

Abstract
Although bioinformatic analysis of the increasing numbers of diverse genome sequences and amount of functional data has provided insight into the evolution of signaling networks, bioinformatics approaches have limited application for understanding the evolution of highly divergent protein families. We used biochemical analyses to determine the in vitro properties of selected divergent components of the heterotrimeric guanine nucleotide–binding protein (G protein) signaling network to investigate signaling network evolution. In animals, G proteins are activated by cell-surface seven-transmembrane (7TM) receptors, which are named G protein–coupled receptors (GPCRs) and function as guanine nucleotide exchange factors (GEFs). In contrast, the plant G protein is intrinsically active, and a 7TM protein terminates G protein activity by functioning as a guanosine triphosphatase–activating protein (GAP). We showed that ancient regulation of the G protein active state is GPCR-independent and “self-activating,” a property that is maintained in Bikonts, one of the two fundamental evolutionary clades containing eukaryotes, whereas G proteins of the other clade, the Unikonts, evolved from being GEF-independent to being GEF-dependent. Self-activating G proteins near the base of the Eukaryota are controlled by 7TM-GAPs, suggesting that the ancestral regulator of G protein activation was a GAP-functioning receptor, not a GEF-functioning GPCR. Our findings indicate that the GPCR paradigm describes a recently evolved network architecture found in a relatively small group of Eukaryota and suggest that the evolution of signaling network architecture is constrained by the availability of molecules that control the activation state of nexus proteins.
INTRODUCTION
Cells transduce extracellular stimuli to intracellular responses with complex signaling molecule systems. Whereas research continues to expand the known edges of these networks, at the core of any signaling system are conserved signaling elements that affect sensitivity, rate, and amplitude limits. These molecules typically serve as the nexus of multiple protein-protein contacts and therefore are more evolutionarily constrained than are peripheral signaling elements. The intrinsic properties of the core proteins in these interactions determine the signaling outcome and therefore constrain evolution.
The evolution and expansion of major signaling networks within an organism affect cell and organism physiology. Take, for example, a heterotrimeric guanine nucleotide–binding protein (G protein)–coupled receptor (GPCR) and its cognate G protein complex (Fig. 1, A and B). The G protein can be viewed as an enzyme that performs hydrolysis of guanosine triphosphate (GTP) in two elementary steps: the exchange of guanosine diphosphate (GDP) for GTP and the subsequent hydrolysis of GTP to GDP (1). Each of these steps occurs at a certain intrinsic rate in vitro. Regulatory molecules, often cell-surface, seven-transmembrane domain (7TM) receptors, can act on one or both of these steps to modify the activity of the G protein. The 7TM cell-surface receptor, upon binding to its ligand, activates the G protein by catalytically removing a tightly bound GDP from the Gα subunit to enable diffusion-limited GTP binding to bring about G protein activation (1, 2) (Fig. 1B). In this system, nucleotide exchange is the rate-limiting step in G protein activation. Now, imagine a structurally equivalent G protein that spontaneously exchanges nucleotides (3, 4), rendering GPCR-modulated activation unnecessary (Fig. 1C). Consequently, some other type of regulatory molecule is needed for self-activating G proteins. In the course of this thought experiment, it becomes clear how the intrinsic properties, including guanine nucleotide exchange factor (GEF) function and self-activation, of two of the core signaling elements can affect the evolutionary trajectory of the system as a whole.
Fig. 1. Distribution of G protein components among eukaryotes.
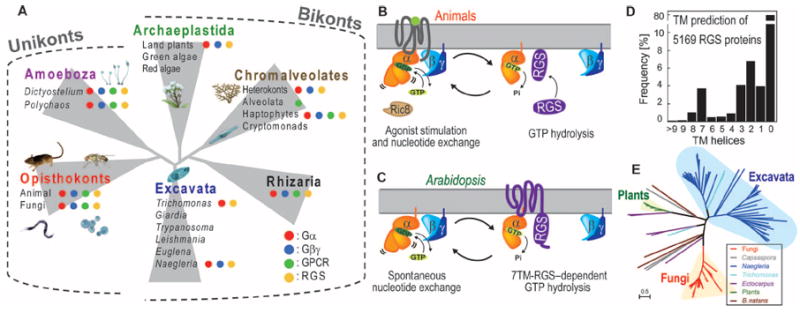
(A) The indicated taxa are representative genomes. The presence of G protein elements in the indicated species or lineages is represented by red, blue, green, and yellow dots for genes encoding Gα, Gβγ, Opisthokont GPCRs, and RGS proteins, respectively. Lack of a dot signifies that those genes were not found. We organized the eukaryotes into six supergroups: Opisthokonta (containing C. owczarzaki and H. sapiens), Amoebozoa (containing D. discoideum), Archaeplastida (containing A. thaliana), Excavata (containing T. vaginalis), Chromalveolata (containing E. siliculosus), and Rhizaria. (B) Regulation of G protein activation in animals. Ligand-bound GPCR accelerates the dissociation of GDP from the G protein α subunit by changing the orientation of its helical domain. Gα hydrolyzes GTP, thereby inactivating itself. GTP hydrolysis is promoted by an RGS or other GAP protein. Nonreceptor GEFs, such as the protein Ric8 (resistance to inhibitors of cholinesterase), act as noncanonical and cytosolic GEFs. (C) Regulatory model of G protein signaling in Arabidopsis. The Arabidopsis Gα protein AtGPA1 rapidly releases its GDP as a result of spontaneous fluctuations between its Ras domain and helical domain. AtGPA1 slowly hydrolyzes its bound GTP; however, the membrane-localized 7TM-RGS protein AtRGS1 constitutively promotes GTP hydrolysis or acts as a GDI. (D) Frequency of TM helices in RGS domain–containing sequences among the 5169 sequences queried (see Materials and Methods). The TM helices were predicted with the membrane prediction software program SOSUI. (E) Distribution of 7TM-RGS proteins in Eukaryotes by a maximum-likelihood (ML) tree of 7TM-RGS proteins. Individual trees of the 7TM and RGS domains are shown in fig. S4 and were generated as described in Materials and Methods. The single genus Naegleria has 50 7TM-RGS proteins.
The bound nucleotide of the Gα subunit determines whether the G protein complex is active (GTP-bound) or inactive (GDP-bound) (Fig. 1B) (1, 2). In animals, the rate of intrinsic GTP hydrolysis is much faster than the rate of basal GDP exchange (1, 2, 5). It follows that the animal G protein forms an inactive heterotrimer and is regulated at the step of exchange by a 7TM-GEF, the GPCR (Fig. 1B). In contrast, G proteins in plants readily exchange GDP for GTP without GPCRs (3, 4, 6-8). Rather, the plant G protein is regulated by a putative 7TM receptor–regulator of G protein signaling (7TM-RGS) protein (4, 7, 9, 10) that accelerates the hydrolysis of GTP by Gα, inactivating the heterotrimer and thus acting as a guanosine triphosphatase (GTPase)–activating protein (GAP). Upon lig- and stimulation, the 7TM-RGS is proposed to be inhibited, enabling the Gα subunit to exchange GDP for GTP (Fig. 1C) (7, 9, 11). Although RGS proteins are found in animals, none has a 7TM domain, and none are regulated by a ligand (12). The divergent intrinsic properties of these two signaling elements, the 7TM receptor and the G protein α subunit, profoundly affect the mechanism of G protein activation; in one case, a ligand stimulates a stimulatory element (receptor-GEF, Fig. 1B), and in the other, the ligand inhibits an inhibitory element (receptor-GAP, Fig. 1C).
Here, we show that the regulation of ancestral G proteins is GPCR-independent, or “self-activating,” and that this property is inherited throughout the Bikonts but not by the Unikonts. Rather than relying strictly on in silico analyses, we purified an informative subset of these proteins and analyzed their physical properties. We found that all G proteins from bikonta species had the trait of GPCR-independent activation, which was characterized by rapid GDP-GTP exchange on Gα and slow GTP hydrolysis. In contrast, none of the unikonta G proteins had the self-activating property. Other Bikonts, such as Arabidopsis, had self-activating G proteins and were selectively regulated at the GTP hydrolysis step by a 7TM-RGS protein, a regulatory mechanism monophyletic to Bikonta. We propose that the main regulatory point of ancestral G proteins is not at the activation step, but rather at the inactivation step, which counters the GPCR-dominant system in animals and fungi. The loss of the intrinsic trait of the self-activating G protein correlates with the expansion of GPCR-encoding genes in Unikonts. Together, our findings provide a new glimpse into the evolutionary mechanism of how signaling molecules change their intrinsic properties as interactions with new partners emerge.
