

Abstract
Autism is a complex neurodevelopmental disorder with unknown etiology. One hypothesis regarding etiology in autism is the “opioid peptide excess” theory that postulates that excessive amounts of exogenous opioid-like peptides derived from dietary proteins are detectable in urine and that these compounds may be pathophysiologically important in autism. A selective LC–MS/MS method was developed to analyze gliadinomorphin, β-casomorphin, deltorphin 1, and deltorphin 2 in urine. The method is based on on-line SPE extraction of the neuropeptides from urine, column switching, and subsequent HPLC analysis. A limit of detection of 0.25 ng/mL was achieved for all analytes. Analyte recovery rates from urine ranged between 78% and 94%, with relative standard deviations of 0.2–6.8%. The method was used to screen 69 urine samples from children with and without autism spectrum disorders for the occurrence of neuropeptides. The target neuropeptides were not detected above the detection limit in either sample set.
Keywords: Autism, Neuropeptides, β-Casomorphin, Gliadinomorphin, Opioid peptide excess theory, On-line SPE–HPLC–MS/MS
Introduction
Autism spectrum disorders (ASD) are a behaviorally defined group of neurodevelopmental disorders characterized by impairments in social interaction and communication, and repetitive, overly focused behaviors. Autism is usually diagnosed during early childhood in the first 3–5 years [1] and is a lifelong disability. A gender difference is observed, with boys being affected about four times more often than girls [2]. In recent years an increased prevalence of autism has been reported. Statistics in the USA showed a prevalence of 20 per 10,000 for ASD in 1990, while recent surveys estimate 60 per 10,000 [3]. Although the increased prevalence is often attributed to greater awareness and better diagnostic tools, improvement in the detection of autism alone is not felt to sufficiently explain the persistent increase in prevalence. While the syndrome has been shown to be highly heritable [4–6], various theories have been presented suggesting both genetic and environmental factors, such as dietary or chemical exposures.
A widespread hypothesis used to explain autistic symptoms is the “opioid peptide excess” theory. This hypothesis postulates that excessive amounts of endogenous or exogenous opioid peptides, derived from dietary proteins, may be pathophysiologically important in autism [7, 8]. Milk (casein) and wheat (gluten) proteins are thought to be incompletely broken down in the intestine due to deficient enzyme activity [9, 10]. As a result, short neuroactive peptides—such as β-casomorphin (Tyr-Pro-Phe-Pro-Gly-Pro-Ile) derived from casein or gliadinomorphin (Tyr-Pro-Gln-Pro-Gln-Pro-Phe) derived from gluten—are formed and have been detected in urine [11, 12].
Many children with ASD show gastrointestinal symptoms such as inflammation, diarrhea, gastroesophageal reflux, abdominal pain and increased intestinal permeability [13–18]. Increased gut permeability caused, for example, by gluten sensitivity could explain how neuroactive peptides enter the blood circulation, cross the blood brain barrier and exert an “opioid-like” effect in the brain [12]. Reichelt et al. have reported abnormal peptide concentrations in urine samples from autistic children [11]. They also studied patients with celiac disease and reported that the excretion of peptides was increased compared with an age- and gender-matched reference group. Pavone et al. evaluated 120 patients with celiac disease for autistic symptoms as well as 11 autistic patients for gluten specific antibodies and could not verify the hypothesis that a link between autism and celiac disease existed [19].
However, the analytical technique used by Reichelt et al. to test for hyperpeptiduria is rather nonspecific for the target analytes. An urine aliquot equivalent to 250 nmol of creatinine was injected on a standard C18 reversed phase column, chromatographic separation was carried out using gradient elution, and detection was performed by UV light absorption at 215 and 280 nm [11]. Unfortunately, no quantitative results using any type of calibration were provided, which would allow an instrument-independent comparison of results obtained by different research groups. Instead, the part of the chromatogram after the elution of hippuric acid was classified as the “peptide elution area,” and a sum integration of the peak areas was performed to compare groups. However, urine is a very complex biological matrix and absorption of UV light is rather nonspecific in that it is not just peptides that are detected.
