

Abstract
Matricellular proteins mediate pleiotropic effects during tissue injury and repair. CCN1 is a matricellular protein that has been implicated in angiogenesis, inflammation, and wound repair. In this study, we identified CCN1 as a gene that is differentially up-regulated in alveolar mesenchymal cells of human subjects with rapidly progressive idiopathic pulmonary fibrosis (IPF). Elevated levels of CCN1 mRNA were confirmed in lung tissues of IPF subjects undergoing lung transplantation, and CCN1 protein was predominantly localized to fibroblastic foci. CCN1 expression in ex vivo IPF lung fibroblasts correlated with gene expression of the extracellular matrix proteins, collagen (Col)1a1, Col1a2, and fibronectin as well as the myofibroblast marker, α-smooth muscle actin. RNA interference (RNAi)-mediated knockdown of CCN1 down-regulated the constitutive expression of these profibrotic genes in IPF fibroblasts. TGF-β1, a known mediator of tissue fibrogenesis, induces gene and protein expression of CCN1 via a mothers against decapentaplegic homolog 3 (SMAD3)-dependent mechanism. Importantly, endogenous CCN1 potentiates TGF-β1-induced SMAD3 activation and induction of profibrotic genes, supporting a positive feedback loop leading to myofibroblast activation. In vivo RNAi-mediated silencing of CCN1 attenuates fibrogenic responses to bleomycin-induced lung injury. These studies support previously unrecognized, cooperative interaction between the CCN1 matricellular protein and canonical TGF-β1/SMAD3 signaling that promotes lung fibrosis.—Kurundkar, A. R., Kurundkar, D., Rangarajan, S., Locy, M. L., Zhou, Y., Liu, R.-M., Zmijewski, J., Thannickal, V. J. The matricellular protein CCN1 enhances TGF-β1/SMAD3-dependent profibrotic signaling in fibroblasts and contributes to fibrogenic responses to lung injury.
Keywords: matrix remodeling, wound repair, myofibroblasts, idiopathic pulmonary fibrosis
Matricellular proteins are secreted extracellular matrix (ECM) proteins that regulate diverse cell functions via its interaction with, and integration of, integrin and growth factor signaling (1, 2). The complexity of matricellular proteins is evident by their cell type- and context-dependent actions, which may sometimes elicit contrasting cellular phenotypes or fates. Matricellular proteins play major roles in development and tissue injury repair responses (3, 4). CCN1 (or cysteine-rich protein 61) belongs to the CCN family of matricellular proteins that regulate a number of biologic processes such as inflammation, angiogenesis, wound healing, and fibrosis (5, 6). The CCN acronym derives from the first 3 members of the 6-member family, namely cysteine-rich protein 61, connective tissue growth factor, and nephroblastoma overexpressed gene. CCN1, akin to other matricellular proteins, mediates pleiotropic cellular effects and regulates a wide range of biologic processes.
CCN1 was identified as a secreted protein encoded by a growth factor-inducible immediate–early gene that induces angiogenesis and promotes tumor growth (7). CCN1 is an essential regulator of vascular development and CCN1-null mice suffer embryonic death due to loss of vascular integrity and impaired placental development (8). CCN1 is highly induced in granulation tissue during wound healing of the skin and activates a set of genes involved in angiogenesis, inflammation, and matrix remodeling (9). CCN1 has been shown to facilitate normal wound healing by inducing senescence of fibroblasts and limiting fibrosis (10). Additionally, exogenous CCN1 or genetic overexpression resulted in regression of established fibrosis in the liver (11, 12). However, in some contexts, CCN1 appears to mediate proinflammatory and profibrotic effects, for example, following ischemic kidney injury (13) or unilateral ureteral obstruction (14). In the lung, CCN1 overexpression exacerbates lung injury and causes neutrophilic alveolitis and obstructive bronchiolitis in mice (15, 16). CCN1 expression is increased in various models of experimental lung fibrosis (15, 17, 18); however, its precise role in physiologic wound healing vs. pathologic tissue remodeling responses to lung injury is not well understood. Idiopathic pulmonary fibrosis (IPF) is a progressive fibrotic remodeling disorder of the lung (19). In this study, we evaluated a potential role for CCN1 in profibrotic signaling and phenotype of ex vivo IPF lung fibroblasts, as well as in an in vivo model of bleomycin-induced lung fibrosis.
MATERIALS AND METHODS
Cell culture
Primary lung fibroblasts were isolated and cultured from lung explants of human subjects undergoing lung transplantation with IPF or failed donors (controls), as previously described (20). Alveolar mesenchymal cells were isolated and cultured from bronchoalveolar lavage of human subjects with IPF, as previously described (21). All studies were approved by the Institutional Review Boards at the University of Michigan and the University of Alabama at Birmingham. Human fetal lung fibroblasts [Institute of Medical Research (IMR)-90 cells] were purchased from Coriell Cell Repositories (Camden, NJ, USA). All cells were cultured in DMEM (Life Technologies, New York, NY, USA) supplemented with 10% fetal bovine serum (Hyclone Laboratories, Logan, UT, USA), penicillin (100 U/ml), streptomycin (100 μg/ml), and amphotericin B (1.25 μg/ml) at 37°C in 5% CO2.
Reagents
Porcine platelet-derived TGF-β1 was purchased from R&D Systems (Minneapolis, MN, USA). The protein kinase inhibitors PD98059, SB600125, Y27632, SU6656, and PP2 (AG1879) were obtained from Calbiochem (San Diego, CA, USA). The Alk-5 inhibitor, SB431542 was obtained from Tocris (Bristol, United Kingdom). Sources and dilutions of antibodies used for the study are provided in Table 1.
TABLE 1.
Antibodies
| Antibody | Manufacturer | Catalog no. | Dilution used |
|---|---|---|---|
| α-SMA | American Research Products | 31-B70000 | 1:3000 (WB), 1:100 (IHC) |
| β-actin | Sigma-Aldrich | A2228 | 1:4000 (WB) |
| Col1a1 | Abcam | ab138492 | 1:1000 (WB) |
| Col1a2 | Santa Cruz Biotechnology | sc-166572 | 1:500 (WB) |
| FN | Sigma-Aldrich | F0916 | 1:4000 (WB) |
| CCN1 | Santa Cruz Biotechnology | sc-374129 | 1:1000 (WB), 1:100 (IHC) |
| SMAD3 | Santa Cruz Biotechnology | sc-101154 | 1:1000 (WB) |
| pSMAD3 | Cell signaling | C25A9 | 1:1000 (WB) |
IHC, immunohistochemistry; WB, Western blot.
RNA interference
Both human and murine CCN1 small interfering RNA (siRNA) and nontargeting (NT) siRNA duplexes (Dharmacon Inc., Pittsburgh, PA, USA) were used for experimental studies (Table 2). For cell culture studies, ∼1.5 × 105 cells/well in 6-well plates were plated in medium containing DMEM with 10% fetal bovine serum (FBS), without antibiotics or fungizone. Following overnight incubation, cells were transfected with NT siRNA and CCN1 siRNA using Lipofectamine 2000 (catalog no. 1668-027; Life Technologies), thus giving minimal time for CCN1 deposition within the pericellular matrix. The day after transfection, medium was changed to DMEM with 10% FBS for 24 h and then to serum-free medium for an additional 48 or 72 h. Efficiency of knockdown was assessed by Western blotting and RT-PCR. For in vivo RNA interference (RNAi) studies, mouse CCN1 siRNA or NT siRNA were reconstituted in PBS and administered to the lungs of mice by intranasal delivery (50 µg in 30 µl PBS) every other day starting from d 8 to 20 postbleomycin injury.