RESULTS
The eukaryotic repertoire of G protein components and 7TM-RGS proteins
Genome sequencing projects have shown the conservation, diversity, and phylogenetic relationships of genes that encode G protein components and 7TM-RGS proteins across organisms. Notably, animal and yeast G proteins, which are primarily regulated by GPCRs (1, 13), are included in Opisthokonts (14, 15), whereas plant G proteins, which do not use GPCRs, separated early from Opisthokonts and constitute Archaeplastida (Fig. 1A) (14-16). To guide the selection of an informative set of divergent G proteins for biochemical testing, we compiled conserved G protein components and their regulators. All eukaryotic organisms are currently classified into six evolutionary supergroups (Fig. 1A) (14, 16). The genes encoding Gα, Gβ, Gγ, and RGS proteins were found in all six supergroups (Fig. 1A and fig. S1), and their sequences were conserved in each organism within each supergroup, which suggests that these four proteins constitute a functional signaling module throughout the eukaryotes. On the other hand, genes homologous to those encoding opisthokonta GPCRs, including class A, B, and C GPCRs, Frizzled/Smoothened-family GPCRs, and yeast GPCRs, or genes homologous to the slime mold 7TM receptor cyclic adenosine monophosphate receptor 1 (cAR1) were found throughout eukaryotes, even in green algae and alveolata (17), despite these taxa lacking genes encoding Gα and Gβγ subunits (figs. S1 to S3) (18). Phylogenetic (fig. S2) (17) and network (fig. S3) analyses of these receptors in bikonta genomes suggest that the products of these homologous GPCR-encoding genes have G protein–independent functions in Bikonts. Furthermore, some eukaryote genomes that have genes encoding G protein components lack genes encoding canonical GPCRs (fig. S1), suggesting that the regulation of their G proteins is GPCR-independent.
We assembled a set of 7TM-RGS proteins with two independent membrane topology programs (Fig. 1D) (19, 20). The 7TM-RGS proteins are distributed in several independent lineages, including fungi, Filasterea (Capsaspora owczarzaki) (21), land plants, Excavata (Trichomonas vaginalis and Naegleria gruberi), Rhizaria (Bigelowiella natans), and brown algae (Ectocarpus siliculosus) (Fig. 1, D and E, and fig. S4). Therefore, we speculate that 7TM-RGS receptor GAPs are extant in organisms whose G proteins exhibit intrinsic, GEF-less activation and are regulated at the step of GTP hydrolysis. The wide distribution of the 7TM-RGS architecture in eukaryotes suggests the potential receptor-GAP systems across supergroups. The 7TM-RGS architecture was retained (for example, within Fungi) or individually invented (for example, in C. owczarzaki) in some evolutionary clades (figs. S1 and S4). The lack of 7TM domain–containing RGS proteins in metazoans, in conjunction with the massive radiation of GPCR-like proteins, indicates when the GPCR-dependent signaling system became dominant.
Nucleotide exchange and hydrolysis rates of evolutionarily informative eukaryotes
To understand the evolutionary process by which GEF-dependent and GEF-independent Gα proteins emerged, we purified Gα subunits from representative eukaryotes and analyzed their intrinsic properties in vitro. With the radioactive nonhydrolyzable GTP analog [35S]GTPγS, we measured the rates of nucleotide exchange of T. vaginalis Gα proteins (TvGα1, TvGα2, TvGα4, and TvGα5), Dictyostelium discoideum Gα4 (DdGα4), Arabidopsis thaliana Gα (AtGPA1), E. siliculosus (EsGPA5), Homo sapiens Gαi1, and C. owczarzaki Gα3 (CoGα3) (Fig. 2 and Table 1). This selection covers the Eukaryota at informative nodes. The GTP binding rates of the TvGα proteins ranged from 0.20 to 1.15 min−1 (Fig. 2B), making the binding rates of TvGα proteins two to three orders of magnitude faster than that of HsGαi1 (Fig. 2E, kobs = 0.006 min−1) and comparable to that of AtGPA1 (Fig. 2C, kobs = 1.15 min−1). DdGα4 and CoGα3 bound GTP at a rate of 0.029 and 0.020 min−1, respectively (Fig. 2, E and F), comparable to that of HsGαi1. Intrinsic tryptophan fluorescence (22), an alternative method to analyze intrinsic rates of GTP binding, confirmed that T. vaginalis G protein subunits have rapid nucleotide exchange (kobs = 1.62 to 6.53 min−1). Notably, plant Gα proteins reach and maintain a high plateau of fluorescence in the presence of hydrolyzable GTP (6, 7), indicating that their exchange rate exceeds their hydrolysis rate. Similar to plant Gα, T. vaginalis Gα proteins exhibited increased fluorescence in the presence of GTP and maintained a plateaued signal (fig. S5), indicating that T. vaginalis Gα proteins favor the GTP-bound state in the absence of GEFs.
Fig. 2. Analysis of nucleotide exchange and hydrolysis by representative Gα subunits indicates that fast nucleotide exchange is an ancestral property.
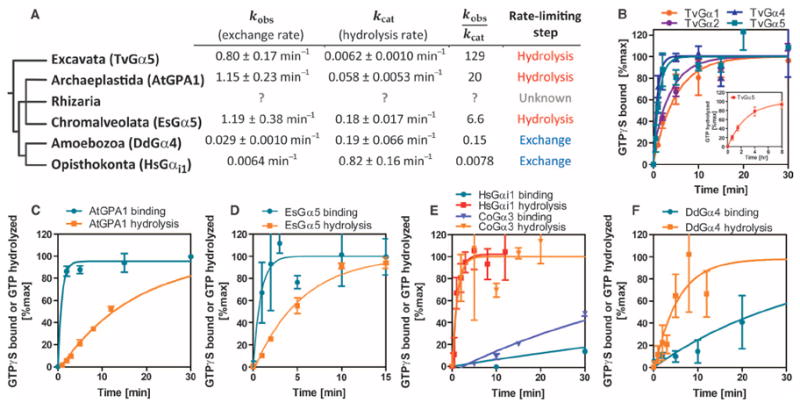
(A) Phylogeny showing the rates of nucleotide exchange and GTP hydrolysis of representative Gα subunits from each of the eukaryotic supergroups: T. vaginalis (Excavata), D. discoideum (Amoebozoa), C. owczarzaki (non-animal Opisthokonta), H. sapiens (Opisthokonta), and A. thaliana (Archae-plastida). See Fig. 1 and figs. S1 to S4 for the genes encoding GPCRs and 7TM-RGS proteins that are found in each clade. No information is available for Rhizaria because the genome was only recently released. Note that the origin of the nucleotide exchange–limited G cycle appears to have come after the split between the Amoebozoa and Opisthokonta and the Rhizaria, Chromalveolata, and Archaeplastida. The rates ± SE are computed from more than 12 data points shown in (B) to (F). A cutoff value of 1.0 in the kobs/kcat indicates the rate-limiting step. (B to F) Superimposed time courses of [35S]GTPγS binding and single-turnover [γ-32P]GTP hydrolysis in room temperature reactions containing 500 nM Gα proteins from C. owczarzaki, D. discoideum, A. thaliana, E. siliculosus, and H. sapiens. The [γ-32P]GTP hydrolysis data from the H. sapiens Gα protein are presented as means ± SEM of four independent experiments. The [35S]GTPγS binding data for T. vaginalis Gα proteins are presented as means ± SEM of three (TvGα1, TvGα2, and TvGα4) or seven (TvGα5) independent experiments. Nucleotide exchange and hydrolysis data for EsGα5, HsGαi1, AtGPA1, and CoGα3 and hydrolysis data for TvGα5 are calculated from means of at least two replicates, and the variation is expressed as SD.
Table 1. Rates of GTPγS binding and GTP hydrolysis.
Rates of GTPγS binding were determined by in vitro [35S]GTPγS binding assays calculated from the data shown in Fig. 2. Rates of GTP binding were determined by intrinsic fluorescence measurements. Rates of GTP hydrolysis were measured by the [γ-32P]GTP hydrolysis assays shown in Figs. 2 and 3. The values of GTPγS binding and GTP hydrolysis (single-turnover) are shown as min−1. The steady-state hydrolysis values are given as amount of GTP hydrolyzed/(min × Gα). G proteins for which no kinetic data were obtained are marked with an “x.”
| TvGα1* | TvGα2 | TvGα4 | TvGα5* | AtGPA1 | EsGPA5 | CoGα3 | DdGα4 | HsGαi1 | |
|---|---|---|---|---|---|---|---|---|---|
| GTPγS binding | 0.20 ± 0.04 | 0.25 ± 0.07 | 1.15 ± 0.37 | 0.80 ± 0.17 | 1.15 ± 0.23 | 1.19 ± 0.38 | 0.020 ± 0.0071 | 0.029 ± 0.010 | 6.4 × 10−3 ± 3.1 × 10−4 |
| Intrinsic fluorescence binding | 1.62 ± 0.10 | x | 1.82 ± 0.21 | 6.53 ± 0.68 | 1.97 ± 0.25 | 0.95 ± 0.14 | x | x | x |
| GTP hydrolysis | 0.018 ± 0.0005 | 0.010 ± 0.001 | 3.8 × 10−3 ± 1.9 × 10−3 | 6.2 × 10−3 ± 1.0 × 10−3 | 0.058 ± 0.0053 | 0.18 ± 0.02 | 0.86 ± 0.54 | 0.19 ± 0.065 | 0.82 ± 0.16 |
| Rate-limiting step† | Hydrolysis | Hydrolysis | Hydrolysis | Hydrolysis | Hydrolysis | Hydrolysis | Binding | Binding | Binding |
The nucleotide exchange rates for TvGα1 and TvGα5 varied between the two methods, likely because of the inhibitory effect of Lubrol-PX on GDP dissociation in the [35S] GTPγS assay (42).