Nevertheless, the “opioid peptide excess” theory has gained the public’s attention and as a consequence many families put their children diagnosed with ASD on gluten- and casein-free diets. This diet is difficult to follow, expensive, time-consuming, and the rigorous restriction of the diet may also create nutritional risk [20, 21]. Moreover, the “opioid peptide excess” theory has not been replicated by other researchers using chromatographic techniques in combination with more selective mass spectrometric detection [22]. Therefore, accurate information about the relationship between gastrointestinal symptoms and metabolic evidence of gluten sensitivity is very important for families of children with ASD, requiring a reliable, sensitive analytical method that determines the exogenous peptides in question selectively.
We have developed a selective and quantitative method for the analysis of four peptides in urine samples. The opioid peptides gliadinomorphin and β-casomorphin are used as lead exogenous neuropeptides. In addition, the endogenous peptides deltorphin 1 and deltorphin 2 are quantitatively analyzed to check for potential urinary excretion of neuroactive peptides. Sample preparation has been coupled directly to high-performance liquid chromatographic separation using on-line solid-phase extraction (SPE). Detection is carried out using tandem mass spectrometry. On-line SPE is a rapidly developing method within the field of sample preparation techniques. A sample aliquot is injected directly onto a SPE column, which is then washed to remove salts and other matrix components. Following the wash step, the SPE column is eluted onto the analytical HPLC column. Since on-line SPE requires only minimal sample handling, it increases method automation, laboratory throughput, and minimizes analyte losses. Application of tandem mass spectrometry provides highly selective detection of the target analytes with low detection limits. The analytical technique described has been applied to 69 first morning urine samples obtained from children with ASD and age- and gender-matched normally developing children.
Experimental
Chemicals
Deltorphin 1, deltorphin 2, and β-casomorphin were purchased from Sigma-Aldrich (St. Louis, MO, USA). Gliadinomorphin is not commercially available and was synthesized in-house. In addition, deuterium-labeled gliadinomorphin was synthesized by incorporating l-tyrosine-β,β-d2. Acetonitrile used for HPLC separation was purchased from Fisher Scientific (Pittsburgh, PA, USA). All solvents were HPLC-grade.
Synthesis of giladinomorpin and gliadinomorphin-d2
Wang (p-benzyloxybenzyl alcohol) resin and N-α-Fmoc-(l)-amino acids were purchased from Novabiochem (San Diego, CA, USA). L-Tyrosine-β,β-d2 and di-tert-butyl-dicarbonate were purchased from Sigma-Aldrich. Analytical HPLC was performed on an Alltech (Deerfield, IL, USA) Alltima™ C18 column (5 µm, 250×4.6 mm), eluted with a linear gradient of 0.075% CF3COOH/CH3CN (v/v) (solvent B) and 0.1% CF3COOH/H2O (v/v) (solvent A) at a flow rate of 1 mL/min. Semi-preparative HPLC was performed on an Alltech Alltima™ C18 column (5 µm, 250×22 mm), eluted with a linear gradient of solvent B and solvent A at a flow rate of 6 mL/min. The effluent was monitored by detecting its optical absorption at 280 nm. ESI–MS was performed on a Thermo Finnigan (San Jose, CA, USA) LCQ™ Deca instrument. Thin layer chromatography was performed with EM Science (Gibbstown, NJ, USA) silica gel 60 F254 plastic-backed plates (cat. no. 5735).
Peptides were synthesized on Wang resin (240 mg, 1.2 mmol/g) via Fmoc solid-phase peptide synthetic techniques. Initial loading of the N-α-Fmoc-l-Phe-OH amino acid onto the resin was accomplished via symmetric anhydride activation. N-α-Fmoc-l-Phe-OH (5 equiv. to resin, 0.465 g) was dissolved in anhydrous CH2Cl2 (10 mL), DIPCDI (2.5 equiv., 95 µL) was added, and the mixture was stirred for 20 min at 0 °C on ice. The CH2Cl2 was removed by evaporation under reduced pressure, the residue resuspended in DMF (4.5 mL) and added along with 0.1 M DMAP in DMF solution (0.05 equiv., 0.024 mL) to pre-swollen resin; the mixture was agitated by bubbling Ar gas through the resin slurry at room temperature for 2 h. After washing and drying the resin, the loading yield was estimated by measuring the amount of Fmoc released from the resin upon piperidine treatment. Two aliquots of resin (about 1–2 mg each) were weighed into test tubes and 20% piperidine in DMF (3 mL) was added to each. After agitation for 3 min, the absorbance at 290 nm of each solution was compared to the 20% piperidine reference solution and loading yield was estimated at 0.478 mmol/g from the equation: mmol/g = (Abssample − Absref)/(1.65 × mg of resin). The remaining hydroxyl functionality on the resin was blocked by reaction with acetic anhydride (2.5 equiv.) and 0.1 M DMAP/DMF solution (0.05 equiv.) in DMF.
