fact that dogs carry clinically and genetically close counterparts of rare human diseases that could be better utilized to advance molecular research and to develop efficient preclinical trials in large animals with spontaneous diseases [13, 14]. Many human rare disease models exist in dogs, and encouraging private pet owners to participate in clinical trials could facilitate the validation of human treatment approaches, while also benefitting canine health.
Identification of the canine CMO variant implicates a sugar-phosphate transporter in hyperostosis
Our study unraveled a physiological function of SLC37A2 and provided new insights into infantile swelling diseases, which may be related to disturbances in the intracellular glucose homeostasis during bone development. We identified an unusual leaky splicing defect in the SLC37A2 gene in CMO-affected dogs. CMO is a self-limiting hyperostosis in multiple bones in young dogs. The most prominent sign in the affected puppies included painful swelling of the jaw, leading to dysphagia and difficulty in opening the mouth. CMO is clinically equivalent to human infantile cortical hyperostosis [10, 15]. A common missense variant in COL1A1 has been found in several patients with an autosomal dominant condition with incomplete penetrance [16, 17]. Interestingly, the mechanism how the defected collagen leads to self-limiting hyperostotic bone lesions is still unknown. Early molecular diagnosis of Caffey patients would avoid invasive procedures, however, the molecular etiology remains unknown in many cases.
SLC37A2 represents a new functional candidate gene. It belongs to the SLC37 family of four ER-associated glucose-phosphate transporters [18]. SLC37A2 is ubiquitously expressed, but transcript and protein levels are particularly high in bone-related tissues such as bone marrow and hematopoietic cell linages such as osteoclasts and macrophages [19, 20]. Murine Slc37a2 was shown to be one of the genes strongly involved in the osteoclast differentiation, suggesting that it plays a role in osteoclast function and differentiation [20]. Therefore, SLC37A2 may play a central role in glucose homeostasis in the key cell types that participate in osteogenesis. For example, an impaired function of SLC37A2 due to a truncating splice variant might disturb proper glucose supply in the osteoclasts, decreasing their overall activity, which in turn would result in an imbalance between osteoblastic and osteoclastic functions in the developing bones eventually leading to hyperostosis. Defects in SLC37A4, glucose-6-phosphate transporter (G6PT), have been associated with glycogen storage disease 1b and 1c, characterized by recurrent infections and neutropenia due to disturbed blood glucose metabolism [21–23]. Our results link SLC37A2 to bone physiology and disease, and we propose SLC37A2 as an excellent candidate for genetic screening in Caffey patients. Meanwhile, the affected dogs provide unique resources for future experiments to address SLC37A2-related mechanisms in osteogenesis biology. A recent study in hematopoietic cells identified SLC37A2 as a primary vitamin D target with a conserved vitamin D receptor-binding site [24]. This may open investigations to study the opportunity to use vitamin D as a therapeutic booster to regulate diminished expression of wild type expression of SLC37A2 in the affected dogs to alleviate clinical signs.
Caffey disease is an autosomal dominant disease with incomplete penetrance, although rare cases of recessively inherited Caffey disease have also been reported [25]. Corresponding canine diseases exist in several terrier breeds with the highest frequency in West Highland White Terriers. The determination of the exact mode of inheritance in dogs is not straightforward due to the nature of the leaky splice variant and mild self-limiting phenotype that may remain unobserved and prevent retrospective diagnosis. We found some dogs that were homozygous for the variant but had no reported clinical signs. However, we observed a considerable level of the wild-type SLC37A2 transcript in homozygous dogs in the peripheral blood due to the splicing leakage, suggesting that the leaky expression is sufficient to avoid a clinical phenotype in some cases. We also found several heterozygous dogs that had developed CMO. We found that heterozygous dogs had lower levels of wild-type SLC37A2 transcript compared to the unaffected dogs with individual variation of expression between dogs. This result suggested a dominant disease with incomplete penetrance that could help to explain the reported differences in the severity and duration of CMO among the affected dogs, although alternative models of inheritance cannot be completely ruled out yet. The dominant phenotype could be due to a dominant-negative effect, but this hypothesis requires further experimental validation to better understand the details of the gene, its regulation and protein function, including potential pairing with other proteins as described for SLC37A4/G6Pase complexes [18]. The in vivo function of SLC37A4 has been shown to depend upon its ability to couple functionally with either G6Pase-a or G6Pase-b [18, 26].