The rate-limiting step was found by comparing the rate of single-turnover (or estimated single-turnover) of GTP hydrolysis to the rate of GTPγS binding. The GTP hydrolysis rates of TvGα1, TvGα2, and TvGα4 were inferred from steady-state turnover rates.
Because a GEF-independent or GEF-dependent classification system implies a relationship between rates of nucleotide exchange and GTP hydrolysis, we also determined the rates of intrinsic GTP hydrolysis by measuring the steady-state consumption and single turnover of 32P-labeled free phosphate hydrolyzed by Gα subunits (Fig. 2 and Table 1). The steady-state rate represents the continuous cycling rate of GDP exchange and GTP hydrolysis of Gα subunits, whereas the single-turnover rate shows a single GTP hydrolysis event for Gα subunits but does not account for GDP exchange. The single-turnover hydrolysis rates of HsGαi1 (kcat = 0.82 min−1), DdGα4 (kcat = 0.19 min−1), and CoGα3 (kcat = 0.86 min−1) were much faster than their respective rates of nucleotide exchange. The hydrolysis rate of AtGPA1 (kcat = 0.053 min−1) was similar to that previously reported (5, 7). TvGα proteins had rates of steady-state GTP hydrolysis ranging from 3.8 × 10−3 to 0.018 GTP/(min × Gα), indicating that their rates of GTP hydrolysis were 11- to 300-fold slower than their rates of nucleotide exchange. Thus, these data suggest that GDP nucleotide exchange is the rate-limiting step in the G protein cycle in Unikonts, which includes animals, fungi, and amoeba, whereas GTP hydrolysis is the rate-limiting step in the G cycle in Bikonts, which includes plants, algae, and protists.
Specific regulation of the G cycle of T. vaginalis by a 7TM-RGS protein
In both animal and plant G protein systems, regulatory molecules act at the rate-limiting steps (1, 7). Because T. vaginalis has genes encoding 7TM-RGS proteins (Fig. 1) and the rate-limiting step of TvGα is GTP hydrolysis (Fig. 2), we hypothesized that the T. vaginalis Gα was likely regulated by 7TM-RGS proteins. The T. vaginalis genome contains genes encoding predicted 5TM-RGS and 7TM-RGS proteins (fig. S6), whereas it lacks genes encoding canonical GPCRs (17, 18). We purified RGS domains from a 5TM-RGS and a 7TM-RGS protein, designated as TvRGS1 and TvRGS2, respectively, and tested them for GAP activity by measuring steady-state GTP hydrolysis by TvGα (Fig. 3). TvRGS2 selectively accelerated the GTPase activity of TvGα5 threefold (Fig. 3, E and F), but it did not affect GTP hydrolysis by TvGα1, TvGα2, or TvGα4 (Fig. 3, B to D). TvRGS1 did not change the rates of GTP hydrolysis of any TvGα protein (Fig. 3, B to F). These results implicate a 7TM-RGS protein as a membrane-bound modulator of another GEF-less G protein system in T. vaginalis.
Fig. 3. Specific regulation of Gα subunits by a 7TM-RGS supports the notion of coevolution of Gα subunits with receptor GAPs.
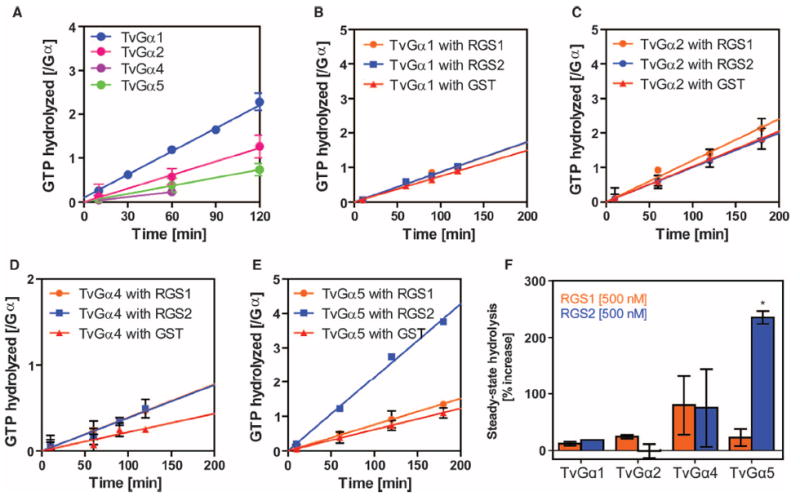
(A) Steady-state hydrolysis of GTP by the indicated wild-type T. vaginalis Gα (TvGα) subunits. Purified Gα subunits (250 or 500 nM) were incubated with 10 μM [γ-32P]GTP for the indicated times before hydrolyzed 32PO4 was extracted with charcoal and quantified. All of the wild-type T. vaginalis Gα subunits displayed relatively slow rates of intrinsic hydrolysis. Data show the relative amounts of GTP hydrolyzed per mole of Gα subunit from two independent experiments. (B to F) Effects of RGS proteins on the GTP hydrolysis rates of TvGα subunits. The indicated Gα subunit (250 nM) and the indicated RGS proteins (500 nM) were incubated over a (B and D) 2-hour or (C and E) 3-hour period. Steady-state [γ-32P]GTP hydrolysis rates were calculated for (B) TvGα1, (C) TvGα2, (D) TvGα4, and (E) TvGα5 in the presence of 500 nM RGS1, RGS2, or glutathione S-transferase (GST). (F) Summary. Enhancement of the GTPase activities of the indicated TvGα subunits by RGS1 or RGS2 relative to those in the presence of the GST control. GAP activity was only seen with RGS2 on TvGα5. *P < 0.01, analysis of variance (ANOVA) followed by Tukey’s test. All other combinations led to statistically insignificant changes in GTPase activity. All data are representative of at least two experiments. Error bars represent SD.
Nonconvergent emergence of the GEF-independent properties of two Gα proteins from basal organisms
To illustrate a limitation of sequence analysis for phylogenetic construction of the G proteins, the amino acid sequence of the plant Gα subunit is only ~33% identical to that of a human G protein subunit, yet the root mean square deviation between the crystal structures of both proteins is 1.8 Å, suggesting that these two divergent Gα subunits have essentially the same three-dimensional (3D) structure despite divergent primary sequences (4). Sequences of genes encoding Gα subunits among the Bikonts (A. thaliana and T. vaginalis) are also only ~30% identical (figs. S6 and S7). Therefore, to address whether the GEF-independent property of Gα subunits was monophyletic (of single origin) or convergent (of independent origins) without relying entirely on sequences, we took a comparative biochemistry approach. We determined whether 3D structural requisites for self-activation were shared between plants and protists, which also are poorly conserved at the level of gene sequence (fig. S6). Our rationale was that if two highly divergent proteins shared the same 3D structural requisite for a complex biochemical property, then they likely shared evolutionary history.
Gα subunits consist of two domains, a GTPase (or Ras) domain and a helical domain (23). Interactions with guanine nucleotides, Gβγ, and GPCRs occur in the Ras domain (23-25). The helical domain has been considered as a cap, shielding the nucleotide-binding pocket from the extracellular environment (23-25). Upon contact with an activated GPCR, there is a large outward swing of the helical domain, presumably exposing GDP and enabling nucleotide exchange (3, 4, 24). A similar mechanism was suggested for the self-activation property of the Arabidopsis Gα subunit in which frequent spontaneous fluctuations of the helical domain constantly expose the nucleotide-binding pocket, enabling rapid exchange of GDP for GTP (3, 4), whereas mutational analyses identified residues that determine the rates of GDP dissociation in a nucleotide-binding motif and others (26-28). It was unknown, however, whether the structural basis for the self-activation of the plant Gα subunit was applicable to T. vaginalis Gα subunits, which branched from the other eukaryotic supergroups earlier in evolution. Given the vast possibilities to evolve spontaneous nucleotide exchange, if Trichomonas G proteins and AtGPA1 used the same 3D structure to achieve self-activation, it follows that this property is likely monophyletic.
Therefore, we created chimeric proteins (Fig. 4, A to D) by exchanging the helical domain of human Gαi1 for that of TvGα5 to generate TvGα5αi helical. Likewise, we substituted the helical domain of TvGα5 with that of human Gαi1 to generate HsGαi1α5 helical. When we analyzed these chimeric proteins for nucleotide exchange rates by measuring intrinsic tryptophan fluorescence, we found that TvGα5αi helical spontaneously exchanged nucleotide at a rate (kobs = 0.37 min−1) that was more than an order of magnitude slower than that of the wild-type TvGα5 (kobs = 6.5 min−1) (Fig. 4E). Intrinsic fluorescence assays demonstrated a low value of intrinsic exchange of GDP for GTP (kobs = 8.2 × 10−3 min−1) in HsGαi1, but when its helical domain was swapped with that of TvGα5, HsGαi1 α5 helical had a fast rate of nucleotide exchange (kobs = 0.25 min−1) (Fig. 4F). Likewise, [35S]GTPγS binding assays showed that the helical domain of TvGα5 was sufficient to confer rapid activation on HsGαi1α5 helical (Fig. 4, G and H, and Table 2).
