

ABSRTACT
Coxiella burnetii is an obligate intracellular bacterium that causes Query (Q) fever, a zoonotic disease. It requires a functional type IV secretion system (T4SS) which translocate bacterial effector proteins into the host cell cytoplasm and thereby facilitates bacterial replication. To date, more than 130 effector proteins have been identified, but their functions remain largely unknown. Recently, we demonstrated that one of these proteins, CaeA (CBU1524) localized to the host cell nucleus and inhibited intrinsic apoptosis of HEK293 or CHO cells. In the present study we addressed the question whether CaeA also affects the extrinsic apoptosis pathway.
Ectopic expression of CaeA reduced extrinsic apoptosis and prevented the cleavage of the executioner caspase 7, but did not impair the activation of initiator caspase 9. CaeA expression resulted in an up-regulation of survivin (an inhibitor of activated caspases), which, however, was not causal for the anti-apoptotic effect of CaeA. Comparing the sequence of CaeA from 25 different C. burnetii isolates we identified an EK (glutamic acid/ lysine) repetition motif as a site of high genetic variability. The EK motif of CaeA was essential for the anti-apoptotic activity of CaeA. From these data, we conclude that the C. burnetii effector protein CaeA interferes with the intrinsic and extrinsic apoptosis pathway. The process requires the EK repetition motif of CaeA, but is independent of the upregulated expression of survivin.
KEYWORDS: apoptosis, bacterial pathogenesis, Coxiella burnetii, surviving, type IV secretion system
Introduction
C. burnetii is a Gram-negative, obligate intracellular bacterium which is the causative agent of Q fever, a zoonotic disease with worldwide prevalence except for New Zealand.1 Humans get infected by inhalation of infectious material transmitted from domestic livestock. Infection by a single bacterium can result in disease.2 Most frequently, Q-fever remains asymptomatic or causes only a mild flu-like illness. However, the infection can also lead to interstitial pneumonia, hepatitis or severe chronic disease, which usually presents in form of an endocarditis.1 Due to the increase in worldwide Q-fever cases over the last decade C. burnetii is considered as an emerging pathogen.3
Once C. burnetii has entered the human body by inhalation it is taken up by mononuclear phagocytes into a phagosome.4 The C. burnetii-containing vacuole (CCV) matures to an acidic, phagolysosomal-like parasitophorous vacuole that is permissive for bacterial replication.5-7 C. burnetii requires a functional Dot/Icm type IV secretion system (T4SS) for the establishment of the replicative CCV.8-10 The T4SS translocate bacterial virulence factors, termed effector proteins, into the host cell cytoplasm.11 Importantly, it was shown that C. burnetii requires the T4SS to prevent host cell death.9 To date, more than 130 putative C. burnetii effector proteins have been identified,12 some of which have anti-apoptotic activity,13-15 whereas the majority still awaits functional characterization.
Apoptosis, a form of programmed cell death, is part of the intrinsic immune defense.16 It allows removal of damaged or infected cells in the absence of inflammation 17 and is a central mechanism of peripheral immune tolerance.18 Two main pathways lead to apoptosis induction: The extrinsic and the intrinsic apoptosis pathway. The extrinsic pathway is triggered by ligand binding to death receptors and adaptor proteins, which activates caspases (cysteinyl aspartate proteases). The intrinsic pathway involves activation of Bax and Bak, which is regulated by the Bcl-2 protein family.19,20 This protein family comprises both positive (BH3-only) and negative (Bcl-2-like) regulators of apoptosis. The ratio of positive and negative regulators expressed in a cell determines whether Bax and Bak are activated. Once activated, Bax and Bak oligomerize and permeabilize the mitochondrial membrane, resulting in the release of small molecules like cytochrome c and activation of caspase 9 through assembly of the apoptosome.21 Activated caspase 9 leads to cleavage and, thus, activation of the key downstream executors of apoptosis: caspase 3 and caspase 7.22 As proteolysis is irreversible, activation of caspases must be tightly regulated. One known mechanism deployed to protect the cell from death receptor and mitochondrial apoptosis is the inhibition of activated caspases by inhibitor of apoptosis proteins (IAPs). In humans the IAPs comprise 8 family members: NAIP, cIAP1, cIAP2, XIAP, survivin, Bruce, ML-IAP and ILP2.23 While it was first believed that all of the IAPs can bind caspases, we now know that in vivo only XIAP functions as a physiological inhibitor of caspases.24,25 The other IAPs require the interaction with cooperative partners to inhibit caspases and thereby apoptosis.24
Several pathogens have evolved mechanisms to modulate host cell apoptosis, which has emerged as a crucial determinant of virulence. Some bacteria actively induce host cell death to escape from an microbicidal, phagosomal environment or to overcome barriers.26,27 In contrast, obligate intracellular pathogens frequently inhibit apoptosis (e. g. by use of T3SS or T4SS effector proteins) to prevent premature host cell death and to generate a habitat for replication.28,29 C. burnetii is no exception as it utilizes its T4SS to inhibit host cell apoptosis.9 So far, 3 anti-apoptotic effector proteins (AnkG, CaeA and CaeB) have been identified.14,15,30 However, their precise molecular activity has still to be unraveled.
The C. burnetii effector protein CaeA localizes to the nucleus when produced in mammalian cells 8 and displays anti-apoptotic activity.14 In this study we found that CaeA does not interfere with the activation of caspase 9 but prevents the cleavage of caspase 7, indicating that CaeA interferes with the apoptotic cascade between the initiator caspase 9 and the executioner caspase 7. Interestingly, CaeA expression resulted in an upregulation of survivin, an IAP known to inhibit activated caspases. However, siRNA experiments revealed that the anti-apoptotic activity of C. burnetii was not mechanistically linked to the increased level of survivin. By comparing the sequence of CaeA from 25 C. burnetii isolates we identified an EK repetition motif, consisting of alternating glutamic acid and lysine residues, as a site of genetic variability. The EK motif of CaeA was required for anti-apoptotic activity, but not for the intracellular localization of CaeA.
Results
CaeA expression prevents apoptosis at the executioner caspase level
An increase in the anti-apoptotic Bcl-2-like steady-state protein level results in cell survival. Therefore, we analyzed whether the expression of CaeA (derived from the C. burnetii Nine Mile [NM] II strain) results in an altered protein level of anti-apoptotic Bcl-2-family members. Thus, HEK293 cells stably expressing GFP or GFP-CaeA were treated with different UV-light intensities. UV-light induces DNA damage and subsequently intrinsic apoptosis.31 Immunoblot analysis revealed a similar relative abundance of the anti-apoptotic Bcl-2-like proteins Bcl-2 and Bcl-xL under all conditions tested (Fig. 1A). In contrast, we detected decreased Mcl-1 protein levels after UV-light treatment, which, however was also true for both GFP-CaeA expressing and GFP control cells. Next, we analyzed whether CaeA inhibits activation of initiator and executioner caspases after UV-light treatment. Apoptosis-induction was measured by assaying the presence of cleaved poly ADP-ribose polymerase (PARP). Proteolytic cleavage of nuclear PARP inactivates DNA repair activity and is a marker for the terminal stages of apoptosis.32 As shown in Figure 1B, GFP-expressing cells treated with UV-light displayed caspase 9 and caspase 7 cleavage as well as PARP cleavage. In contrast, cells expressing GFP-CaeA were refractory to UV-induced proteolytic cleavage of caspase 7 and PARP, but not to proteolytic cleavage of caspase 9. This result suggests that CaeA interferes with the apoptotic cascade downstream of caspase 9 activation and upstream of caspase 7 activation.
Figure 1.