TABLE 2.
Sense sequences of siRNA used for knockdown
| Target gene | siRNA sequence, 5′-3′ |
|---|---|
| NT | UAAGGCUAUGAAGAGAUAC |
| CCN1 (human) | CCAGAAAUGUAUUGUUCAA |
| CCN1 (mouse) | CCAGAAAUGCAUCGUUCAG |
Western immunoblotting and immunoprecipitation
Cell lysates were prepared in RIPA buffer and subjected to SDS-PAGE under reducing conditions and Western immunoblotting was performed as previously described (22).
Densitometric analyses were performed using ImageQuant TL (version 8.1; GE Healthcare Life Sciences, Pittsburgh, PA, USA).
Real-time PCR
Total RNA from cells was isolated using the RNeasy Mini Kit (Qiagen, Germantown, MD, USA), and reverse transcribed to cDNA using Bio-Rad iScript cDNA synthesis kit (catalog no. 1708890; Bio-Rad, Hercules, CA, USA) according to the manufacturer’s protocol. Primers used for the study are given in Table 3. Real-time PCR procedure and analysis was performed as described previously (23).
TABLE 3.
Primer sequences
| Target gene (human) | Primer sequence, 5′-3′ |
|
|---|---|---|
| Forward | Reverse | |
| Col1a1 | ACGAAGACATCCCACCAATCACCT | AGATCACGTCATCGCACAACACCT |
| Col1a2 | GGTTACGATGGAGACTTCTACAGG | CAGGAGTAAGAAGGGTCTCAATCTG |
| Col3a1 | ACAGCCTCCAACTGCTCCTA | GTCACCATTTCTCCCAGGAA |
| FN | TCCACAAGCGTCATGAAGAG | CTCTGAATCCTGGCATTGGT |
| α-SMA | TCCTCATCCTCCCTTGAGAA | ATGAAGGATGGCTGGAACAG |
| CCN1 | CTCCCTGTTTTTGGAATGGA | TGGTCTTGCTGCATTTCTTG |
| CCN2 | GGCTTACCGACTGGAAGACA | CATCCCACAGGTCTTGGAAC |
| CCN3 | CAAAACCATCCAGGCAGAGT | AGTTGGTGTGACAGGTGCAG |
| CCN4 | CTCCAATGTTAACGCCCAGT | AAGTTCATGGATGCCTCTGG |
| CCN5 | ATATTGAGGCTGCAGCAGGT | AAACTCCAGAAAAGGCAGCA |
| CCN6 | CAGATGCACCTCAGCGTAAA | ATCCACAGCCATCTCTCACC |
| 18S | GTCTGCCCTATCAACTTTCG | ATGTGGTAGCCGTTTCTCAG |
Bleomycin lung injury model and in vivo siRNA
Two-month-old female C57BL/6 mice (The Jackson Laboratory, Bar Harbor, ME, USA) were anesthetized with intraperitoneal injection of ketamine (100 mg/kg) and xylazine (10 mg/kg). Bleomycin (1.25 U/kg) or saline (uninjured control) was administered intratracheally to induce lung injury as described previously (20). Mice were euthanized by CO2 inhalation and lung tissues harvested for biochemical and histologic assays. All procedures involving animals were approved by the Institutional Animal Care and Use Committees at the University of Alabama at Birmingham.
Hydroxyproline assay
Lung tissues were dried in an oven at 70°C for 48 h, and dry lung weights were measured. Tissues were hydrolyzed using 6 N HCl at 95°C for 20 h. Hydroxyproline assay was performed according to manufacturer’s instructions (QuickZyme BioSciences, Leiden, The Netherlands) using hydroxyproline as a standard.
Lung histology and immunohistochemical staining
We processed paraffin-embedded tissue sections for lung histology and immunohistochemical staining as previously described (23).
Statistical analysis
Statistical analysis was performed using GraphPad software (Prism 5; GraphPad Software, La Jolla, CA, USA). Results are expressed as means ± sem. Student’s t tests and ANOVA statistical analysis were performed to evaluate difference between groups. Significance was set at P < 0.05.
RESULTS
CCN1 expression in IPF
IPF is a fibrosing disorder of the lungs with variable rates of disease progression (24). Previous studies from our group have shown that alveolar mesenchymal cells isolated from lungs of human subjects are tissue-resident (21), and their phenotype may serve to inform clinical outcomes following lung injury (25). We isolated alveolar mesenchymal cells from a cohort of patients with IPF with progressive vs. stable lung function, and assessed their gene expression profiles by Affymetrix analysis. Progressive and stable IPF were defined by declines in functional vital capacity ≥ 10% and < 5%, respectively, over the preceding 6 mo. We found that expression of CCN1 mRNA was significantly higher in progressive vs. stable IPF (n = 3 per group; P = 0.02) (Fig. 1A). Next, we determined whether whole lung expression of CCN1 mRNA was increased in IPF subjects. RNA was isolated from whole lung homogenates of explants of patients undergoing lung transplantation for IPF (n = 11) and from control subjects (failed donor lungs; n = 9). Despite heterogeneity among the IPF cohort, these patients demonstrated significantly higher levels of CCN1 mRNA compared with controls (P = 0.038; Fig. 1B). To determine the localization of CCN1 in human IPF, we performed immunofluorescence staining of formalin-fixed, paraffin-embedded lung tissues from control subjects and patients with IPF. CCN1 expression was markedly low in control lung as compared with IPF lung tissue section (Fig. 1C). In IPF lung, CCN1 was found to be highly expressed in areas of active fibrosis, specifically in α-smooth muscle actin (α-SMA)-expressing myofibroblasts and the overlying epithelium of fibroblastic foci (Fig. 1C). These data indicate that CCN1 is highly expressed in at least a subset of IPF subjects and suggest that this matricellular protein may play an active role in patients with an accelerated disease phenotype.
Figure 1.

CCN1 expression in IPF. A) Fold change in mRNA of CCN1 in alveolar mesenchymal cells from stable (n = 3) vs. progressive (n = 3) IPF. B) Relative mRNA expression of CCN1 in lung tissue from controls (n = 9) and IPF (n = 11) normalized to average expression of controls. C) Immunofluorescence staining of IPF lung tissue for CCN1 (green) and α-SMA expression (red; ×40 magnification). Two-tailed unpaired Student’s t test was performed between groups. *P < 0.05. Scale bars, 50 µm.