A SCARF2 truncation in a canine model of van den Ende-Gupta syndrome
We identified a 2-bp deletion in SCARF2 in dogs with severe mandibular prognathia and other skeletal abnormalities and established a canine model for van den Ende-Gupta syndrome (VDEGS). VDEGS is a very rare disease with less than 30 reported cases [27]. It is characterized by a heterogeneous variety of craniofacial and skeletal abnormalities including blepharophimosis, a flat and wide nasal bridge, narrow and beaked nose, hypoplastic maxilla with or without cleft palate and everted lower lip, prominent deformed ears, down-slanting eyes, arachnodactyly, and camptodactyly. Patients may present congenital joint contractures that improve without intervention, and have normal growth and development. Enlarged cerebellum is an infrequent finding yet intelligence is normal. Some patients experience respiratory problems due to laryngeal abnormalities. Human and canine VDEGS patients share many similarities including hypoplastic maxilla, dislocated radial head, patellar dislocation, and deviated nasal septum. Both have small eyes.
It remains unknown how the loss of function of SCARF2 leads to VDEGS. SCARF2 is a poorly characterized member of the scavenger receptor type F family [28]. Besides epidermis, Scarf2 is expressed in branchial arches, mandible, maxilla and urogenital ridge tissue of developing mouse embryos [29, 30]. It is a single-pass transmembrane protein with homology to calmodulin (CaM)-like Ca2+-binding protein genes. The extracellular domain contains several putative epidermal growth factor-like (EGF) domains, and it has a number of positively charged residues within the intracellular domain, suggesting a role in intracellular signaling. The 2-bp deletion of the canine SCARF2 gene in one of the extracellular EGF domains leads to a severely truncated protein that completely lacks the transmembrane and intracellular domains. The lack of a transgenic mouse model and scarcity of human patients highlight the role of affected dogs as a novel resource to understand SCARF2 functions and molecular pathology. As some of the affected dogs survive past 10 years, they could potentially serve also as preclinical models.
A missense variant in FAM20C identifies a canine model of Raine syndrome
Whole genome sequencing of a family of several affected dogs that suffered from a severe dental wear and loss of teeth revealed a recessive missense variant in the kinase domain of the FAM20C gene. FAM20C defects cause autosomal recessive osteosclerotic bone dysplasia (Raine syndrome) in humans. This rare syndrome with less than 40 reported cases was originally described to be neonatal lethal, but recently there have been several reports of cases surviving into childhood with variable severity and clinical heterogeneity [31–38]. Typical characteristics in Raine syndrome include craniofacial anomalies, such as exophthalmos, abnormal and hypomineralized teeth, midface hypoplasia, microcephaly and cleft palate, as well as gingival hyperplasia, generalized osteosclerosis and intracerebral calcifications. Variable extent of hypophosphatemia has been observed, sometimes as the primary diagnosis [32, 35, 38]. Canine findings in our cohorts were more limited to severe hypomineralization of teeth, leading to extensive wear and inflammation as prominent features. We did not observe some of the typical gross changes described in Raine patients such as hypophosphatemia and craniofacial anomalies. However, there is a significant clinical heterogeneity in the symptoms between human patients and more detailed radiographic analyses should be performed in dogs to observe potential mild changes outside the dental phenotype.
FAM20C is a Golgi casein kinase that phosphorylates secretory proteins such as FGF23 and SIBLING (Small Integrin-Binding Ligand, N-linked Glycoprotein) family [39, 40]. Fam20c is significantly expressed in mouse teeth and bone and transgenic mice studies have indicated a role in differentiation and mineralization of odontoblasts, ameloblasts, osteoblasts and osteocytes during tooth and bone development. FAM20C-deficient mice also have a prominent dental phenotype [41–43]. Ablation of the Fam20c gene in conditional knockout mice affects tooth and bone development by downregulation of SIBLING family of proteins such as DMP1 and DSPP and by increasing FGF23 in serum and promoting phosphate excretion and hypophosphatemia [44]. Our FAM20C-deficient dogs have a dental phenotype similar to mice and humans and provide a new research and preclinical model for this rare human bone disease. Unlike rodents, dogs have dental physiology similar to human, having both deciduous and permanent dentition.
In summary, we describe here the clinical and genetic characteristics of three new canine models for rare human bone disorders. While highlighting clinical and genetic similarities between canine and human conditions, our study have several implications; it indicates new physiological functions for the identified genes and provides new candidate genes to rare human diseases, establishes potential preclinical models, and finally enables the development of genetic tests for veterinary diagnostics and breeding purposes.