Fig. 4. Nucleotide binding kinetics of wild-type and chimeric Gα subunits support a monophyletic relationship for the property of fast nucleotide exchange.
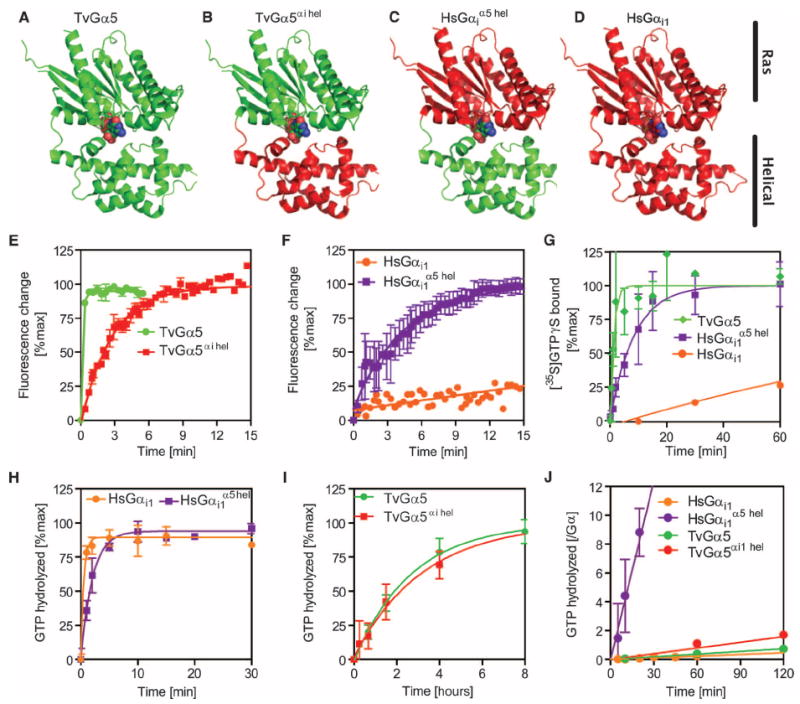
(A to D) Cartoon representations of wild-type and chimeric G proteins with the Ras and helical domains indicated: (A) the T. vaginalis Gα5 subunit structure (green), (B) the T. vaginalis TvGα5αi hel chimera containing the T. vaginalis Ras (green) and human helical (red) domains, (C) the human chimera HsGαi1α5 hel containing the human Ras (red) and the T. vaginalis helical (green) domains, and (D) the human Gαi1 subunit (all red). (E and F) Purified (E) TvGα5 and TvGα5αi hel subunits (400 nM) and (F) HsGαi1α5 hel and HsGαi1 subunits (400 nM) were incubated at 25°C before nonhydrolyzable GTPγS was added, and the change in intrinsic fluorescence was monitored over time. GTP binding manifests as a positive change in intrinsic fluorescence. (G) Purified TvGα5, HsGαi1, or HsGαi1α5 hel chimeric proteins (500 nM) were incubated with the radio-nucleotide [35S]GTPγS for the indicated times before bound radionucleotide was quantified. (H and I) Single-turnover GTP hydrolysis assays in which the indicated purified Gα proteins (800 nM) were preloaded with hydrolyzable [γ-32P]GTP for the indicated times and then incubated in an excess of non-hydrolyzable GTPγS before charcoal extraction and quantification of 32PO4 were performed. (J) Steady-state GTP hydrolysis of chimeric proteins. The procedure followed was the same as that for the wild-type TvGα proteins in Fig. 3. Note that wild-type HsGαi1, TvGα5, and TvGα5αi1 hel displayed slow steady-state hydrolysis, whereas HsGαi1α5 hel displayed faster steady-state hydrolysis. All data are representative of at least two experiments. Error bars represent SD. GTPγS binding and GTP hydrolysis data for HsGαi1 and TvGa5 are as shown in Fig. 2.
Table 2. Rates of GTPγS binding and GTP hydrolysis in wild-type and chimeric Gα subunits.
Rates of GTPγS binding were determined by in vitro [35S]GTPγS binding assays. Rates of GTP binding were determined by intrinsic fluorescence measurements. Rates of GTP hydrolysis were measured by [γ-32P]GTP hydrolysis assay. The values of GTPγS binding and GTP hydrolysis (single-turnover) are shown as min−1. The steady-state hydrolysis values are given as GTP hydrolyzed/(min × Gα), where the amount of active Gα was determined by its ability to bind to GTPγS. Experiments for which no data were obtained are marked with an “x.”
| HsGαi1 | HsGαi1α5 hel | TvGα5αi1 hel | TvGα5 | |
|---|---|---|---|---|
| GTPγS binding | 6.4 × 10−3 ± 3.1 × 10−4 | 0.12 ± 0.03 | x | 0.80 ± 0.17 |
| Intrinsic fluorescence binding | 8.2 × 10−3 ± 5.2 × 10−3 | 0.25 ± 0.01 | 0.37 ± 0.02 | 6.53 ± 0.68 |
| GTP hydrolysis (single-turnover) | 0.82 ± 0.16 | 0.49 ± 0.05 | 0.0052 ± 0.001 | 6.2 × 10−3 ± 1.0 × 10−3 |
| Steady-state hydrolysis | 3.9 × 10−3 ± 5.8 × 10−4 | 0.41 ± 0.03 | 0.013 ± 0.002 | 6.1 × 10−3 ± 5.8 × 10−4 |
| Rate-limiting step* | Binding | Binding | Hydrolysis | Hydrolysis |
The rate-limiting step was found by comparing the rate of single-turnover (or estimated single-turnover) GTP hydrolysis to the rate of GTPγS binding. The rates shown in this table were computed from more than 12 data points of at least two experiments. Variability is represented by the SEM.
Next, we measured the rates of GTP hydrolysis of the chimeric proteins with single-turnover or steady-state [γ-32P]GTP hydrolysis assays (Fig. 4, H to J, and Table 2). The TvGα5αi helical chimera, which had a slow GTP-hydrolyzing Ras domain and a slow nucleotide-exchanging helical domain, had a markedly slow rate of steady-state GTP hydrolysis [kcat = 0.013 GTP/(min × Gα)]. Conversely, the HsGαi1α5 helical chimera, which had a fast nucleotide-exchanging helical domain and a fast GTP-hydrolyzing Ras domain, showed the fastest rate of steady-state GTP hydrolysis [kcat = 0.41 GTP/(min × Gα)]. The wild-type TvGα5 and HsGαi1 proteins showed slow rates of steady-state GTP hydrolysis [kcat = 6.1 × 10–3 GTP/(min × Gα) and kcat = 3.9 × 10−3 GTP/(min × Gα), respectively], likely as a result of slow rates of nucleotide exchange or GTP hydrolysis (Table 2). Evidence from the experiments with chimeric proteins suggests that the helical domain is necessary and sufficient to confer properties of self-activation on a G protein α subunit, and that the Ras domain determines the rate of GTP hydrolysis, which is the same structural basis for activation that was shown in Gα subunits from plants and mammals.
DISCUSSION
To understand primitive regulatory systems, we comprehensively analyzed the biochemical properties of a divergent and informative set of heterotrimeric G proteins. We identified GEF-independent Gα subunits in plants, brown algae, and T. vaginalis and GEF-dependent Gα subunits in ancient animals and amoeba. Furthermore, we demonstrated that a 7TM-RGS protein from a basal eukaryote, T. vaginalis, is selective for T. vaginalis Gα proteins. On this basis, we propose a new canon in the evolution of signaling molecules and regulatory molecules whereby the intrinsic properties of a signaling molecule are constrained by its cognate regulatory element.
Ancestral G protein regulation
Genes encoding G proteins are broadly conserved in eukaryotes (Fig. 1) (18) but not in prokaryotes and Archaea. The RGS domain is also found in primitive eukaryotes (Fig. 1), and GAP activity is conserved (Fig. 3) (6). The evidence suggests that regulation of the nucleotide-bound state of G proteins by RGS proteins has existed since the ancient eukaryotes but that regulation by GPCRs is a recent event. In general, functionally paired genes create genetic constraints to keep both genes in the genome. The evidence that RGS-encoding genes are conserved only in organisms that have Gα subunits indicates that the function of RGS proteins is linked to G protein signaling (figs. S1 and S4) (18). In contrast to the apparent evolutionary linkage of genes encoding Gα and RGS proteins, genes encoding GPCR-like proteins were found in eukaryotes (figs. S1 to S3) even in alveolata (17) and green algae, which lack genes encoding Gα, Gβ, and Gγ proteins (fig. S1) (18). Although those divergent GPCR-like proteins share similarity with the GPCRs of Opisthokonts, they have G protein–independent functions. We propose that 7TM receptors functionally coupled to G proteins in Unikonts, and that the cognate Gα subunit evolved from a GEF-independent, spontaneous nucleotide exchanger to a GEF-dependent, slow nucleotide exchanger, and that the 7TM receptor co-evolved to become a GPCR.
