

Abstract
Rett syndrome (RTT) is a neurodevelopmental disorder caused by mutations in Methyl-CpG-binding protein 2 (MECP2). Severe breathing abnormalities are common in RTT and are reproduced in mouse models of RTT. Previously, we found that removing MeCP2 from the brainstem and spinal cord in mice caused early lethality and abnormal breathing. To determine whether loss of MeCP2 in functional components of the respiratory network causes specific breathing disorders, we used the Cre/LoxP system to differentially manipulate MeCP2 expression throughout the brainstem respiratory network, specifically within HoxA4-derived tissues, which include breathing control circuitry within the nucleus tractus solitarius and the caudal part of ventral respiratory column but do not include more rostral parts of the breathing control circuitry. To determine whether respiratory phenotypes manifested in animals with MeCP2 removed from specific pons medullary respiratory circuits, we performed whole-body plethysmography and electrophysiological recordings from in vitro brainstem slices from mice lacking MeCP2 in different circuits. Our results indicate that MeCP2 expression in the medullary respiratory network is sufficient for normal respiratory rhythm and preventing apnea. However, MeCP2 expression within components of the breathing circuitry rostral to the HoxA4 domain are neither sufficient to prevent the hyperventilation nor abnormal hypoxic ventilatory response. Surprisingly, we found that MeCP2 expression in the HoxA4 domain alone is critical for survival. Our study reveals that MeCP2 is differentially required in select respiratory components for different aspects of respiratory functions, and collectively for the integrity of this network functions to maintain proper respiration.
SIGNIFICANCE STATEMENT Breathing abnormalities are a significant clinical feature in Rett syndrome and are robustly reproduced in the mouse models of this disease. Previous work has established that alterations in the function of MeCP2, the protein encoded by the gene mutated in Rett syndrome, within the hindbrain are critical for control of normal breathing. Here we show that MeCP2 function plays distinct roles in specific brainstem regions in the genesis of various aspects of abnormal breathing. This provides insight into the pathogenesis of these breathing abnormalities in Rett syndrome, which could be used to target treatments to improve these symptoms. Furthermore, it provides further knowledge about the fundamental neural circuits that control breathing.
Keywords: breathing, HoxA4, HoxB1, MECP2, Rett
Introduction
Rett syndrome (RTT) is a neurodevelopmental disorder affecting ∼1:10,000 live female births (Laurvick et al., 2006). Mutations in the X-linked transcription regulator methyl-CpG-binding protein 2 (MECP2) cause 90%–95% of all typical cases of RTT. Girls and women with RTT and disease-causing mutations in MECP2 have normal development until 6–18 months of age and then display developmental regression that includes the loss of speech and hand skills (Neul et al., 2010). Subsequently, affected individuals develop a variety of autonomic problems, including severe breathing irregularities (Kerr et al., 1997; Julu et al., 2001; Weese-Mayer et al., 2006; Rohdin et al., 2007). In contrast, boys with RTT-causing mutations in MECP2 have a dramatically more severe clinical phenotype, congenital encephalopathy, and often die in the first years of life due to severe cardiorespiratory dysfunction (Kankirawatana et al., 2006, Schule et al., 2008).
Mice lacking MeCP2 function reproduce many of the clinical features of RTT and are thus useful to understand the pathophysiological mechanisms that underlie these symptoms (Chen et al., 2001; Guy et al., 2001; Viemari et al., 2005; Ogier et al., 2007; Voituron et al., 2009, 2010a, 2010b; Ward et al., 2011; Bissonnette et al., 2014). In RTT mouse models, cellular and network-level abnormalities are found in the respiratory network (Viemari et al., 2005; Stettner et al., 2007; Medrihan et al., 2008), but the origins of breathing dysfunctions remain unclear. Brainstem respiratory networks are composed of functionally interacting components extending from the pons to the medulla (Cohen, 1979; Bianchi et al., 1995; Feldman and Smith, 1995; Richter, 1996). Smith et al. (2007). Previous work revealed that pontine respiratory group (PRG), Bötzinger complex (BötC), and pre-Bötzinger complex (preBötC) play critical roles in central respiratory rhythm generation. Other components of the network refine this rhythmic activity in response to homeostatic drives and environmental stimuli (Mulkey et al., 2004; Depuy et al., 2011; Smith et al., 2013).
Our previous work showed that restoring MeCP2 function from the caudal pons through the spinal cord, while excluding the PRG, rescued hypoxic ventilation response and life span, but not proper basal breathing rate (Ward et al., 2011). This finding established the feasibility of using anatomically restricted conditional genetics to dissect the role of MeCP2 within components of the respiratory network. To further dissect the role of MeCP2 in different components of the respiratory control network, we generated a new HoxA4–Cre line (Huang et al., 2012) that drives Cre expression in the caudal medulla, which includes important components of the breathing circuitry, such as the nucleus tractus solitarius (NTS) and the caudal portion of the ventral respiratory group. Using reciprocal conditional knock-out and conditional rescue approaches, we sought to examine the consequences of loss or restoration of MeCP2 function within the various components of the respiratory network, notably the known rhythm-generating components. Here we determine that specific breathing phenotypes, such as apnea and irregular breathing, are related to loss of MeCP2 function within these components, whereas other phenotypes, such as abnormal baseline breathing rate and hypoxic ventilation response (HVD), are caused by loss of MeCP2 in other distinct components of the respiratory network. Our results demonstrate that MeCP2 function is broadly required throughout the distributed breathing circuitry and that this function is necessary in specific components of the distributed circuit for precise control of different aspects of breathing. In addition, adaptation and progressive loss of function among the respiratory network cause age-dependent changes of breathing patterns in Mecp2-deficient animals.
Materials and Methods
Animals used in experiments.
