

Abstract
Blood can become hypercoagulable by shear-induced platelet activation and generation of microparticles. It has been reported that non-physiological shear stress (NPSS) could induce shedding of platelet receptor glycoprotein (GP) Ibα, which may result in an opposite effect to hemostasis. The aim of this study was to investigate the influence of the NPSS on platelets and von Willebrand factor (vWF). Human blood was exposed to two levels of NPSS (25Pa, 125Pa) with an exposure time of 0.5 sec., generated by using a novel blood shearing device. Platelet activation (P-selectin expression, GPIIb/IIIa activation and generation of microparticles) and shedding of three platelet receptors (GPIbα, GPVI, GPIIb/IIIa) in sheared blood were quantified using flow cytometry. Aggregation capacity of sheared blood induced by ristocetin and collagen was evaluated using an aggregometer. Shear-induced vWF damage was characterized with western blotting. Consistent with the published data, the NPSS caused significantly more platelets to become activated with increasing NPSS level. Meanwhile, the NPSS induced the shedding of platelet receptors. The loss of the platelet receptors increased with increasing NPSS level. The aggregation capacity of sheared blood induced by ristocetin and collagen decreased. There was a loss of high molecular weight multimers (HMWM) of vWF in sheared blood. These results suggest that the NPSS induced a paradoxical effect. More activated platelets increase the risk of thrombosis while the reduction in platelet receptors and the loss of HMWM-vWF increased the propensity of bleeding. The finding might provide a new perspective to understand thrombosis and acquired bleeding disorder in patients supported with blood contacting medical devices.
Keywords: Non-physiological high shear stress, blood contacting medical devices, thrombosis, bleeding, receptor shedding
INTRODUCTION
Cardiovascular disease causes nearly one million deaths per year.1, 2 In order to treat or relieve symptoms of this disease, a variety of blood contacting medical devices (BCMDs) have been developed in the past five decades. Clinicians have routinely used these devices, such as total artificial hearts, ventricular assist devices (VADs), mechanical heart valves (MHVs), vascular grafts, stents, circulatory assist devices, blood oxygenators, respiratory assist devices, cardiopulmonary bypass support systems and hemodialyzers, as standard therapies or treatment tools for various forms of cardiovascular, respiratory and renal diseases, and saved or extended the lives of many patients with otherwise hopeless medical conditions. However, the benefits of using these devices are not without risks. During the use of these devices, a complex anticoagulation therapy is recommended to avoid the thrombosis due to device-induced platelet and coagulation activation and artificial surfaces. However, the anticoagulation therapy does not completely eliminate the risk of thromboembolic events, but rather, increases the risk for hemorrhage and other adverse anti-coagulation events.3, 4 Although the surface-related biocompatibility issue of these devices is very important for initiating protein deposition and cellular adhesion, leading to device thrombosis and subsequent thromboembolism, blood trauma induced by the NPSS in some blood circulating devices, such as total artificial hearts, VADs, MHVs and cardiopulmonary bypass, can’t be ignored. Device-induced blood trauma has been suggested to be a central mechanism for hemostatic dysfunction.5, 6
Platelet receptors play very important roles in physiological hemostasis and pathophysiological thrombosis. Platelet receptor-ligand interactions result in a sequence of bidirectional intracellular signals, leading to the subsequent platelet activation and their aggregation as well as the recruitment of circulating platelets to exposed subendothelial extracellular matrix at sites of vascular injury. Activated platelets are joined by vWF due to its ability to bind the activated GPIIb/IIIa complex. Shear stress also plays an important role in this process. It has been shown that shear stress could induce platelet activation and the binding of platelet with vWF, initiating the signal pathway and irreversible aggregation in the absence of any exogenous agonist.7–10
In the context of VADs and MHVs, the NPSS commonly exists in some regions within a device, such as in the blade region of VADs and hinge region of MHVs. It is a general belief that the activation of platelets by the NPSS may increase the risk of thrombosis. Thus, anticoagulant therapies are often prescribed for patients supported by BCMDs. It has been reported that the NPSS could induce shedding of platelet receptors glycoprotein (GP) Ibα, glycoprotein (GP) VI11–13 and fragmentation of vWF.14 Platelet receptor shedding provides a mechanism for down-regulating surface expression of platelet receptors, resulting in a loss of ligand binding, decreasing the surface density, affecting receptor cross linking and signaling, thus ultimately leading to platelet dysfunction. The loss of GPIbα and GPVI receptors on the platelet surface may result in the reduced capacity of platelet adhesion and aggregation to vWF and collagen substrates, leading to an opposite effect to hemostasis and increasing the risk of bleeding.13, 15–17 However, these studies were performed in a relatively low shear stress range with much longer exposure time (seconds to minutes) compared to those shear stress ranges in severe stenotic blood vessels, VADs or MHVs.18, 19 Thus it remains unknown whether the NPSS with short exposure time relevant to BCMDs with high shear stresses for short exposure times could induce the same effect on platelets. It has been reported in several clinical investigations that patients supported with continuous flow VADs (CF-VAD) or extracorporeal membrane oxygenation (ECMO) exhibited the acquired von Willebrand syndrome (AVWS) characterized by loss of the high molecular weight multimers of vWF (HMWM-vWF).20, 21 The loss of HMWM-vWF has been suggested to contribute to bleeding complications.
Because of the importance of platelet receptors and vWF in hemostasis, excessive activation or damage of them induced by the NPSS might lead to dysfunction of the blood coagulation system, causing thrombosis or bleeding.22 In this study, the effects of the NPSS with a short exposure time on platelets and vWF were investigated. An in-vitro blood shearing system was constructed and human blood donated from healthy donors was exposed to the two levels of NPSS for a short exposure time. The NPSS-induced platelet activation and shedding of three major platelet receptors (GPIbα, GPVI, GPIIb/IIIa) were quantified. Aggregation capacity of sheared blood induced by ristocetin and collagen were evaluated. Shear-induced vWF damage was also characterized.