After the initial loading, peptides were extended in a batch-wise manner: Fmoc was removed from resin-bound N-terminal amines with 20% piperidine in DMF, and amide couplings were performed by the addition of a solution containing 3 molar equivalents of protected amino acid, HOBt, PyBOP, and DIEA in DMF (10 mL/g resin). Removal of the Fmoc group and completion of the conjugation reaction were monitored by the quantitative ninhydrin test [23].
Upon addition of the penultimate amino acid, the resin was divided into two aliquots and the final amino acid was added as either N-α-Fmoc-Tyr-OH or N-α-Boc-Tyr-β,β-d2-OH. The Boc-protected form of the deuterated tyrosine was prepared by dissolving 183 mg of l-tyrosine-β,β-d2 (1 mmol) in dioxane, adding (Boc)2O (1 equiv., 174 mg) and NaHCO3 (250 mg), and sonicating the mixture for 6 h, after which the solids were filtered and the remaining solution brought to dryness under vacuum [24]. The residue was dissolved in 10 mL of distilled water, acidified by the addition of a saturated solution of KHSO4 until the pH was 2.0, and the solution was extracted with five portions of Et2O. The organic phase was subsequently dried with NaSO4 and evaporated under vacuum to yield 266 mg N-α-Boc-Tyr-β,β-d2-OH (94% yield). The N-α-Boc-Tyr-β,β-d2-OH was >99% pure by HPLC and was used without further purification in the peptide synthesis.
The peptide resin was washed thoroughly and dried under reduced pressure overnight. Cleavage and deprotection of both the native and deuterated forms of the gliadinomorphin peptides was afforded by treatment of the resin with a mixture of 95% CF3COOH, 2.5% H2O, and 2.5% TIS (10 mL/g peptide resin) in a stoppered flask left to stand at room temperature for 2.5 h with occasional swirling. The resin was removed by filtration and rinsed twice with cleavage mixture; the filtrates were combined and CF3COOH was evaporated under reduced pressure. Cold Et2O was added to the residue and the obtained precipitate was extracted into water. Crude peptides were >85% pure by analytical HPLC. Following purification by semi-preparative HPLC, peptides of >98% purity were lyophilized and stored at −80 °C.
On-line SPE–HPLC–tandem MS conditions
The scheme used for the analytical instrumentation with the on-line SPE setup is shown in Fig. 1. Chromatographic separation was performed using a Shimadzu ASP10 HPLC system (Shimadzu, Pleasanton, CA, USA) equipped with a ten-port valve and a two-channel UV detector. The HPLC system consists of three pumps. Pumps A and B are used for binary high-pressure gradient mixing, and pump C is delivering an independent purge solvent flow. Biofluid extraction was performed using an Oasis HLB SPE column (20 mm × 2.1 mm I.D., Waters, Milford, MA, USA). Analytes were separated on a reversed phase HPLC column (4u Proteo 90 Å, 150 mm, 4.6 mm I.D., 4 µm; Phenomenex, Torrance, CA, USA) using gradient elution with a water–0.1% formic acid (solvent A)/acetonitrile–0.1% formic acid (solvent B) solvent system. A flow rate of 0.300 mL/min was used. In valve position A (see Fig. 1), the sample is injected on the SPE column (Oasis HLB), and then a wash step is performed to remove salts and proteins. The wash fluid is directed to waste while pump C independently delivers solvent (acetonitrile–0.1% formic acid) with a flow of 0.1 mL/min to the mass spectrometer to maintain a stable spray in the electrospray source during the sample extraction step. Subsequently, the valve is switched to position B, the solvent flow through the SPE column is reversed, directed onto the analytical reversed phase peptide column, and the gradient is started. The analytical HPLC column is not flushed during the load and wash steps, but stays well-equilibrated in the starting gradient conditions for the next run. Valve switching events as well as the gradient program are summarized in Table 1. The injection volume was 20 µL. The samples were kept at 10 °C in the autosampler.
Fig. 1.

Experimental setup for on-line SPE–HPLC–MS/MS analysis of urinary neuropetides
Table 1.