GEF-dependent regulation of G proteins emerged in Unikonts, which include animals, fungi, and amoeba (1, 13, 29). These three evolutionarily distant organisms, whose Gα proteins are activated independently of GEFs (Fig. 2), are included in Bikonts, which separated early from Unikonts (fig. S1) (14-16). This suggests that the canonical model of G protein exchange, in which the activation state of the Gα protein is modulated by GEFs, emerged within one evolutionary clade, the Unikonts. This is consistent with claims that the GPCR-encoding genes of H. sapiens originated from a common ancestor to both animals and amoeba within the Unikonts (30, 31). Because the root of Eukaryotes is predicted to lie within Excavata or between Excavata and the other supergroups (16), a self-activating G protein is likely the ancestral state, with the paradigmatic mammalian Gα that has slow nucleotide exchange evolving later within the Unikonts and radiating greatly within the Opisthokonts.
G protein regulation by the 7TM-RGS protein in T. vaginalis: Evidence for convergent evolution
Like that of other Bikonts, the G cycle of T. vaginalis is also regulated by the GEF-independent activation of Gα and by its modulator, a 7TM-RGS protein (Fig. 3). One of the three 7TM-RGS proteins in T. vaginalis specifically stimulates the GTPase activity of TvGαs, which indicates specific receptor-Gα coupling. Phylogenetic analyses indicate a complex evolutionary history of the 7TM and the RGS domain (fig. S4). Indeed, for certain nodes, the gene trees conflict with the species tree, which suggests the independent origin of the 7TM-RGS signaling system in the eukaryotic supergroup (fig. S4).
In contrast to the convergent evolution of the 7TM-RGS protein topology, the conserved structural basis for nucleotide exchange rate in T. vaginalis and A. thaliana (Fig. 4) strongly suggests the existence of a common Gα ancestor shared by Unikonts and Bikonts. The helical domain is unconstrained by effector coupling (2), leaving it free to evolve faster or slower intrinsic nucleotide exchange properties through a series of stepwise mutations in response to previously unencountered modulator couplings (17, 31). This reveals a structural basis for the evolutionary plasticity seen in the G protein signaling network architecture, and provides a partial answer to how such an apparently highly constrained system can display such diversity in regulation across evolutionary time and space.
How the Trichomonas G protein is regulated in vivo is unknown; however, the intrinsic property prompts the hypothesis that either a GAP (for example, a 7TM-RGS protein) or a GDI (GDP dissociation inhibitor) functions to maintain inactive TvGα proteins at steady state. A noncanonical GEF similar to animal Ric8 (32-34) might function to promote GTP loading onto TvGα proteins. This happens in animal systems in which G proteins are regulated by both a GEF (for example, GPCRs and Ric8) and a GAP (for example, RGS proteins) (35), although the rate-limiting step and the primary regulation point are nucleotide exchange (1). Future in vivo studies may identify such a system that complements the 7TM-RGS regulatory system that we identified here, but our work suggests that the primary locus of G protein regulation is at the step of nucleotide hydrolysis. In either case of GDIs or GEFs, the unidentified regulators of Trichomonas G proteins likely have structures and actions that are distinct from those found in the mammalian G protein system because the Trichomonas genome does not encode canonical GDIs (for example, Gβγ-encoding genes) or GEFs (for example, GPCRs and Ric8).
Conclusion and future perspectives
Functional residues, such as those within the catalytic sites of enzymes, are highly constrained and thus well conserved across organisms, because mutations within these sites are normally deleterious (36). We extend this concept of constraint from the level of the primary sequence to the functional traits of signaling proteins, and we propose that an intrinsic functional property of a signaling molecule, which is often not evident in 3D structures, is also evolutionally constrained by the binding regulatory partners (for example, an RGS protein for a Gα subunit). Although Gα proteins showed low sequence similarity across supergroups (26 to 37% similarity in sequence, figs. S2 and S3), four tested plants (6, 7), the Chromalveolate E. siliculosus, and four T. vaginalis Gα proteins show properties of GEF-independent activation (Fig. 2) (6, 7). In comparison, the Gα proteins of animals (7), fungi (37), C. owczarzaki, and D. discoideum are dependent on GEFs for their activation (Fig. 2).
To create a type of network architecture, signaling pathways are reconstituted with new connections (or disconnections) between signaling molecules. Once a protein-protein interaction is fixed, the molecular trait (for example, a binding surface) becomes constrained. Here, we showed that the origins of the GEF-dependent G protein correlated with the expansion in the number of GPCR-encoding genes (Fig. 2 and fig. S1) (17, 31). The slow nucleotide exchange property of the Gα subunit was likely constrained by the dominant regulator, Opisthokonta GPCRs, in that clade. Whereas understanding the divergent G protein signaling pathways and the mechanisms involved provides a fascinating look into evolutionary time, it may also prove to have practical implications in the development of new pharmaceutical treatments. For example, although trichomoniasis caused by the protist T. vaginalis has efficacious clinical treatment (38), other GEF-independent relatives, including the “brain-eating amoeba” Naegleria fowleri, the lethal causative agent in primary amoebic meningoencephalitis, lack viable therapies (39). Given that there are 50 genes encoding 7TM-RGS proteins in N. fowleri (Fig. 1), the bikont G protein pathway may prove to be an important target for future anti-protozoan pharmaceuticals.
MATERIALS AND METHODS
Cloning and protein purification of wild-type proteins
Complementary DNAs (cDNAs) encoding the T. vaginalis Gα subunits TvGα1, TvGα2, TvGα5, and TvGα6 were amplified by polymerase chain reaction (PCR) assay from the genomic DNA of T. vaginalis strain G3. cDNAs encoding TvGα4 and the E. siliculosus Gα subunit EsGα5 (accession no. D8LTN0) were amplified by PCR from an Escherichia coli–optimized synthetic gene made by CellTek. TvRGS1CTerm consists of residues 230 to 362 of TvRGS1, TvRGS2CTerm of residues 294 to 436 of TvRGS2, TvRGS3CTerm of residues 284 to 427 of TvRGS3, and TvRGS4CTerm of residues 295 to 429 of TvRGS4. A sequence encoding a His6 tag was added to those encoding the TvGα and TvRGS proteins with the Gateway TOPO cloning system (pENTR to pDEST17). cDNAs encoding the C. owczarzaki Gα subunit CoGα3 (accession no. EFW45256) and the D. discoideum Gα subunit DsGα4 (XP_638196.1) were amplified from their cDNA libraries and cloned into the pENTR-TOPO vector. The Gα-encoding genes were subcloned into pDEST17, which expresses N-terminal His6-tagged proteins in E. coli. The expression vectors for A. thaliana GPA1 and H. sapiens Gαi1 were as described previously (19). The Gα proteins were expressed in ArcticExpress RP cells (Agilent Technologies) at 12°C. The ArcticExpress RP cells were transformed with the pDEST17 constructs, plated onto LB agar containing carbenicillin (100 μg/ml) and gentamicin (30 μg/ml), and grown at 37°C overnight. A single colony was then isolated and cultured overnight in 5 ml of LB supplemented with ampicillin (100 μg/ml) and gentamicin (30 μg/ml). This overnight culture was then used to inoculate 2 liters of TB broth containing ampicillin (10 μg/ml), and the culture was grown at 37°C with shaking at 225 rpm until the absorbance at 600 nm (A600) reached a value of 1.00. Cultures were then chilled to 4°C and incubated at 12°C with shaking at 150 rpm for 30 min to induce expression of the ArcticExpress chaperonin. Isopropyl-β-d-thiogalactopyranoside was added to a final concentration of 0.5 mM to induce protein expression. The cultures were incubated at 12°C with shaking at 150 rpm overnight (for 16 hours). Cells were pelleted by centrifugation at 3000g at 4°C for 45 min and stored at −20°C until ready for use. The pellet was allowed to thaw in a room temperature water bath. Cold N1 buffer [25 mM tris-HCl (pH 7.6), 100 mM NaCl, 5% (v/v) glycerol, 10 mM imidazole, 5 mM MgCl2, 50 µM GDP, 5 mM β-mercaptoethanol] was added to a thawed pellet to a final volume of 40 ml. Phenylmethylsulfonyl fluoride and leupeptin were added to final concentrations of 1 mM and 1 μg/ml, respectively. The pellet was fully resuspended by a minimal amount of low-intensity sonication. Lysozyme (from chicken egg white) and MgCl2 were then added to final concentrations of 0.25 mg/ml and 10 mM, respectively. The suspension was rocked at 4°C for 20 min, at which point Lubrol-PX detergent was added to a final concentration of 0.1%. The solution was rocked for an additional 30 min. The cells were then fully lysed by sonication with a 550 Sonic Dismembrator (Fisher Scientific) attached to a 20-kHz ultrasonic converter. A pulsing program (0.5 s on, 0.5 s off) at the max microtip setting was applied for a total of 6 min. An ice water bath was used to maintain the samples at 4°C. NaCl was then added to a final concentration of 300 mM, and the solution was allowed to rock at 4°C for 20 min. The solution was then centrifuged at 47,800g for 35 min in an SS-34 rotor (Sorvall Instruments). The soluble fraction was mixed with the pellet (100 μl/g) of prewashed TALON resin (Clontech Laboratories, catalog no. 635504). This suspension was rocked at 4°C for 90 min. The tubes were centrifuged briefly to pellet the resin, which was removed to a prechilled column, in which it was washed with a combination of 10 ml of N1-300 (N1 buffer containing 200 mM NaCl) in five separate 2-ml additions and 6 ml of N1 buffer in three separate 2-ml additions. N2 buffer [200 mM tris-HCl (pH 7.6), 100 mM NaCl, 5% (v/v) glycerol, 300 mM imidazole (pH 7.5, filtrated), 10 mM MgCl2, 5 μM GDP, 5 mM β-mercaptoethanol] was used to elute the protein in 500-μl aliquots. The Bradford assay (Bio-Rad) was used to determine protein concentrations, and SDS–polyacrylamide gel electrophoresis analysis was performed to determine protein purity. Peak fractions were combined and dialyzed into imidazole-free buffer.