MATERIALS AND METHODS
Blood-shearing device
A centrifugal flow-through Couette device, adapted from the CentriMag blood pump (Thoratec, Pleasanton, CA, USA), was used as the blood-shearing device in the study (Figure 1). The rotor was magnetically suspended with the bearingless motor technology. A narrow gap with a uniform gap of 150 μm and a length of 2.5 mm was created between the housing and the rotor in the shearing device while the flow gaps in other regions were 10 times larger than the narrow gap. Blood would be exposed to a uniform shear region in the narrow gap when it was driven to flow through the shearing device23. When the stable laminar flow is achieved in the narrow gap of this blood-shearing device23, the average shear stress τ can be determined:
Figure 1.
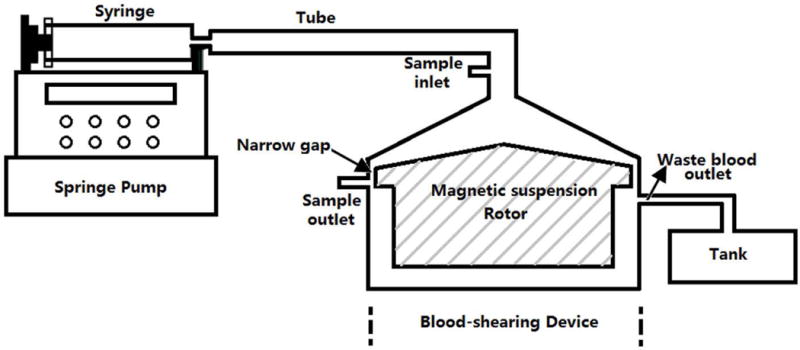
The blood shearing system; it consisted of a syring pump used to control the flow rate, 140ml syringe, blood shearing device (Hemolyzer-L) including 150μm narrow gap between the inner rotor and the outer housing, waste container, and tubing.
Here, ω is the rotational speed of the rotor, R is the radius of the rotor at the narrow gap region, μ is the dynamic viscosity, h is the width of the narrow gap. The corresponding shear stress range was from 21 to 212Pa for the blood with a typical viscosity of 0.0036 Pa·s. A syringe pump (PHD 2000, Harvard Apparatus, Holliston, MA) used to control the flow rate of blood through the narrow gap was connected with the shearing device. Whole blood was pressure-driven to pass through the narrow gap between the magnetically suspended rotor and the stationary outer housing in the axial direction. The high shear stress and short exposure time through the narrow gap region could be controlled by adjusting the rotor rotational speed and the axial flow rate.23
Experimental procedure
Five healthy human donors (3 men and 2 women, age from 20 ~ 50 years old) were recruited to donate blood for the study. Fresh blood (250 ml) drawn from these donors by antecubital vena puncture was mixed with anticoagulant acid citrate dextrose (ACD-A) with the volume ratio of 9 to 1. All the blood donors did not take aspirin or other platelet active medication two weeks prior to the blood donation. The study and all procedures involving the blood collection were approved by the Institutional Review Board (IRB) of the University of Maryland and the University of Louisville. All the blood volunteers gave their written, informed consent after the purpose of the study was explained.
Before the blood shearing experiment, a semi-micro viscometer (Cannon Instrument Company, State College, PA) was used to measure the blood viscosity. The rotor rotational speed and axial flow rate of the blood-shearing system were decided by the desired shear stress and exposure time based on the blood viscosity.23 In order to investigate the effect of the NPSS, which is relevant to BCMDs, on platelets and vWF, two levels of the NPSS (25 Pa and 125 Pa) were chosen. These levels of shear stresses represent the conditions from pathological status (25Pa) in sever stenotic vessels to non-physiological status (125Pa) commonly encountered within VADs, ECMO or MHVs18, 24. Since the exposure time of blood to the NPSS in these BCMDs is relatively short,18, 24 the exposure time of 0.5 sec was chosen for the present study. During each experiment run, the blood from the inlet of the shearing device and the NPSS exposed blood at the outlet were collected as the baseline control sample and the sheared sample for analysis, respectively. The blood shearing experiments were repeated 5 times for each shear condition with blood from the five donors. The platelet counts had no significant change with the increase of shear stress in all the samples.
Flow cytometric assays of platelet activation and shedding of platelet receptors
To quantify the shear-induced platelet activation in the baseline and sheared blood samples, three platelet activation markers (expression of surface P-selectin25, activation of GPIIb/IIIa complex15 and generation of platelet-derived microparticles (PMPs)) were used for analysis with flow cytometry.
The platelet population was identified by scatter gating (gating on forward scatter [FSC] and side scatter [SSC]) of flow cytometry data (BD Bioscience, San Jose, CA). The level of P-selectin was determined by phycoerythrin (PE) conjugated anti-CD62P antibody (BioLegend, San Diego, CA). The activated level of the platelet receptor GPIIb/IIIa was determined by fluorescein isothiocyanate (FITC) labeled PAC-1 (BD Bioscience, San Jose, CA). FITC conjugated IgMK antibody (BioLegend, San Diego, CA) was used to serve as the negative control. A mixture containing 25 μl HEPES (10mM) buffer, 20μl PAC-1 and 10μl anti-CD62P antibody was prepared first. Then 5μl whole blood sample was added to the mixed buffer and incubated at room temperature in the dark for 30 min. In parallel, a 5 μl whole blood sample was incubated with 25 μl HEPES buffer mixed with 10μl anti-CD62P antibody and 20μl IgMK and used as the negative control. The NPSS-induced platelet activation was expressed as the percentage of CD62P or PAC-1 positive platelets in the total platelet population.