All research and animal care procedures were approved by the Baylor College of Medicine Institutional Animal Care and Use Committee and housed in the Association for Assessment and Accreditation of Laboratory Animal Care-approved animal facility at Baylor College of Medicine. Mecp2Tm2Bird mice (Guy et al., 2007) were obtained from The Jackson Laboratory. Mecp2TM1Jae animals (Chen et al., 2001) were acquired from the University of California–Davis Mouse Mutant Resource Center. HoxB1-cre mice (Arenkiel et al., 2003) were crossed to Mecp2Tm2Bird mice to generate male experimental animals. HoxA4-cre transgenic mice were crossed to Mecp2Tm2Bird or Mecp2TM1Jae mice to generate male experimental animals. All strains were maintained on C57BL6/J background.
Generation of the HoxA4-Cre transgenic mice.
A plasmid containing the HoxA4 minimal promoter (Behringer et al., 1993) was obtained from Dr. Richard Behringer as a gift. The generation of the final construct is described by Huang et al., (2012). Briefly, the fragments were cloned into the pDONR223 vector (Invitrogen) to yield a transgene consisting of the HoxA4 enhancer/promoter, the Cre coding sequence fused in-frame with the HoxA4 promoter sequence at exon 2, and a polyadenylation signal. The transgene was excised with KpnI and used in a pronuclear injection to generate FVB transgenic mice according to standard procedures. Twelve pups were generated, and four of them carried the HoxA4-Cre transgene. The expression pattern of HoxA4-Cre transgene was determined by crossing founder animals to ROSA26-lacZ reporter animals. One stable transgenic line was mated to C57BL6/J wild-type (WT) mice for >10 generations to generate C57BL6/J congenic animals before using them to generate experimental animals.
Unrestrained whole-body plethysmography.
Whole-body plethysmography was recorded as described previously (Ward et al., 2011; Pitcher et al., 2013, 2015; Herrera et al., 2015). Briefly, male mice were placed within unrestrained whole-body plethysmography chambers (Buxco, DSI) of ∼500 ml in volume with a continuous flow rate of 0.5 L/min flushing the chambers with fresh air. Mice were acclimated in chambers 1 h a day for 2 d before the experiment. During recording, mice were allowed to acclimate for 50 min, and baseline breathing was then recorded for 60 min. To determine responses to hypoxic gas (10% O2, balance with N2), the chamber was flushed with hypoxic gas for 15 min. Apnea during hypoxic challenge is sampled from the first 5 min. Breathing rate, inspiratory time, expiratory time, and apnea were determined using a customized algorithm written in MATLAB (The MathWorks) using the plethysmography signal files captured with Ponemah3 (DSI) software. To reduce the artifacts from excessive movement and sniffing behavior, breaths that exhibited an inspiratory time <0.025 s, an expiratory time >10 s, or exhaled tidal volumes >150% or <50% of calculated inhaled tidal volume were excluded; apnea was defined as breathing interval >0.5 s, longer than 2× segment average interval, and longer than 2× local average; breaths were then filtered with intervals excluded if they possessed >10% of their breaths >500 breaths per minute (sliding 200 breath windows centered on the current breath). Plethysmography data were collected from mice at the indicated ages. Breathing parameters for each animal during baseline and hypoxic challenge were determined as the average instantaneous value over the recorded interval and then averaged across trials. Respiratory parameters were then compared across genotypes.
Immunohistology.
The methods of perfusion and tissue collection were performed as described previously (Ward et al., 2011). Briefly, mice were deeply anesthetized by intraperitoneal injection with Avertin at a dose of 0.04 ml/g and then fixed by transcardiac perfusion with PBS followed by 4% PFA in PBS. Tissues for histological analysis were harvested immediately after perfusion. The tissues were then fixed 30 min in 4% PFA in PBS and cryopreserved by overnight incubations in increasing concentrations of sucrose (up to 30% sucrose). Tissues were embedded in OCT compound (Sakura) and sectioned. For brain samples, 50 μm sections were collected with a cryostat and stained as floating sections. For DRG, carotid body, and superior cervical ganglion samples, 25 μm sections were collected with a cryostat and stained as mounted sections on polylysine-coated slides. Sections were blocked for 1 h in a PBS solution containing 10% serum (matched to the host used for the secondary antibodies) and 0.3% Triton X-100. Primary antibody incubation was performed in the blocking solution overnight at 4°C for floating sections, and at room temperature in a humidity chamber for slide-mounted sections. Secondary antibody incubation was performed in the blocking solution with 0.1% Triton X-100 for floating sections and in a humidity chamber for slide-mounted sections at room temperature for 6 h. Sections were washed between incubations with PBS containing 0.1% Triton X-100. DAPI was included in the penultimate wash. Primary antibodies were used at the following dilutions: chicken anti-MeCP2 1:1000 (Millipore), chicken anti-MeCP2 1:1000 (Prosci), mouse anti-tyrosine hydroxylase 1:1000 (Sigma), chicken anti-GFP 1:1000 (Abcam), mouse anti-NeuN 1:1000 (Millipore), and rabbit anti-neurokinin 1 receptor (Nk1r) 1:500 (Sigma). Secondary antibodies conjugated to DyLight 488, 549, or 649 were used at a dilution of 1:500 and raised in goat or donkey (Jackson ImmunoResearch Laboratories). Sections were mounted with Prolong mounting medium (Invitrogen) and imaged via epifluorescent microscopy (Zeiss M1 with ApoTome2 and ZEN2 software). NTS and ventral respiratory column (VRC) were identified by using Nk1r expression as described previously (Ward et al., 2011).
preBötC electrophysiology.
The preparation of respiratory brainstem slices was performed as described by Ramirez et al. (1996). Briefly, male mice (P12-P14) were deeply anesthetized with isoflurane and decapitated at the C3-C4 spinal level. The brain and upper cervical spinal cord were isolated in ice-cold ACSF bubbled with Carbogen (95% O2/5% CO2). After transecting transversely of the neuroaxis at the level of the inferior colliculus, the cerebellum was removed to isolate the brainstem. The brainstem was glued vertically with its rostral end up and its dorsal side attached to an agar block. Serial slices were made from rostral to caudal using a Leica VT1200 vibratome until the anatomic landmarks associated with the rostral boundary of preBötC were observed. The rhythmic area brain slice was routinely contained within the next 550–600 μm of tissue. This slice was immediately transferred into the recording chamber and submerged under a stream of ACSF (temperature, 29°C; flow rate, 5 ml/min). Spontaneous extracellular population activity was recorded using an ACSF-filled glass electrode (300–500 kOhm tip resistance) that was positioned on the slice surface over VRC area containing the preBötC. Data were analyzed using Spike2 bursts script (CED) and Sigma Plot (Systat Software).