HPLC conditions
| Time (min) | Solvent A: H2O, 0.1% formic acid |
Solvent B: ACN, 0.1% formic acid |
Valve position |
|---|---|---|---|
| 0 | 99% | 1% | A: Load sample, wash |
| 2.2 | 99% | 1% | B: Elution |
| 20.0 | 70% | 30% | B: Elution |
| 25.0 | 50% | 50% | B: Elution |
| 27.0 | 0% | 100% | B: Elution |
| 32.0 | 0% | 100% | B: Elution |
| 35.0 | 99% | 1% | B: Elution |
Pump C: Constant flow of 0.1 mL/min ACN, 0.1% formic acid
Analytes were detected by electrospray ionization (in positive mode) tandem quadrupole mass spectrometry (in multiple reaction monitoring mode, MRM) using a Quattro Premier tandem quadrupole mass spectrometer (Waters/Micromass, Manchester, UK). Nitrogen gas flow rates were fixed with a cone gas flow of 25 L/h and a desolvation gas flow of 700 L/h. A source temperature of 125 °C and a desolvation temperature of 300 °C were applied. Electrospray ionization was performed with a capillary voltage fixed at 3.00 kV. Cone voltages were optimized for each analyte individually. Tune parameters including capillary voltage, cone voltage and extraction cone were optimized in an infusion experiment (data not shown). An extraction cone voltage of 5 V was selected. An offset value of 1 was used for the ion energies applied to quadrupole 1 and 2. Quadrupole resolution in MRM mode was set to an offset value of 12, resulting in a peak width at half height of 1 Da. A multiplier voltage of 650 V was applied. Argon was used as collision gas and the optimum gas pressure was experimentally determined as 4.19×10−3 Torr. Optimum collision voltages were determined experimentally by acquiring product ion spectra (Table 2). These spectra were used to select a dominant product ion to set up the transition monitored in the MRM mode.
Table 2.
ESI-MS/MS detection parameters
| Analyte | Transition | Cone voltage (V) |
Collision voltage (V) |
|---|---|---|---|
 |
769.2 > 695.0 (b6) | 50.0 | 27.0 |
 |
783.1 > 709.2 (b6) | 50.0 | 25.0 |
 |
790.3 > 383.1 (y4) | 50.0 | 30.0 |
 |
876.4 > 487.8 (y4) | 60.0 | 35.0 |
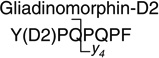 |
878.4 > 487.8 (y4) | 60.0 | 35.0 |
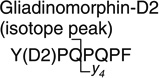 |
879.4 > 487.8(y4) | 60.0 | 35.0 |
Calibration standards containing the neuropetides in increasing concentrations were prepared in water. All standards contained gliadinomorphin-d2 as internal standard at a concentration of 600 ng/mL. The linearity of the calibration curves for the target neuropeptides was tested over the range 0.25–893 ng/mL. Evaluation of the on-line SPE extraction procedure was first performed by determining recovery rates of neuropetides from aqueous solution at seven concentrations. Each sample was analyzed in triplicate. Calibration curves obtained by injecting the standards directly onto the HPLC column (without on-line SPE extraction) were used to determine neuropeptide concentrations in the on-line SPE-extracted aqueous samples, and recovery rates were calculated.
Control urine samples were spiked with neuropeptides in order to evaluate potential matrix effects caused by ion suppression. Urine samples from three different individuals were spiked with standard neuropeptide solutions, resulting in three different concentrations of 250, 428, and 643 ng/mL, and analyzed as described.
Sample acquisition and preparation
First morning urine samples were obtained from 54 children (45 males and 9 females) 2–6 years of age with confirmed diagnoses of autistic disorder, using the Autism Diagnostic Interview—Revised [25] and Autism Diagnostic Observation Schedules [26], and 15 age-matched control children without an autism spectrum diagnosis who participated in a larger study at the M.I.N.D. Institute, University of California, Davis. The Institutional Review Board at University of California, Davis approved the study protocol. Urine samples were collected in standard sterile collection cups and kept at −20 °C until analysis. Upon analysis, samples were thawed and centrifuged. A 500-µL urine aliquot was spiked with 3 µL of internal standard (deuterated gliadinomorphin) and subjected to analysis.