The PMPs were determined by the forward scatter characteristics (FSC) of all CD41 positive events (the CD41 data was obtained from the following receptor shedding analysis) according to the method described in the reference.26 Briefly, the platelet population with CD41 positive events was divided into subpopulations according to their FSC corresponding to their size. In the baseline blood sample, the 2.5% smallest particles were defined as PMPs. In sheared blood samples, the PMPs were determined by the corresponding gate obtained from 2.5% smallest particles of the baseline blood sample. The generation of the NPSS-induced PMPs was expressed by the percentage of PMPs events in all CD41 positive events.
To quantify the shear-induced platelet receptor shedding in the baseline and sheared blood samples, three important platelet functional receptors on the platelet surface (GPIbα, GPVI and GPIIb/IIIa) were analyzed with flow cytometry. For the GPIbα shedding, the platelet population was identified by PE conjugated anti-CD41 antibody (CD41-PE) (BioLegend, San Diego, CA). The level of expression of platelet receptor GPIbα was determined by FITC conjugated anti-CD42b antibody (BioLegend, San Diego, CA). FITC conjugated IgG1K antibody (BioLegend, San Diego, CA) were used to serve as negative control. A mixture containing 25 μl HEPES (10mM) buffer, 10μl anti-CD41 antibody and 10μl anti-CD42b antibody was prepared first. Then 5 μl whole blood sample was added to the mixed buffer and incubated at room temperature in the dark for 30 min. In parallel, a 5 μl whole blood sample was added with 25 μl HEPES buffer mixed with 10μl anti-CD41 antibody and 10μl IgG1K antibody and used as the negative control.
For the GPVI shedding, CD41-PE was used to identify the platelet population. The level of expression of platelet receptor GPVI was determined by eFluor 660 conjugated anti-human GPVI (eBioscience, San Diego, CA). eFluor 660 conjugated Mouse IgG1K Isotype (eBioscience, San Diego, CA) was used as the negative control. In this test, 5μl anti-CD41 antibody, 5μl anti-GPVI antibody and 5μl Mouse IgG1K were used. The method of sample incubated with antibodies was the same as the GPIbα shedding test.
For the GPIIb/IIIa shedding, the platelet population was identified by FITC conjugated anti-CD41 antibody (BioLegend, San Diego, CA). The level of the platelet GPIIb/IIIa receptors was determined by PE conjugated anti-CD41/61 antibody (BioLegend, San Diego, CA). PE conjugated IgG2aK antibody (BioLegend, San Diego, CA) was used as the negative control. In this test, 5µl anti-CD41 antibody, 5µl anti-CD41/61 and 5µl IgG2aK antibody were used. The other labeling steps were the same as the GPIbα shedding test.
After the above labeling steps, 1 ml of 1% paraformaldehyde (PFA) in PBS was used to fix the samples for 30 min at 4 degree in the dark. The flow cytometric data collection of the blood samples was performed with a four color flow cytometer (FACS Calibur, BD Bioscience, San Jose, CA). The data were analyzed offline using the software FCS Express 4.0 (De Novo Software, Los Angeles, CA).
Platelet Aggregation test
The aggregation capacity of the baseline and sheared blood samples was evaluated by adding the agonists of collagen and ristocetin into blood using an aggregometer (Chrono-Log, Havertown, PA). When collagen was added into normal blood, it directly bound to the platelet receptor GPVI to cause an activation step leading to platelet secretion, recruitment of additional platelets, and aggregation. When ristocetin was added to normal blood, it caused vWF to bind the platelet receptor GPIbα, resulting platelet aggregation.
According to the manufacturer’s instruction, 0.5ml whole blood sample was added into the preheated polystyrene cuvette containing 0.5ml 0.9% saline and a stir bar. 1 ml mixed blood sample was heated to 37°C in the aggregometer and stirred at a stirring rate of 1200 rpm. Then 5μl collagen or 8μl ristocetin was added into the mixed blood sample to have the final concentration of 4μg/ml or 1mg/ml, respectively. After adding agonists, the platelets were stimulated to adhere on two electrodes. This adhesion would cause the increase of impedance between the electrodes. Once the agonist reagent was added, the time course of the impedance was recorded for 6 minutes. The integral of the impedance curve (area under the impedance curve, AUC) which indicates the overall metric for platelet aggregation was used to represent the agonist induced platelet aggregation in this study.
Western blot assay of the loss of HMWM-vWF
The loss of the HMWM-vWF in blood samples after exposed to the NPSS was analyzed by western blotting. 2ml baseline and sheared blood samples were centrifuged at 160g for 15minutes at 20°C firstly, and then the supernate was collected for second centrifugation with 14000 rpm for 15minutes at 4 °C. The plasma samples for western blot were obtained after the second centrifugation. The plasma of the baseline and sheared blood samples was mixed with 2X laemmli sample buffer (Bio-Rad, Hercules, CA) with the volume ratio of 1:50, resolved with agarose gel electrophoresis. The protein bands were transferred onto a polyvinylidene difluoride (PVDF) 0.45 μ membrane (Immobilon-P; Millpore Corporation, Bedford, MA, USA). vWF was detected using a primary antibody against vWF (polyclonal rabbit anti-human vWF antibody, Dako North America, Inc. Carpinteria, CA, USA) and a secondary antibody (donkey anti-rabbit IgG H&L conjugated to horseradish peroxidase; AbD Serotec, Kidlington, UK). The primary and secondary antibodies were diluted 1:4000 and 1:10000, respectively, in Tris-buffered saline/Tween-20 (TBS-T). A sheet of HyBlot CL® Autoradiography film (Denville Scientific Inc. MA, USA) was placed over the membrane and exposed for times varying from 5 to 120 seconds in the dark room. The optical density of bands in the film was quantitated with UN-SCAN-IT Gel 6.1 analysis software (Silk Scientific, Orem, USA). The detail steps for this experiment could be found in the work of Krizek, et al.27
Statistical analysis
The data are presented as mean ± SE (standard Error) and statistically analyzed using SPSS statistical software (Statistical Package for Social Sciences for windows, release 18.0; SPSS Inc., Chicago, IL, USA). Statistical differences were determined by using Student’s t-test. Statistical significance was assigned at P< 0.05.
