

Abstract
Purpose
To report on the variability and progression of clinical presentation in three family members with spinocerebellar ataxia type 7 (SCA7) including early recognizable features on retinal imaging and magnetic resonance imaging (MRI).
Methods
Retrospective case series
Results
The proband, Patient 1 (Mother) presented at age 26 with Light Perception vision. Initial examination was significant for optic disc pallor, vascular attenuation and central macular atrophy. Two years later, her vision declined to No Light Perception and fundus exam demonstrated marked progression of macular atrophy and peripheral bone spicule formation. Seven years after the onset of vision loss, neurologic exam demonstrated ataxia, dysarthria, and slowed saccades. Genetic testing of ATXN7 identified heterozygous 61-CAG trinucleotide repeat expansion confirming the diagnosis of SCA7. Patient 2 (Son) presented at age 11 with visual acuity of 20/300 bilaterally and decreased color vision. Fundoscopic exam was notable for disc pallor, vascular attenuation and peripheral pigmentary changes. Electroretinography (ERG) demonstrated diminished rod and cone function, and Goldmann visual field testing revealed paracentral scotoma. Patient 3 (Daughter) presented at age 14 with visual acuity of 20/50 bilaterally and minimal fundoscopic changes. The only significant ophthalmic finding was retinal thinning with atrophy of the outer nuclear layer and subfoveal ellipsoid zone on optical coherence tomography. Early cerebellar volume loss was also noted on MRI.
Conclusion
The clinical presentation of SCA7 can vary widely even within the same family. In individuals with vision loss and normal fundus exam, careful evaluation of OCT and brain MRI facilitates early diagnosis and genetic testing.
Keywords: Cone-Rod Dystrophy, Inherited Retinal Dystrophy, Spinocerebellar Ataxia Type 7
Introduction
Spinocerebellar ataxia type 7 (SCA7) is an autosomal dominant disorder characterized by progressive cerebellar ataxia and a predominant association with retinal degeneration and vision loss. It is caused by CAG-trinucleotide repeat expansion in the ATXN7 gene leading to polyglutamine expansion in the protein ataxin-71. We report three family members with SCA7 that presented with varying degrees of retinopathy and visual impairment.
Patient 1
Patient 1, the proband, is the mother of Patients 2 and 3 (Figure 1, Pedigree). She presented to the eye clinic at age 26 with a complaint of worsening vision over one year. Visual acuity was Light Perception in both eyes. Funduscopic exam revealed optic disc pallor, vascular attenuation, central macular atrophy and peripheral pigmentary changes. Full field electroretinography (ERG) demonstrated extinguished rod and cone response bilaterally, and she was diagnosed with advanced retinitis pigmentosa. When she returned to clinic two years later, her vision had declined to No Light Perception in both eyes. Figure 2 demonstrates the marked progression of macular atrophy and peripheral bone spicules over 6 years.
Figure 1.

Family pedigree. Family members with a reported diagnosis of retinitis pigmentosa are marked in blue. Members with a reported diagnosis of ataxia are marked with dashed lines. Patients 1,2, and 3 are labelled.
Figure 2.

Patient 1. Fundus photos demonstrate optic nerve pallor and vascular attenuation. There is progression of macular atrophy and bone spicule formation from age 26 (top row) to 28 (middle) and 32 (bottom).
Approximately 7 years after the onset of vision loss, she complained of difficulty with her balance. She reported a dominant family history of vision loss, diagnosed as retinitis pigmentosa (RP), and ataxia including her mother, maternal aunt and two brothers. Family pedigree (Figure 1) demonstrates family members with vision loss and/or ataxia according to patient report. Neurologic exam revealed dysarthria, ataxia, spasticity, and slowed saccades. She was referred for genetic evaluation which revealed strong clinical suspicion for SCA7. Genetic testing of ATXN7 identified heterozygous 61-CAG trinucleotide repeat expansion confirming the clinical diagnosis. Her neurologic function has since declined, and she is no longer able to ambulate without assistance.
Patient 2
The son of Patient 1 was referred for evaluation of possible retinitis pigmentosa at age 11 due to a strong family history of RP and his progressive vision loss. He had no neurologic complaints. Visual acuity was 20/300 OU with poor color vision bilaterally. Funduscopic exam demonstrated bilateral optic nerve pallor and vascular attenuation (Figure 3). Subtle macular changes were noted on fundus autofluorescence (Figure 4). No bone spicules were present. His ERG revealed diminished rod and cone responses. Goldmann visual field testing revealed bilateral paracentral scotoma (Figure 5). His neurologic exam was notable for only mild dysmetria, a form of ataxia characterized by overshooting and undershooting limb movements.
Figure 3.
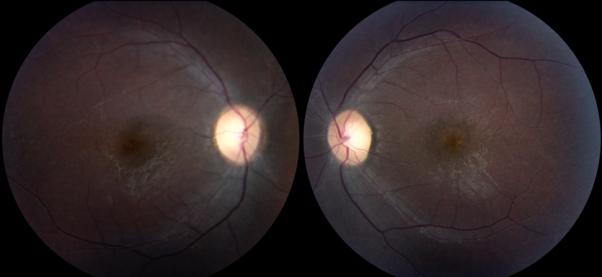
Patient 2. Fundus photos demonstrate bilateral optic nerve pallor and vascular attenuation.
Figure 4.