Pulse oximetry.
Heart rate data were collected by pulse oximetry with the Mouse Ox (Starr Life Sciences). Measurements were collected from the tail of male mice. Animals were placed within a restraint tube allowing access to the tail. After the signal stabilized, the mice were recorded for 5 min, and their heart rate defined as the average over the valid recording. Heart rate data of HoxA4 RESCUE and conditional knock-out (CKO) animals were collected once per week at the indicated ages.
Determination of the rescue efficiency.
Efficiency of HoxA4-Cre induced expression of MeCP2 in the brainstem was quantified by stereological assessment of immunofluorescent histology. We collected brainstem samples from either WT or HoxA4-Cre RESCUE animals (N = 3 each) and sectioned them by cryostat at a thickness of 50 μm and arranged in rostral to caudal sequence. After immunostaining, images were taken by Zeiss M1 epifluorescent microscopy with ApoTome2 and ZEN2 software as described previously. We defined regions within NTS and VRC based on morphology compared with reference images in Paxinos and Franklin (2008); qualitative assessment of costaining with Nk1r and NeuN was also used to help define the anatomic regions contained within the sections. The coordinates of the analogous regions from Paxinos and Franklin (2008) were used when presenting results. For quantification, a slide sampling frequency of 1/3 and an area sampling fraction of 1/10 were used. Cells were sampled in counting frames 77.49 μm on a side (6005 μm2), spaced using grid steps 245.05 μm in each direction (60,050 μm2). Counting was performed using ImageJ software with the cell counter plugin (National Institutes of Health). We quantified the relative efficiency of MeCP2 expression as the fraction of sampled NeuN-positive cells that were also positive for MeCP2. The HoxA4-Cre RESCUE results were normalized to those obtained from WT.
Statistics.
All statistical analyses were performed using IBM SPSS Statistics version 20 on a PC. Parametric statistics were performed using ANOVA with genotype as a factor. For conditions in which the genotype factor had more than two levels, formalized post hoc testing was performed using Tukey's HSD correction to detect pairwise differences, with the exception of age-dependent baseline breathing rates using LSD correction. Survival analysis was performed using Kaplan–Meier survival analysis, with application of the Tarone–Ware method to detect differences in survival between genotype groups. For analysis of brainstem electrophysiological recordings, intervals between spontaneous rhythmic bursts were measured over a 20 to 65 min period. Data were then summarized as an average interburst interval, irregularity score (Garcia et al., 2011), and summarized in a cumulative probability plot. Differences between genotype groups were analyzed using ANOVA with a Holm–Sidak multiple-comparison procedure and two-sample Kolmogorov–Smirnov test.
Results
MeCP2 expression is restored in caudal medulla, spinal cord, and part of the peripheral nervous system by HoxA4-Cre
Our previous work showed that MeCP2 expression within HoxB1-derived tissue (brainstem and spinal cord) is important for various cardiorespiratory functions and survival (Ward et al., 2011). In regards to specific breathing phenotypes, basal hyperventilation appears to be dependent on MeCP2 function in the regions rostral to the HoxB1-defined tissues, whereas the abnormal response to hypoxic challenge is dependent on MeCP2 function within HoxB1-defined tissues. To further dissect the requirement of MeCP2 in the respiratory network, we generated a transgenic animal that expresses the Cre recombinase driven by HoxA4 promoter (Huang et al., 2012). This Cre line allows manipulation of MeCP2 expression only in the caudal part of the HoxB1 domain. Based on the expression pattern of the HoxA4-LacZ that was used as the template for the construction for HoxA4-Cre (Behringer et al., 1993), we expected to identify HoxA4-Cre expression in the region caudal to the base of fourth ventricle. Using an engineered loxP-flanked STOP allele of Mecp2, Mecp2tm2Bird (Guy et al., 2007), we generated mice with MeCP2 specifically expressed in the HoxA4 domain (HoxA4 RESCUE animals) and littermate control animals and found that MeCP2 expression was in the caudal hindbrain as expected (Fig. 1). To identify key components of the breathing control circuitry, specifically the VRC, we used Nk1r expression as a molecular landmark (Alheid and McCrimmon, 2008) (Fig. 1A–C). Additionally, because norepinephrine and epinephrine neurons in the hindbrain are important for various aspects of physiological control, we identified the A1/C1 and A2/C2 cell groups using an antibody against tyrosine hydroxylase, which also identifies the O2-sensing cells within the carotid body (Fig. 1).
Figure 1.