RESULTS
Figure 2A shows typical flow cytometry dot plots of platelet activation indicated by both the expression of surface P-selectin and activation of GPIIb/IIIa receptors in the baseline blood and two sheared blood samples (0.5s/25Pa and 0.5s/125Pa). Figures 2B and 2C show the percentage of activated platelets. As shown in Figure 2, the number of activated platelets indicated by the expression of surface P-selectin and activation of glycoprotein IIb/IIIa increased with increasing level of NPSS. Compared with the baseline blood sample, the increase in the number of activated platelets in the sheared blood samples became significant (P<0.05) when the NPSS increased to 25 Pa and when P-selectin was used as the platelet activation marker. Figure 3A shows typical flow cytometry dot plots of PMPs in the baseline and sheared blood samples (0.5s/25pa and 0.5s/125Pa). Figure 3B shows the average percentage of PMPs out of total CD41-positive events in the baseline blood sample and the sheared blood samples by the two levels of NPSS. The results indicated that the generation of PMPs was insignificant when the blood was exposed to the NPSS of 25 Pa compared to the baseline samples. However, when the NPSS increased to 125 Pa, the generation of PMPs became significant. Overall, the three platelet activation markers (expression of P-selectin, GPIIb/IIIa activation and the generation of PMPs) indicated that the NPSS with short exposure time could induce platelet activation.
Figure 2.
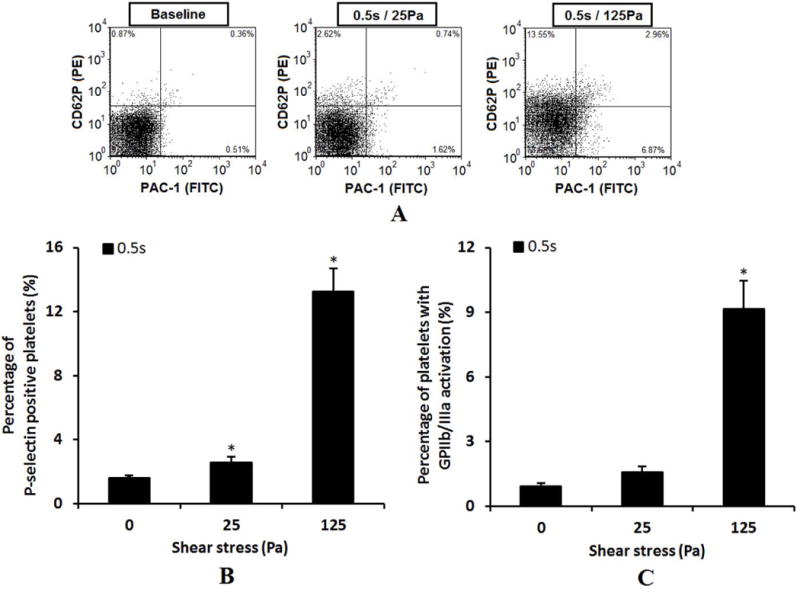
Comparison of P-selectin expression and GP IIb/IIIa activation among baseline blood sample and the sheared samples (0.5s/25pa and 0.5s/125Pa) using flow cytometry. A: Typical flow cytometry dot plots. The platelets population were identified by scatter gating (gating on forward scatter [FSC] and side scatter [SSC]) of flow cytometer. The percent of platelets with P-selectin expression is indicated by the antibody of PE conjugated anti-CD62P and the percent of platelets with GP IIb/IIIa activation is indicated by the antibody of FITC labeled PAC-1. B: Average percentage of activated platelets indicated by the P-selectin surface expression (n=5). C: Average percentage of activated platelets indicated by the GP IIb/IIIa activation (n=5).
Figure 3.
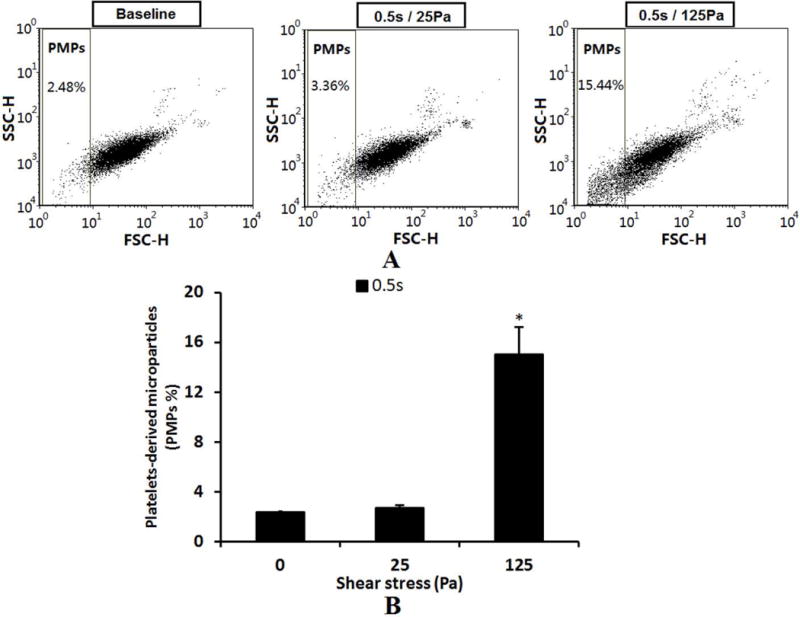
A: The flow cytometry dot plots of comparing of PMPs among base sample and sheared samples (0.5s/25pa and 0.5s/125Pa). The PMPs were determined by the FSC characteristics of all CD41 positive events. B: The comparison of average percentage of PMPs (n=5).