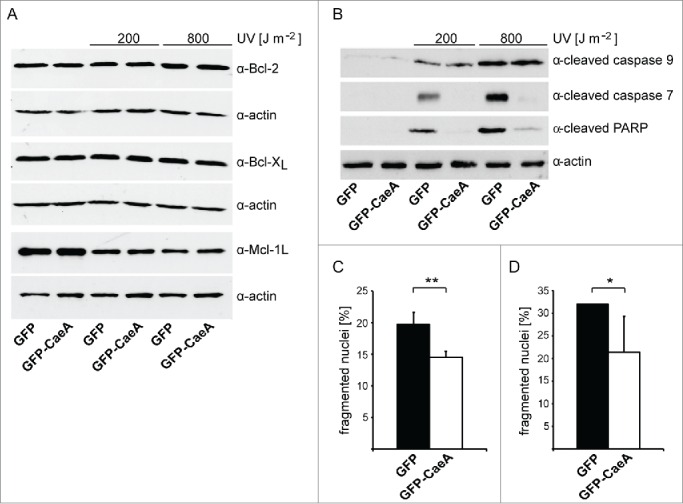
CaeA prevents activation of executioner caspase 7. HEK293 cells stably expressing GFP or GFP-CaeA were exposed to UV-light (200 J/m2 or 800 J/m2) and incubated for 6 h at 37°C in 5% CO2. Proteins were separated by SDS-PAGE, transferred to a PVDF membrane and probed with antibodies against (a) Bcl-2, Bcl-xL, Mcl-1 and actin as loading control or (b) cleaved caspase 9, cleaved caspase 7, cleaved PARP and actin as loading control. The result of one representative experiment out of 3 independent experiments with similar results is shown. (c) HeLa cells were transiently transfected with plasmids encoding GFP as control or GFP-CaeA followed by treatment with TNF (20 ng/ml) and cycloheximide (6 µg/ml) for 5 h at 37°C in 5% CO2. The cells were fixed, permeabilized and the nuclei were stained with DAPI. The nuclear morphology of GFP-expressing cells was scored. Data represent average values ± SD of 100 nuclei counted per sample from GFP-expressing cells from 3 independent experiments. (d) HeLa-Fas cells were transiently transfected with plasmids encoding GFP as control or GFP-CaeA followed by treatment with 0.125 µg/ml anti-Fas IgG for 5 h. The cells were fixed, permeabilized and the nuclei were stained with DAPI. The nuclear morphology of GFP-expressing cells was scored. Data represent average values ± SD of 100 nuclei counted per sample from GFP-expressing cells from 3 independent experiments.
CaeA also inhibits extrinsic apoptosis
As our data suggested that CaeA inhibits apoptosis at the executioner caspase level, we assumed that CaeA not only inhibits intrinsic apoptosis but also extrinsic apoptosis. Fas-ligand and tumor necrosis factor (TNF) in combination with cycloheximide (CHX) are known inducers of the extrinsic apoptosis pathway.33 Thus, we induced extrinsic apoptosis with TNF and CHX in HeLa cells ectopically producing GFP or GFP-CaeA and measured nuclear fragmentation visualized by DAPI staining. This assay was used as it is a fast and robust assay. Importantly, we used the TUNEL assay to validate our nuclear fragmentation assay. As shown in Figure 1C the expression of GFP-CaeA protected the cells significantly from TNF plus CHX-induced apoptosis compared to cells expressing GFP alone. To determine whether CaeA expression also inhibits Fas-ligand induced apoptosis, HeLa cells stably expressing the Fas receptor (HeLa-Fas) and transiently expressing GFP or GFP-CaeA were treated with Fas-ligand. As expected, CaeA expression significantly inhibited Fas-ligand-induced extrinsic apoptosis visualized by DAPI staining (Fig. 1D). Importantly, expression of GFP or GFP-CaeA without cell death induction did not influence viability of HeLa and HeLa-Fas cells (data not shown). Together these data indicate that CaeA protects cells from both intrinsic and extrinsic apoptosis.
CaeA expression results in an up-regulation of survivin
Apoptotic cell death can be prevented by inhibitor of apoptosis proteins (IAPs). Therefore, we assessed the steady state level of the IAPs XIAP and survivin. XIAP can bind directly to caspase 3, 7 and 9, protecting them from proteolytic cleavage.34 Survivin is the smallest member of the IAP family 35 and regulates several cellular processes, including apoptosis.36 We also aimed to analyze the expression level of the other members of the IAP family. However, probably due to low expression level we were unable to detect these IAP at protein and mRNA level.
Thus, HEK293 cells stably expressing GFP or GFP-CaeA were treated with different UV-light intensities. Immunoblot analysis revealed an upregulation of survivin in GFP-CaeA expressing cells as compared to GFP expressing cells, while XIAP was not altered (Fig. 2A). This effect of CaeA was detected during steady-state conditions as well as after UV exposure of the cells (Fig. 2A and 2B).
Figure 2.
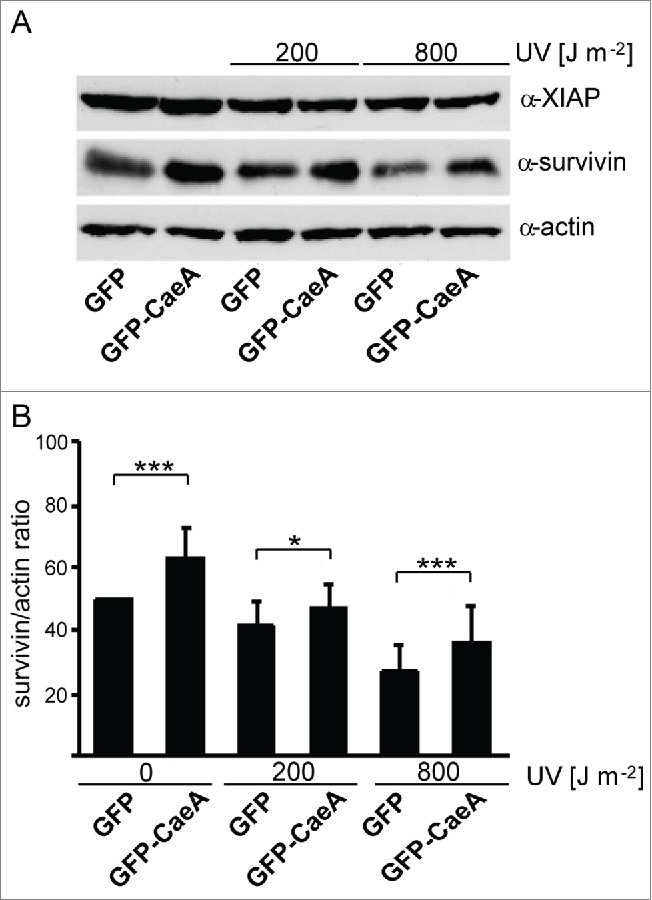
CaeA leads to survivin up-regulation. HEK293 cells stably expressing GFP or GFP-CaeA were exposed to UV-light (200 J/m2 or 800 J/m2) and incubated for 6 h. (a) Proteins were separated by SDS-PAGE, transferred to a PVDF membrane and probed with antibodies against survivin, XIAP and actin as loading control. The result of one representative experiment out of 3 independent experiments with similar results is shown. (b) Densitometric analysis of the survivin/actin ratio was performed using ImageJ. The data shown represent average values ± SD from at least 3 independent experiments. *P < 0.05 ***P < 0.001
CaeA-induced inhibition of apoptosis is not dependent on survivin up-regulation
As CaeA expression caused inhibition of apoptosis and upregulation of survivin, we investigated whether the increased protein expression of survivin accounts for the anti-apoptotic effect of CaeA. To this end, we transfected HEK293 cells stably expressing GFP or GFP-CaeA with survivin siRNA, which efficiently depleted the cells of survivin mRNA (Fig. 3A) and survivin protein (Fig. 3B). After exposure of the cells to UV light, CaeA was equally effective in protecting the cells from PARP cleavage and apoptosis in the presence and in the absence of survivin (Fig. 3B). From these data we conclude that inhibition of apoptosis by CaeA is not mediated by the upregulation of survivin.
Figure 3.