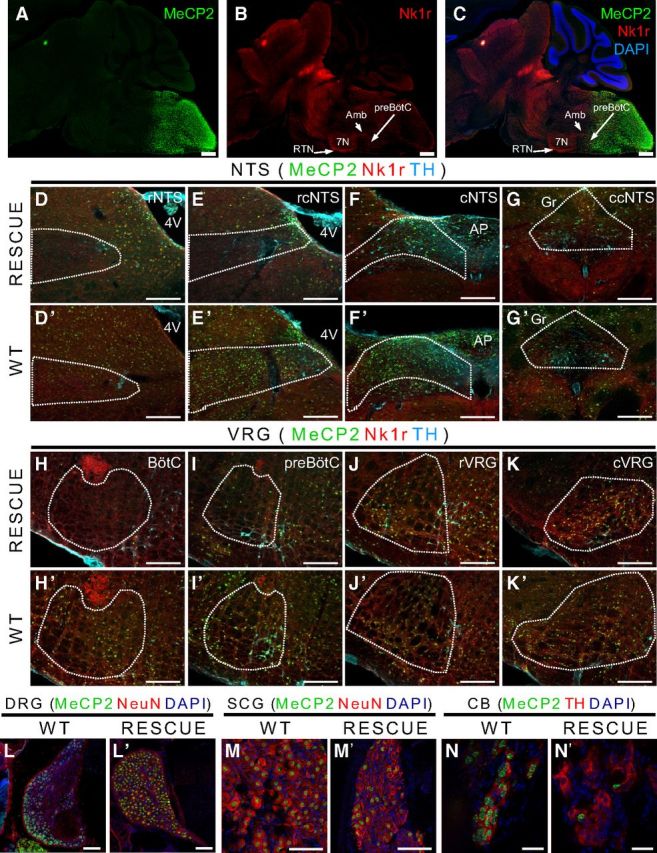
HoxA4-Cre-dependent expression of MeCP2 in caudal medulla and spinal cord. A set of images with staining of a parasagittal section through the brainstem of a HoxA4 RESCUE animal shows the induced MeCP2 protein expression in the caudal medulla (A–C). MeCP2 (green) staining shows MeCP2 expression in caudal medulla but not in rostral regions (A). With Nk1r staining (red) to show the structure of ventral respiratory group (B), a boundary with sparse MeCP2-positive cells can be observed next to preBötC (C). Two important regions regulating the respiratory function are partially targeted by HoxA4-Cre, including NTS (D–G, D′–G′) and VRC (H–K, H′–K′). Both regions are separated into four parts from rostral to caudal (NTS: rNTS, rcNTS, cNTS, ccNTS; VRC: BötC, preBötC, rVRG, cVRG). In HoxA4 RESCUE animals, the rostral regions of the NTS contains no or few MeCP2-expressing cells compared with NTS of WT animals (D, E, D′, E′). MeCP2-expressing cells can be observed among the region in the caudal part of NTS of HoxA4 RESCUE animals (F, G, F′, G′). A gradient of MeCP2 expression between rostral and caudal regions can also be observed in VRC of HoxA4 RESCUE animals. BötC is not targeted by HoxA4-Cre and with no MeCP2-positive cells (H, H′). The preBötC is next to the boundary and with few MeCP2-positive cells (I, I′). rVRG and cVRG show robust MeCP2 expression (J, K, J′, K′). MeCP2 is also expressed in the DRGs of WT (L) and HoxA4 RESCUE (L′) animals. However, HoxA4 RESCUE animals had low ratio of MeCP2-positive cells in superior cervical ganglion (SCG) (M′) and carotid body (CB) (N′) compared with the WT (M, N). Scale bars: A–C, 500 μm; D–G, D′–G′, H–K, H′–K′, 200 μm; L, M, L′, M′, 100 μm; N, N′, 20 μm.
The boundary of HoxA4-Cre dependent MeCP2 expression is immediately adjacent to preBötC, a region proposed to act as the pacemaker of breathing circuit, and absent from both the retrotrapezoid nucleus (RTN) and the BötC (Fig. 1A–C). Sagittal sections revealed that the HoxA4 domain includes the caudal parts of VRC and NTS (Fig. 1A–C). Both VRC and NTS are composed of spatially distinct anatomical compartments with different functions. The caudal part of VRC is composed of the rostral and caudal ventral respiratory group (rVRG and cVRG) that contain premotor neurons responsible for transmitting rhythmic respiratory activity to motorneurons. The caudal solitary tract nucleus (NTS) is a coordination center of cardiorespiratory reflex. To examine the rescue efficiency across different rostral-to-caudal regions of the NTS and VRC, we performed serial coronal brainstem sections across NTS and VRC to determine the exact region in which MeCP2 expression was restored with HoxA4-Cre. Figure 1D–K′ presents representative images of specific NTS or VRC regions, and Figure 2 shows a schematic representation of a sagittal section through the brainstem with the spatially distinct functional components of the respiratory network identified in relation to the boundaries of HoxB1 domain (red dashed line). The rescue efficiency of each region in HoxA4 RESCUE animals was normalized to the ratio of the same region in WT animals and presented in charts in Figure 2 (dorsal at top, ventral at bottom).
Figure 2.
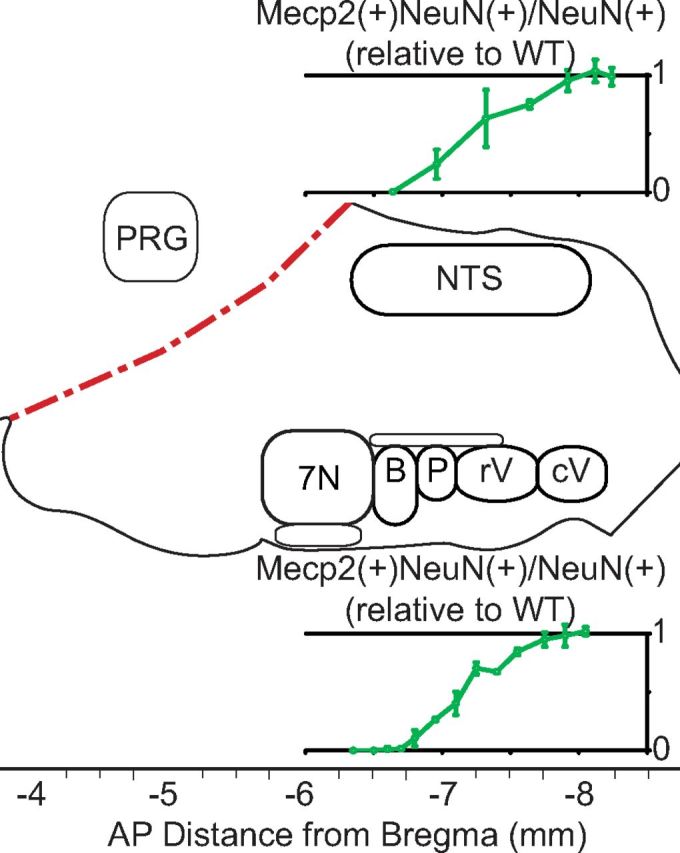
Stereological cell counting of the gradient of rescue efficiency around the HoxA4 boundary in the medulla of HoxA4 RESCUE animals. Different components of the respiratory network are presented in the relative locations of a sagittal representation of the brainstem with the indication of AP distance to bregma. Dotted red lines indicate the boundaries defined by HoxB1-Cre. Top, Ratios of MeCP2(+) NeuN(+) cells/NeuN(+) cells in HoxA4 RESCUE animals normalized to WT animals in different regions of the NTS. Bottom, Ratios in the VRC (n = 3 in each group). 7N, Facial nuclei; B, Bötzinger complex; P, preBötC; rV, rVRG; cV, cVRG. Error bars indicate SEM.