Figures 4A and 4B show typical histograms of the mean fluorescence of the platelet GPIbα expression and the quantification of the platelet GPIbα expression (FITC mean fluorescence) in the baseline blood sample and the two sheared blood samples by the two level of NPSS with the exposure time of 0.5 sec respectively. The results show that for the short exposure time of 0.5s, the reduction of GPIbα expression in platelet surface increased with shear stress level and the loss of GPIbα in platelet surface became significant when exposed to the NPSS of 125 Pa compared to the baseline sample.
Figure 4.
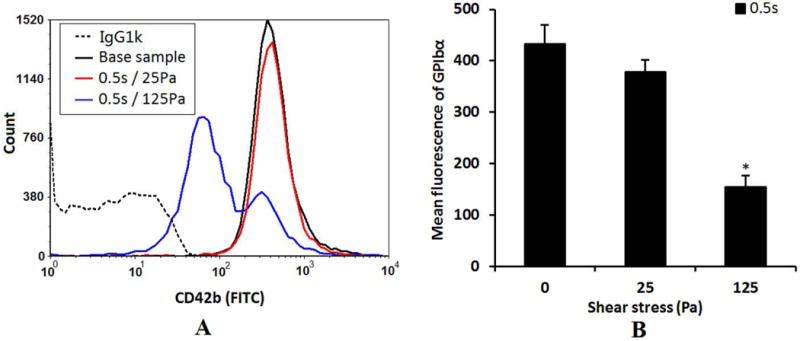
A: The typical histograms of the mean fluorescence of the platelet receptor GPIbα expression (FITC conjugated CD42b) in the baseline blood and two sheared blood samples (0.5s/25Pa and 0.5s/125Pa). B: The quantification comparison of the platelet receptor GPIbα expression (mean fluorescence) (n=5).
Figures 5A shows typical histograms of the mean fluorescence of the platelet GPVI expression in the baseline and two sheared samples (25Pa, 125Pa). Based on this histograms, there was an obvious shift in the GPVI surface expression in the platelets of the sheared blood sample when the NPSS increased to 125Pa. This suggests that the population of platelets with a low fluorescence intensity increased, indicating the loss of the GPVI receptors. Figure 5B shows the quantification of the platelet GPVI surface expression and the reduction in mean fluorescent intensity of the GPVI expression of the platelets in the sheared blood sample by the NPSS of 125 Pa.
Figure 5.
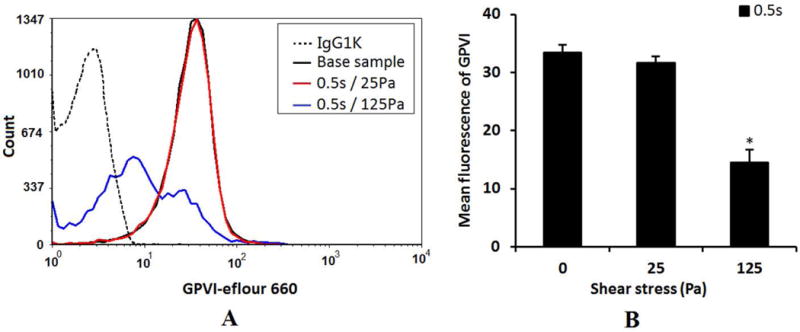
A: The typical histograms of the mean fluorescence of the platelet receptor GPVI expression (eflour660) in the baseline blood and two sheared blood samples (0.5s/25Pa and 0.5s/125Pa). B: The quantification comparison of the platelet receptor GPVI expression (mean fluorescence) (n=5).
Figures 6A and 6B show typical histograms of mean fluorescence of the platelet GPIIb/IIIa complex and the quantification of the platelet GPIIb/IIIa expression (PE mean fluorescence), respectively, in the baseline blood sample and the two sheared blood samples (25Pa and 125Pa). Compared to the baseline sample, the mean fluorescence of the platelet GPIIb/IIIa in the sheared sample by the NPSS of 25Pa with an expousre time of 0.5 sec was close to that of the baseline blood sample. However, a significant shift to the lower level (reduction) in the mean fluorescence of the platelet GPIIb/IIIa was noted when the blood was exposed to the NPSS of 125Pa with the same exposure time, indicating that the number of GPIIb/IIIa receptor available for binding to anti GPIIb/IIIa antibody was significantly reduced.
Figure 6.
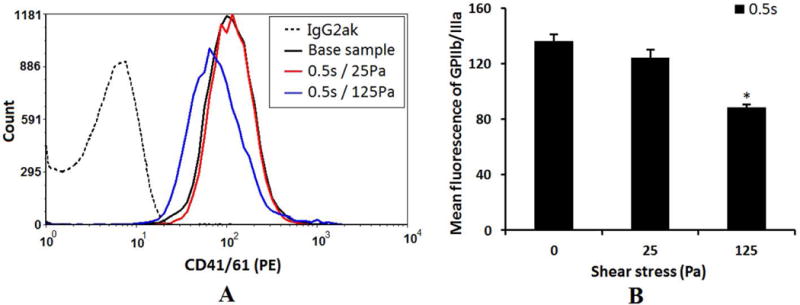
A: The typical histograms of the mean fluorescence of the platelet receptor GP IIb/IIIa (PE conjugated anti-CD41/61) in the baseline blood and two sheared blood samples (0.5s/25Pa and 0.5s/125Pa). B: The quantification comparison of the platelet receptor GP IIb/IIIa (mean fluorescence) (n=5).