Results and discussion
Gliadinomorphin (Tyr-Pro-Gln-Pro-Gln-Pro-Phe), the neuropetide derived from gluten, was not commercially available as a standard and was therefore synthesized in our lab using standard protocols. In addition, a deuterium-labeled analog containing l-tyrosine-β,β-d2 was prepared as internal standard. After purification, peptides of >98% purity were obtained. Peptide identities were confirmed by ESI–MS: gliadinomorphin (C43H58N9O11), calculated (M+H)+ m/z 876.43, found (M+H)+ 876.42; gliadinomorphin-d2 (C43H56D2N9O11), calculated (M+H)+ m/z 878.44, found (M+H)+ 878.44.
Since the intention was to develop a selective method with low detection limits for the analysis of urinary neuropeptides, multiple reaction monitoring was chosen as the operating mode for the tandem-mass spectrometer. In a first experiment, the optimum cone voltage for the formation of the precursor ion during electrospray ionization was determined experimentally by an infusion experiment. Further experiments were performed to determine the fragmentation behavior of these precursor ions and the corresponding optimum collision energy. The dominant fragment ions seen in the product ion spectra were used to set up the MRM transition parameters as described in Table 2. After the MS parameters were set, the chromatographic analysis was optimized using standard solutions. Figure 2 shows a representative chromatogram of a standard solution containing gliadinomorphin, β-casomorphin, deltorphin 1, and deltorphin 2. Figure 2 indicates that sufficient chromatographic resolution was achieved for all analytes.
Fig. 2.

Extracted ion chromatograms for gliadinomorphin, β-casomorphin, deltorphin 2, and deltorphin 1, as obtained by direct injection of 20 µL of a standard solution (143 ng/mL) and HPLC–MS/MS analysis
In order to evaluate the utility of the Oasis HLB SPE column for extracting neuropeptides from aqueous samples, recovery experiments were performed by analyzing aqueous solutions containing neuropeptide concentrations ranging from 25 ng/mL up to 428 ng/mL. Direct injection of the standards onto the analytical column was used as a reference to calculate recovery rates. Each sample was analyzed in triplicate. The average recovery rates are shown in Fig. 3. Average recovery rates were above 80% at all seven spike levels for gliadinomorphin, β-casomorphin, deltorphin 1, and deltorphin 2, with an overall average of 103%, 94%, 92% and 90%, respectively. Average relative standard deviations (RSD) across all spike levels ranged between 2.2 and 6.4%. Average RSD for the primary target analytes, gliadinomorphin and β-casomorphin, were excellent: 2.5% and 2.2%, respectively.
Fig. 3.

Evaluation of the on-line SPE extraction, achieved by determining the recovery rates of neuropetides from aqueous solution at seven spike levels. Each sample was analyzed in triplicate. Calibration curves generated by injecting the standards directly onto the HPLC column were used to calculate the recovery rates
A calibration using on-line SPE analysis of aqueous standard solutions was performed, which allowed the quantification of neuropeptides in urine samples (see Table 3). Gliadinomorphin-d2 was used as an internal standard at a concentration of 600 ng/mL after it had been confirmed that the compound, or an interfering artifact, was not already present in urine. In the internal standard, only two protons were exchanged for deuterium atoms, giving a 2-Da difference in mass from the analyte. At very high analyte concentrations, the overlap of the analyte’s second isotopic peak transition and that of the pseudo-molecular ion of the internal standard cannot be completely excluded. Therefore, in addition to monitoring the internal standard’s pseudo-molecular ion transition, the first isotope peak was also recorded and can be used to construct the calibration curve. The limits of detection, defined as the smallest concentration that gave a signal-to-noise ratio greater than three, was 250 pg/mL using an injection volume of 20 µL (see Table 2). Linear calibration curves were observed for all analytes up to the highest measured concentration of 893 ng/mL. The limit of quantification for an extended calibration was set to 2.5 ng/mL for all analytes. However, since the opiod peptide theory is based on data obtained via HPLC with UV detection, it was expected that the neuropeptide would be detected at higher concentrations. Therefore, in order to increase sample throughput, a limited calibration in the range of 25–893 ng/mL was routinely performed, which could be extended if necessary.
Table 3.