Materials and Methods
Ethics statement
Sample collection in Finland was ethically approved by the Animal Ethics Committee of State Provincial Office of Southern Finland (Finland, ESAVI/6054/04.10.03/ 2012). The collection of blood samples in Switzerland was approved by the “Cantonal Committee For Animal Experiments” (Canton of Bern; permit 23/10).
Study cohorts
EDTA-blood and tissue samples were collected from privately owned dogs in Finland, US and Switzerland. The samples were stored at -20°C until genomic DNA was extracted using the semi-automated Chemagen extraction robot (PerkinElmer Chemagen Technologie GmbH). DNA concentration was determined either with the NanoDrop ND-1000 UV/Vis Spectrophotometer or Qubit 3.0 Fluorometer (Thermo Fisher Scientific Inc.). Pedigrees were drawn by the GenoPro genealogy software (http://www.genopro.com/), and utilizing the public dog registry by the Finnish Kennel Club (http://jalostus.kennelliitto.fi).
Clinical examinations for each condition are described in detail in the results section. CMO cases were diagnosed by radiography by local veterinarians. The developmental disorder in Wire Fox Terriers was investigated by radiography and CT. Neurological examination was performed for three and ophthalmoscopy for two of the affected dogs. A specialized dental veterinarian examined two of the affected Border Collies with dental hypomineralization, while the others were examined by local veterinarians. Clinical phenotype information was not available for dogs used as population controls.
Genome-wide association study and fine mapping
Genome-wide association studies were performed in the CMO and VDEGS projects. For CMO, altogether 51 dogs including 10 affected and 41 control dogs, were genotyped using Illumina’s CanineSNP20 BeadChip of 22,362 validated SNPs. For VDEGS, a total of 15 dogs including 4 affected and 11 unaffected Wire Fox Terriers, were genotyped using Illumina’s HD array. The genotype data in both projects was filtered with a SNP call rate of >95%, array call rate of >95% and minor allele frequency of >5%. No individual dogs were removed for low genotyping and no SNPs were removed because of significant deviations from the Hardy-Weinberg equilibrium (p ≤ 0.0001). After frequency and genotyping pruning, 14,835 and 69,694 SNPs remained for analyses for CMO and VDEGS data, respectively. Basic case-control association test was performed by PLINK [45]. Genome-wide significance was ascertained with phenotype permutation testing (n = 10,000 for CMO and n = 100,000 for VDEGS). Fine mapping of the identified CMO locus was performed with 105 selected SNPs from a 1.9-Mb region (7,764,955–9,707,794 bp) on CFA5 (based on CanFam3.1). The SNPs were selected using Broad Institute SNP collection CanFam2.0. Genotyping was performed using the Sequenom (San Diego, CA, USA) iPLEX methodology at our local core facility in the FIMM Technology Centre, University of Helsinki, Finland. A total of 88 samples were genotyped including 8 cases and 8 controls in Cairn Terriers, 9 cases and 8 controls in Scottish Terriers and 29 cases and 26 controls in WHWTs. Association analysis was performed with PLINK using a single-marker association analysis.
Targeted capture and resequencing
We performed a targeted sequence capture and next generation sequencing to identify the pathogenic variants. We used NimbleGen’s in-solution capture technology to enrich the target regions for sequencing (Roche NimbleGen, Madison, WI, USA). We captured 1.8-Mb region of CMO associated locus at position CFA5: 10,750,000–12,550,000 using two WHWT cases and controls with opposite haplotypes. The haplotypes were assessed manually using SNP genotype data. The same targeting experiment also contained samples from our other targeting projects including 8 Border Terriers, 12 Duck Tolling Retrievers, 8 Schipperkes and 4 Brazilian Terriers and these samples were used as additional controls. For target enrichment and sequencing of associated locus in Wire Fox Terriers, we captured a 3.3-Mb region at CFA26: 29,030,700–32,328,700 using two affected and two controls with opposite haplotypes and one obligate carrier. The filtered case-specific variants were further checked from 169 additional dogs in our variant database. Probes in the target regions were designed by Roche NimbleGen (Roche NimbleGen). Target enrichment, alignment and variant calling pipeline were performed as previously described [8]. Further data analysis was performed using open source R language and environment (http://www.r-project.org). Canine genome build CanFam3.1 was used as a reference sequence.