Expression of CCN1 in IPF fibroblasts correlates with profibrotic gene expression
Fibroblasts are key effectors of the fibrotic process and synthesize the vast majority of ECM proteins in fibrotic disorders (26). We compared the gene expression of CCN1 in fibroblasts isolated from IPF lung explants and controls (failed donors); similar to results of whole lung expression of CCN1 mRNA (Fig. 1B), we detected significantly higher levels of CCN1 in IPF lung fibroblasts (P = 0.04; Fig. 2A). Based on the observed heterogeneity in CCN1 expression in control and IPF lung fibroblasts, we determined whether the baseline expression of CCN1 correlates with constitutive expression of ECM genes and the myofibroblast marker, α-SMA. Linear logistic analysis revealed significant positive correlation between CCN1 mRNA with gene expression of collagens (Col)1a1 (Fig. 2B), Col1a2 (Fig. 2C), and fibronectin (FN; Fig. 2D), as well as with α-SMA (Fig. 2E). These data show that higher levels of CCN1 expression are associated with enhanced expression of profibrotic genes in lung fibroblasts, supporting a potential role for CCN1 in mediating the profibrotic phenotype of lung fibroblasts.
Figure 2.

Expression of CCN1 in IPF fibroblasts correlates with profibrotic gene expression. Control fibroblasts (n = 3) and IPF fibroblasts (n = 5) isolated from lung explants were serum starved overnight at 70–80% confluence. RNA was isolated and gene expression analyzed by RT-PCR. A) CCN1 mRNA expression relative to average of controls. Two-tailed unpaired Student’s t test was performed between groups. *P < 0.05. B–E) Linear regression analysis was done to compare the mRNA levels of CCN1 to mRNA levels of Col1a1, Col1a2, FN, and α-SMA. Correlation graphs with mRNA levels CCN1 vs. Col1a1 (B), Col1a2 (C), FN (D), and α-SMA (E). Values represents means of 2 technical duplicates relative to average control and are color-coded as controls in red and IPF in black. R2 is coefficient of determination.
Silencing of CCN1 down-regulates constitutive expression of profibrotic genes in IPF fibroblasts
Next, we determined whether endogenous CCN1 regulates the constitutive expression of profibrotic genes in IPF lung fibroblasts. We analyzed lung fibroblasts from 4 different patients with IPF and confirmed that RNAi-mediated silencing of CCN1 efficiently knocks down constitutive CCN1 protein expression (Fig. 3A, representative blots; Fig. 3B, densitometry analysis of n = 4, P = 0.0056). Importantly, CCN1 silencing resulted in significant decreases in expression of the profibrotic proteins, Col1a1, Col1a2, and α-SMA protein, in IPF fibroblasts (Fig. 3C; representative Western blots are shown in Fig. 3A). Expression of FN protein was more variable and significant changes were not detected across different IPF fibroblasts (Fig. 3A, C).
Figure 3.

Silencing of CCN1 down-regulates constitutive expression of profibrotic proteins in IPF fibroblasts. IPF fibroblasts were subjected to NT and CCN1 siRNA (final concentration = 100 nM) overnight, then medium was changed to 10% FBS medium for 24 h, and then medium was changed to serum free for 3 d. A) Representative Western blots of showing expression of CCN1, Col1a1, Col1a2, FN, α-SMA, and β-actin with CCN1 knockdown in 3 IPF cell lines. B, C) Densitometric analyses of CCN1 expression (B) and Col1a1, Col1a2, FN, and α-SMA expression (C) relative to β-action on 4 IPF fibroblasts. Data represent means ± sem (n = 4). Two-way ANOVA was performed between NT and CCN1 siRNA groups for densitometric analyses. *P < 0.05.
To determine whether changes in protein expression are related to modulation of gene expression, we assessed mRNA expression by real-time PCR of an array of profibrotic genes. CCN1 silencing resulted in significant decreases in gene expression of Col1a1, Col1a2, and Col3a1 (Fig. 4A–D). Although there was a trend toward a decrease in α-SMA gene expression with CCN1 knockdown, statistical significance was not reached, suggesting additional posttranslational mechanisms (Fig. 4E). Expression of FN mRNA did not change significantly with CCN1 knockdown (Fig. 4F). Taken together, these data demonstrate that endogenous CCN1 regulates the constitutive expression of Cols, at the level of mRNA, in IPF fibroblasts.
Figure 4.

Silencing of CCN1 down-regulates constitutive expression of profibrotic genes in IPF fibroblasts. IPF fibroblasts were subjected to NT and CCN1 siRNA (final concentration = 100 nM) for overnight, then medium was changed to 10% FBS medium for 24 h, and then medium was changed to serum free for 3 d. Graphs represent fold change in mRNA expression of CCN1 (A),Col1a1 (B), Col1a2 (C), Col3a1 (D), α-SMA (E), and FN (F) relative to NT siRNA-treated fibroblasts. n.s., not significant. Values represent means ± sem (n = 3). Two-tailed unpaired Student's t test was performed between groups. *P < 0.05.
TGF-β1 up-regulates CCN1 in lung fibroblasts
TGF-β1 is a key fibrogenic cytokine that is overexpressed in IPF and mediates myofibroblast differentiation of lung fibroblasts (27–29). We examined whether TGF-β1 regulates the expression of CCN family members in human lung fibroblasts. We found that TGF-β1 markedly induces CCN1 gene expression in IMR-90 fibroblasts when analyzed by transcriptomic Affymetrix analysis (7.6-fold induction; n = 3, P = 0.0003; Fig. 5A); there was a trend toward induction of CCN2 and CCN4, although not statistically significant in this data set (1.9-fold, P = 0.10 and 1.52-fold, P = 0.2, respectively). Other members of the CCN gene family were not significantly affected at the mRNA level. To validate the effects of TGF-β1 on the regulation of CCN1 gene expression, we examined the effects of TGF-β1 stimulation on CCN1 mRNA (by real-time PCR) at various times after treatment of IMR-90 fibroblasts. TGF-β1 induced a time-dependent increase in CCN1 gene expression, with maximal effect at 24 h (Fig. 5B). Furthermore, in primary adult lung fibroblasts, CCN1, among all CCN family members, was most significantly induced by TGF-β1 (Supplemental Fig. 1A). Similarly, we observed TGF-β1-induced expression of CCN1 protein (by Western immunoblotting) in primary adult lung fibroblasts (Fig. 5C). Together, these data indicate that TGF-β1 robustly induces CCN1 expression in lung fibroblasts in a time-dependent manner, both at the level of mRNA and protein.
Figure 5.

TGF-β1 up-regulates CCN1 in lung fibroblasts. A) Relative mRNA expression of genes of CCN family in IMR-90 fibroblasts treated with or without TGF-β1 (2 ng/ml), as analyzed by whole-genome Affymetrix microarray for 24 h. B) RT-PCR showing fold change in mRNA levels of CCN1 compared with control at indicated intervals in IMR-90 fibroblasts treated with TGF-β1 (2.5 ng/ml) after serum starved for 24 h. C) Western blot showing expression of CCN1 and Col1a1 in primary adult lung fibroblasts treated with TGF-β1 (2 ng/ml) for indicated periods. *P < 0.05.