Calibration parameters
| Analyte | Calibration curve equation | R2 | LOD [ng/mL] (S/N>3/1) |
LOQ [ng/mL] |
|---|---|---|---|---|
| Gliadinomorphin | 5.135x+158.497 | 0.998 | 0.25 | 2.5 |
| Casomorphin | 146.192x+2088.649 | 0.996 | 0.25 | 2.5 |
| Deltorphin 1 | 9.889x+250.463 | 0.994 | 0.25 | 2.5 |
| Deltorphin 2 | 22.963x+432.206 | 0.990 | 0.25 | 2.5 |
The efficiency and reproducibility of the on-line SPE extraction method was also analyzed with spiked urine samples after it had been confirmed that the analytes were not already present in those reference urine samples. Urine samples from three different individuals were spiked with standard solutions, resulting in three different concentrations of 250, 428, and 643 ng/mL, and the samples were analyzed in triplicate as described. The recovery rates obtained in the urine spike experiment are shown in Fig. 4. Data are also summarized in Table 4. Recovery rates were calculated as the average values from the analysis of three different urine samples. Recoveries ranged between 78% and 94%, with relative standard deviations (RSD) of between 0.2 and 6.8%. The good recovery rates and the good reproducibility show that the method can be applied to the analysis of urinary neuropeptides. The good recoveries of the neuropeptides in the urine spike experiments also indicate that ion suppression caused by matrix effects is negligible.
Fig. 4.

Recovery rates of neuropetides from spiked urine samples (250, 428, 643 ng/mL) after on-line SPE–HPLC tandem MS analysis. Recovery rates are average values from the analysis of three different urine samples, and each sample was analyzed in triplicate
Table 4.
Mean recovery rates of neuropetides from spiked urine samples from three different individuals after on-line SPE–HPLC tandem MS analysis
| Recovery [%] ± SD | Donor 1 | Donor 2 | Donor 3 | Average |
|---|---|---|---|---|
| Spike 1: 250 ng/mL | ||||
| Gliadinomorphin | 75.3±0.8 | 82.6±4.2 | 81.1±1.6 | 79.7±3.9 |
| Casomorphin | 93.3±2.8 | 91.6±2.4 | 88.5±2.4 | 91.1±2.5 |
| Deltorphin 2 | 94.7±0.3 | 94.3±3.8 | 90.7±3.3 | 93.2±2.2 |
| Deltorphin 1 | 76.3±1.8 | 81.9±3.6 | 76.1±2.9 | 78.1±3.3 |
| Spike 2: 428 ng/mL | ||||
| Gliadinomorphin | 82±2.3 | 82.5±1.9 | 83.7±2.8 | 82.7±0.9 |
| Casomorphin | 98.7±1.7 | 90.2±1 | 86±0.3 | 91.7±6.5 |
| Deltorphin 2 | 96.8±1.5 | 95±0.7 | 88.1±1.5 | 93.3±4.6 |
| Deltorphin 1 | 82±2.5 | 82.8±1.5 | 75.9±0.9 | 80.2±3.8 |
| Spike 3: 643 ng/mL | ||||
| Gliadinomorphin | 85.2±2.2 | 84.7±2.7 | 84.7±0.5 | 84.8±0.3 |
| Casomorphin | 98.1±0.5 | 89.7±1.5 | 84.6±1.5 | 90.8±6.8 |
| Deltorphin 2 | 98.7±0.4 | 94.9±0.2 | 88±0.9 | 93.9±5.4 |
| Deltorphin 1 | 84.3±0.7 | 83.1±2.3 | 74.9±1.7 | 80.8±5.1 |
Mean recovery is given in percent with standard deviation (n=3).
69 first morning urine samples were quantitatively analyzed for urinary peptides. The sample set contained 54 samples from children with autistic disorders and 15 control samples. In order to control the stability of the analytical system during the analysis of the samples, six calibration checks were analyzed within the sample batch. Recovery rates for the analytes were in the range 96–107%, with RSD values of 1.5–9.6%.
The target neuropeptides gliadinomorphin, β-casomorphin, deltorphin 1, and deltorphin 2 were not detected above the limit of detection within the 69 samples analyzed. This result is in agreement with the data reported by Hunter et al. [22]. Compared to the data provided by Hunter et al., a much larger sample cohort was analyzed in the study presented here, and a standard for the quantification of gliadinomorphin was synthesized. In addition, Hunter et al. acquired full-scan mass spectral data, while the MRM mode used here should provide lower detection limits and more selective detection. Nevertheless, the opioid peptide excess theory was also not validated in our study.