Fundus Autofluorescence of the right eye demonstrates perifoveal hypoautofluorescent pigment clumping (white arrows) and a peripheral ring of hyperautofluorescence. Similar findings were seen in the left eye.
Figure 5.

Patient 2. Goldmann visual fields of the left (A) and right (B) eyes demonstrate bilateral paracentral scotoma.
Patient 3
Patient 3 was examined at age 14 with only mild visual complaints. She had visual acuity of 20/50 OU. Color vision testing revealed a blue-yellow deficit in both eyes. At that time, funduscopic exam was felt to be unremarkable with only blunting of the normal foveal light reflex. A full-field ERG was normal. Two years later, visual acuity had decreased to 20/70 OU, and she had developed fine granular pigmentary macular changes. She did not exhibit any neurological abnormalities. Optical coherence tomography (OCT) imaging of the macula was obtained and demonstrated bilateral outer retinal atrophy with loss of the subfoveal outer nuclear layer and ellipsoid zone (Figure 6). MRI of the brain revealed early cerebellar volume loss (Figure 7).
Figure 6.

Patient 3. Normal appearing fundus photos (top row) and fundus autofluorescence (middle row). Retinal thinning and loss of the outer nuclear layer and subfoveal ellipsoid zone are seen on OCT (bottom row).
Figure 7.