Whole genome sequencing
The genetic causes of CMO in WHWTs and tooth attrition in Border Collies were studied by whole genome sequencing. In the CMO study, we performed whole genome sequencing of one affected WHWT dog and used 188 other available whole genomes as controls (S2 Table). A fragment library was prepared with a 290 bp insert size and collected a single lane of Illumina HiSeq2000 paired-end reads (2 x 100 bp). The reads were mapped to the dog reference genome using the Burrows-Wheeler Aligner (BWA) version 0.5.9-r16 with default settings. The Picard tools (http://sourceforge.net/projects/picard/) were used to sort the mapped reads by the sequence coordinates and to label the PCR duplicates. The Genome Analysis Tool Kit (GATK version v2.3–6) was used to perform local realignment and to produce a cleaned BAM file. Variant calls were then made using the unified genotyper module of GATK. Variant data was obtained in variant call format (version 4.0) as raw calls for all samples and sites flagged using the variant filtration module of GATK. Variant calls that failed to pass the following filters were labeled accordingly in the call set: (i) Hard to Validate MQ0 ≥ 4 & ((MQ0 / (1.0 * DP)) > 0.1); (ii) strand bias (low Quality scores) QUAL < 30.0 || (Quality by depth) QD < 5.0 || (homopolymer runs) HRun > 5 || (strand bias) SB > 0.00; (iii) SNP cluster window size 10. The SnpEff software together with the CanFam 3.1 annotation was used to predict the functional effects of detected variants. In addition to the SNP and short indel variant calling, large deletions contained in the candidate region were searched by visual inspection of the BAM file using the Integrative Genomics Viewer (IGV). In the Border Collie study, we whole genome sequenced altogether nineteen dogs, including three affected dogs, two carriers and fourteen unaffected Border Collies, in the Science for Life Laboratory in Stockholm, Sweden. The reads were processed using speedseq align module available in SpeedSeq suite to produce a duplicate-marked, sorted and indexed BAM file. The Genome Analysis Tool kit (version = 3.3.0-g37228af) was used to perform realignment around potential indel sites and base quality score recalibration using the known SNP variation available at the Broad Institute (https://www.broadinstitute.org/ftp/pub/vgb/dog/trackHub/canFam3/variation). Dual algorithms, Samtools mpileup (version samtools-1.2) and GATK haplotype caller module were used to detect variants and the variants from both algorithms were merged into variant call format (VCFv4.1). Annovar and SnpEff tools were used to annotate the variants to Ensembl, NCBI and Broad annotation databases to predict the functional effects of the variants. We identified on average ~6 million variants per sample and the sequencing coverage varied between 22-49x. The three affected dogs shared ~1.5 million homozygous variants. Filtering under recessive model against two carriers and fourteen controls left 2690 homozygous variants in total, of which five variants were in the predicted coding regions (1 indel, 2 non-synonymous, 1 synonymous).
Genomic DNA analysis
Large numbers of dogs were genotyped for the identified variants using various protocols. Genotyping of individual dogs for the CMO variant was performed either by TaqMan assay (Applied Biosystems) or by sequencing a 786-bp PCR product using a forward primer (5-GGCTCCAGTCTAAGCCAGGT-3) and a reverse primer (5-AAGGAGTGCGCTCAAGACAG-3) flanking the SLC37A2 SNP. The PCR products were amplified with AmpliTaqGold360Mastermix (Life Technologies), and the products were directly sequenced using the PCR primers on an ABI 3730 capillary sequencer (Life Technologies) after treatment with exonuclease I (New England Biolabs) and rapid alkaline phosphatase (Roche). The sequence data were analyzed using Sequencher 5.1 (GeneCodes). Potential exonic splice enhancer (ESE) motifs were detected with ESEfinder 3.0 [46, 47].
Pathogenicity of the FAM20C c.899C>T variant was evaluated using web-based bioinformatic prediction tools SIFT [48] and PolyPhen-2 (genetics.bwh.harvard.edu/pph2) [49] was applied to evaluate the pathogenic effect of the mutation. SIFT score ranges from 0 to 1. The amino acid substitution is predicted to be damaging if the score is smaller than 0.05. PolyPhen-2 score ranges from 0 to 1. The amino acid substitution is predicted to be damaging if the score is bigger than 0.85.
Genotyping of FAM20C and SCARF2 variants was performed by standard PCR with the following primers: FAM20C: 5-GCTTCTATGGCGAGTGTTCC-3 and 5-CCGGGATGTCTGAGTAAGGA-3; SCARF2:5-CAATCCCCGAGTGCTCTCC-3 and 5-AGGAAACTGCCCCCAAAGAG-3.