However, in order to completely eliminate the applicability of this theory as a contributing factor in the development of autism, an even larger number of samples will need to be analyzed to verify these findings. The developed method allows for the relatively fast and accurate analysis of the samples without time-consuming sample preparation steps. There is also the potential to apply the method to celiac disease in order to monitor urinary gliadinomorphin in gluten-sensitive patients.
Conclusions
An analytical method combining on-line SPE extraction and HPLC–tandem MS analysis was developed to analyze gliadinomorphin, β-casomorphin, deltorphin 1, and deltorphin 2 in urine. Gliadinomorphin was not commercially available and was therefore synthesized in-house together with deuterium-labeled gliadinomorphin used as internal standard. A limit of detection of 0.25 ng/mL was obtained for all analytes, and analyte recovery rates from urine ranged between 78% and 94%, with relative standard deviations between 0.2 and 6.8%. The application of the method to screen 69 urine samples from 54 children with confirmed autistic disorders and 15 age-matched controls did not detect any of the target neuropeptides. The opioid peptide excess theory was not validated using this small sample set. Our findings are in contrast to earlier work published on urinary neuropeptides using HPLC with UV detection, but are in agreement with other, more recent, LC–MS-based work in this area. However, a much larger sample set needs to be analyzed to be able to verify the data. The method could also be a useful tool for analyzing gliadinomorphin in patients with celiac disease. Furthermore, the principle of combining on-line SPE with HPLC–ESI–tandem MS analysis can be adapted to analyze other urinary metabolites.
Acknowledgments
This work was supported by grants from NIEHS Grant R37 ES02710, NIEHS Superfund Basic Research Program Grant P42 ES04699, National Institute of Environmental Health Sciences Center, R01 ES013933, P30 ES05707, and NIEHS Center for Children’s Environmental Health & Disease Prevention Grant P01 ES11269, the UC Davis M.I.N.D. Institute, and in part by the German Research Foundation and Baygene.
Abbreviations
- ASD
autistic spectrum disorder
- DIEA
N,N-diisopropylethylamine
- DIPCDI
N,N′-diisopropylcarbodiimide
- DMAP
4-dimethylaminopyridine
- DMF
N,N-dimethylformamide
- Fmoc
9-fluorenylmethoxycarbonyl
- HOBt
N-hydroxybenzotriazole
- PyBOP
benzotriazole-1-yl-oxy-tris-pyrrolidino-phosphonium hexafluorophosphate
- SPE
solid-phase extraction
- TIS
triisopropylsilane
- Trt
trityl
Contributor Information
K. Dettmer, Email: katja.dettmer@klinik.uni-regensburg.de, Institute of Functional Genomics, University of Regensburg, Josef-Engert-Str. 9, 93053 Regensburg, Germany; Department of Entomology, University of California at Davis, 1 Shields Avenue, Davis, CA 95616, USA.
D. Hanna, Department of Entomology, University of California at Davis, 1 Shields Avenue, Davis, CA 95616, USA
P. Whetstone, Department of Entomology, University of California at Davis, 1 Shields Avenue, Davis, CA 95616, USA
R. Hansen, Department of Pediatrics, M.I.N.D. Institute, University of California Davis School of Medicine, 2825 50th Street, Sacramento, CA 95817, USA
B. D. Hammock, Department of Entomology, University of California at Davis, 1 Shields Avenue, Davis, CA 95616, USA
References
- 1.Wiggins LD, Baio J, Rice C. J Dev Behav Pediatr. 2006;27:S79–S87. doi: 10.1097/00004703-200604002-00005. [DOI] [PubMed] [Google Scholar]