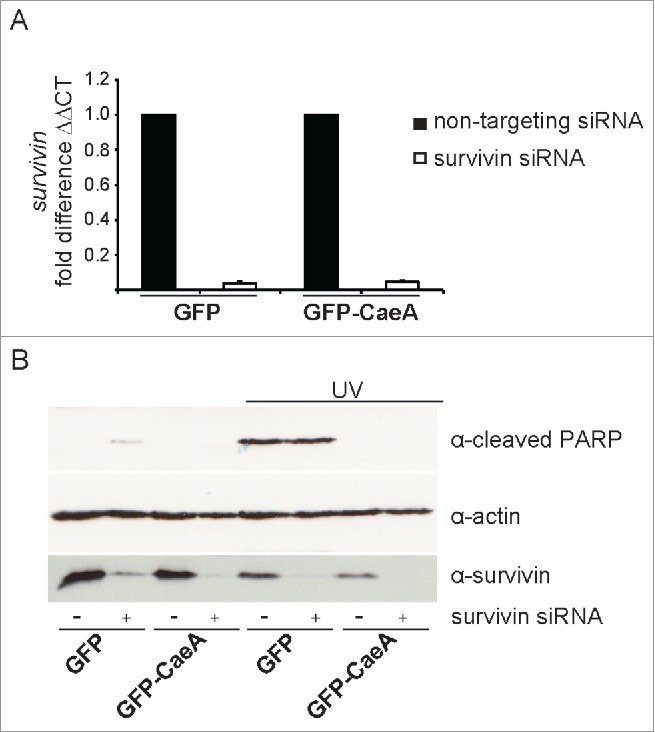
Survivin knock-down does not affect CaeA-induced apoptosis-inhibition. HEK293 cells stably expressing GFP or GFP-CaeA were transfected with 50nM non-targeting siRNA (−) or human survivin siRNA (+). (a) 48 hours post-transfection total isolated RNAs were reverse transcribed using SuperScript II reverse transcriptase according to the manufacturer's protocol and a qRT-PCR was performed with oligonucleotides specific for survivin and hpbgd as a house keeping gene. The ΔΔCt values were calculated for the fold difference of survivin siRNA-treated cells to non-targeting siRNA-treated cells using the 2−ΔΔ Ct method. (b) 48 hours post-transfection cells were treated with or without UV light (800 J/m2) and incubated for 6h at 37°C in 5% CO2. Proteins were separated by SDS-PAGE, transferred to a PVDF membrane and probed with antibodies against cleaved PARP, survivin and actin as loading control. The result of one representative experiment out of 3 independent experiments with similar results is shown.
Sequence analysis of C. burnetii strains reveals a genetic variability of CaeA
CaeA has a molecular weight of 25.1 kDa and contains a predicted coiled-coil region that spans from aa 25 to 117 and 2 nuclear localization signals (NLS).8,14 Comparison of genomes from different C. burnetii isolates pointed to a significant plasticity in the repertoire of T4SS effector proteins.8 With respect to single effector proteins, differences in the DNA sequence might help to elucidate their function. Therefore, we analyzed the sequence of caeA from 25 different strains of C. burnetii (Table 1). As shown in Figure 4, the sequence analysis revealed a striking similarity of all strains except for a 5′ gene locus starting from base pair 148 that contains GAA AAG/A repeats. This segment, which is part of the coiled-coil region, encodes alternating glutamic acid (E) and lysine (K) residues. It is present in all strains analyzed, but the number of EK repeats varied between 3 and 13. In addition, 2 single base deletions occurred. Twenty of the sequenced strains showed a single base deletion that provoked a frame shift and the introduction of a premature ochre stop codon at base pairs 61-63. An additional second single base deletion, downstream of this stop codon, was present in 8 of these isolates. Consequently, caeA represents a presumptive pseudogene in 20 of the 25 isolates analyzed in this study. Based on these in silico analyses only the C. burnetii strain NM I, which contains 6 EK repeats, and 4 further isolates, which all contain 3 EK repeats, are predicted to express a functional CaeA molecule. These five isolates originated from different regions and host species. While strain F-3 was obtained in France from a patient with endocarditis and therefore appears to be associated with chronic Q-fever, strain F-4 was isolated in France from an acute Q-fever patient 37 and the NM strain originated from a tick in Montana, USA.1 The strain Namibia was obtained from an infected goat in Namibia and the strain Z3574-1/92 was derived from milk of an infected ewe in Germany; in both cases there was no information available on the clinical status of the host animals.
Table 1.
Coxiella burnetii strains used in this study.
| Cluster | Isolate | Host | Material | Clinic | Origin | Year |
|---|---|---|---|---|---|---|
| I | F-3 | human | mitral valve | endocarditis | Lyon, France | 1978 |
| I | F-4 | human | blood, mitral valve | endocarditis | Marseille, France | 1978 |
| I | Namibia | goat | — | — | Windhoek, Namibia | 1991 |
| I | Z 3574-1/92 | sheep | milk | — | Berlin, Germany | 1992 |
| II | 19/34 | goat | fetus, afterbirth | — | Bissingen/Teck, Germany | 2010 |
| II | 23/2 | sheep | fetus | — | Jork, Germany | 2011 |
| II | 30/14 | sheep | afterbirth | — | Jork, Germany | 2011 |
| II | W-3 | fallow deer | afterbirth | abort | Gäufelden, Germany | 1997 |
| II | W-4 | sheep | afterbirth | abort | Aidlingen, Germany | 1996/97 |
| II | Z 104/94 | sheep | afterbirth | — | Dillenburg, Germany | 1994 |
| II | Z 346/99 | sheep | afterbirth | abort | Sarstedt, Germany | 1999 |
| II | Z 3464/92 | goat | afterbirth | abort | Pohlheim, Germany | 1992 |
| II | Z 3468-5/92 | sheep | placenta | — | Niedersachsen, Germany | 1992 |
| II | Z 3478/92 | sheep | organs lamb | abort | Pohlheim, Germany | 1992 |
| II | Z 4485/93 | sheep | afterbirth | abort | Dillenburg, Germany | 1993 |
| III | NM II | tick | — | — | Montana, USA | 1938 |
| IV | Z 66/96 | goat | organs lamb | abort | Driedorf, Germany | 1996 |
| V | Z 232-3/02 | cattle | placenta | fertility disorder in livestock | Bobingen, Germany | 2002 |
| V | Z 502/99 | cattle | placenta | — | — | 1999 |
| V | Z 3568/92 | cattle | organs calf | abort | Niedersachsen, Germany | 1992 |
| VI | 71/3 | goat | afterbirth | — | Altkirchen, Germany | 2010 |
| VI | 98/2 | cattle | organ material | — | Altenberga, Germany | 2010 |
| VI | Z 488/94 | cattle | amniotic liquor, afterbirth | abort | Lauterbach, Germany | 1994 |
| VI | Z 3351/92 | cattle | placenta | abort | Hannover, Germany | 1992 |
| VII | Z 3567/92 | cattle | afterbirth, maw | abort | Niedersachsen, Germany | 1992 |
Figure 4.

Genomic plasticity in the caeA gene. Genomic DNA of 25 C. burnetii isolates was prepared, the gene caeA and flanking regions was amplified and sequenced, and a multiple sequence alignment was performed. The illustrated alignment includes 63 bp upstream of the start codon (underlined) and the first 182–240 bp of the caeA open reading frame. Nucleotides matching in all isolates are marked by a dot and single base deletions are indicated by a dash in a red box. Non-displayed regions (positions 7–48 and 62–129) are identical in all isolates. Within the sequence of caeA each isolate contains a GAA AAG/A (EK) motif with a different number (3 to 13) of repetitions (blue box). Strains were grouped into 7 clusters (I to VII) according to their EK-repetition motif profile. The first base deletion (nt∼55) results in a frame shift and premature stop codon ‘TAA’ at bp 61 to 63, the second base deletion (nt∼136) similarly provokes a ‘TAA’ stop codon immediately downstream of the GAA AAG/A motif.
Taken together, the caeA gene of C. burnetii shows a considerable genetic polymorphism in a region which encodes an EK-repetition motif.