In HoxA4 RESCUE animals, low levels of MeCP2 expression (ratio: 0.006 ± 0.0 to 0.242 ± 0.125) are observed in the rostral part of NTS (Figs. 1D,D′, 2, top; AP: −6.64 to −6.96 mm), and the rescue efficiency increases to more than half (0.634 ± 0.244) in the middle part of the NTS (Figs. 1E,E′, 2, top; AP: −7.32 mm). Robust MeCP2 expression (0.752 ± 0.035 to 0.99 ± 0.076) can be observed in the caudal part of NTS (Figs. 1F,G,F′,G′, 2, top; AP: −7.64 to −8.24 mm). MeCP2 rescue efficiency across the NTS of HoxA4 RESCUE animals gradually increases from rostral to caudal.
The VRC is composed of four anatomic regions with different functions arranged along the rostral-caudal axis (BötC, preBötC, rVRG, and cVRG). BötC is rostral to the HoxA4 domain, and MeCP2-expressing cells are rarely found in this region (0 to 0.104 ± 0.068; Figs. 1H,H′, 2, bottom; AP: −6.5 to −6.8 mm). MeCP2 expression within the preBötC (Figs. 1I,I′, 2, bottom; AP: −6.8 to −7.1 mm) ranges from very low at the rostral end (0.104 ± 0.068), to low in the middle (0.264 ± 0.014, AP: −6.95 mm), and finally increasing slightly at the very caudal end where the preBötC transitions into the rVRG (0.402 ± 0.1). In rVRG and cVRG of HoxA4 RESCUE animals, intense MeCP2-expressing cells (ratio in rVRG: 0.402 ± 0.1 to 0.845 ± 0.029; ratio in cVRG: 0.951 ± 0.061 to 1.02 ± 0.037) similar to WT animals were observed (Figs. 1J,K,J′,K′, 2, bottom; AP: −7.1 to −7.55 mm, and −7.75 to −8.05 mm). We also found that MeCP2 is expressed in the DRG of RESCUE animals (Fig. 1L,L′), and partially restored in the superior cervical ganglion (Fig. 1M,M′) and carotid body (Fig. 1N,N′), similar to what was observed in HoxB1 RESCUE animals (Ward et al., 2011). Our results suggest that HoxA4-Cre causes a gradient of recombination efficiency around the expression boundary of HoxA4, but within the caudal part of the medulla MeCP2 rescue efficiency is at WT levels. This allows us to specifically manipulate the MeCP2 expression within the breathing circuits in the caudal medulla.
MeCP2 expression in the HoxA4 domain is necessary but not sufficient for maintaining normal breathing rate
Breathing disorders are common in people with RTT and in RTT mouse models and have been proposed as a contributor to early lethality in a subset of individuals with RTT (Kerr et al., 1997; Julu et al., 2001; Viemari et al., 2005; Weese-Mayer et al., 2006; Ogier et al., 2007; Rohdin et al., 2007; Voituron et al., 2010a; Ward et al., 2011; Ren et al., 2012; Bissonnette et al., 2014; Pitcher et al., 2015). Previously, we found that MeCP2 expression in the HoxB1 domain is necessary and sufficient for the normal hypoxic ventilation response but not basal breathing rate (Ward et al., 2011); however, the previous study did not examine other breathing abnormalities found in RTT, such as apneas and breathing irregularity, and did not formally characterize changes to breathing patterns during development. To explore this, we measured breathing parameters of HoxB1 RESCUE animals from young adult to the age of 35 weeks. As in the previous study, MeCP2 was reexpressed within the HoxB1 domain using a HoxB1-Cre transgene (Ward et al., 2011). Restoring MeCP2 expression in the HoxB1 domain (HoxB1 RESCUE animals) did not rescue the increased breathing rate (Fig. 3A), and surprisingly the breathing rate progressively increased from 7 to 20 weeks of age. HoxB1 RESCUE animals showed both decreased inspiratory and decreased expiratory times (Fig. 3B,C). Importantly, in contrast to NR animals, HoxB1 RESCUE animals did not develop increased breathing irregularity with aging (Fig. 3D).
Figure 3.