- 2.Association AP. Diagnostic and statistical manual of mental disorders IV—Test Revision. Washington, DC: American Psychiatric Association; 2000. [Google Scholar]
- 3.Fombonne E. JAMA. 2003;289:87–89. doi: 10.1001/jama.289.1.87. [DOI] [PubMed] [Google Scholar]
- 4.Muhle R, Trentacoste SV, Rapin I. Pediatrics. 2004;113:e472–e486. doi: 10.1542/peds.113.5.e472. [DOI] [PubMed] [Google Scholar]
- 5.Bailey A, Le Couteur A, Gottesman I, Bolton P, Simonoff E, Yuzda E, Rutter M. Psychol Med. 1995;25:63–77. doi: 10.1017/s0033291700028099. [DOI] [PubMed] [Google Scholar]
- 6.Bailey A, Palferman S, Heavey L, Le Couteur A. J Autism Dev Disord. 1998;28:369–392. doi: 10.1023/a:1026048320785. [DOI] [PubMed] [Google Scholar]
- 7.Panksepp J. Trends Neuroscience. 1979;2:174–177. [Google Scholar]
- 8.Knivsberg AM, Reichelt KL, Hoien T, Nodland M. Nutr Neurosci. 2002;5:251–261. doi: 10.1080/10284150290028945. [DOI] [PubMed] [Google Scholar]
- 9.Kidd PM. Altern Med Rev. 2002;7:472–499. [PubMed] [Google Scholar]
- 10.Kidd PM. Altern Med Rev. 2002;7:292–316. [PubMed] [Google Scholar]
- 11.Reichelt KL, Knivsberg AM, Nodland M, Lind G. Dev Brain Dysfunct. 1994;7:71–85. [Google Scholar]
- 12.Reichelt KL, Knivsberg AM. Nutr Neurosci. 2003;6:19–28. doi: 10.1080/1028415021000042839. [DOI] [PubMed] [Google Scholar]
- 13.Horvath K, Perman JA. Curr Gastroenterol Rep. 2002;4:251–258. doi: 10.1007/s11894-002-0071-6. [DOI] [PubMed] [Google Scholar]
- 14.Wakefield AJ, Puleston JM, Montgomery SM, Anthony A, O’Leary JJ, Murch SH. Aliment Pharmacol Ther. 2002;16:663–674. doi: 10.1046/j.1365-2036.2002.01206.x. [DOI] [PubMed] [Google Scholar]
- 15.D’Eufemia P, Celli M, Finocchiaro R, Pacifico L, iozzi L, Zaccagnini M, Cardi E, Giardini O. Acta Paediatr. 1996;85:1076–1079. doi: 10.1111/j.1651-2227.1996.tb14220.x. [DOI] [PubMed] [Google Scholar]
- 16.Ashwood P, Anthony A, Pellicer AA, Torrente F, Walker-Smith JA, Wakefield AJ. J Clin Immunol. 2003;23:504–517. doi: 10.1023/b:joci.0000010427.05143.bb. [DOI] [PubMed] [Google Scholar]
- 17.Jyonouchi H, Sun S, Itokazu N. Neuropsychobiology. 2002;46:76–84. doi: 10.1159/000065416. [DOI] [PubMed] [Google Scholar]
- 18.Horvath K, Papadimitriou JC, Rabsztyn A, Drachenberg C, Tildon JT. J Pediatr. 1999;135:559–563. doi: 10.1016/s0022-3476(99)70052-1. [DOI] [PubMed] [Google Scholar]
- 19.Pavone L, Fiumara A, Bottaro G, Mazzone D, Coleman M. Biol Psychiatry. 1997;42:72–75. doi: 10.1016/S0006-3223(97)00267-9. [DOI] [PubMed] [Google Scholar]
- 20.Arnold GL, Hyman SL, Mooney RA, Kirby RS. J Autism Dev Disord. 2003;33:449–454. doi: 10.1023/a:1025071014191. [DOI] [PubMed] [Google Scholar]
- 21.Bowers L. J Hum Nutr Diet. 2002;15:141–144. doi: 10.1046/j.1365-277x.2002.00345.x. [DOI] [PubMed] [Google Scholar]
- 22.Hunter LC, O’Hare A, Herron WJ, Fisher LA, Jones GE. Dev Med Child Neurol. 2003;45:121–128. [PubMed] [Google Scholar]
- 23.Kaiser E, Colescott RL, Bossinger CD, Cook PI. Anal Biochem. 1970;34:595–598. doi: 10.1016/0003-2697(70)90146-6. [DOI] [PubMed] [Google Scholar]
- 24.Einhorn J, Einhorn C, Luche JL. Synlett. 1991;1:37–38. [Google Scholar]
- 25.Rutter M, Couteur AL, Lord C. Autism diagnostic interview—revised, WPS edition. Los Angeles, CA: Western Psychological Services; 2003. [Google Scholar]
- 26.Lord C, Rutter M, DiLavore PC, Risi S. Autism diagnostic observation schedules, WPS edition. Los Angeles, CA: Western Psychological Services; 1999. [Google Scholar]