The EK repetition motif is required for the anti-apoptotic activity of CaeA
To assess the functional role of the EK motif, a HEK293 cell line that stably expressed GFP-CaeA lacking the EK repeat (GFP-CaeAΔEK) was established. This cell line was analyzed by flow cytometry together with the already established HEK293 cells stably expressing GFP or GFP-CaeA. As shown in Figure 5A, around 90% of the cells in all 3 stable cell lines expressed GFP. Next, the anti-apoptotic activity of CaeA lacking the EK repeats was tested. While HEK293-GFP cells showed caspase 7 and PARP cleavage after UV-light exposure, HEK293-GFP-CaeA cells were protected from caspase 7 and PARP cleavage (Fig. 5B and 5C), confirming the results in Figure 1B. In contrast, HEK293-GFP-CaeAΔEK showed increased levels of caspase 7 and PARP cleavage after UV-light treatment, suggesting that CaeA lacking the EK repeats was unable to protect cells from apoptosis. Importantly, ectopically expressed GFP-CaeAΔEK still exhibited a proper intracellular localization under steady-state conditions and after apoptosis induction. As depicted in Figure 6, both GFP-CaeA and GFP-CaeAΔEK showed nuclear speckle-like localization with and without apoptosis-induction. Thus, the EK repetition motif enables CaeA to interfere with the apoptosis cascade, but is not required for the intracellular localization of CaeA.
Figure 5.
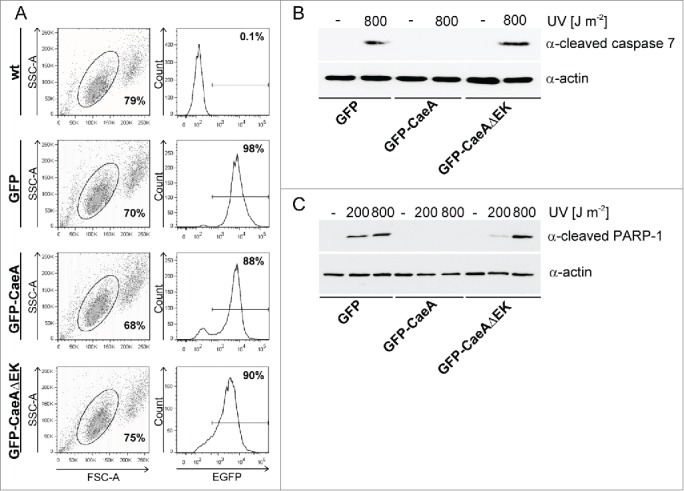
The EK repetition motif is required for apoptosis inhibition. (a) HEK293 cells and HEK293 cells stably expressing GFP, GFP-CaeA and GFP-CaeAΔEK were analyzed by flow cytometry. One representative experiment out of at least 3 independent experiments with similar results is depicted. (b and c) HEK293 cells stably expressing GFP, GFP-CaeA or GFP-CaeAΔEK were exposed to the UV-light intensities indicated and incubated for 6 h at 37°C in 5% CO2. Proteins were separated by SDS-PAGE, transferred to a PVDF membrane and probed with antibodies against (b) cleaved caspase 7, cleaved PARP (c) and actin as loading control. The result of one representative experiment out of at least 3 independent experiments with similar results is shown.
Figure 6.

Deletion of the EK repetition motif does not alter intracellular localization of CaeA. Representative immunofluorescence micrographs show CHO-FcR cells transiently transfected with GFP, GFP-CaeA and GFP-CaeAΔEK. The cells were treated with or without 1 µM staurosporine for 6 h at 37°C in 5% CO2 followed by fixation, permeabilization and staining of the nuclei with DAPI (blue).
CaeA-induced apoptosis inhibition varies with the number of EK repeats
Sequence analyses of 25 different isolates of C. burnetii showed that CaeA contains between 3 and 13 EK repeats. As stated above, only CaeA molecules containing either 3 or 6 EK repeats are expressed as functional effector proteins. To determine how many repetitions of the EK motif in CaeA are minimally required for efficient inhibition of apoptosis, we tested CHO cells ectopically expressing CaeAΔEK with 2 (CaeAΔEK+2EK) or 4 EK (CaeAΔEK+4EK) repeats. CHO cells expressing wild-type NM II-derived CaeA were significantly protected from apoptosis, whereas cells expressing CaeAΔEK were not (Fig. 7), confirming the results obtained in HEK293 cells (Fig. 5B and 5C). Importantly, expression of CaeAΔEK+2EK also inhibited apoptosis, but significantly less than CaeA, while expression of CaeAΔEK+4EK protected the cells as efficiently as wild-type NM II CaeA, which contains 6 EK repeats. From these findings we conclude that at least 4 EK repetitions are required for full anti-apoptotic activity of CaeA.
Figure 7.
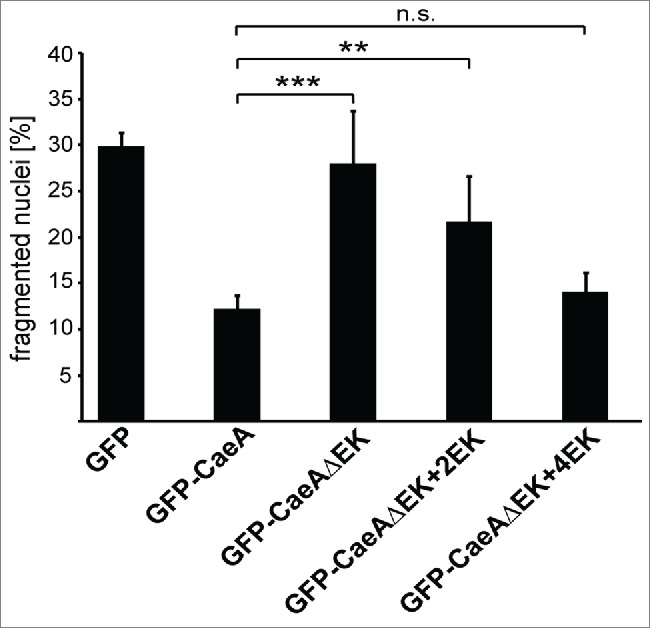
Four EK repeats are sufficient for inhibition of apoptosis. CHO-FcR cells transiently transfected with GFP, GFP-CaeA, GFP-CaeAΔEK, GFP-CaeAΔEK+2EK and GFP-CaeAΔEK+4EK were treated with 1µM staurosporine for 6 h at 37°C in 5% CO2. The cells were fixed, permeabilized and the nuclei were stained with DAPI. The nuclear morphology of GFP-expressing cells was scored. Data represent average values ± SD of 100 nuclei counted per sample from GFP-expressing cells from 5 independent experiments. **P < 0.01, ***P < 0.001.
Discussion
Apoptosis of infected host cells has been identified as an important element of the innate immune response contributes to the clearance of microorganisms cleared from the body. Especially obligate intracellular pathogens have evolved mechanisms to impede the process of host cell apoptosis, but the underlying molecular mechanisms are still incompletely understood. This is also the case for C. burnetii, for which inhibition of host cell apoptosis was first described in 2007.38,39 The anti-apoptotic function of the C. burnetii effector proteins AnkG, CaeA and CaeB 14,15 suggests that proteins transported by the T4SS are important regulators of host cell survival.
Due to a re-annotation of C. burnetii Nine Mile phase I (RSA493) CaeA and CaeB are now annotated as pseudogenes. CaeA (former CBU1524) is now part of CBU1523 and CaeB (former CBU1532) is now part of CBU1531. However, RT-PCR analysis confirmed transcription of CaeA and CaeB and the presence of a functional T4SS translocation signal at its C-terminus.8 Whether CaeA is only a fragment of CBU1523 or whether CBU1523 and CaeA (CBU1524) are 2 independent proteins is unknown and has not been experimentally tested. However, we hypothesize that CaeA is not a fragment of CBU1523 but rather a discrete protein, as the CBU1524 gene contains a potential Shine Dalgarno sequence upstream of the start codon, and an 114bp intergenic stretch, which separates the stop codon of CBU1523 and the start codon of CBU1524. This intergenic stretch is enough sequence space to contain transcriptional and translational start sites for CBU1524.