TGF-β1 mediated CCN1 up-regulation is ALK5/SMAD3-dependent in lung fibroblasts
TGF-β1 signals via both the canonical SMAD2/3 pathway, as well as SMAD-independent activation of protein kinase pathways, including MAPKs (30, 31). TGF-β1 induces SMAD3 interactions with the CCN1 promoter to enhance transcriptional activity in breast cancer cells (32). To determine the role of SMAD-dependent and -independent pathways on TGF-β1-induced CCN1 expression in lung fibroblasts, we employed inhibitors of the type 1 TGF-β receptor (TβR1/ALK5; SB431542, 1 µM), ERK (PD98059, 10 µM), p38 MAPK (SB203580, 6 µM), JNK (SP600125, 100 nM). The doses of these inhibitors were chosen based on published data and their ability to block their respective kinase activities in lung fibroblasts (22, 33). Lung fibroblasts were serum starved overnight, and then treated with or without inhibitors 45 min before TGF-β1 (2.5 ng/ml) for 24 h. ALK5 inhibition blocked the up-regulation of CCN1 by TGF-β1, but the other kinase inhibitors had minimal effect (Fig 6A and Supplemental Fig. 2A), suggesting that the TβR1/ALK5 pathway is likely involved in CCN1 induction.
Figure 6.

TGF-β1 mediated CCN1 up-regulation is ALK5/SMAD3-dependent in lung fibroblasts. A) Primary adult lung fibroblasts were pretreated with or without chemical inhibitors type 1 TGF-β receptor (TβR1/ALK5; SB431542, 1 µM), ERK (PD98059, 10 µM), MAPK (SB203580, 6 µM), and cJNK (SP600125, 100 nM) for 45 min and then treated with or without TGF-β1 (2 ng/ml) for 24 h. B) Primary adult lung fibroblasts were transfected with NT and SMAD3 siRNA (100 nM) for 2 d, then serum starved for 24 h and treated with TGF-β1 (2 ng/ml) for 24 h. Expression of CCN1 and tSMAD3 was evaluated by immunoblotting.
Most of the profibrotic effects of canonical TGF-β1/ALK5 signaling have been attributed to SMAD3 (34). To determine whether SMAD3 mediates TGF-β1-induced CCN1 expression and to confirm the results of pharmacologic inhibition, we performed RNAi studies targeting of SMAD3. Lung fibroblasts were transfected with NT siRNA and SMAD3 siRNA followed by treatment with or without TGF-β1. SMAD3 silencing resulted in a failure of TGF-β1 to up-regulate CCN1 (Fig. 6B and Supplemental Fig. 2B), indicating that activation of SMAD3 is required for TGF-β1-mediated CCN1 up-regulation in lung fibroblasts.
CCN1 knockdown impairs TGF-β1-mediated activation of IPF fibroblasts
Based on our findings that CCN1 regulates constitutive profibrotic gene expression and that TGF-β1 induces CCN1, we examined whether CCN1 is required for TGF-β1-induced profibrotic activity. IPF fibroblasts were transfected with NT or CCN1 siRNA for 72 h, and treated with TGF-β1 for 24 h prior to assessment for protein and mRNA expression of profibrotic genes. CCN1 knockdown significantly attenuated TGF-β1-mediated induction of Col1a1, Col1a2, FN, and α-SMA as assessed by Western blotting (Fig. 7A and Supplemental Fig. 3A–E). Similarly, there was decreased induction of α-SMA and Col1a1 in myofibroblasts by immunofluorescence in CCN1-silenced cells (Fig. 7B). We next sought to determine whether CCN1 knockdown attenuates TGF-β1-mediated induction of profibrotic genes at the mRNA levels. TGF-β1-induced expression of CCN1, Col1a1, Col1a2, Col3a1, FN, and α-SMA were significantly reduced with CCN1 knockdown (Fig. 8A–F). These results suggest that, in addition to regulation of constitutive profibrotic gene expression, CCN1 is required for optimal induction of TGF-β1-regulated profibrotic gene activation in IPF lung fibroblasts.
Figure 7.

CCN1 knockdown impairs TGF-β1-induced expression of profibrotic proteins in IPF fibroblasts. IPF fibroblasts (n = 5) were subjected to NT and CCN1 siRNA (final concentration = 100 nM) for 2 d, then medium was changed to serum free for 24 h, and then fibroblasts were treated with TGF-β1 (2.5 ng/ml) for 24 h. A) Representative Western blot (n = 3) showing expression of CCN1, Col1a1, Col1a2, FN, and α-SMA. B) Immunofluorescence staining showing expression of α-SMA (magenta) and Col1a1 (red; ×20 magnification). Scale bars, 50 µm.
Figure 8.

CCN1 knockdown impairs TGF-β1-induced expression of profibrotic genes in IPF fibroblasts. IPF fibroblasts (n = 3) were subjected to NT and CCN1 siRNA (final concentration = 100 nM) for 2 d, then medium was changed to serum free for 24 h, and then fibroblasts were treated with TGF-β1 (2.5 ng/ml) for 24 h. Graphs represent fold change in mRNA expression of CCN1 (A), Col1a1 (B), Col1a2 (C), Col3a1 (D), FN (E), and α-SMA (F). Data represent means ± sem (n = 3) of 1 IPF cell line. Similar results were seen in 2 additional IPF cell lines. Two-way ANOVA was performed between groups. *P < 0.05.
CCN1 knockdown attenuates SMAD3 signaling
Activation of SMAD3 is a critical regulatory pathway mediating profibrotic actions of TGF-β1 (35). To determine whether impaired activation of this pathway may explain the observed attenuation of profibrotic gene induction in the context of CCN1 deficiency, we assessed TGF-β1-mediated SMAD3 phosphorylation/activation in CCN1-silenced cells. We found that SMAD3 phosphorylation induced by TGF-β1 is attenuated with CCN1 knockdown in IPF fibroblasts (n = 3) (Fig. 9A, B). Together, these data indicate that endogenous CCN1 is required for activation of SMAD3 signaling in IPF lung fibroblasts.
Figure 9.
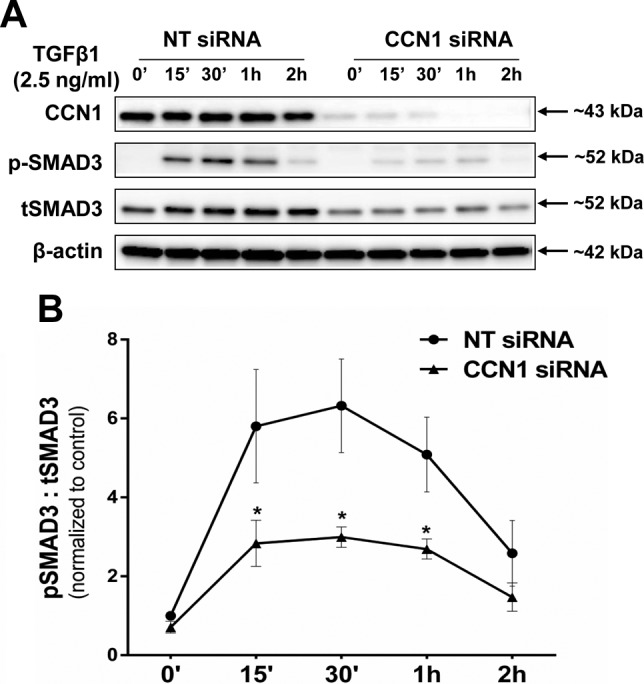
CCN1 knockdown inhibits SMAD3 phosphorylation in IPF fibroblasts. IPF fibroblasts (n = 3) were subjected to NT and CCN1 siRNA (final concentration = 100 nM) for 2 d, then medium was changed to serum free for 24 h, and then fibroblasts were treated with TGF-β1 (2.5 ng/ml) for indicated time periods. A) Representative Western blot of independent experiments on 3 different IPF fibroblasts showing expression of CCN1, phospho-SMAD3 (Ser423/425), and total SMAD3 (tSMAD3). B) Densitometric analyses of pSMAD3:tSMAD3 in IPF fibroblasts (n = 3). Two-way ANOVA was performed between groups. *P < 0.05.