Cloning and purification of chimeric proteins
The helical domains of TvGα5 and HsGαi1 were swapped by site-directed mutagenesis. A multiple sequence alignment of TvGα5, HsGαi1, and AtGPA1 was used to identify the conserved residues linking the helical and Ras domains. Large primers were constructed for the beginning and end of the helical domain, but with about half of the primer (whichever portion was upstream of the polymerase from the desired helical domain) consisting of the Ras domain–encoding sequence from the gene with the desired retained Ras domain. For example, a forward primer for the generation of a TvGα5αi1 helical cDNA began within ~90 bases from the end of the region encoding the N-terminal Ras domain of TvGα5 and ended ~90 nucleotides from the region encoding the beginning of the helical domain of HsGαi1. KOD Hot Start DNA Polymerase (Novagen) was used in a PCR to amplify the region encoding the desired helical domain, with “sticky ends” on either side corresponding to the sequence of the region encoding the desired Ras domain. PCR products were resolved on a 1% agarose gel and purified. A QuikChange Lightning Mutagenesis Kit (Agilent Technologies) was then used to perform a PCR in which entry vectors containing the gene with the desired Ras domain were amplified with the linear DNA of the opposite helical domain with sticky ends, termed a megaprimer. The Dpn I restriction enzyme, which cleaves only methylated DNA, was used to cleave any remaining template vectors. Transformation of DH5α cells with the amplified vectors and subsequent miniprep was followed by gene sequencing as described earlier. DNAs containing helical domain swaps with the correct sequence were subcloned into vectors expressing His6-tagged protein. All purification steps were performed at 4°C. Pellets of ArcticExpress RP cells were isolated in the manner described earlier, and then were thawed, crushed, and resuspended mechanically with a pipette tip in ~20 ml of resuspension solution [25 mM tris (pH 7.6), 100 mM NaCl, 5% (v/v) glycerol, 20 mM imidazole, 2 mM MgCl2, 1 mM dithiothreitol (DTT), and 25 μM GDP]. One EDTA-free protease inhibitor cocktail tablet (Roche) was added for every 50 ml of buffer. The volume was adjusted to 40 ml, and a high-pressure homogenizer (Avestin EmulsiFlex-C5) was used to emulsify the suspension, which was then centrifuged for 30 min at 27,000g. The supernatant was transferred to a new tube and mixed with prewashed Clontech nickel resin beads. The solution was incubated with shaking for 90 min and then washed three times at 4°C with resuspension buffer. The beads were then transferred to a 1.5-ml tube and centrifuged before being resuspended in 500 μl of elution buffer (resuspension buffer adjusted to 10 μM GDP, 200 mM imidazole). After centrifuging the beads, the supernatant was withdrawn as fraction 1. Two more fractions were taken by an identical method, and their concentrations were determined by photospectroscopy.
Intrinsic tryptophan fluorescence measurements
The tryptophan fluorescence of a given Gα subunit was used as an indicator of its activation state (22). TvGα proteins have the critical tryptophan residue in their switch II region, which shifts from an aqueous to a hydrophobic environment upon activation, resulting in increased fluorescence in the activated state (22). This shift enabled measurement of the relative portions of the sample that were in activated versus inactivated states. Fluorescence was measured at 20°C in 100 mM tris-HCl (pH 7.6), 100 mM NaCl, 1 mM EDTA, 10 mM MgCl2, and 5% glycerol. The Gα protein was added to a final concentration of 400 to 500 nM (active concentration varied). After establishment of the baseline fluorescence of the subunit, the reaction solution was spiked with the indicated amount of nucleotide (in proportion to the concentration of the active Gα protein). Excitation and emission wavelengths were 284 and 340 nm, respectively, and the excitation and emission slit widths were 3.0 and 4.0 nm, respectively. Data were collected at the shortest possible interval, usually every 20 s. The observed rate constants, kobs, were estimated by the one-phase association equation in GraphPad Prism 5.
Steady-state and single-turnover GTP hydrolysis assays
For steady-state measurements, 800 nM purified Gα protein was prepared in HEL buffer [50 mM Hepes-NaOH (pH 7.0), 1 mM EDTA, 0.1% Lubrol-PX, and 1 mM DTT] with an equal volume of [γ-32P]GTP buffer (HEL buffer containing 10 mM MgCl2, 2 µM GTP, and [γ-32P]GTP at ~5000 to 10,000 cpm/pmol) to start the hydrolysis reaction. At a given time point, the reaction was stopped by quenching a 100-μl aliquot in ice-cold 50 mM HPO4 buffer (pH 2.0) containing 5% (w/v) charcoal. After quenching and charcoal extraction, which denatures proteins and removes organic compounds, the amount of 32PO4 hydrolyzed was quantified by Cherenkov radiation counting of supernatants. For single-turnover reactions, 900 nM purified Gα subunit was preloaded with a mixture of 3 mM GTP and [γ-32P]GTP for 10 to 30 min at room temperature before the reaction was moved to ice for 5 min. The hydrolysis reaction was then started by the addition of 10 mM MgCl2 and 100 μM GTPγS. Reactions were quenched, extracted, and processed as described earlier. kcat values observed in single-turnover experiments were estimated from the one-phase association model. Because single-turnover GTP hydrolysis was not successfully measured for some of the Trichomonas Gα proteins, kcat values (min−1) for TvGα1, TvGα2, and TvGα4 were inferred from the steady-state rates of hydrolysis and from the specific activities of the TvGα proteins.
[35S]GTPγS binding assay
Purified Gα (1 µM) in assay buffer [50 mM tris-HCl (pH 7.0), 1 mM EDTA, 0.1% Lubrol-PX, 1 mM DTT] was mixed with an equal volume of [35S]GTPγS GTP buffer (HEL buffer containing 8 µM GTPγS and [35S]GTPγS at ~2000 cpm/pmol) to start the reaction. At each time point, an aliquot of the reaction mixture was removed and quenched in 1.5 ml of ice-cold wash buffer [20 mM tris-HCl (pH 7.5), 100 mM NaCl, 10 mM MgCl2]. At the earliest possible time, the quenched reaction mixtures were filtered through nitrocellulose filters (type HA Millipore) at −5 mmHg, washed three times with 3 ml of cold wash buffer, and then dried. A Cherenkov radiation count of the nitrocellulose filters with ScintiSafe scintillation cocktail was performed. Counts per minute at a particular time were taken to be directly proportional to the amount of GTPγS bound at that time. kobs was calculated in the same way as described for the intrinsic fluorescence assay.
Identification of G protein–encoding genes
Amino acid sequences for Gα, Gγ, and RGS proteins were downloaded from the National Center for Biotechnology Information (NCBI) conserved domain database (CDD) with the following accession numbers: SM00275 for Gα, SM00224 for Gγ, and SM00315 for RGS. To further identify Gα-, Gγ-, and RGS-encoding genes, we downloaded Pfam seed alignment files (Gα: PF00503; Gγ: PF00631; and RGS: PF00615) and used them for a hidden Markov model (HMM)–based search (http://hmmer.janelia.org/) to nonredundant protein data set from the GenBank or UniProtKB database. To identify Gβ proteins, we used an aligned sequence file of H. sapiens and land plant Gβ proteins as a template. Matches showing high sequence similarity were downloaded, and duplicates were removed. Remaining matches were checked for residues critical for the functions of Gα (nucleotide-binding residues and helical domain insertion), Gβ (N-terminal coiled-coil), or Gγ (coiled-coil and C-terminal CAAX motif) proteins. Those candidates that lacked these sites were eliminated. The proteome data sets for B. natans CCMP2755 v1.0, Guillardia theta CCMP2712 v1.0, Emiliania huxleyi CCMP1516 v1.0, and Cyanophora paradoxa were retrieved from the Joint Genome Institute database (JGI, http://www.jgi.doe.gov/) or the C. paradoxa genome database (http://cyanophora.rutgers.edu/cyanophora/) and were used for the HMM-based search. Those candidates that remained were declared G protein candidates and were targeted for cloning and purification. To identify Trichomonas RGS protein sequences, we used the NCBI RefSeq database.