In this study, we investigated the determinants of the anti-apoptotic function of the C. burnetii effector protein CaeA. CaeA was originally described as an inhibitor of the intrinsic apoptosis pathway when endogenously expressed in HEK293 or CHO cells.14 Our present results suggest that CaeA also protects from the induction of extrinsically triggered apoptosis (Fig. 1C and D). This indicates that CaeA might act at the intersection of both apoptosis pathways. In agreement with this assumption, cleavage of the effector caspase 7 was completely inhibited in presence of CaeA whereas cleavage of the upstream initiator caspase 9 was not (Fig. 1B). IAPs are known to inhibit activated caspases and to interfere at this late step of the apoptotic pathway.24 Some pathogens exploit the anti-apoptotic function of IAPs in order to manipulate the host apoptotic pathway and to promote host cell survival. Importantly, CaeA expression resulted in up-regulation of the steady-state protein level of the IAP survivin (Fig. 2). Several intracellular pathogens have been described to elicit an upregulation of survivin which subsequently improved host cell survival.40-42 For instance, the intracellular protozoan Cryptosporidium parvum caused an increased expression of cIAP1, cIAP2, XIAP as well as survivin in HCT-8 epithelial colon cells, and survivin was shown to be important for apoptosis inhibition by the pathogen.41 These observations contrast with our findings. While CaeA expression led to higher levels of survivin protein in the host cell, CaeA-mediated inhibition of apoptosis was independent of survivin (Fig. 3).
Sequence analysis of caeA from 25 different C. burnetii isolates revealed a noticeable difference in a region consisting of a variable number of GAA AAG/A (EK) codons (Fig. 4). The EK repetition motif of CaeA represents a short tandem repeat (STR) with the 6 bases ‘GAA AAG/A’. In general, STRs or ‘microsatellites’ are widely distributed in the genomes of eukaryotes and prokaryotes and are characterized by a sequentially repeated unit of one to 6 bp reaching up to 100 nt. Tandem repeats are hot spots for mutational events contributing to gene diversity and the evolutionary process. The high mutation rate and variability of tandem repeats is mainly explained by slipped-strand mispairing and a malfunctioning repair system during replication.43 Consistently, the presence of diverse numbers of EK repeats among the analyzed strains is most likely due to events of slipped-strand mispairing during the replication of the hexanucleotide GAA AAG/A. Besides the variability in the number of GAA AAG/A repetitions, 2 single base deletions were found to contribute to the sequence polymorphism of caeA in the different strains. In isolates encoding 5, 8, 10, 11 or 13 GAA AAG/A repetitions a single base deletion occurred leading to a frameshift and premature stop. In these strains caeA presumably represents a pseudogene. Interestingly, in addition to this first single base deletion, only in isolates encoding a higher number of repetitions, i.e. 10 to 13 GAA AAG/A units, a second single base deletion was found. In the absence of the first single base deletion the second base deletion would result in a frameshift altering the EK motif to an irregular succession of lysine and arginine residues and provoking a premature stop codon directly downstream. Thus, the second base deletion would similarly ensure the expression of a truncated protein. However, every isolate harboring the second base deletion, already showed the first base deletion at position ∼55 which per se is sufficient to cause the truncation of CaeA. Remarkably, in addition to NM only 4 of all 25 analyzed isolates are also capable to express a non-truncated protein. All these isolates of C. burnetii contain 3 EK repeat units and include 2 human, one goat and one sheep isolate. Thus, one might speculate the presence of a low number of EK repeats is a prerequisite for the expression of a non-truncated CaeA.
In prokaryotic genomes STRs exhibit a high mutation rate of up to 10−1 44 which contributes to phenotypic variability and fast adaptation to environmental or host conditions as has been shown especially for pathogenic bacteria. Indeed, human pathogens such as Neisseria gonorrhoeae or Haemophilus influenzae have been shown to undergo phase variation and to regulate protein expression similar to a switch on and off mechanism by alteration of the number of 5 and 4 base pair repetition units, respectively.45 Thus, the expression of H. influenzae licA, a gene that is participates in the synthesis of lipoteichoic acid (LTA), undergoes a phase variation that is mediated by the varying number of a 4 base pair repetition unit.46 In contrast, variations of the number of tri- or hexanucleotides (such as the CaeA EK repetition motif) do not influence the frame of a sequence and alter gene expression per se. Tri- or hexanucleotide tandem repeats seem to rather influence the function of a protein as variations of the repeat number may alter structural characteristics of a protein.47 For instance, the activity of the Salmonella enterica serovar Typhimurium and Escherichia coli DNA mismatch repair protein MutL is lost by alteration of the number of the 3 hexanucleotide repeat units encoded in the ATP binding pocket of MutL.48,49 As shown by Shaver et al. the deletion as well as the addition of one unit of the hexanucleotide repeat presumably interferes with ATP binding resulting in the inhibition of the ATP-dependent MutL function and an increased mutation rate.49
Regarding CaeA, the EK repetition lies within a predicted coiled-coil region, a domain that is organized in α-helical bundles facilitating protein-protein interactions. The negatively and positively charged amino acids of the EK motif account for a charged region with hydrophilic character and presumably lead to the exposition of the domain to the protein surface. Thus, the EK motif might provide a protein interaction site within the coiled-coil region which accounts for the interaction with specific protein partners. Similar to the aforementioned MutL mutation, variation of the EK motif might influence the interaction and the function of the protein. In accordance with this hypothesis the EK repetition motif was shown to be essential for CaeA-mediated inhibition of apoptosis (Fig. 5B and 5C).
Taken together, we have demonstrated that the effector protein CaeA of C. burnetii is an efficient inhibitor of the extrinsic pathway of apoptosis. Furthermore, we defined the EK repetition motif of CaeA as a structural component that is crucial for the anti-apoptotic function of CaeA. Further studies will aim to identify the exact interaction partners of CaeA in the host cell.
Materials and methods
Reagents, cell lines and bacterial strains
Unless otherwise noted, chemicals were purchased from Sigma Aldrich. Complete Protease inhibitor cocktail mixture and X-tremeGENE 9 Transfection Reagent were from Roche. Staurosporine was from Cell Signaling. Cell lines were cultured at 37°C in 5% CO2 in media containing 5% heat-inactivated fetal bovine serum (Biochrom). Chinese hamster ovary (CHO) fibroblasts were grown in minimal essential medium α medium (Invitrogen) and stable human embryonic kidney (HEK293) cells were maintained in Dulbecco´s modified Eagle medium (Invitrogen) supplemented with 1.5 mg/ml G418 (Roth). To construct HEK293 cells stably expressing GFP-CaeAΔEK the corresponding plasmid was transfected into HEK293 cells using X-tremeGENE 9 and selected by culturing in media supplemented with 1.5 mg/ml G418 (Roth) for 7 days. Single GFP-positive cells were sorted and grown for additional 10 to 14 days. C. burnetii strains used in this study are listed in Table 1. To generate plasmids encoding CaeA fusion proteins, genes were amplified by PCR, using C. burnetii Nine Mile phase II clone 4 as template and primers designed according to an earlier annotation of RSA493 (C. burnetii Nine Mile phase I).
siRNA knock-down
The protocol used for siRNA transfection was adapted from Dharmacon's HeLa cell transfection protocol. One volume of siRNA buffer containing 50nM of non-targeting or survivin siRNA (Dharmacon) was incubated with one volume of serum-free DMEM high glucose containing 5 μl/ml DharmaFECT-1 transfection reagent for 20 min at room temperature. Two volumes of DMEM high glucose supplemented with 20 % FBS containing 5 × 104 HeLa cells were added. The cells were seeded in a 12-well plates and incubated at 37°C with 5% CO2. The transfection mix was replaced with 500 μl of fresh media (DMEM high glucose supplemented with 10% FBS) 24 h post transfection. 3 days post transfection, the cells were washed with PBS and exposed to UV light (800 J/m2) in a transilluminator box (Stratagene). After adding fresh media, the cells were incubated for 6 h at 37°C in 5% CO2. The samples were separated by SDS-PAGE and transferred to a PVDF membrane (Millipore). The membranes were probed with antibodies against survivin (2808, Cell Signaling), cleaved PARP (611038, BD Biosciences) and actin (A2066, Sigma-Aldrich). The proteins were visualized by using horseradish peroxidase-conjugated secondary antibodies (Dianova) and a chemiluminescence detection system (Thermo Scientific or Millipore).