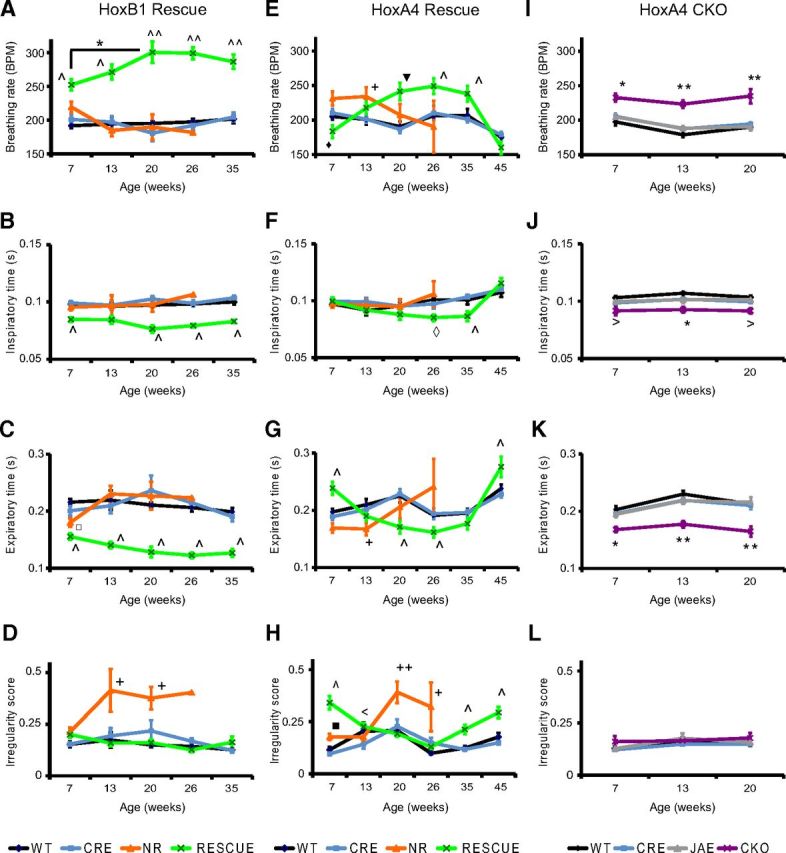
Distinct patterns of age-dependent changes in basal breathing in animals with hindbrain regional alterations in MeCP2 expression. Basal breathing was recorded in animals with MeCP2 selectively expressed within the HoxB1 domain (A–D, HoxB1 Rescue), in animals with MeCP2 selectively expressed in HoxA4 domain (E–H, HoxA4 Rescue), and in animals selectively lacking MeCP2 in the HoxA4 domain (I–L, HoxA4 CKO). Data were collected over a variety of ages and analyzed for breathing rate (A, E, I), inspiratory time (B, F, J), expiratory time (C, G, K), and irregularity score (D, H, L). Complete absence of MeCP2 (NR, orange) caused increased breathing rate compared with control animals (WT, black; CRE, blue) in young adults, but the breathing rate drops with aging (A, E). Early hyperventilation seen in NR animals is driven by decreased expiratory time rather than changes in inspiratory time (B, C, F, G), and breathing irregularity progressively increases with age in NR animals (D, H). HoxB1 RESCUE animals (RESCUE, green) have hyperventilation that worsens with age (A), driven by both decreased inspiratory and expiratory times (B, C). HoxB1 RESCUE animals, unlike NR mice, show no increase in breathing irregularity (D). HoxA4 RESCUE animals (RESCUE, green) have a dynamic pattern of hyperventilation with age, with relatively normal breathing rate at young and old ages but hyperventilation at middle ages (E), which is partially driven by decreased inspiratory time (F) but strongly driven by changes in expiratory time (G). These animals also showed increased breathing irregularity at young and old ages (H). HoxA4 CKO animals (CKO, purple) have increased breathing rates at all ages (I) compared with Cre only (CRE, blue), Flox only (JAE, gray), and WT animals (WT, black), which is driven predominantly by decreased expiratory time (K) with a minor contribution from decreased inspiratory time (J). HoxA4 CKO animals do not have irregular breathing at any age (L). Numbers of animals with different genotypes in the group of HoxB1 rescue experiment at 5 different time points (from 7 to 35 weeks) are as follows: WT: n = 8, 8, 7, 6, 6. CRE: n = 5, 5, 5, 5, 4. NR: n = 7, 4, 4, 1, 0. RESCUE: n = 12, 12, 9, 9, 3. The numbers of animals with different genotypes in the group of HoxA4 rescue experiment at 6 different time points (from 7 to 45 weeks) are as follows: WT: n = 11, 7, 11, 11, 11, 11. CRE: n = 15, 12, 12, 12, 11, 11. NR: n = 15, 8, 4, 3, 0, 0. RESCUE: n = 16, 13, 12, 12, 12, 9. Numbers of animals with different genotypes in the group of HoxA4 CKO experiment at 3 different time points (from 7 to 20 weeks) are as follows: WT: n = 10, 10, 9. CRE: n = 10, 10, 9. JAE: n = 10, 10, 10. CKO: n = 12, 9, 3. *p < 0.05 (one-way ANOVA with genotype as factor). **p < 0.001 (one-way ANOVA with genotype as factor). *p < 0.05 (Tukey post hoc pairwise comparisons, but LSD pairwise comparisons for breathing rate). *NR and RESCUE versus WT and CRE, or CKO versus WT, CRE, and JAE. +NR versus WT, CRE, and RESCUE. ∧RESCUE versus WT, CRE, and NR. ▾ RESCUE versus WT and CRE. ‡RESCUE versus NR versus WT and CRE. ♢ represents RESCUE versus WT and NR. ♦ represents RESCUE versus CRE and NR. □ represents NR versus WT. ■ represents NR versus CRE. > represents CKO versus WT. < represents RESCUE versus CRE. Error bars indicate SEM.
To further determine the role of MeCP2 in the respiratory network, we then measured and compared the breathing parameters in HoxA4 RESCUE, HoxA4 CKO (HoxA4-CreTg/+; Mecp2Jae/y animals), and littermate control animals at different ages to characterize the dynamic breathing abnormalities from early symptomatic periods to the end stage of the disease. Previous data indicated that NULL mice showed age-dependent changes in breathing frequency with increased irregularity (Ren et al., 2012). Similarly, NR mice showed significantly increased breathing rates at 13 weeks of age compared with control littermates, and the average breathing rate dropped to normal ranges after 20 weeks of age (Fig. 3E). In contrast, HoxA4 RESCUE animals exhibited dramatic changes of breathing rate at different ages. Initially, HoxA4 RESCUE animals showed slightly decreased breathing rate at 7 weeks of age, but the rate gradually increased with age until it was significantly higher than WT littermate control animals at 26 weeks of age (Fig. 3E). The elevated breathing rate in the HoxA4 RESCUE animals persisted until 45 weeks of age when it dropped below normal rates. The increased breathing rate in NR animals at 13 weeks of age was driven by decreased expiratory time (Fig. 3F,G), similar to prior observations in NULL animals (Ward et al., 2011). In contrast, the relative contribution of inspiratory and expiratory time to breathing rate was more complicated in HoxA4 RESCUE animals. At 7 weeks of age, the observed decreased rate is driven by increased expiratory time with normal inspiratory time (Fig. 3F,G); however, the expiratory time eventually declined below WT levels and was the major factor leading to the observed increased rate between 20 and 35 weeks of age (Fig. 3F,G). In contrast to the normal inspiratory time observed at all ages in NR animals, HoxA4 RESCUE animals showed decreased inspiratory times at 26 and 35 weeks of age compared with control littermates, which normalized at 45 weeks of age (Fig. 3F). As observed at 7 weeks of age, expiratory time was increased at 45 weeks of age in HoxA4 RESCUE animals, driving the decreased breathing rate observed at the age (Fig. 3G).