In vivo RNAi-mediated silencing of CCN1 attenuates the fibrogenic response to bleomycin lung injury
We determined the in vivo relevance of CCN1 as a potential profibrotic mediator in a murine model of intratracheal bleomycin-induced lung injury and fibrosis, which is known to be TGF-β1/SMAD3 dependent (36, 37). We achieved in vivo knockdown of CCN1 in the lung utilizing intranasal delivery of CCN1 siRNA, as previously described by our group (20) and others (38). We first confirmed that the mouse CCN1 siRNA homologous to human CCN1 siRNA knocks down CCN1 expression in murine mesenchymal cells in vitro (Table 2 and Fig. 10A). CCN1 siRNA or NT siRNA was administered every other day by intranasal delivery to the lungs of bleomycin-treated mice, specifically during postinflammatory fibrogenic phase (d 8–20) for a total of 7 treatments. We confirmed the in vivo knockdown by analyzing expression of CCN1 protein in mouse lung tissue isolated from NT/CCN1 siRNA-treated mice postbleomycin injury (Fig. 10B, C). There was significantly lower expression of activated SMAD3 (pSMAD3) in lung tissue with CCN1 knockdown, and there was significant correlation between the extent of CCN1 knockdown and phospho-SMAD3 expression (Fig. 10B, D, E). CCN1 knockdown attenuated fibrogenic responses as determined by changes in dry lung weights (Fig. 10F), biochemical analyses of hydroxyproline content (Fig. 10G), histopathology with trichrome staining for Col, and immunofluorescence staining for α-SMA (Fig. 11A–C). These data support an in vivo profibrotic role for CCN1 in contributing to the fibrogenic response to lung injury in mice.
Figure 10.

In vivo RNAi-mediated silencing of CCN1 attenuates fibrogenic response to bleomycin injury. A) Mouse lung fibroblasts were transfected with NT and CCN1 siRNA (final concentration = 100 nM) for 2 d, then were serum free for 24 h, and then were treated with TGF-β1 (2.5 ng/ml) for 24 h. Graph shows the mRNA expression of CCN1 relative to NT control. Data represent means ± sem (n = 3). Two-way ANOVA analyses were performed between groups. B–G) C57BL/6 mice were subjected to acute lung injury by airway (intratracheal) administration of bleomycin or saline/control on d 0. NT siRNA or CCN1 siRNA (50 µg/mouse) was administered by intranasal delivery every other day for 2 wk, starting from d 8 to 20 postinjury. B–D) Whole lung harvested at the 3 wk following bleomycin injury and were analyzed for expression of CCN1 and pSMAD3 by Western blotting (B) and densitometric analyses (C, D). Unpaired Student’s t test was performed for densitometric analyses between 2 groups. E) Correlation graph of protein expression of CCN1 and pSMAD3 in lung tissue of NT and CCN1 siRNA-treated mice post–bleomycin injury. Linear regression analysis was done to compare the protein expression of CCN1 and pSMAD3 in lung tissue in NT/CCN1 siRNA-treated mice post–bleomycin injury (R2 = 0.86; P = 0.001; n = 4 in each group). Values represent densitometric analyses of CCN1 and pSMAD3 (normalized to β-actin; relative to NT average) and are color-coded as NT siRNA-treated in red and CCN1 siRNA-treated in black. F, G) Severity of fibrosis was assessed by measuring dry lung weights (F) and quantitative hydroxyproline assay (G). Data are expressed as increase in micrograms of hydroxyproline/whole lung. Values represent means ± sem; n = 4–6 per group. Two-way ANOVA was performed between groups. *P < 0.05.
Figure 11.

In vivo RNAi-mediated silencing of CCN1 attenuates fibrogenic response to bleomycin injury. Lung tissue sections from NT/CCN1 siRNA-treated mice (n = 2 each group) were used for immunohistochemistry, including hematoxylin and eosin staining (A), Masson’s trichrome staining (B), and immunofluorescence staining for α-SMA (magenta) (C). Scale bars, 50 µm.
DISCUSSION
The ECM is a dynamic microenvironment that undergoes continuous remodeling during the process of injury and repair (39). Matricellular proteins are emerging as essential regulators of inflammation and repair (4, 6). CCN1, a matricellular protein, is highly expressed in injury repair and mediates varied and divergent cellular responses in cell-type and context-dependent manner (40). Recent reports suggest that CCN1 mediates profibrotic responses in the kidney (13, 14), whereas it ameliorates skin (10) and liver fibrosis (11, 12). Studies of the role of CCN1 in the lung are limited, although overexpression of CCN1 mediates neutrophilic alveolitis and acute lung injury in mice (15). The role of endogenous CCN1 in the fibrogenic response to lung injury and potential roles in regulating the profibrotic phenotype of IPF lung fibroblasts have not been elucidated. Here, we show that: 1) CCN1 is expressed in areas of active fibrosis within the IPF lung, and whole lung and/or fibroblast-specific expression of CCN1 may be indicative of a progressive phenotype; 2) constitutive levels of CCN1 in IPF lung fibroblast correlates positively with and is, at least in part, essential for the basal expression of profibrotic genes; 3) the profibrotic mediator, TGF-β1, induces CCN1 via a ALK5/SMAD3-dependent mechanism; 4) CCN1 enhances TGF-β1-induced SMAD3 signaling and profibrotic gene expression; and 5) endogenous CCN1 is required for mediating fibrogenic responses to lung injury in mice (summarized in Fig. 12).
Figure 12.

Schematic of proposed model of CCN1 and TGF-β1 interactions in profibrotic signaling. Our studies indicate that endogenous CCN1 is required for constitutive and TGF-β1-induced SMAD3 activation and profibrogenic gene expression. TGF-β1 induces CCN1 expression via a SMAD3-dependent pathway, supporting a positive feedback loop. Abrogation of CCN1 during the active fibrogenic phase of lung injury repair protects from fibrosis in an in vivo model of lung fibrosis. The precise mechanism by which CCN1 cooperates with TGF-β receptors and/or integrins to activate SMAD3 requires further investigation.