Prediction of TM RGS–containing proteins
RGS protein sequences were retrieved from the NCBI CDD (RGS superfamily: cl02565), JGI, and Broad Institute (http://www.broadinstitute.org/) databases for membrane prediction. The prediction of TM helices was made with the online programs SOSUI (20) and TMHMM2.0 (19). Those sequences that were predicted by at least either SOSUI or TMHMM2.0 to have a heptahelical portion with an extracellular N terminus were declared 7TM-RGS candidates. Those sequences that had an N-terminal 7TM domain and a C-terminal RGS domain were used for further phylogenetic analysis.
Phylogenetic analyses
The phylogenetic trees were constructed on 108 7TM-RGS proteins with the MEGA5.0 software package (40). The amino acid sequences were aligned with ClustalW using the standard settings: gap opening penalty and gap extension penalty for initial pairwise alignment: 10 and 0.1, respectively; gap opening penalty and gap extension penalty for multiple alignment: 10 and 0.2, respectively; Gonnet protein weight matrix; residue-specific penalties: on; hydrophilic penalties: on; gap separation distance: 4; end gap separation: off; use negative matrix: off. The 7TM domains or the RGS domains were individually extracted from the aligned sequences. The nonaligned portions were manually deleted, and the remaining sequences were used for phylogenetic analysis. The ML trees were created with the Jones-Taylor-Thornton (JTT) substitution model with a bootstrap of 100. The tree for 7TM-RGS proteins was made with the combined sequences of the 7TM domains and the RGS domains. Three sequences were removed before the phylogenetic analysis because of long deletions in the aligned sequences. For a phylogenetic analysis of GPCRs, the seed sequences of GPCRs (PF00001, PF00002, PF00003, and PF05462) were downloaded from the Pfam26.0 database. The bikonta GPCRs (PF00001, PF00002, PF00003, PF01534, and PF05462) were collected from NCBI CDD or the JGI database. The bikonta sequences were queried with TMHMM2.0 (19) or SOSUI (20) online programs to predict TM helices. Those bikonta GPCRs having five to eight TM regions were aligned with the Pfam GPCR sequences with ClustalW implemented in MEGA5.0 (40). Those aligned sites containing more than 80% gap were removed. The ML tree was created with a JTT+F substitution model of amino acids with a bootstrap of 50. A phylogenetic network was created with a packaged program, Splitstree4.0 (41). Distances between sequences were obtained with the ML method, and the minimum spanning network was computed.
Statistical analysis
Statistical significance was evaluated by one-way ANOVA followed by Tukey’s test with GraphPad Prism 5.
Supplementary Material
www.sciencesignaling.org/cgi/content/full/6/276/ra37/DC1
Fig. S1. Genes encoding heterotrimeric G protein components found in eukaryotic genomes.
Fig. S2. Phylogeny of unikonta GPCRs.
Fig. S3. Minimum spanning network for unikonta GPCRs.
Fig. S4. ML trees of the 7TM-RGS proteins show the distinct origin of 7TM-RGS proteins.
Fig. S5. Intrinsic fluorescence measurements of the nucleotide exchange and hydrolysis activities of Gα proteins support the radionucleotide assay–based measurements.
Fig. S6. Identity among T. vaginalis G protein components.
Fig. S7. Multiple sequence alignment of Gα subunits showing conserved residues.
Table S1. Accession numbers of genes encoding G proteins, RGS proteins, and GPCRs.
References (43–47)
Acknowledgments
We thank J. Yang, J. Xi, and R. Kula for assistance with experiments; J. C. Jones and B. Temple for their helpful discussions; and J. Sondek for use of the Perkin-Elmer fluorimeter. We also thank I. Ruiz-Trillo for sharing the cDNA encoding C. owczarzaki, M. Hobbs for sharing genomic DNA of T. vaginalis, and J. Hadwiger for sharing D. discoideum cDNAs. The icons for species in Fig. 1A were provided by National Bioscience Database Center Japan.
Funding: This work was supported by grants from the National Institute of General Medical Sciences (R01GM065989) and NSF (MCB-0723515 and MCB-1158054) to A.M.J. The Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences of the U.S. Department of Energy through grant DE-FG02-05er15671 to A.M.J. funded technical support.
Footnotes
Author contributions: W.B. and A.B. prepared experimental materials, acquired and analyzed data, and wrote the manuscript; J.M. and C.P. made experimental materials and acquired and analyzed data; D.U. designed and managed this project, prepared materials, acquired and analyzed data, and wrote the manuscript; and A.M.J. designed and managed this project and wrote the manuscript.
Competing interests: The authors declare that they have no competing interests.
References and Notes
- 1.Gilman AG. G proteins: Transducers of receptor-generated signals. Annu Rev Biochem. 1987;56:615– 649. doi: 10.1146/annurev.bi.56.070187.003151. [DOI] [PubMed] [Google Scholar]
- 2.Sprang SR. G protein mechanisms: Insights from structural analysis. Annu Rev Biochem. 1997;66:639–678. doi: 10.1146/annurev.biochem.66.1.639. [DOI] [PubMed] [Google Scholar]
- 3.Jones JC, Jones AM, Temple BR, Dohlman HG. Differences in intradomain and interdomain motion confer distinct activation properties to structurally similar Gα proteins. Proc Natl Acad Sci U S A. 2012;109:7275–7279. doi: 10.1073/pnas.1202943109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jones JC, Duffy JW, Machius M, Temple BR, Dohlman HG, Jones AM. The crystal structure of a self-activating G protein α subunit reveals its distinct mechanism of signal initiation. Sci Signal. 2011;4:ra8. doi: 10.1126/scisignal.2001446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ferguson KM, Higashijima T, Smigel MD, Gilman AG. The influence of bound GDP on the kinetics of guanine nucleotide binding to G proteins. J Biol Chem. 1986;261:7393–7399. [PubMed] [Google Scholar]
- 6.Urano D, Jones JC, Wang H, Matthews M, Bradford W, Bennetzen JL, Jones AM. G protein activation without a GEF in the plant kingdom. PLoS Genet. 2012;8:e1002756. doi: 10.1371/journal.pgen.1002756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnston CA, Taylor JP, Gao Y, Kimple AJ, Grigston JC, Chen JG, Siderovski DP, Jones AM, Willard FS. GTPase acceleration as the rate-limiting step in Arabidopsis G protein-coupled sugar signaling. Proc Natl Acad Sci U.S.A. 2007;104:17317–17322. doi: 10.1073/pnas.0704751104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jones JC, Temple BRS, Jones AM, Dohlman HG. Functional reconstitution of an atypical G protein heterotrimer and regulator of G protein signaling protein (RGS1) from Arabidopsis thaliana. J Biol Chem. 2011;286:13143–13150. doi: 10.1074/jbc.M110.190355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen JG, Willard FS, Huang J, Liang J, Chasse SA, Jones AM, Siderovski DP. A seven-transmembrane RGS protein that modulates plant cell proliferation. Science. 2003;301:1728–1731. doi: 10.1126/science.1087790. [DOI] [PubMed] [Google Scholar]
- 10.Urano D, Phan N, Jones JC, Yang J, Huang J, Grigston J, Taylor JP, Jones AM. Endocytosis of the seven-transmembrane RGS1 protein activates G-protein-coupled signalling in Arabidopsis. Nat Cell Biol. 2012;14:1079–1088. doi: 10.1038/ncb2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Booker KS, Schwarz J, Garrett MB, Jones AM. Glucose attenuation of auxin-mediated bimodality in lateral root formation is partly coupled by the heterotrimeric G protein complex. PLoS One. 2010;5:e12833. doi: 10.1371/journal.pone.0012833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kimple AJ, Bosch DE, Giguere PM, Siderovski DP. Regulators of G-protein signaling and their Gα substrates: Promises and challenges in their use as drug discovery targets. Pharmacol Rev. 2011;63:728–749. doi: 10.1124/pr.110.003038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dohlman HG. G proteins and pheromone signaling. Annu Rev Physiol. 2002;64:129–152. doi: 10.1146/annurev.physiol.64.081701.133448. [DOI] [PubMed] [Google Scholar]