Figure 7 shows the reduction in the collagen-(Fig.7A) or ristocetin-(Fig.7C) induced aggregation capacity of the sheared blood samples by the two levels of the NPSS compared to that of the baseline blood sample and the quantitative comparison of the collagen-(Fig.7B) or ristocetin-(Fig.7D) induced platelet aggregation among the baseline blood sample and the two sheared blood samples. As the data suggested, both the levels of NPSS at 25Pa and 125 Pa with the exposure time of 0.5s caused the reduction in collagen-induced and ristocetin-induced aggregation. However, only when the level of NPSS increased to 125 Pa, the reduction in the agonist-induced aggregation become significant. These results further confirmed the reduction in the available number of the GPVI receptors for collagen binding or the GPIbα receptors for ristocetin to cause vWF binding on the platelet surface of the sheared blood samples after being exposed to the NPSS.
Figure 7.
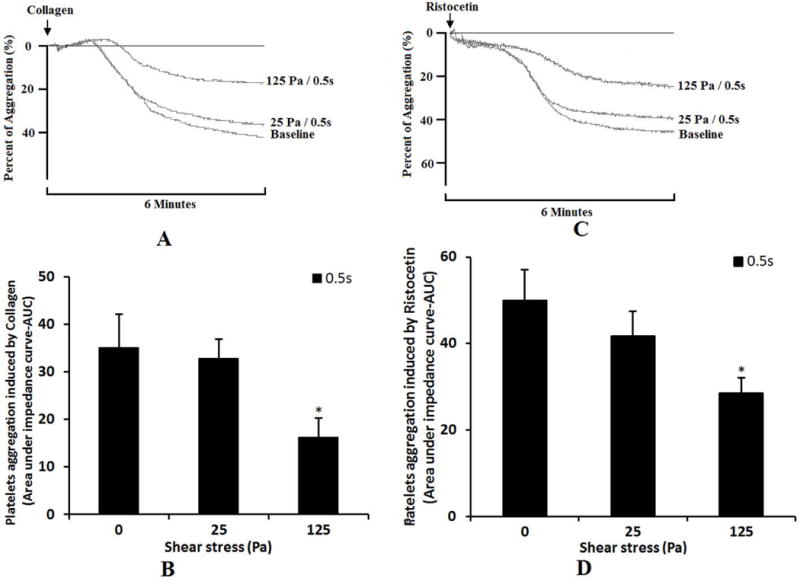
Comparison of platelet aggregation ability among baseline blood sample and the sheared samples (0.5s/25pa and 0.5s/125Pa) using the aggregometer. A: The changing of impedance curve (aggregation curve) of the platelets aggregation induced by collagen. The platelet aggregation is indicated by area under the impedance curve. The time course of the platelets aggregation curve changing was recorded for 6 minutes. B: The average comparing of platelet aggregation induced by collagen (n=5). C: The changing of impedance curve (aggregation curve) of the platelets aggregation induced by ristocetin. D: The average comparing of platelet aggregation induced by ristocetin (n=5).
Figure 8 shows representative images of the western blots of plasma vWF multimeric distribution in the baseline blood sample and the two sheared samples by the NPSS (Figure 8A) and the quantitative comparison of the image optical density ratio of HMWM-vWF of the sheared samples and the baseline sample (Figure 8B). After the blood was exposed to the NPSS, the HMWM-vWF progressively disappeared as the level of the NPSS increased. Although the slightly eleveated NPSS (25 Pa) caused a reduction in HMWM-vWF in the sheared blood sample, the reduction was not significant compared with the baseline blood sample. The loss of HMWM-vWF became significant when the NPSS increased to 125Pa.
Figure 8.

A: The representative images of plasma vWF multimers in the baseline blood and two sheared blood samples (0.5s/25Pa and 0.5s/125Pa). B: The quantitative comparison of plasma HMWM-vWF images optical density ratio (n=5).
The loss of the HMWM-vWF and the reduction in the GPIbα receptors on the platelet surface all can lead to impaired binding of vWF to platelets. This was further verified by the reduced ristocetin-induced aggregation capacity of the sheared blood samples (Figure 7C and 7D). The ristocetin induced-platelet aggregation occurs by stimulating the binding of platelet receptor GPIbα with vWF. The decrease of ristocetin induced-platelet aggregation can be caused by either the shear-induced shedding of the platelet receptor GPIbα or the shear-induced loss of HMWM-vWF. In the present study, it was found that the NPSS could induce both the shedding of the platelet receptor GPIbα and the loss of HMWM-vWF.
DISCUSSION
Many studies have been carried out to characterize the effect of shear stress on platelets under various clinical situations or experimental conditions. It has been established that the NPSS can induce a variety of procoagulant activities to platelets. Shear-induced platelet activation, platelet aggregation, generation of procoagulant microparticles and shear-induced vWF unfolding are frequently referenced to the prothrombotic tendency of platelets and vWF caused by the NPSS. There is a close relationship between the level and exposure time of the NPSS and the prothrombotic propensity by these indicators. Thus, it is commonly believed that the elevated shear stress would be pro-thrombogenic. Interestingly, most of these previous studies have been conducted under a relatively low shear stress range (<75 Pa) with a relatively long exposure time (seconds to minutes) generated using viscometers or parallel flow chambers.
The emergence of some BCMDs or severe stenosis in circulation would subject blood to NPSS. However, the exposures to NPSS are often very brief.16, 23 The effect of shear stress on platelets and vWF under high NPSS with short exposure time has not been well studied. Platelets and vWF are two important factors for hemostasis. Their integrity is essential for the normal hemostasis. Altough the shear-induced platelet activation increases the thrombosis tendency, the shear-induced receptor shedding may induce platelet dysnfuction, leading to bleeding due to a reduced capacity to adhere to their respective counterpart substrate.5, 13, 15, 16 In the present study, the effect of the NPSS with short exposure time on platelets and vWF was investigated.