The studies described here support a profibrotic role for CCN1 in tissue injury repair, in contrast to the elegant studies by Jun and Lau (10) and Kim et. al. (12), CCN1 as a critical factor in terminating the wound healing process by mediating fibroblast senescence. The development of a conceptual framework on how this matricellular protein functions in injury repair should take into account tissue-specific factors, influence of age, and the specific models utilized. Indeed, similar to our findings, CCN1 appears to mediate profibrotic effects in in vivo models of tissue injury involving the kidney and the liver (13, 14, 41). In the latter report, it was postulated that the paradoxical effect of CCN1 is mediated by differential expression/activation of CCN1-binding integrins, α6β1 and αvβ5/αvβ3 (41). We have not specifically studied the expression of these integrins in the lung or their relative roles in regulating profibrotic gene expression in IPF lung fibroblasts. However, our results clearly demonstrate that IPF lung fibroblasts are dependent on endogenous CCN1 for robust activation of a profibrotic gene expression profile. Most of the antifibrotic effects of CCN1 reported in the literature involve young animals and nonsenescent fibroblasts (10–12, 41). It would be interesting to test the effects of CCN1 abrogation/silencing in aging models of injury fibrosis that may not be as dependent on proliferation as much as senescence to promote fibrotic remodeling (23). Interestingly, we have found higher levels of CCN1 expression with replicative senescence of lung fibroblasts (data not shown), a finding similar to the constitutive phenotype of IPF lung fibroblasts. In our studies, these high CCN1-expressing fibroblasts are more responsive to CCN1 knockdown in down-regulating profibrotic genes, although similar effects were observed in nonsenescent and nonfibrotic control lung fibroblasts (data not shown). With respect to animal models of injury repair, most of the current models represent resolving fibrosis rather than progressive disease, analogous to that more commonly encountered in clinical contexts. It is possible that CCN1 may be beneficial in normal wound healing and tissue repair in contexts of resolving fibrosis but contribute to fibrosis in models of progressive fibrosis. Although the bleomycin model of lung fibrosis is known to be a slowly resolving model, we attempted to test the influence of targeting CCN1 by administering siRNA during the active fibrotic phase (d 8–20). Future studies should address the effects of CCN1 in preclinical animal models of progressive fibrosis.
The pleiotropic activities of matricellular proteins are dictated by their ability to interact with growth factors, their receptors, other ECM proteins, and integrins. TGF-β1 has been reported to up-regulate CCN1 gene in cancer cells and epithelial cells (32, 42). However, the regulation of TGF-β1/SMAD3 pathway by CCN1 has not been explored. A key finding in our studies is the essential role of CCN1 in mediating profibrotic gene expression via enhanced activation of the canonical TGF-β1/SMAD3 pathway. This suggests that endogenous CCN1 is, either directly or indirectly, activating the TβR1/ALK5 receptor; alternatively, it may cooperatively augment signaling via this pathway by its binding to integrin receptors. Integrins are known to regulate TGF-β1 signaling through trans-activation of TGF-β1 receptors and downstream signaling (43–45). CCN1 binds to integrin α αvβ3 and αvβ5, which are known to enhance TGF-β signaling by physical association with TGF-βRII (46–49). We speculate that CCN1 binding to these integrins may be necessary for enhanced TGF-β1/SMAD3 signaling. Similar to findings of our study with CCN1, there are reports of crosstalk of the TGF-β/SMAD pathway with other matricellular proteins, specifically secreted protein acidic and rich in cysteine (50), and connective tissue growth factor (connective tissue growth factor; CCN2) (51, 52).
IPF fibroblasts are known to maintain a profibrotic phenotype in ex vivo culture (53), and express constitutively higher nuclear SMAD3 (54, 55). There have been very few studies that have investigated the regulatory mechanisms of constitutively active IPF fibroblasts (53, 55–57). The observation that disease-associated fibroblasts from lungs of patients with IPF express higher levels of CCN1 and that this is mechanistically linked to heightened expression of profibrotic genes has potential therapeutic implications. Therapeutic strategies that target CCN1 may be more feasible and safer than directly targeting TGF-β in the context of adult fibrotic diseases; additionally, it affords the opportunity to develop a personalized/precision approach to treatment of fibrosis in patients with high levels of CCN1 expression/signaling.
Supplementary Material
Acknowledgments
This work was supported, in whole or in part, by U.S. National Institutes of Health Grants P01 HL114470 (National Heart, Lung, and Blood Institute), R01 AG046210 (National Institute on Aging), and U.S. Veterans Affairs Merit Grant I01BX003056. The authors declare no conflicts of interest.
Glossary
- α-SMA
α-smooth muscle actin
- Col
collagen
- ECM
extracellular matrix
- FBS
fetal bovine serum
- FN
fibronectin
- IMR
Institute of Medical Research
- IPF
idiopathic pulmonary fibrosis
- NT
nontargeting
- RNAi
RNA interference
- siRNA
small interfering RNA
- SMAD3
mothers against decapentaplegic homolog 3
- IMR-90
Institute of Medical Research-90
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
REFERENCES
- 1.Bornstein P., Sage E. H. (2002) Matricellular proteins: extracellular modulators of cell function. Curr. Opin. Cell Biol. 14, 608–616 [DOI] [PubMed] [Google Scholar]
- 2.Murphy-Ullrich J. E., Sage E. H. (2014) Revisiting the matricellular concept. Matrix Biol. 37, 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Murphy-Ullrich J. E. (2001) The de-adhesive activity of matricellular proteins: is intermediate cell adhesion an adaptive state? J. Clin. Invest. 107, 785–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kyriakides T. R., Bornstein P. (2003) Matricellular proteins as modulators of wound healing and the foreign body response. Thromb. Haemost. 90, 986–992 [DOI] [PubMed] [Google Scholar]
- 5.Kubota S., Takigawa M. (2007) CCN family proteins and angiogenesis: from embryo to adulthood. Angiogenesis 10, 1–11 [DOI] [PubMed] [Google Scholar]
- 6.Kular L., Pakradouni J., Kitabgi P., Laurent M., Martinerie C. (2011) The CCN family: a new class of inflammation modulators? Biochimie 93, 377–388 [DOI] [PubMed] [Google Scholar]