In addition to altered breathing rate, NR animals also demonstrated increased breathing irregularity at 7 weeks and then again beyond 20 weeks of age (Fig. 3H) as in late-stage NULL mice (Stettner et al., 2007; Ren et al., 2012). In HoxA4 RESCUE animals, increased breathing irregularity was observed in 7-week-old animals (Fig. 3H) but gradually ameliorated and reached a normal range at 20–26 weeks of age, and then increased again after 35 weeks of age. Unlike NR mice that showed a linear progression of breathing dysfunction, breathing parameters in HoxA4 RESCUE animals dynamically changed, with early-age abnormalities followed by relative normalization, but eventually becoming abnormal again at older ages (Fig. 3H).
We next examined the effects of removal of MeCP2 function from the HoxA4 domain. HoxA4 CKO mice showed increased breathing rates from 7 to 20 weeks of age (Fig. 3I). The increased breathing rate was relatively stable, and no individual showed a decline in breathing rate before death, as was observed in NR (Fig. 3A,E) and in NULL animals (Ward et al., 2011). All HoxA4 CKO animals died before breathing could be measured at 26 weeks. Decreased inspiratory and expiratory times were observed in HoxA4 CKO mice at different ages (Fig. 3J,K), and the decreased expiratory time was about twice the decreased inspiratory time. Although hyperventilation was observed in HoxA4 CKO animals, there was no increase in breathing irregularity (Fig. 3L).
These results demonstrated that loss of MeCP2 function in components of the respiratory network alter different aspects of the breathing regulation. We observed a subtle difference in the onset of the breathing abnormalities in the NR animals between the HoxB1 and the HoxA4 Rescue experiments, which we believe may be the result of small genetic differences between the Cre lines, although they are both maintained on a C57BL6/J background. Our results also suggested that MeCP2 expression in either the rostral or the HoxA4 domain is necessary for the regulation of breathing rate. However, restoring MeCP2 expression in the HoxA4 domain was neither sufficient for maintaining the stability of the respiratory rhythm generation nor for full rescue of the abnormal breathing rate.
MeCP2 expression in rostral medullary respiratory circuits prevents apnea
Breath holds or apneic events are present in people with RTT (Weese-Mayer et al., 2006) and have also been found in MeCP2-deficient animals (Ogier et al., 2007; Voituron et al., 2009, 2010a, 2010b). Similar to observations in NULL animals, we found that NR animals also showed increased apneas that worsen with age (Fig. 4A). To determine the possible role of MeCP2 within the HoxB1 domain in the apnea phenotype, we evaluated apneas in HoxB1 RESCUE animals. HoxB1 RESCUE animals show no increase of apneic events compared with control littermates at any age (Fig. 4A). Thus, restoring MeCP2 function in the medullary respiratory network is sufficient to prevent the development of apneas, although PRG and suprapontine structures are lacking MeCP2 in these animals.
Figure 4.

MeCP2 expression within the rostral VRC is critical for apnea phenotypes. Incidences of apnea in NR animals increased to extremely high level from 7 weeks old (A), but HoxB1 RESCUE animals did not have increased apnea compared with control littermates at all ages (A). HoxA4 RESCUE animals had increased apnea at 7, 13, 35, and 45 weeks of age, but WT levels at 20–26 weeks of age (B). In contrast, HoxA4 CKO animals did not have increased apnea compared with control littermates at all ages (C). Three specific types of apnea were defined in NR and HoxA4 RESCUE animals: apnea after a sudden deep inhale or exhale as “post-sigh” (D), apnea after progressively shallower respiration as “weak breathing” (E), and apnea after a short period of sudden faster breathing as “quick breathing” (F). The relative fraction of the different types of apneas in old (20 weeks) NR, and young (7 weeks) and old (45 weeks) HoxA4 RESCUE animals were compared (G). The percentages of apnea longer than 1 s increased with age in both NR and HoxA4 RESCUE animals (H). HoxB1 RESCUE did not show increased long apneas at any age (I). Animal numbers and colors in A–C, G, and H are the same as Figure 4. *p < 0.05 (one-way ANOVA with genotype as factor). **p < 0.001 (one-way ANOVA with genotype as factor). *p < 0.05 (Tukey post hoc pairwise comparisons). *NR and RESCUE versus WT and CRE, or CKO versus WT, CRE, and JAE. +NR versus WT, CRE, and RESCUE. ∧RESCUE versus WT, CRE, and NR. ▾ RESCUE versus WT and CRE. < RESCUE versus CRE. NS, Not significant. Error bars indicate SEM.
We next examined the apneic events in HoxA4 RESCUE and CKO animals. Surprisingly, restoring MeCP2 in the HoxA4 domain increased apnea frequency in the HoxA4 RESCUE animals at 7 weeks compared with both WT and NR animals (Fig. 4B). Similar to the pattern observed in the breathing irregularity (Fig. 3H), a dynamic age-dependent change in the amount of apneas was observed in HoxA4 RESCUE animals. These animals showed a decreasing pattern of apnea frequency from 7 to 26 weeks of age. In contrast to the high frequency of apnea in NR mice after 20 weeks age, HoxA4 RESCUE animals showed a decrease in apneas from 20 to 26 weeks of age to a level similar to WT littermates. The frequency of apneas then increased in HoxA4 RESCUE animals at 35 and 45 weeks of age (Fig. 4B).