Transcript analysis
Expression analyses were performed for the CMO and VDEGS projects. Blood samples were collected into PAXgene tubes (PreAnalytix) and RNA was isolated using PAXgene Blood RNA Kit (Qiagen). The cDNA synthesis was performed using SuperScriptIII enzyme (Invitrogen) with an oligo d(T)24V primer according to manufacturer’s instructions. To investigate possible aberrant splicing events, we amplified cDNAs from the junction of exons 7 and 8 to the junction of exons 17 and 18 of the SLC37A2 gene (810 bp) using SequalPrep Long Polymerase (Invitrogen) with a forward primer GAATACCCAGAAGACGTGGAC and a reverse primer CCTCTGTCTCTGTTCAGGAATG in 16 WHWTs. B2M was used as a loading control. The identity of the amplicons was confirmed by Sanger sequencing. In the VDEGS project, the possible effect of the 2-bp deletion on the stability of the SCARF2 transcript was investigated by RT-PCR. Total RNAs were isolated from skin samples from one affected and unaffected dog obtained in the postmortem autopsies. The cDNA synthesis was performed as described above. Forward 5-CAACCACGTCACTGGCAAGT-3 and reverse 5-TTACAGTGGGG CCCGTGG-3 primers were designed to amplify a 188-bp region between exon 6 and exon 8 of SLC37A2. Semi-quantitative analysis of the expression was determined and visualized by electrophoresis.
Histological examination of teeth
Permanent and deciduous incisor and premolar teeth were removed for therapeutic reasons from two Border Collie bitches with clinically affected teeth. The dogs were from the same litter. Three teeth, two incisors and one premolar, were obtained from one dog. One incisor and the premolar were processed to ground sections and the other incisor to paraffin sections. From the other dog, four teeth, two incisors and two premolars, were obtained. One incisor and one premolar were processed to ground sections and the other incisor and the other premolar to paraffin sections. For comparison, deciduous teeth obtained from a healthy Border Collie were studied. One incisor was processed to ground sections and one tooth to paraffin sections. The teeth to be processed to ground sections (a procedure preserving the enamel with a proportionally high mineral content and sparse organic matrix) were fixed with 10% neutral buffered formalin, dehydrated and embedded in liquid methylmethacrylate monomer. After complete polymerization, started up by benzoylperoxide (2 g/l), the teeth were serially cut to 100–150 μm thick longitudinal sections with a rotating diamond-coated saw microtome, let dry and mounted unstained with DePex (Gurr, BDH, Poole, UK). For preparation to paraffin sections, the teeth were fixed with formalin, demineralized with EDTA (0.33 mol/l, pH 7.2), which leads to the loss of the enamel, and embedded in paraffin. A representative series of sections were longitudinally cut at 7 μm and stained with haematoxylin and eosin (HE).
Supporting Information
Dogs that were whole genome sequenced are indicated by open red box. Full segregation of the FAM20C c.899C>T variant with the disease according to recessive model is indicated.
(PDF)
(DOCX)
(XLSX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
Acknowledgments
We are grateful to Reetta Hänninen, Ranja Eklund, Sini Karjalainen, Marjatta Kivekäs and Minna Virta for technical assistance. Kirsi Sainio is thanked for encouraging breeders to participate in the CMO and VDEGS studies. We thank all dog breeders and owners who contributed samples and participated in this study.
Data Availability
The genome data is freely available under accession no. PRJEB13723 (http://www.ebi.ac.uk/ena/data/view/PRJEB13723) at the European Nucleotide Archive and accession no. SRP074053 at the NCBI Sequence Read Archive.