- 7.Babic A. M., Kireeva M. L., Kolesnikova T. V., Lau L. F. (1998) CYR61, a product of a growth factor-inducible immediate early gene, promotes angiogenesis and tumor growth. Proc. Natl. Acad. Sci. USA 95, 6355–6360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mo F. E., Muntean A. G., Chen C. C., Stolz D. B., Watkins S. C., Lau L. F. (2002) CYR61 (CCN1) is essential for placental development and vascular integrity. Mol. Cell. Biol. 22, 8709–8720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen C. C., Mo F. E., Lau L. F. (2001) The angiogenic factor Cyr61 activates a genetic program for wound healing in human skin fibroblasts. J. Biol. Chem. 276, 47329–47337 [DOI] [PubMed] [Google Scholar]
- 10.Jun J. I., Lau L. F. (2010) The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat. Cell Biol. 12, 676–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Borkham-Kamphorst E., Schaffrath C., Van de Leur E., Haas U., Tihaa L., Meurer S. K., Nevzorova Y. A., Liedtke C., Weiskirchen R. (2014) The anti-fibrotic effects of CCN1/CYR61 in primary portal myofibroblasts are mediated through induction of reactive oxygen species resulting in cellular senescence, apoptosis and attenuated TGF-β signaling. Biochim. Biophys. Acta 1843, 902–914 [DOI] [PubMed] [Google Scholar]
- 12.Kim K. H., Chen C. C., Monzon R. I., Lau L. F. (2013) Matricellular protein CCN1 promotes regression of liver fibrosis through induction of cellular senescence in hepatic myofibroblasts. Mol. Cell. Biol. 33, 2078–2090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lai C. F., Lin S. L., Chiang W. C., Chen Y. M., Wu V. C., Young G. H., Ko W. J., Kuo M. L., Tsai T. J., Wu K. D. (2014) Blockade of cysteine-rich protein 61 attenuates renal inflammation and fibrosis after ischemic kidney injury. Am. J. Physiol. Renal Physiol. 307, F581–F592 [DOI] [PubMed] [Google Scholar]
- 14.Lai C. F., Chen Y. M., Chiang W. C., Lin S. L., Kuo M. L., Tsai T. J. (2013) Cysteine-rich protein 61 plays a proinflammatory role in obstructive kidney fibrosis. PLoS One 8, e56481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grazioli S., Gil S., An D., Kajikawa O., Farnand A. W., Hanson J. F., Birkland T., Chen P., Duffield J., Schnapp L. M., Altemeier W. A., Matute-Bello G. (2015) CYR61 (CCN1) overexpression induces lung injury in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 308, L759–L765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Raissadati A., Nykänen A. I., Tuuminen R., Syrjälä S. O., Krebs R., Arnaudova R., Rouvinen E., Wang X., Poller W., Lemström K. B. (2015) Systemic overexpression of matricellular protein CCN1 exacerbates obliterative bronchiolitis in mouse tracheal allografts. Transpl. Int. 28, 1416–1425 [DOI] [PubMed] [Google Scholar]
- 17.Matute-Bello G., Wurfel M. M., Lee J. S., Park D. R., Frevert C. W., Madtes D. K., Shapiro S. D., Martin T. R. (2007) Essential role of MMP-12 in Fas-induced lung fibrosis. Am. J. Respir. Cell Mol. Biol. 37, 210–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Königshoff M., Kramer M., Balsara N., Wilhelm J., Amarie O. V., Jahn A., Rose F., Fink L., Seeger W., Schaefer L., Günther A., Eickelberg O. (2009) WNT1-inducible signaling protein-1 mediates pulmonary fibrosis in mice and is upregulated in humans with idiopathic pulmonary fibrosis. J. Clin. Invest. 119, 772–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thannickal V. J., Zhou Y., Gaggar A., Duncan S. R. (2014) Fibrosis: ultimate and proximate causes. J. Clin. Invest. 124, 4673–4677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hecker L., Vittal R., Jones T., Jagirdar R., Luckhardt T. R., Horowitz J. C., Pennathur S., Martinez F. J., Thannickal V. J. (2009) NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat. Med. 15, 1077–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lama V. N., Smith L., Badri L., Flint A., Andrei A. C., Murray S., Wang Z., Liao H., Toews G. B., Krebsbach P. H., Peters-Golden M., Pinsky D. J., Martinez F. J., Thannickal V. J. (2007) Evidence for tissue-resident mesenchymal stem cells in human adult lung from studies of transplanted allografts. J. Clin. Invest. 117, 989–996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Horowitz J. C., Lee D. Y., Waghray M., Keshamouni V. G., Thomas P. E., Zhang H., Cui Z., Thannickal V. J. (2004) Activation of the pro-survival phosphatidylinositol 3-kinase/AKT pathway by transforming growth factor-beta1 in mesenchymal cells is mediated by p38 MAPK-dependent induction of an autocrine growth factor. J. Biol. Chem. 279, 1359–1367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hecker L., Logsdon N. J., Kurundkar D., Kurundkar A., Bernard K., Hock T., Meldrum E., Sanders Y. Y., Thannickal V. J. (2014) Reversal of persistent fibrosis in aging by targeting Nox4-Nrf2 redox imbalance. Sci. Transl. Med. 6, 231ra47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Raghu G., Collard H. R., Egan J. J., Martinez F. J., Behr J., Brown K. K., Colby T. V., Cordier J. F., Flaherty K. R., Lasky J. A., Lynch D. A., Ryu J. H., Swigris J. J., Wells A. U., Ancochea J., Bouros D., Carvalho C., Costabel U., Ebina M., Hansell D. M., Johkoh T., Kim D. S., King T. E. Jr., Kondoh Y., Myers J., Müller N. L., Nicholson A. G., Richeldi L., Selman M., Dudden R. F., Griss B. S., Protzko S. L., Schünemann H. J.; ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis (2011) An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 183, 788–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Horowitz J. C., Cui Z., Moore T. A., Meier T. R., Reddy R. C., Toews G. B., Standiford T. J., Thannickal V. J. (2006) Constitutive activation of prosurvival signaling in alveolar mesenchymal cells isolated from patients with nonresolving acute respiratory distress syndrome. Am. J. Physiol. Lung Cell. Mol. Physiol. 290, L415–L425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hinz B., Phan S. H., Thannickal V. J., Prunotto M., Desmoulière A., Varga J., De Wever O., Mareel M., Gabbiani G. (2012) Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am. J. Pathol. 180, 1340–1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baughman R. P., Lower E. E., Miller M. A., Bejarano P. A., Heffelfinger S. C. (1999) Overexpression of transforming growth factor-alpha and epidermal growth factor-receptor in idiopathic pulmonary fibrosis. Sarcoidosis Vasc. Diffuse Lung Dis. 16, 57–61 [PubMed] [Google Scholar]
- 28.Evans R. A., Tian Y. C., Steadman R., Phillips A. O. (2003) TGF-beta1-mediated fibroblast-myofibroblast terminal differentiation-the role of Smad proteins. Exp. Cell Res. 282, 90–100 [DOI] [PubMed] [Google Scholar]
- 29.Thannickal V. J., Lee D. Y., White E. S., Cui Z., Larios J. M., Chacon R., Horowitz J. C., Day R. M., Thomas P. E. (2003) Myofibroblast differentiation by transforming growth factor-beta1 is dependent on cell adhesion and integrin signaling via focal adhesion kinase. J. Biol. Chem. 278, 12384–12389 [DOI] [PubMed] [Google Scholar]
- 30.Heldin C. H., Miyazono K., ten Dijke P. (1997) TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature 390, 465–471 [DOI] [PubMed] [Google Scholar]