Plasmids and primers
Plasmid and primers used in this study are listed in.Tables 2–4
Table 2.
Plasmids used in this study.
| no | Plasmid | Reference |
|---|---|---|
| 1 | pCMV-HA-CaeA | Klingenbeck et al. |
| 2 | pCMV-HA-CaeAΔEK | this study |
| 3 | pEGFP-CaeAΔEK | this study |
| 4 | pCMV-HA-CaeAΔEK+2EK | this study |
| 5 | pCMV-HA-CaeAΔEK+4EK | this study |
| 6 | pEGFP-CaeAΔEK +2EK | this study |
| 7 | pEGFP-CaeAΔEK +4EK | this study |
Table 3.
PCR Primers used in this study.
| no | Oligonucleotide | Sequence 5′-3′ |
|---|---|---|
| 1 | caeAΔEK_fwd | TTAACACAGCAACTCACCGAAGAACAGGAGCGTTC |
| 2 | caeAΔEK_rev | AAGGTCTGTGCTTTTTTCTTCGAGTCGCTGATAGTCATTG |
| 3 | caeAΔEK+2_rev | TTCCTTTTCCTTTTCAAGGTCTGTGCTTTTTTCTTC |
| 4 | ceaAΔEK+4_rev | TTCCTTTTCCTTTTCCTTTTCCTTTTCAAGGTCTGTGCTTTTTTCTTC |
| 5 | caeA_3_fwd | AACTCGCAACCACATCCTCAAACC |
| 6 | caeA_4_rev | GAAGAAAAAGCGGCCATCCCTAAT |
Table 4.
Sequencing Primers used in this study.
| no | Oligonucleotide | Sequence 5′-3′ |
|---|---|---|
| 1 | caeA_S1_rev | CTCCTGAAATTGCATCGCAA |
| 2 | caeA_S2_fwd | CCTTGAGCAAGTAATTAACC |
Plasmid construction
pEGFP-CaeAΔEK was generated by mutagenic PCR with primers 1 and 2 (Table 3) amplified from pCMV-HA-CaeA. Ligation of PCR product resulted in pCMV-HA-CaeAΔEK. This vector was restricted with BglII and KpnI, followed by ligation with like-wise restricted pEGFP-C2.
pEGFP-CaeAΔEK+2EK and pEGFP-CaeAΔEK+4EK were generated by mutagenic PCR with primers 1 and 3 or 1 and 4 (Table 3) amplified from pCMV-HA-CaeAΔEK. Ligation of the PCR product resulted in pEGFP-CaeAΔEK+2EK and pEGFP-CaeAΔEK+4EK. The vectors were restricted with BglII and KpnI, followed by ligation with like-wise restricted pEGFP-C2.
Apoptosis assays
Nuclear fragmentation assay
CHO cells were plated on coverslips in 24-well dishes at a density of 2.5 × 104 cells/well. After an overnight incubation, cells were transfected with the plasmids indicated. 18 h post-transfection, the cells were incubated with staurosporine (1μg/ml) for 6 h at 37°C in 5% CO2. The cells were fixed with 4 % paraformaldehyde (Alfa Aeser) in PBS (Biochrom) for 20min at room temperature, permeabilized with ice-cold methanol for 30sec and quenched with 50 mM NH4Cl (Roth) in PBS for 15min at room temperature. The cells were mounted using ProLong Gold with DAPI (Invitrogen) to visualize the nucleus.
Immunoblotting
HEK293 cells stably expressing GFP, GFP-CaeA or GFP-CaeAΔEK were seeded in a 12-well plate at a density of 3 × 105 cells/well. The cells were washed with PBS before exposure with the indicated UV-light intensities (Stratagene). After 6h incubation at 37°C in 5%CO2 samples were separated by SDS-PAGE and transferred to a PVDF membrane (Millipore). The membranes were probed with antibodies directed against Bcl-2 (2870), Bcl-xL (2764), Mcl-1 (4572), cleaved caspase 7 (9491), cleaved caspase 9 (9501), XIAP (2045) and survivin (2808) all from Cell Signaling, cleaved PARP (611038, BD Biosciences) and actin (A2066, Sigma-Aldrich). The proteins were visualized by using horseradish peroxidase-conjugated secondary antibodies (Dianova) and a chemiluminescence detection system (Thermo Scientific or Millipore).
TNF + CHX–induced extrinsic apoptosis
HeLa cells were plated on coverslips in 24-well dishes at a density of 3.5 × 104 cells/well and were transfected with the plasmids indicated. 40 h post-transfection, the cells were incubated with 20 ng/ml TNF (Calbiochem) and 6 µg/ml cycloheximide (Sigma) for 5 h at 37°C in 5% CO2. The cells were fixed with 4 % paraformaldehyde (Alfa Aeser) in PBS (Biochrom) for 20 min at room temperature, permeabilized with ice-cold methanol for 30 sec and quenched with 50 mM NH4Cl (Roth) in PBS for 15 min at room temperature. The cells were mounted using ProLong Gold with DAPI (Invitrogen) to visualize the nucleus.
Fas-ligand-induced extrinsic apoptosis
HeLa-Fas cells 50 were plated on coverslips in 24-well dishes at a density of 4 × 104 cells/well. After an overnight incubation, cells were transfected with the plasmids indicated. 18 h post-transfection, the cells were incubated with 0.125 µg/ml anti-Fas IgG (Millipore) for 5 h at 37°C in 5% CO2. The cells were fixed with 4 % paraformaldehyde (Alfa Aeser) in PBS (Biochrom) for 20 min at room temperature, permeabilized with ice-cold methanol for 30 sec and quenched with 50 mM NH4Cl (Roth) in PBS for 15 min at room temperature. The cells were mounted using ProLong Gold with DAPI (Invitrogen) to visualize the nucleus.
Confocal microscopy
2 × 104 CHO cells were plated on coverslips and were transfected with the plasmids indicated. The cells were fixed with 4% paraformaldehyde (Alfa Aeser) in PBS (Biochrom), permeabilized with ice-cold methanol and quenched with 50mM NH4Cl (Roth) in PBS. The cells were mounted using ProLong Gold with DAPI (Invitrogen) to visualize the nucleus. Fluorescence was detected using a Carl Zeiss LSM 700 Laser Scan Confocal Microscope and a 64x objective. Images were analyzed using the ZEN2009 software (Jena, Germany).
FACS analysis
Protein expression levels of HEK293 cells stably expressing GFP, GFP-CaeA and GFP-CaeAΔEK were analyzed by flow cytometry.
PCR, sequencing and sequence analysis
PCR primers 5 and 6 (Table 3) for caeA were designed with Primer Select software implemented in Lasergene 9.0 (DNASTAR, Madison, WI). caeA was amplified using 1x OptiBuffer (Bioline, London, United Kingdom), 1.4 mM MgCL2, 200µM deoxynucleoside triphosphates (Bioline), 25pmol of each primer, 1.5 U Bio-X-Act Short DNA polymerase (Bioline), and 5 µl of DNA, in a final volume of 25 µl. The PCR was run with the following thermocycling profile: 5 min at 95°C; 1 min at 58°C; 35 cycles, each consisting of 1 min and 15 s at 72°C, 30 s at 95°C, and 30 s at 58°C; and a final elongation step of 10 min at 72°C. Cycle sequencing was performed using BigDye v1.1 chemistry according to the manufacturer's instructions (Applied Biosystems) (see Sequencing primers in Table 4), purified with DyeEx 96 plates (Qiagen) and electrophoresed on a 3130XL Genetic Analyzer (Applied Biosystems). Sequence analysis and polymorphism evaluation was performed using Lasergene 9.0 (DNASTAR, Madison, WI) and MEGA5 (Tamura et al., 2011).