- 14.Burki F, Shalchian-Tabrizi K, Minge M, Skjaeveland A, Nikolaev SI, Jakobsen KS, Pawlowski J. Phylogenomics reshuffles the eukaryotic supergroups. PLoS One. 2007;2:e790. doi: 10.1371/journal.pone.0000790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hampl V, Hug L, Leigh JW, Dacks JB, Lang BF, Simpson AG, Roger AJ. Phylogenomic analyses support the monophyly of Excavata and resolve relationships among eukaryotic “supergroups”. Proc Natl Acad Sci U.S.A. 2009;106:3859–3864. doi: 10.1073/pnas.0807880106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cavalier-Smith T. Kingdoms Protozoa and Chromista and the eozoan root of the eukaryotic tree. Biol Lett. 2010;6:342–345. doi: 10.1098/rsbl.2009.0948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krishnan A, Almen MS, Fredriksson R, Schioth HB. The origin of GPCRs: Identification of mammalian like Rhodopsin, Adhesion, Glutamate and Frizzled GPCRs in fungi. PLoS One. 2012;7:e29817. doi: 10.1371/journal.pone.0029817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anantharaman V, Abhiman S, de Souza RF, Aravind L. Comparative genomics uncovers novel structural and functional features of the heterotrimeric GTPase signaling system. Gene. 2011;475:63–78. doi: 10.1016/j.gene.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sonnhammer EL, von Heijne G, Krogh A. A hidden Markov model for predicting transmembrane helices in protein sequences. Proc Int Conf Intell Syst Mol Biol. 1998;6:175–182. [PubMed] [Google Scholar]
- 20.Hirokawa T, Boon-Chieng S, Mitaku S. SOSUI: Classification and secondary structure prediction system for membrane proteins. Bioinformatics. 1998;14:378–379. doi: 10.1093/bioinformatics/14.4.378. [DOI] [PubMed] [Google Scholar]
- 21.Torruella G, Derelle R, Paps J, Lang BF, Roger AJ, Shalchian-Tabrizi K, Ruiz-Trillo I. Phylogenetic relationships within the Opisthokonta based on phylogenomic analyses of conserved single-copy protein domains. Mol Biol Evol. 2012;29:531–544. doi: 10.1093/molbev/msr185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Higashijima T, Ferguson KM, Sternweis PC, Smigel MD, Gilman AG. Effects of Mg2+ and the βγ-subunit complex on the interactions of guanine nucleotides with G proteins. J Biol Chem. 1987;262:762–766. [PubMed] [Google Scholar]
- 23.Noel JP, Hamm HE, Sigler PB. The 2.2 Å crystal structure of transducin-a complexed with GTPγS. Nature. 1993;366:654–663. doi: 10.1038/366654a0. [DOI] [PubMed] [Google Scholar]
- 24.Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, Mathiesen JM, Shah ST, Lyons JA, Caffrey M, Gellman SH, Steyaert J, Skiniotis G, Weis WI, Sunahara RK, Kobilka BK. Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature. 2011;477:549– 555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lambright DG, Sondek J, Bohm A, Skiba NP, Hamm HE, Sigler PB. The 2.0 Å crystal structure of a heterotrimeric G protein. Nature. 1996;379:311–319. doi: 10.1038/379311a0. [DOI] [PubMed] [Google Scholar]
- 26.Majumdar S, Ramachandran S, Cerione RA. Perturbing the linker regions of the α-subunit of transducin: A new class of constitutively active GTP-binding proteins. J Biol Chem. 2004;279:40137–40145. doi: 10.1074/jbc.M405420200. [DOI] [PubMed] [Google Scholar]
- 27.Marin EP, Krishna AG, Sakmar TP. Rapid activation of transducin by mutations distant from the nucleotide-binding site: Evidence for a mechanistic model of receptor-catalyzed nucleotide exchange by G proteins. J Biol Chem. 2001;276:27400–27405. doi: 10.1074/jbc.C100198200. [DOI] [PubMed] [Google Scholar]
- 28.Iiri T, Herzmark P, Nakamoto JM, van Dop C, Bourne HR. Rapid GDP release from Gsa in patients with gain and loss of endocrine function. Nature. 1994;371:164–168. doi: 10.1038/371164a0. [DOI] [PubMed] [Google Scholar]
- 29.Klein PS, Sun TJ, Saxe CL, III, Kimmel AR, Johnson RL, Devreotes PN. A chemoattractant receptor controls development in Dictyostelium discoideum. Science. 1988;241:1467–1472. doi: 10.1126/science.3047871. [DOI] [PubMed] [Google Scholar]
- 30.Wilkie TM, Kinch L. New roles for Gα and RGS proteins: Communication continues despite pulling sisters apart. Curr Biol. 2005;15:R843–R854. doi: 10.1016/j.cub.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 31.Nordström KJ, Sällman Almén M, Edstam MM, Fredriksson R, Schiöth HB. Independent HHsearch, Needleman–Wunsch-based, and motif analyses reveal the overall hierarchy for most of the G protein-coupled receptor families. Mol Biol Evol. 2011;28:2471–2480. doi: 10.1093/molbev/msr061. [DOI] [PubMed] [Google Scholar]
- 32.Gabay M, Pinter ME, Wright FA, Chan P, Murphy AJ, Valenzuela DM, Yancopoulos GD, Tall GG. Ric-8 proteins are molecular chaperones that direct nascent G protein α subunit membrane association. Sci Signal. 2011;4:ra79. doi: 10.1126/scisignal.2002223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chan P, Gabay M, Wright FA, Tall GG. Ric-8B is a GTP-dependent G protein as guanine nucleotide exchange factor. J Biol Chem. 2011;286:19932–19942. doi: 10.1074/jbc.M110.163675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tall GG, Krumins AM, Gilman AG. Mammalian Ric-8A (synembryn) is a heterotrimeric Gα protein guanine nucleotide exchange factor. J Biol Chem. 2003;278:8356–8362. doi: 10.1074/jbc.M211862200. [DOI] [PubMed] [Google Scholar]
- 35.Mukhopadhyay S, Ross EM. Rapid GTP binding and hydrolysis by Gq promoted by receptor and GTPase-activating proteins. Proc Natl Acad Sci U S A. 1999;96:9539–9544. doi: 10.1073/pnas.96.17.9539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nei M. Selectionism and neutralism in molecular evolution. Mol Biol Evol. 2005;22:2318–2342. doi: 10.1093/molbev/msi242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Apanovitch DM, Slep KC, Sigler PB, Dohlman HG. Sst2 is a GTPase-activating protein for Gpa1: Purification and characterization of a cognate RGS–Gα protein pair in yeast. Biochemistry. 1998;37:4815–4822. doi: 10.1021/bi9729965. [DOI] [PubMed] [Google Scholar]
- 38.Lofmark S, Edlund C, Nord CE. Metronidazole is still the drug of choice for treatment of anaerobic infections. Clin Infect Dis. 2010;50(suppl. 1):S16–S23. doi: 10.1086/647939. [DOI] [PubMed] [Google Scholar]
- 39.John DT. Primary amebic meningoencephalitis and the biology of Naegleria fowleri. Annu Rev Microbiol. 1982;36:101–123. doi: 10.1146/annurev.mi.36.100182.000533. [DOI] [PubMed] [Google Scholar]
- 40.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huson DH, Bryant D. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol. 2006;23:254–267. doi: 10.1093/molbev/msj030. [DOI] [PubMed] [Google Scholar]
- 42.Brandt DR, Ross EM. GTPase activity of the stimulatory GTP-binding regulatory protein of adenylate cyclase, Gs. Accumulation and turnover of enzyme-nucleotide intermediates. J Biol Chem. 1985;260:266–272. [PubMed] [Google Scholar]
- 43.Chasse SA, Flanary P, Parnell SC, Hao N, Cha JY, Siderovski DP, Dohlman HG. Genome-scale analysis reveals Sst2 as the principal regulator of mating pheromone signaling in the yeast Saccharomyces cerevisiae. Eukaryot Cell. 2006;5:330–346. doi: 10.1128/EC.5.2.330-346.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li L, Wright SJ, Krystofova S, Park G, Borkovich KA. Heterotrimeric G protein signaling in filamentous fungi. Annu Rev Microbiol. 2007;61:423–452. doi: 10.1146/annurev.micro.61.080706.093432. [DOI] [PubMed] [Google Scholar]
- 45.Prabhu Y, Eichinger L. The Dictyostelium repertoire of seven transmembrane domain receptors. Eur J Cell Biol. 2006;85:937–946. doi: 10.1016/j.ejcb.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 46.Simon MI, Strathmann MP, Gautam N. Diversity of G proteins in signal transduction. Science. 1991;252:802– 808. doi: 10.1126/science.1902986. [DOI] [PubMed] [Google Scholar]
- 47.Temple BR, Jones CD, Jones AM. Evolution of a signaling nexus constrained by protein interfaces and conformational states. PLoS Comput Biol. 2010;6:e1000962. doi: 10.1371/journal.pcbi.1000962. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
www.sciencesignaling.org/cgi/content/full/6/276/ra37/DC1
Fig. S1. Genes encoding heterotrimeric G protein components found in eukaryotic genomes.
Fig. S2. Phylogeny of unikonta GPCRs.
Fig. S3. Minimum spanning network for unikonta GPCRs.
Fig. S4. ML trees of the 7TM-RGS proteins show the distinct origin of 7TM-RGS proteins.
Fig. S5. Intrinsic fluorescence measurements of the nucleotide exchange and hydrolysis activities of Gα proteins support the radionucleotide assay–based measurements.
Fig. S6. Identity among T. vaginalis G protein components.
Fig. S7. Multiple sequence alignment of Gα subunits showing conserved residues.
Table S1. Accession numbers of genes encoding G proteins, RGS proteins, and GPCRs.
References (43–47)