The results from the present study clearly show that the NPSS induced a paradoxical phenomenon. On the one hand, the NPSS induced the elevated P-selectin expression, GPIIb/IIIa activation and PMPs generation. More activated platelets would enhance platelet adhesion and aggregation, leading to thrombosis. On the other hand, the NPSS induced the shedding of three major platelet receptors (GPIba, GPVI and GPIIb/IIIa) which are essential for initiation of platelet adhesion and formation of firm clot. The reduced aggregation capacity of the sheared blood samples was verified by collagen- and ristocetin-induced platelet aggregation. The platelet aggregation experiment further confirmed the loss of GPIba and GPVI on the platelet surface of the sheared blood samples. It is believed that the increase in shear stress can cause vWF unfolding to increase vWF binding sites with GPIbα and to activate vWF’s adhesion potential. In the present study, it was shown that the NPSS could fragment vWF, resulting in the loss of HMWM-vWF. The degree of loss of HMWM-vWF increased with increasing the level of the NPSS. The loss of HMWM-vWF has been attributed to bleeding complication associated with vWF diseases. The reduction of the platelet functional receptors (GPIba, GPVI and GPIIb/IIIa) and the loss of HMWM-vWF all would lower the adhesion and aggregation capacity of the platelets for the normal hemostasis and increase the risk of bleeding.
The results form the present study data show that these opposite effects occur at the same time. When blood was subjected to NPSS, shedding of platelet receptors occurred accompanying with platelet activation. This phenomenon might be used to explain thrombosis and bleeding that are two opposite pathological complications occurring in patients implanted with some BCMDs (VADs, ECMO or MHVs).17, 28 Therefore, not only the shear-induced platelet activation but also shear-induced shedding of platelet receptors and loss of HMWM-vWF should be considered during the development of BCMDs.
Our shearing device was designed to generate shear conditions occurring in some BCMDs. In our previous work23, we found that the flow within the narrow gap region is stable and laminar at all of the experimental conditions because the Taylor number of this device is below the critical value, where flow shifts from stable to unstable region. Therefore, the stable uniform non-physiological shear stress region created in this test was reasonable. In addition, the CFD simulation results in that work23 supported that the NPSS in the gap varied in a very small range, which is acceptable for this comparing study.
In this article, although it is premature to make a conclusion based solely on an in vitro study. Animal experiments should be carried out to validate this paradoxical phenomenon. We had put forward this paradoxical phenomenon for the first time, which might be useful for optimization of some BCMDs (VADs or MHVs).
CONCLUSION
This study found that NPSS could not only increase the number of activated platelets, but also cause reduction of platelet receptors and loss of HMWM-vWF. This appears to be a paradoxical effect. The data of this study may be useful for the design and development of BCMDs such as VADs, ECMO system and MHVs.
Acknowledgments
This work was partially supported by the National Institutes of Health (Grant numbers: R01 HL 088100, R01HL124170) and the International Postdoctoral Exchange Fellowship Program (No. 20130028).
Footnotes
CONFLICT OF INTEREST STATEMENT
The authors declared that there are no conflicts of interests.
References
- 1.Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006;3(11):e442. doi: 10.1371/journal.pmed.0030442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Blaha MJ, et al. Heart disease and stroke statistics–2014 update: a report from the american heart association. Circulation. 2014;129(3):e28–e292. doi: 10.1161/01.cir.0000441139.02102.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Butchart EG, Ionescu A, Payne N, Giddings J, Grunkemeier GL, Fraser AG. A new scoring system to determine thromboembolic risk after heart valve replacement. Circulation. 2003;108:II68–74. doi: 10.1161/01.cir.0000087383.62522.1e. [DOI] [PubMed] [Google Scholar]
- 4.Eckman PM, John R. Bleeding and thrombosis in patients with continuous-flow ventricular assist devices. Circulation. 2012;125:3038–47. doi: 10.1161/CIRCULATIONAHA.111.040246. [DOI] [PubMed] [Google Scholar]
- 5.Bluestein D. Research approaches for studying flow-induced thromboembolic complications in blood recirculating devices. Expert Rev Med Devices. 2004;1(1):65–80. doi: 10.1586/17434440.1.1.65. [DOI] [PubMed] [Google Scholar]
- 6.Steinlechner B, Dworschak M, Birkenberg B, Duris M, Zeidler P, Fischer H, et al. Platelet dysfunction in outpatients with left ventricular assist devices. Ann Thorac Surg. 2009;87(1):131–7. doi: 10.1016/j.athoracsur.2008.10.027. [DOI] [PubMed] [Google Scholar]
- 7.Kroll MH, Hellums JD, McIntire LV, Schafer AI, Moake JL. Platelets and shear stress. Blood. 1996;88(5):1525–41. [PubMed] [Google Scholar]