The sequences of caeA gene are available at GenBank under the accession numbers KT828680-KT828703.
Isolation of genomic Coxiella burnetii DNA
Genomic DNAs of 25 paraformaldehyde-fixed or heat-killed C. burnetii isolates was prepared according to the manufacturer´s protocol with the illustra bacteria genomic Prep Mini Spin Kit (GE Health Care), with 3×10 min incubation at 55°C in the beginning and a final elution with 30 µl H2O.
Statistical analysis
An unpaired Student´s t-test or Chi-square test was used for statistical analysis.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Harald Wajant, Würzburg, Germany and Carsten Lüder, Göttingen, Germany for kindly providing Fas/CD95-overexpressing HeLa cells. We also thank Didier Soulat and Christian Berens for critically reading the manuscript and Martha Ölke for excellent technical assistance. AL is grateful to Prof. Dr. Christian Bogdan for his mentorship and his altruistic support.
Funding
This work was supported by the Deutsche Forschungsgemeinschaft (SFB796 project B8) to AL and by the ERA-NET PathoGenoMics 3rd call to AL and JPG.
References
- [1].Maurin M, Raoult D. Q fever. Clin Microbiol Rev 1999; 12:518-53; PMID:10515901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Baca OG, Paretsky D. Q fever and Coxiella burnetii: a model for host-parasite interactions. Microbiol Rev 1983; 47:127-49; PMID:6348504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Enserink M. Infectious diseases. Questions abound in Q-fever explosion in the Netherlands. Science 2010; 327:266-7; PMID:AMBIGUOUS [DOI] [PubMed] [Google Scholar]
- [4].Stein A, Louveau C, Lepidi H, Ricci F, Baylac P, Davoust B, Raoult D. Q fever pneumonia: virulence of Coxiella burnetii pathovars in a murine model of aerosol infection. Infect Immun 2005; 73:2469-77; PMID:15784593; http://dx.doi.org/ 10.1128/IAI.73.4.2469-2477.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Hackstadt T, Williams JC. Biochemical stratagem for obligate parasitism of eukaryotic cells by Coxiella burnetii. Proc Natl Acad Sci U S A 1981; 78:3240-4; PMID:6942430; http://dx.doi.org/ 10.1073/pnas.78.5.3240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Howe D, Mallavia LP. Coxiella burnetii exhibits morphological change and delays phagolysosomal fusion after internalization by J774A.1 cells. Infect Immun 2000; 68:3815-21; PMID:10858189; http://dx.doi.org/ 10.1128/IAI.68.7.3815-3821.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Beron W, Gutierrez MG, Rabinovitch M, Colombo MI. Coxiella burnetii localizes in a Rab7-labeled compartment with autophagic characteristics. Infect Immun 2002; 70:5816-21; PMID:12228312; http://dx.doi.org/ 10.1128/IAI.70.10.5816-5821.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Carey KL, Newton HJ, Luhrmann A, Roy CR. The Coxiella burnetii Dot/Icm system delivers a unique repertoire of type IV effectors into host cells and is required for intracellular replication. PLoS Pathog 2011; 7:e1002056; PMID:21637816; http://dx.doi.org/ 10.1371/journal.ppat.1002056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Beare PA, Gilk SD, Larson CL, Hill J, Stead CM, Omsland A, Cockrell DC, Howe D, Voth DE, Heinzen RA. Dot/Icm type IVB secretion system requirements for Coxiella burnetii growth in human macrophages. MBio 2011; 2:e00175-11; PMID:21862628; http://dx.doi.org/ 10.1128/mBio.00175-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Chen C, Banga S, Mertens K, Weber MM, Gorbaslieva I, Tan Y, Luo ZQ, Samuel JE. Large-scale identification and translocation of type IV secretion substrates by Coxiella burnetii. Proc Natl Acad Sci U S A 2010; 107:21755-60; PMID:21098666; http://dx.doi.org/ 10.1073/pnas.1010485107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Christie PJ, Vogel JP. Bacterial type IV secretion: conjugation systems adapted to deliver effector molecules to host cells. Trends Microbiol 2000; 8:354-60; PMID:10920394; http://dx.doi.org/ 10.1016/S0966-842X(00)01792-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Moffatt JH, Newton P, Newton HJ. Coxiella burnetii: turning hostility into a home. Cell Microbiol 2015; 17:621-31; PMID:25728389; http://dx.doi.org/ 10.1111/cmi.12432 [DOI] [PubMed] [Google Scholar]
- [13].Eckart RA, Bisle S, Schulze-Luehrmann J, Wittmann I, Jantsch J, Schmid B, Berens C, Luhrmann A. Antiapoptotic activity of Coxiella burnetii effector protein AnkG is controlled by p32-dependent trafficking. Infect Immun 2014; 82:2763-71; PMID:24733095; http://dx.doi.org/ 10.1128/IAI.01204-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Klingenbeck L, Eckart RA, Berens C, Luhrmann A. The Coxiella burnetii type IV secretion system substrate CaeB inhibits intrinsic apoptosis at the mitochondrial level. Cell Microbiol 2013; 15:675-87; PMID:23126667; http://dx.doi.org/ 10.1111/cmi.12066 [DOI] [PubMed] [Google Scholar]
- [15].Luhrmann A, Nogueira CV, Carey KL, Roy CR. Inhibition of pathogen-induced apoptosis by a Coxiella burnetii type IV effector protein. Proc Natl Acad Sci U S A 2010; 107:18997-9001; PMID:20944063; http://dx.doi.org/ 10.1073/pnas.1004380107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lamkanfi M, Dixit VM. Manipulation of host cell death pathways during microbial infections. Cell Host Microbe 2010; 8:44-54; PMID:20638641; http://dx.doi.org/ 10.1016/j.chom.2010.06.007 [DOI] [PubMed] [Google Scholar]
- [17].Gallucci S, Lolkema M, Matzinger P. Natural adjuvants: endogenous activators of dendritic cells. Nat Med 1999; 5:1249-55; PMID:10545990; http://dx.doi.org/ 10.1038/15200 [DOI] [PubMed] [Google Scholar]
- [18].Mueller DL. Mechanisms maintaining peripheral tolerance. Nat Immunol 2010; 11:21-7; PMID:20016506; http://dx.doi.org/ 10.1038/ni.1817 [DOI] [PubMed] [Google Scholar]
- [19].van Delft MF, Huang DC. How the Bcl-2 family of proteins interact to regulate apoptosis. Cell Res 2006; 16:203-13; PMID:16474435; http://dx.doi.org/ 10.1038/sj.cr.7310028 [DOI] [PubMed] [Google Scholar]
- [20].Willis SN, Adams JM. Life in the balance: how BH3-only proteins induce apoptosis. Curr Opin Cell Biol 2005; 17:617-25; PMID:16243507; http://dx.doi.org/ 10.1016/j.ceb.2005.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Adams JM, Cory S. Apoptosomes: engines for caspase activation. Curr Opin Cell Biol 2002; 14:715-20; PMID:12473344; http://dx.doi.org/ 10.1016/S0955-0674(02)00381-2 [DOI] [PubMed] [Google Scholar]
- [22].Cory S, Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer 2002; 2:647-56; PMID:12209154; http://dx.doi.org/ 10.1038/nrc883 [DOI] [PubMed] [Google Scholar]
- [23].Salvesen GS, Duckett CS. IAP proteins: blocking the road to death's door. Nat Rev Mol Cell Biol 2002; 3:401-10; PMID:12042762; http://dx.doi.org/ 10.1038/nrm830 [DOI] [PubMed] [Google Scholar]