Funding Statement
This work was partially supported by the Academy of Finland (1268091), the Sigrid Juselius Foundation, the Jane and Aatos Erkko Foundation, ERCStG (260997), Biocentrum Helsinki, the Morris Animal Foundation (D13CA-403) and the Jenni and Antti Wihuri Foundation. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Scriver CR. (1995) Disease, war, and biology: Languages for medicine—and pediatrics. Pediatr Res 38(6): 819–829. [DOI] [PubMed] [Google Scholar]
- 2.Rabbani B, Mahdieh N, Hosomichi K, Nakaoka H, Inoue I. (2012) Next-generation sequencing: Impact of exome sequencing in characterizing mendelian disorders. J Hum Genet 57(10): 621–632. 10.1038/jhg.2012.91 [DOI] [PubMed] [Google Scholar]
- 3.Tønnessen R, Borge KS, Nødtvedt A, Indrebø A. (2012) Canine perinatal mortality: A cohort study of 224 breeds. Theriogenology 77(9): 1788–1801. 10.1016/j.theriogenology.2011.12.023 [DOI] [PubMed] [Google Scholar]
- 4.Kyöstilä K, Syrjä P, Jagannathan V, Chandrasekar G, Jokinen TS, et al. (2015) A missense change in the ATG4D gene links aberrant autophagy to a neurodegenerative vacuolar storage disease. PLoS Genet 11(4):e1005169 10.1371/journal.pgen.1005169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Drögemüller M, Jagannathan V, Becker D, Drögemüller C, Schelling C et al. (2014) A mutation in the FAM83G gene in dogs with hereditary footpad hyperkeratosis (HFH). PLoS Genet. 10(5):e1004370 10.1371/journal.pgen.1004370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ahonen SJ, Arumilli M, Lohi H. (2013) A CNGB1 frameshift mutation in Papillon and Phalène dogs with progressive retinal atrophy. PLoS One 8(8):e72122 10.1371/journal.pone.0072122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bareil C, Hamel C, Delague V, Arnaud B, Demaille J, et al. (2001) Segregation of a mutation in CNGB1 encoding the β-subunit of the rod cGMP-gated channel in a family with autosomal recessive retinitis pigmentosa. Hum Genet 108: 328–334. [DOI] [PubMed] [Google Scholar]
- 8.Hytönen MK, Arumilli M, Lappalainen AK, Kallio H, Snellman M, et al. (2012) A novel GUSB mutation in brazilian terriers with severe skeletal abnormalities defines the disease as mucopolysaccharidosis VII. PLoS One 7(7): e40281 10.1371/journal.pone.0040281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Littlewort MCG. (1958) Tumor-like exostoses on the bones of the head in puppies. The Veterinary Record 70: 977–978. [Google Scholar]
- 10.Thornburg LP. (1979) Infantile cortical hyperostosis (Caffey-Silverman syndrome). animal model: Craniomandibular osteopathy in the canine. Am J Pathol 95(2): 575–578. [PMC free article] [PubMed] [Google Scholar]
- 11.LaFond E, Breur GJ, Austin CC. (2002) Breed susceptibility for developmental orthopedic diseases in dogs. J Am Anim Hosp Assoc 38(5): 467–477. [DOI] [PubMed] [Google Scholar]
- 12.Padgett GA, Mostosky UV. (1986) The mode of inheritance of craniomandibular osteopathy in West Highland White Terrier dogs. Am J Med Genet 25(1): 9–13. [DOI] [PubMed] [Google Scholar]
- 13.Cideciyan AV, Jacobson SG, Beltran WA, Sumaroka A, Swider M, et al. (2013) Human retinal gene therapy for leber congenital amaurosis shows advancing retinal degeneration despite enduring visual improvement. Proc Natl Acad Sci U S A 110(6): E517–25. 10.1073/pnas.1218933110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Casal ML, Lewis JR, Mauldin EA, Tardivel A, Ingold K, et al. (2007) Significant correction of disease after postnatal administration of recombinant ectodysplasin A in canine X-linked ectodermal dysplasia. Am J Hum Genet 81(5): 1050–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nistala H, Makitie O, Juppner H. (2014) Caffey disease: New perspectives on old questions. Bone 60: 246–251. 10.1016/j.bone.2013.12.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gensure RC, Mäkitie O, Barclay C, Chan C, Depalma SR, et al. (2005) A novel COL1A1 mutation in infantile cortical hyperostosis (Caffey disease) expands the spectrum of collagen-related disorders. J Clin Invest 115(5): 1250–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kitaoka T, Miyoshi Y, Namba N, Miura K, Kubota T, et al. (2014) Two Japanese familial cases of Caffey disease with and without the common COL1A1 mutation and normal bone density, and review of the literature. Eur J Pediatr 173(6):799–804. 10.1007/s00431-013-2252-8 [DOI] [PubMed] [Google Scholar]