- 31.Yamaguchi K., Shirakabe K., Shibuya H., Irie K., Oishi I., Ueno N., Taniguchi T., Nishida E., Matsumoto K. (1995) Identification of a member of the MAPKKK family as a potential mediator of TGF-beta signal transduction. Science 270, 2008–2011 [DOI] [PubMed] [Google Scholar]
- 32.Bartholin L., Wessner L. L., Chirgwin J. M., Guise T. A. (2007) The human Cyr61 gene is a transcriptional target of transforming growth factor beta in cancer cells. Cancer Lett. 246, 230–236 [DOI] [PubMed] [Google Scholar]
- 33.Horowitz J. C., Rogers D. S., Sharma V., Vittal R., White E. S., Cui Z., Thannickal V. J. (2007) Combinatorial activation of FAK and AKT by transforming growth factor-beta1 confers an anoikis-resistant phenotype to myofibroblasts. Cell. Signal. 19, 761–771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roberts A. B., Russo A., Felici A., Flanders K. C. (2003) Smad3: a key player in pathogenetic mechanisms dependent on TGF-beta. Ann. N. Y. Acad. Sci. 995, 1–10 [DOI] [PubMed] [Google Scholar]
- 35.Zhang Y., Feng X. H., Derynck R. (1998) Smad3 and Smad4 cooperate with c-Jun/c-Fos to mediate TGF-beta-induced transcription. Nature 394, 909–913 [DOI] [PubMed] [Google Scholar]
- 36.Zhao J., Shi W., Wang Y. L., Chen H., Bringas P. Jr., Datto M. B., Frederick J. P., Wang X. F., Warburton D. (2002) Smad3 deficiency attenuates bleomycin-induced pulmonary fibrosis in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 282, L585–L593 [DOI] [PubMed] [Google Scholar]
- 37.Higashiyama H., Yoshimoto D., Kaise T., Matsubara S., Fujiwara M., Kikkawa H., Asano S., Kinoshita M. (2007) Inhibition of activin receptor-like kinase 5 attenuates bleomycin-induced pulmonary fibrosis. Exp. Mol. Pathol. 83, 39–46 [DOI] [PubMed] [Google Scholar]
- 38.Bitko V., Musiyenko A., Shulyayeva O., Barik S. (2005) Inhibition of respiratory viruses by nasally administered siRNA. Nat. Med. 11, 50–55 [DOI] [PubMed] [Google Scholar]
- 39.Midwood K. S., Williams L. V., Schwarzbauer J. E. (2004) Tissue repair and the dynamics of the extracellular matrix. Int. J. Biochem. Cell Biol. 36, 1031–1037 [DOI] [PubMed] [Google Scholar]
- 40.Lau L. F. (2011) CCN1/CYR61: the very model of a modern matricellular protein. Cell. Mol. Life Sci. 68, 3149–3163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim K. H., Chen C. C., Alpini G., Lau L. F. (2015) CCN1 induces hepatic ductular reaction through integrin αvβ₅-mediated activation of NF-κB. J. Clin. Invest. 125, 1886–1900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sakamoto S., Yokoyama M., Aoki M., Suzuki K., Kakehi Y., Saito Y. (2004) Induction and function of CYR61 (CCN1) in prostatic stromal and epithelial cells: CYR61 is required for prostatic cell proliferation. Prostate 61, 305–317 [DOI] [PubMed] [Google Scholar]
- 43.Bhowmick N. A., Zent R., Ghiassi M., McDonnell M., Moses H. L. (2001) Integrin beta 1 signaling is necessary for transforming growth factor-beta activation of p38MAPK and epithelial plasticity. J. Biol. Chem. 276, 46707–46713 [DOI] [PubMed] [Google Scholar]
- 44.Chen X., Wang H., Liao H. J., Hu W., Gewin L., Mernaugh G., Zhang S., Zhang Z. Y., Vega-Montoto L., Vanacore R. M., Fässler R., Zent R., Pozzi A. (2014) Integrin-mediated type II TGF-β receptor tyrosine dephosphorylation controls SMAD-dependent profibrotic signaling. J. Clin. Invest. 124, 3295–3310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Margadant C., Sonnenberg A. (2010) Integrin-TGF-beta crosstalk in fibrosis, cancer and wound healing. EMBO Rep. 11, 97–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Scaffidi A. K., Petrovic N., Moodley Y. P., Fogel-Petrovic M., Kroeger K. M., Seeber R. M., Eidne K. A., Thompson P. J., Knight D. A. (2004) alpha(v)beta(3) Integrin interacts with the transforming growth factor beta (TGFbeta) type II receptor to potentiate the proliferative effects of TGFbeta1 in living human lung fibroblasts. J. Biol. Chem. 279, 37726–37733 [DOI] [PubMed] [Google Scholar]
- 47.Kireeva M. L., Lam S. C., Lau L. F. (1998) Adhesion of human umbilical vein endothelial cells to the immediate-early gene product Cyr61 is mediated through integrin alphavbeta3. J. Biol. Chem. 273, 3090–3096 [DOI] [PubMed] [Google Scholar]
- 48.Monnier Y., Farmer P., Bieler G., Imaizumi N., Sengstag T., Alghisi G. C., Stehle J. C., Ciarloni L., Andrejevic-Blant S., Moeckli R., Mirimanoff R. O., Goodman S. L., Delorenzi M., Rüegg C. (2008) CYR61 and alphaVbeta5 integrin cooperate to promote invasion and metastasis of tumors growing in preirradiated stroma. Cancer Res. 68, 7323–7331 [DOI] [PubMed] [Google Scholar]
- 49.Asano Y., Ihn H., Yamane K., Jinnin M., Tamaki K. (2006) Increased expression of integrin alphavbeta5 induces the myofibroblastic differentiation of dermal fibroblasts. Am. J. Pathol. 168, 499–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Francki A., McClure T. D., Brekken R. A., Motamed K., Murri C., Wang T., Sage E. H. (2004) SPARC regulates TGF-beta1-dependent signaling in primary glomerular mesangial cells. J. Cell. Biochem. 91, 915–925 [DOI] [PubMed] [Google Scholar]
- 51.Wahab N. A., Weston B. S., Mason R. M. (2005) Modulation of the TGFbeta/Smad signaling pathway in mesangial cells by CTGF/CCN2. Exp. Cell Res. 307, 305–314 [DOI] [PubMed] [Google Scholar]
- 52.Nakerakanti S. S., Bujor A. M., Trojanowska M. (2011) CCN2 is required for the TGF-β induced activation of Smad1-Erk1/2 signaling network. PLoS One 6, e21911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bocchino M., Agnese S., Fagone E., Svegliati S., Grieco D., Vancheri C., Gabrielli A., Sanduzzi A., Avvedimento E. V. (2010) Reactive oxygen species are required for maintenance and differentiation of primary lung fibroblasts in idiopathic pulmonary fibrosis. PLoS One 5, e14003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rahaman S. O., Grove L. M., Paruchuri S., Southern B. D., Abraham S., Niese K. A., Scheraga R. G., Ghosh S., Thodeti C. K., Zhang D. X., Moran M. M., Schilling W. P., Tschumperlin D. J., Olman M. A. (2014) TRPV4 mediates myofibroblast differentiation and pulmonary fibrosis in mice. J. Clin. Invest. 124, 5225–5238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Roach K. M., Wulff H., Feghali-Bostwick C., Amrani Y., Bradding P. (2014) Increased constitutive αSMA and Smad2/3 expression in idiopathic pulmonary fibrosis myofibroblasts is KCa3.1-dependent. Respir. Res. 15, 155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Uhal B. D., Kim J. K., Li X., Molina-Molina M. (2007) Angiotensin-TGF-beta 1 crosstalk in human idiopathic pulmonary fibrosis: autocrine mechanisms in myofibroblasts and macrophages. Curr. Pharm. Des. 13, 1247–1256 [DOI] [PubMed] [Google Scholar]
- 57.Lundvig D. M., Pennings S. W., Brouwer K. M., Mtaya-Mlangwa M., Mugonzibwa E. A., Kuijpers-Jagtman A. M., Von den Hoff J. W., Wagener F. A. (2015) Curcumin induces differential expression of cytoprotective enzymes but similar apoptotic responses in fibroblasts and myofibroblasts. Exp. Cell Res. 330, 429–441 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.