- [24].Altieri DC. Survivin and IAP proteins in cell-death mechanisms. Biochem J 2010; 430:199-205; PMID:20704571; http://dx.doi.org/ 10.1042/BJ20100814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Eckelman BP, Salvesen GS, Scott FL. Human inhibitor of apoptosis proteins: why XIAP is the black sheep of the family. EMBO Rep 2006; 7:988-94; PMID:17016456; http://dx.doi.org/ 10.1038/sj.embor.7400795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Muller A, Gunther D, Dux F, Naumann M, Meyer TF, Rudel T. Neisserial porin (PorB) causes rapid calcium influx in target cells and induces apoptosis by the activation of cysteine proteases. EMBO J 1999; 18:339-52; PMID:9889191; http://dx.doi.org/ 10.1093/emboj/18.2.339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Zychlinsky A, Prevost MC, Sansonetti PJ. Shigella flexneri induces apoptosis in infected macrophages. Nature 1992; 358:167-9; PMID:1614548; http://dx.doi.org/ 10.1038/358167a0 [DOI] [PubMed] [Google Scholar]
- [28].Raymond B, Young JC, Pallett M, Endres RG, Clements A, Frankel G. Subversion of trafficking, apoptosis, and innate immunity by type III secretion system effectors. Trends Microbiol 2013; 21:430-41; PMID:23870533; http://dx.doi.org/ 10.1016/j.tim.2013.06.008 [DOI] [PubMed] [Google Scholar]
- [29].Laguna RK, Creasey EA, Li Z, Valtz N, Isberg RR. A Legionella pneumophila-translocated substrate that is required for growth within macrophages and protection from host cell death. Proc Natl Acad Sci U S A 2006; 103:18745-50; PMID:17124169; http://dx.doi.org/ 10.1073/pnas.0609012103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Berens C, Bisle S, Klingenbeck L, Luhrmann A. Applying an Inducible Expression System to Study Interference of Bacterial Virulence Factors with Intracellular Signaling. J Vis Exp 2015; 100:e52903; PMID:26168006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kulms D, Schwarz T. Independent contribution of three different pathways to ultraviolet-B-induced apoptosis. Biochem Pharmacol 2002; 64:837-41; PMID:12213577; http://dx.doi.org/ 10.1016/S0006-2952(02)01146-2 [DOI] [PubMed] [Google Scholar]
- [32].Kaufmann SH, Desnoyers S, Ottaviano Y, Davidson NE, Poirier GG. Specific proteolytic cleavage of poly(ADP-ribose) polymerase: an early marker of chemotherapy-induced apoptosis. Cancer Res 1993; 53:3976-85; PMID:8358726 [PubMed] [Google Scholar]
- [33].Jin Z, El-Deiry WS. Overview of cell death signaling pathways. Cancer Biol Ther 2005; 4:139-63; PMID:15725726; http://dx.doi.org/ 10.4161/cbt.4.2.1508 [DOI] [PubMed] [Google Scholar]
- [34].Galban S, Duckett CS. XIAP as a ubiquitin ligase in cellular signaling. Cell Death Differ 2010; 17:54-60; PMID:19590513; http://dx.doi.org/ 10.1038/cdd.2009.81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Srinivasula SM, Ashwell JD. IAPs: what's in a name? Mol Cell 2008; 30:123-35; PMID:18439892; http://dx.doi.org/ 10.1016/j.molcel.2008.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Pavlyukov MS, Antipova NV, Balashova MV, Vinogradova TV, Kopantzev EP, Shakhparonov MI. Survivin monomer plays an essential role in apoptosis regulation. J Biol Chem 2011; 286:23296-307; PMID:21536684; http://dx.doi.org/ 10.1074/jbc.M111.237586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Sekeyova Z, Roux V, Raoult D. Intraspecies diversity of Coxiella burnetii as revealed by com1 and mucZ sequence comparison. FEMS Microbiol Lett 1999; 180:61-7; PMID:10547445; http://dx.doi.org/ 10.1111/j.1574-6968.1999.tb08778.x [DOI] [PubMed] [Google Scholar]
- [38].Luhrmann A, Roy CR. Coxiella burnetii inhibits activation of host cell apoptosis through a mechanism that involves preventing cytochrome c release from mitochondria. Infect Immun 2007; 75:5282-9; PMID:17709406; http://dx.doi.org/ 10.1128/IAI.00863-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Voth DE, Howe D, Heinzen RA. Coxiella burnetii inhibits apoptosis in human THP-1 cells and monkey primary alveolar macrophages. Infect Immun 2007; 75:4263-71; PMID:17606599; http://dx.doi.org/ 10.1128/IAI.00594-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kawakami H, Tomita M, Matsuda T, Ohta T, Tanaka Y, Fujii M, Hatano M, Tokuhisa T, Mori N. Transcriptional activation of survivin through the NF-kappaB pathway by human T-cell leukemia virus type I tax. Int J Cancer 2005; 115:967-74; PMID:15729715; http://dx.doi.org/ 10.1002/ijc.20954 [DOI] [PubMed] [Google Scholar]
- [41].Liu J, Enomoto S, Lancto CA, Abrahamsen MS, Rutherford MS. Inhibition of apoptosis in Cryptosporidium parvum-infected intestinal epithelial cells is dependent on survivin. Infect Immun 2008; 76:3784-92; PMID:18519556; http://dx.doi.org/ 10.1128/IAI.00308-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Zhu Y, Roshal M, Li F, Blackett J, Planelles V. Upregulation of survivin by HIV-1 Vpr. Apoptosis 2003; 8:71-9; PMID:12510154; http://dx.doi.org/ 10.1023/A:1021653119934 [DOI] [PubMed] [Google Scholar]
- [43].Levinson G, Gutman GA. Slipped-strand mispairing: a major mechanism for DNA sequence evolution. Mol Biol Evol 1987; 4:203-21; PMID:3328815 [DOI] [PubMed] [Google Scholar]
- [44].Rando OJ, Verstrepen KJ. Timescales of genetic and epigenetic inheritance. Cell 2007; 128:655-68; PMID:17320504; http://dx.doi.org/ 10.1016/j.cell.2007.01.023 [DOI] [PubMed] [Google Scholar]
- [45].Murphy GL, Connell TD, Barritt DS, Koomey M, Cannon JG. Phase variation of gonococcal protein II: regulation of gene expression by slipped-strand mispairing of a repetitive DNA sequence. Cell 1989; 56:539-47; PMID:2492905; http://dx.doi.org/ 10.1016/0092-8674(89)90577-1 [DOI] [PubMed] [Google Scholar]
- [46].Weiser JN, Shchepetov M, Chong ST. Decoration of lipopolysaccharide with phosphorylcholine: a phase-variable characteristic of Haemophilus influenzae. Infect Immun 1997; 65:943-50; PMID:9038301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Gemayel R, Vinces MD, Legendre M, Verstrepen KJ. Variable tandem repeats accelerate evolution of coding and regulatory sequences. Annu Rev Genet 2010; 44:445-77; PMID:20809801; http://dx.doi.org/ 10.1146/annurev-genet-072610-155046 [DOI] [PubMed] [Google Scholar]
- [48].Le Bars H, Bousarghin L, Bonnaure-Mallet M, Jolivet-Gougeon A. Role of a short tandem leucine/arginine repeat in strong mutator phenotype acquisition in a clinical isolate of Salmonella Typhimurium. FEMS Microbiol Lett 2013; 338:101-6; PMID:23106515; http://dx.doi.org/ 10.1111/1574-6968.12039 [DOI] [PubMed] [Google Scholar]
- [49].Shaver AC, Sniegowski PD. Spontaneously arising mutL mutators in evolving Escherichia coli populations are the result of changes in repeat length. J Bacteriol 2003; 185:6076-82; PMID:14526019; http://dx.doi.org/ 10.1128/JB.185.20.6076-6082.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Wajant H, Haas E, Schwenzer R, Muhlenbeck F, Kreuz S, Schubert G, Grell M, Smith C, Scheurich P. Inhibition of death receptor-mediated gene induction by a cycloheximide-sensitive factor occurs at the level of or upstream of Fas-associated death domain protein (FADD). J Biol Chem 2000; 275:24357-66; PMID:10823821; http://dx.doi.org/ 10.1074/jbc.M000811200 [DOI] [PubMed] [Google Scholar]