- 18.Chou JY, Mansfield BC. (2014) The SLC37 family of sugar-phosphate/phosphate exchangers. Curr Top Membr 73: 357–382. 10.1016/B978-0-12-800223-0.00010-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim JY, Tillison K, Zhou S, Wu Y, Smas CM. (2007) The major facilitator superfamily member Slc37a2 is a novel macrophage- specific gene selectively expressed in obese white adipose tissue. Am J Physiol Endocrinol Metab 293(1): E110–20. [DOI] [PubMed] [Google Scholar]
- 20.Ha BG, Hong JM, Park JY, Ha MH, Kim TH, et al. (2008) Proteomic profile of osteoclast membrane proteins: Identification of Na+/H+ exchanger domain containing 2 and its role in osteoclast fusion. Proteomics 8(13): 2625–2639. 10.1002/pmic.200701192 [DOI] [PubMed] [Google Scholar]
- 21.Veiga-da-Cunha M, Gerin I, Chen YT, de Barsy T, de Lonlay P, et al. (1998) A gene on chromosome 11q23 coding for a putative glucose- 6-phosphate translocase is mutated in glycogen-storage disease types ib and ic. Am J Hum Genet 63(4): 976–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kure S, Suzuki Y, Matsubara Y, Sakamoto O, Shintaku H, et al. (1998) Molecular analysis of glycogen storage disease type ib: Identification of a prevalent mutation among Japanese patients and assignment of a putative glucose-6-phosphate translocase gene to chromosome 11. Biochem Biophys Res Commun 248(2): 426–431. [DOI] [PubMed] [Google Scholar]
- 23.Gerin I, Veiga-da-Cunha M, Achouri Y, Collet JF, Van Schaftingen E. (1997) Sequence of a putative glucose 6-phosphate translocase, mutated in glycogen storage disease type ib. FEBS Lett 419(2–3): 235–238. [DOI] [PubMed] [Google Scholar]
- 24.Saksa N, Neme A, Ryynänen J, Uusitupa M, de Mello VD, et al. (2015) Dissecting high from low responders in a vitamin D3 intervention study. J Steroid Biochem Mol Biol 148: 275–282. 10.1016/j.jsbmb.2014.11.012 [DOI] [PubMed] [Google Scholar]
- 25.Schweiger S, Chaoui R, Tennstedt C, Lehmann K, Mundlos S, et al. (2003) Antenatal onset of cortical hyperostosis (caffey disease): Case report and review. Am J Med Genet A 120A(4): 547–552. [DOI] [PubMed] [Google Scholar]
- 26.Shieh JJ, Pan CJ, Mansfield BC, Chou JY. (2003) A glucose-6-phosphate hydrolase, widely expressed outside the liver, can explain age-dependent resolution of hypoglycemia in glycogen storage disease type IA. J Biol Chem 278(47): 47098–47103. [DOI] [PubMed] [Google Scholar]
- 27.Anastasio N, Ben-Omran T, Teebi A, Ha KCH, Lalonde E, et al. (2010) Mutations in SCARF2 are responsible for van den Ende-Gupta syndrome. Am J Hum Genet 87(4): 553–559. 10.1016/j.ajhg.2010.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ishii J, Adachi H, Aoki J, Koizumi H, Tomita S, et al. (2002) SREC-II, a new member of the scavenger receptor type F family, trans-interacts with SREC-I through its extracellular domain. J Biol Chem 277(42): 39696–39702. [DOI] [PubMed] [Google Scholar]
- 29.Hwang M, Kalinin A, Morasso MI. (2005) The temporal and spatial expression of the novel Ca++-binding proteins, Scarf and Scarf2, during development and epidermal differentiation. Gene Expr Patterns 5(6): 801–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hwang M, Morasso MI. (2003) The novel murine Ca2+-binding protein, SCARF, is differentially expressed during epidermal differentiation. J Biol Chem 278(48): 47827–47833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Takeyari S, Yamamoto T, Kinoshita Y, Fukumoto S, Glorieux FH, et al. (2014) Hypophosphatemic osteomalacia and bone sclerosis caused by a novel homozygous mutation of the FAM20C gene in an elderly man with a mild variant of Raine syndrome. Bone 67: 56–62. 10.1016/j.bone.2014.06.026 [DOI] [PubMed] [Google Scholar]
- 32.Rafaelsen SH, Raeder H, Fagerheim AK, Knappskog P, Carpenter TO, et al. (2013) Exome sequencing reveals FAM20c mutations associated with fibroblast growth factor 23-related hypophosphatemia, dental anomalies, and ectopic calcification. J Bone Miner Res 28(6): 1378–1385. 10.1002/jbmr.1850 [DOI] [PubMed] [Google Scholar]