Patient 3. MRI brain reveals mild cerebellar volume loss with prominence of cerebellar sulci. (A) T1-weighted sagital (B) T2-weighted axial.
Discussion
Spinocerebellar ataxia type 7, caused by trinucleotide expansion in ATXN7, is unique among the autosomal dominant cerebellar ataxia syndromes for its strong association with vision loss due to progressive cone-rod dystrophy. Patients may present initially either with neurologic or visual complaints and diagnosis may be delayed if family history is negative or not well known. In infantile-onset disease, neurologic manifestations often predominate while vision loss tends to precede ataxia in adults1. The individuals in our series illustrate the variable expressivity of the disease.
Patient 1 presented with end-stage retinopathy and was initially diagnosed with RP based on her phenotypic fundus presentation. However, the rapid progression of funduscopic changes is atypical for RP. The underlying cause for her retinopathy did not become apparent until years later when her neurologic symptoms developed. The initial exam, with fundus changes confined mainly to the macula and limited peripheral pigmentary changes, are consistent with previous reports in the literature2. The development of diffuse bone spicules and intraretinal pigmentation over the course of only a few years has not been previously reported in SCA7. Another interesting feature seen in Patient 1 is the slowing of saccades which is typically an early finding in SCA7. As neurologic degeneration progresses, patients may develop ophthalmoplegia3.
The presentation of Patient 3 differed remarkably from her younger brother. She complained of decreased vision bilaterally but had minimal funduscopic changes. Her finding of decreased blue-yellow color discrimination is significant as it has been described as one of the initial findings in SCA7, and may be present in asymptomatic individuals4. However, a diagnosis of SCA7 for Patient 3 would be unlikely to be considered without obtaining a family history. Her complaints might have been dismissed in the setting of a normal funduscopic exam and full-field ERG. In this instance, OCT imaging was essential to diagnosis. Earlier papers have reported retinal thinning on OCT in patients with SCA75,6. The OCT for Patient 3 demonstrates outer retinal atrophy with loss of the outer nuclear layer and ellipsoid zone in the central macula. The loss of the subfoveal ellipsoid zone corresponds to loss of cone photoreceptor cells which may be the earliest clinical sign of cone-rod dystrophy associated with SCA7. With the increased resolution of spectral domain technology, OCT can play a larger role in identifying subtle retinal abnormalities. Finally, MRI confirmed the presence of early cerebellar atrophy in Patient 3 prior to the development of neurologic symptoms.
Early funduscopic changes, as seen in Patient 3, include loss of the normal foveal light reflex and granular macular pigmentary changes. Initial visual complaints are secondary to cone dysfunction and include decreased central visual acuity, central scotomata, and dyschromatopsia. Full-field ERG is abnormal in more advanced disease but may lack the sensitivity to detect early cases, such as Patient 3, in whom early cone dysfunction is confined to the macular region. In these cases, multi-focal ERG may be a more useful test. As retinal degeneration progresses, funduscopic findings become more evident and may include vascular attenuation, optic disc pallor, and peripheral pigmentary changes as seen in Patient 2.
The underlying genetic defect in SCA7 is the expansion of the polyglumatine-tract in ataxin-7 due to CAG-trinucleotide repeats on chromosome 3p. The number of CAG-repeats varies in normal individuals from 7 to 172,7. In affected individuals, the CAG-repeats range from 36 to more than 4508. Like other trinucleotide-expansion disorders, SCA7 demonstrates genetic anticipation. Increase in the number of repeats results in earlier onset and more severe disease.
Treatment options for SCA7 are currently limited. Suppression of mutant gene expression in an inducible SCA7 mouse model resulted in reversibility of behavioral, histological, and neurodegenerative phenotypes and holds promise for SCA7 and other polyglutamine-repeat disorders9. As new treatment strategies emerge, early identification of affected family members is of increased importance. Ancillary testing such as OCT, multi-focal ERG, MRI and fundus autofluorescence can be invaluable screening tests to identify individuals for genetic testing and clinical trial participation.
Summary Statement.
Spinocerebellar ataxia type 7 (SCA7) is an autosomal dominant progressive neurodegenerative condition. It is notable for its predominant association with retinal degeneration. We describe three family members with SCA7 presenting with varying degrees of retinopathy and report on the role of ancillary testing in assisting with early diagnosis.
Acknowledgments
Supported in part by NIH Departmental Core Grant EY006360 and Research to Prevent Blindness, Inc, New York, New York
Footnotes
The authors report no conflicts of interest.
References
- 1.Garden G, Pagon RA, Adam MP, Ardinger HH, et al. GeneReviews® [Internet] University of Washington, Seattle; Seattle, WA: 2012. Spinocerebellar Ataxia Type 7. [Google Scholar]
- 2.McLaughlin ME, Dryja TP. Ocular findings in spinocerebellar ataxia 7. Arch Ophthalmol. 2002;120:655–9. [PubMed] [Google Scholar]
- 3.Thurtell MJ, Fraser JA, Bala E, et al. Two patients with spinocerebellar ataxia type 7 presenting with profound binocular visual loss yet minimal ophthalmoscopic findings. J Neuroophthalmol. 2009;29:187–91. doi: 10.1097/WNO.0b013e3181b41764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guow LG, Kaplan CD, Haines JH, et al. Retinal degeneration characterizes a spinocerebellar ataxia mapping to chromosome 3p. Nat Genet. 1995;10:89–93. doi: 10.1038/ng0595-89. [DOI] [PubMed] [Google Scholar]
- 5.Aleman TS, Cideciyan AV, Volpe NJ, et al. Spinocerebellar ataxia type 7 (SCA7) shows a cone-rod dystrophy phenotype. Exp Eye Res. 2002;74:737–45. doi: 10.1006/exer.2002.1169. [DOI] [PubMed] [Google Scholar]
- 6.Hugosson T, Gränse L, Ponjavic V, et al. Macular dysfunction and morphology in spinocerebellar ataxia type 7 (SCA 7) Ophthalmic Genet. 2009;30:1–6. doi: 10.1080/13816810802454081. [DOI] [PubMed] [Google Scholar]
- 7.David G, Abbas N, Stevanin G, et al. Cloning of the SCA7 gene reveals a highly unstable CAG repeat expansion. Nat Genet. 1997;17:65–70. doi: 10.1038/ng0997-65. [DOI] [PubMed] [Google Scholar]
- 8.Michalik A, Martin JJ, Van Broeckhoven C. Spinocerebellar ataxia type 7 associated with pigmentary retinal dystrophy. Eur J Hum Genet. 2004;12:2–15. doi: 10.1038/sj.ejhg.5201108. [DOI] [PubMed] [Google Scholar]
- 9.Furrer SA, Waldherr SM, Mohanachandran MS, et al. Reduction of mutant ataxin-7 expression restores motor function and prevents cerebellar synaptic reorganization in a conditional mouse model of SCA7. Hum Mol Genet. 2013;22:890–903. doi: 10.1093/hmg/dds495. [DOI] [PMC free article] [PubMed] [Google Scholar]