- 8.Zhang JN, Bergeron AL, Yu Q, Sun C, McIntire LV, López JA, Dong JF. Platelet aggregation and activation under complex patterns of shear stress. Thromb Haemost. 2002;88(5):817–21. [PubMed] [Google Scholar]
- 9.Yin W, Gallocher S, Pinchuk L, Schoephoerster RT, Jesty J, Bluestein D. Flow-induced platelet activation in a St. Jude mechanical heart valve, a trileaflet polymeric heart valve, and a St. Jude tissue valve. Artif Organs. 2005;29(10):826–31. doi: 10.1111/j.1525-1594.2005.29109.x. [DOI] [PubMed] [Google Scholar]
- 10.Bluestein D, Yin W, Affeld K, Jesty J. Flow-induced platelet activation in mechanical heart valves. J Heart Valve Dis. 2004;13(3):501–8. [PubMed] [Google Scholar]
- 11.Furie B, Furie BC. Mechanisms of thrombus formation. N Engl J Med. 2008;359:938–49. doi: 10.1056/NEJMra0801082. [DOI] [PubMed] [Google Scholar]
- 12.Canobbio I, Balduini C, Torti M. Signalling through the platelet glycoprotein Ib-V-IX complex. Cell Signal. 2004;16(12):1329–44. doi: 10.1016/j.cellsig.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 13.Al-Tamimi M, Tan CW, Qiao J, Pennings GJ, Javadzadegan A, Yong ASC, et al. Pathologic shear triggers shedding of vascular receptors: a novel mechanism for down-regulation of platelet glycoprotein VI in stenosed coronary vessels. Blood. 2012;119:4311–20. doi: 10.1182/blood-2011-10-386607. [DOI] [PubMed] [Google Scholar]
- 14.Tsai HM, Sussman II, Nagel RL. Shear stress enhances the proteolysis of von Willebrand factor in normal plasma. Blood. 1994;83(8):2171–9. [PubMed] [Google Scholar]
- 15.Cheng H, Yan R, Li S, Yuan Y, Liu J, Ruan C, et al. Shear-induced interaction of platelets with von Willebrand factor results in glycoprotein Ibα shedding. American Journal of Physiology-Heart and Circulatory Physiology. 2009;297:H2128–H35. doi: 10.1152/ajpheart.00107.2009. [DOI] [PubMed] [Google Scholar]
- 16.Arthur JF, Dunkley S, Andrews RK. Platelet glycoprotein VI-related clinical defects. Br J Haematol. 2007;139(3):363–72. doi: 10.1111/j.1365-2141.2007.06799.x. [DOI] [PubMed] [Google Scholar]
- 17.Hu J, Mondal NK, Sorensen EN, Cai L, Fang HB, Griffith BP, Wu ZJ. Platelet glycoprotein Ibα ectodomain shedding and non-surgical bleeding in heart failure patients supported by continuous-flow left ventricular assist devices. J Heart Lung Transplant. 2014;33(1):71–9. doi: 10.1016/j.healun.2013.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fraser KH, Zhang T, Taskin ME, Griffith BP, Wu ZJ. A quantitative comparison of mechanical blood damage parameters in rotary ventricular assist devices: shear stress, exposure time and hemolysis index. J Biomech Eng. 2012;134(8):081002. doi: 10.1115/1.4007092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Taskin ME, Fraser KH, Zhang T, Wu C, Griffith BP, Wu ZJ. Evaluation of Eulerian and Lagrangian models for hemolysis estimation. ASAIO J. 2012;58(4):363–72. doi: 10.1097/MAT.0b013e318254833b. [DOI] [PubMed] [Google Scholar]
- 20.Heilmann C, Geisen U, Beyersdorf F, Nakamura L, Benk C, Trummer G, et al. Acquired von Willebrand syndrome in patients with extracorporeal life support (ECLS) Intensive Care Med. 2012;38(1):62–8. doi: 10.1007/s00134-011-2370-6. [DOI] [PubMed] [Google Scholar]
- 21.Kalbhenn J, Schmidt R, Nakamura L, Schelling J, Rosenfelder S, Zieger B. Early Diagnosis of Acquired von Willebrand Syndrome (AVWS) is Elementary for Clinical Practice in Patients Treated with ECMO Therapy. J Atheroscler Thromb. 2015;22(3):265–71. doi: 10.5551/jat.27268. [DOI] [PubMed] [Google Scholar]
- 22.Bluestein D. Research approaches for studying flow-induced thromboembolic complications in blood recirculating devices. Expert Rev Med Devices. 2004;1(1):65–80. doi: 10.1586/17434440.1.1.65. [DOI] [PubMed] [Google Scholar]
- 23.Zhang T, Taskin ME, Fang HB, Pampori A, Jarvik R, Griffith BP, et al. Study of Flow-Induced Hemolysis Using Novel Couette-Type Blood-Shearing Devices. Artificial organs. 2011;35:1180–6. doi: 10.1111/j.1525-1594.2011.01243.x. [DOI] [PubMed] [Google Scholar]
- 24.Paul R, Apel J, Klaus S, Schügner F, Schwindke P, Reul H. Shear stress related blood damage in laminar couette flow. Artif Organs. 2003;27(6):517–29. doi: 10.1046/j.1525-1594.2003.07103.x. [DOI] [PubMed] [Google Scholar]
- 25.Ding J, Chen Z, Niu S, Zhang J, Mondal NK, Griffith BP, Wu ZJ. Quantification of Shear-Induced Platelet Activation: High Shear Stresses for Short Exposure Time. Artif Organs. 2015;21 doi: 10.1111/aor.12438. [DOI] [PubMed] [Google Scholar]
- 26.Hagberg IA, Lyberg T. Blood platelet activation evaluated by flow cytometry: optimised methods for clinical studies. Platelets. 2000;11(3):137–50. doi: 10.1080/095371000403071. [DOI] [PubMed] [Google Scholar]
- 27.Krizek DR, Rick ME. A rapid method to visualize von willebrand factor multimers by using agarose gel electrophoresis, immunolocalization and luminographic detection. Thromb Res. 2000;97(6):457–62. doi: 10.1016/s0049-3848(99)00196-6. [DOI] [PubMed] [Google Scholar]
- 28.Heilmann C, Geisen U, Beyersdorf F, Nakamura L, Benk C, Berchtold-Herz M, Trummer G, Schlensak C, Zieger B. Acquired von Willebrand syndrome in patients with ventricular assist device or total artificial heart. Thromb Haemost. 2010;103(5):962–7. doi: 10.1160/TH09-07-0497. [DOI] [PubMed] [Google Scholar]