- 33.Ababneh FK, AlSwaid A, Youssef T, Al Azzawi M, Crosby A, et al. (2013) Hereditary deletion of the entire FAM20C gene in a patient with Raine syndrome. Am J Med Genet A 161A(12): 3155–3160. 10.1002/ajmg.a.36160 [DOI] [PubMed] [Google Scholar]
- 34.Acevedo AC, Poulter JA, Alves PG, de Lima CL, Castro LC, et al. (2015) Variability of systemic and oro-dental phenotype in two families with non-lethal Raine syndrome with FAM20C mutations. BMC Med Genet 16: 8-015-0154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fradin M, Stoetzel C, Muller J, Koob M, Christmann D, et al. (2011) Osteosclerotic bone dysplasia in siblings with a Fam20C mutation. Clin Genet 80(2): 177–183. 10.1111/j.1399-0004.2010.01516.x [DOI] [PubMed] [Google Scholar]
- 36.Raine J, Winter RM, Davey A, Tucker SM. (1989) Unknown syndrome: Microcephaly, hypoplastic nose, exophthalmos, gum hyperplasia, cleft palate, low set ears, and osteosclerosis. J Med Genet 26(12): 786–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Simpson MA, Hsu R, Keir LS, Hao J, Sivapalan G, et al. (2007) Mutations in FAM20C are associated with lethal osteosclerotic bone dysplasia (Raine syndrome), highlighting a crucial molecule in bone development. Am J Hum Genet 81(5): 906–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Simpson MA, Scheuerle A, Hurst J, Patton MA, Stewart H, et al. (2009) Mutations in FAM20C also identified in non-lethal osteosclerotic bone dysplasia. Clin Genet 75(3): 271–276. 10.1111/j.1399-0004.2008.01118.x [DOI] [PubMed] [Google Scholar]
- 39.Ishikawa HO, Xu A, Ogura E, Manning G, Irvine KD. (2012) The Raine syndrome protein FAM20C is a golgi kinase that phosphorylates bio-mineralization proteins. PLoS One 7(8): e42988 10.1371/journal.pone.0042988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tagliabracci VS, Engel JL, Wen J, Wiley SE, Worby CA, et al. (2012) Secreted kinase phosphorylates extracellular proteins that regulate biomineralization. Science 336(6085): 1150–1153. 10.1126/science.1217817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang X, Wang J, Liu Y, Yuan B, Ruest LB, et al. (2015) The specific role of FAM20C in dentinogenesis. J Dent Res 94(2): 330–336. 10.1177/0022034514563334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang X, Jung J, Liu Y, Yuan B, Lu Y, et al. (2013) The specific role of FAM20C in amelogenesis. J Dent Res 92(11): 995–999. 10.1177/0022034513504588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang X, Wang S, Lu Y, Gibson MP, Liu Y, et al. (2012) FAM20C plays an essential role in the formation of murine teeth. J Biol Chem 287(43): 35934–35942. 10.1074/jbc.M112.386862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang X, Wang S, Li C, Gao T, Liu Y, et al. (2012) Inactivation of a novel FGF23 regulator, FAM20C, leads to hypophosphatemic rickets in mice. PLoS Genet 8(5): e1002708 10.1371/journal.pgen.1002708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, et al. (2007) PLINK: A tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81(3): 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Smith PJ, Zhang C, Wang J, Chew SL, Zhang MQ, et al. (2006) An increased specificity score matrix for the prediction of SF2/ASF-specific exonic splicing enhancers. Hum Mol Genet 15(16): 2490–2508. [DOI] [PubMed] [Google Scholar]
- 47.Cartegni L, Wang J, Zhu Z, Zhang MQ, Krainer AR. (2003) ESEfinder: A web resource to identify exonic splicing enhancers. Nucleic Acids Res 31(13): 3568–3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ng PC, Henikoff S. (2001) Predicting deleterious amino acid substitutions. Genome Res 11: 863–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, et al. (2010) A method and server for predicting damaging missense mutations. Nat Methods 7(4): 248–249. 10.1038/nmeth0410-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Dogs that were whole genome sequenced are indicated by open red box. Full segregation of the FAM20C c.899C>T variant with the disease according to recessive model is indicated.
(PDF)
(DOCX)
(XLSX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
Data Availability Statement
The genome data is freely available under accession no. PRJEB13723 (http://www.ebi.ac.uk/ena/data/view/PRJEB13723) at the European Nucleotide Archive and accession no. SRP074053 at the NCBI Sequence Read Archive.