In contrast to the pattern of apneas in HoxA4 RESCUE animals, selective removal of MeCP2 from the HoxA4 domain in HoxA4 CKO animals did not result in increased apnea frequency at any age compared with WT animals (Fig. 4C). These data suggest that MeCP2 expression within the HoxA4 domain is not sufficient to prevent the development of apneas, and the critical region requiring MeCP2 in the origin of apneas should lie in the region rostral to the HoxA4 domain. Furthermore, because apneas were observed in HoxA4 RESCUE but not HoxB1 RESCUE animals, these data suggest that the critical region requiring MeCP2 function to prevent apnea is within the HoxB1 domain but rostral to the HoxA4 domain.
Although the work presented above provides insight about apneas in RTT, previous studies suggest that not only the number, but also the characteristic patterns of apneic events are different in early- and late-stage NULL mice (Voituron et al., 2010a, 2010b; Ren et al., 2012). Different patterns of apneic events represent different types of dysfunction within the respiratory network. We observed progressively increased frequency of apneic events in NR mice, whereas HoxA4 RESCUE animals show an age-dependent, U-shaped pattern of apnea frequency with markedly increased frequency at 7 and 46 weeks of age. An important question is whether the specific type of apneas observed in these HoxA4 RESCUE animals is qualitatively the same at 7 and 46 weeks and whether it was characteristically similar or distinct from the type of apneas observed in NR animals. To address this, we examined breathing patterns surrounding apnea events to categorize the specific apnea patterns. Three major patterns of apnea were observed: (1) a sudden deep inhalation or exhalation preceding the apnea (Post-sigh, Fig. 4D), (2) progressively shallower breaths preceding the apnea (weak breathing, Fig. 4E), and (3) a sudden, short run of fast breathing immediately preceding the apnea (quick breathing, Fig. 4F). Additionally, we observed apneic events that did not have any specific defining pattern and these events were grouped together (Other).
The predominant pattern of classifiable apneas in 20-week-old NR animals is weak breathing, with only a small amount of Post-sigh or quick breathing (Fig. 4G). In contrast, 7-week-old HoxA4 RESCUE animals exhibited more quick breathing apneas than the NR animals (Fig. 4G). Interestingly, as HoxA4 RESCUE animals aged, the percentage of apneas that were classified as quick breathing dropped to the low level seen in 20-week-old NR animals, but the frequency of Post-sigh apneas dramatically increased to levels much higher than observed in either NR animals or young HoxA4 RESCUE animals (Fig. 4G). Thus, although NR and young and old HoxA4 RESCUE animals all showed increased apneas, there are qualitative differences of types of apnea patterns between these three groups. To further identify differences in the apneas observed in these three groups, we also examined the percentage of apneas that were prolonged (>1 s in duration). NR animals show an age-dependent increase in the percentage of prolonged apneas (Fig. 4H). At 7 weeks of age, HoxA4 RESCUE animals show a greater percentage of prolonged apneas compared with NR animals (Fig. 4H). As observed for the frequencies of all apneas (Fig. 4B), in HoxA4 RESCUE animals the percentage of prolonged apneas initially decreased as the mice aged (Fig. 4H); however, by 26 weeks, HoxA4 RESCUE animals showed a similar percentage of prolonged apneas to age-matched NR animals. The percentage of prolonged apneas in HoxA4 RESCUE animals continued to increase with age, independent of the change in the frequency of all apnea (Fig. 4B,H), such that at 45 weeks of age the HoxA4 RESCUE animals exhibited twice the percentage of prolonged apneas observed in the same animals at 7 weeks of age. In contrast, HoxB1 RESCUE animals had a low ratio of prolonged apneas at all ages (Fig. 4I), and this result suggested that MeCP2 expression in the HoxB1 domain is sufficient to prevent the apnea-causing defects at all ages.
Our data indicate that MeCP2 expression in the medullary respiratory network is important for generating nonapneic breathing. Restoring MeCP2 expression in all medullary components within the HoxB1 domain is sufficient to prevent the apnea. However, restoring MeCP2 within the HoxA4 domain only reduced apnea and changed the patterns of phenotypic development. Although HoxA4 RESCUE animals showed similar frequencies of apnea at 7 and 45 weeks of age, the age-dependent variation of apnea patterns suggests that there is still a transition of apnea types during aging, similar to NULL animals. In previous work (Ren et al., 2012), in situ recordings of respiratory rhythms detected regular respiratory patterns from isolated brainstems of NULL mice in early symptomatic stages and patterns with increased central apnea from isolated brainstems of NULL mice in late symptomatic stages, suggesting that the origin of apnea in NULL mice transitions from the state-dependent irregularity to intrinsic defects of central rhythmic activity. Our results further support the existence of a transition of the underlying mechanisms of apnea. In addition, MeCP2 expression in components within the HoxB1 domain, but rostral to HoxA4 domain, is important for the prevention of the development of apneic breathing. However, temporary normalization of the state-dependent irregularity is possible when MeCP2 is expressed solely within the HoxA4 domain.
Stable hypoxic ventilation response requires MeCP2 expression in multiple brain regions
Mice completely lacking MeCP2 function exhibit an abnormal breathing response to hypoxia (Ward et al., 2011). Our previous work indicated that MeCP2 expression in the HoxB1 domain is both necessary and sufficient for normal hypoxic ventilation response. Two anatomical components within the HoxB1 domain, the preBötC and NTS, have been proposed to be critical for normal hypoxic ventilation response (Teppema and Dahan, 2010; Hill et al., 2011). The HoxA4 domain, in contrast, targets the caudal NTS and only sparsely involves the preBötC. Thus, characterization of the hypoxic ventilation response in HoxA4 RESCUE and CKO animals allows the determination of whether the caudal NTS or the preBötC requires MeCP2 function for a normal hypoxic ventilation response.
The hypoxic ventilation response is biphasic: the breathing rate initially rapidly rises until a peak breathing response is reached, followed by a steady decline over the next 20–30 min, referred to as HVD (Easton et al., 1986). As we have previously observed in NULL animals (Ward et al., 2011), 7-week-old NR animals showed normal peak response, but with increased breathing rates at 15 min of hypoxia (Fig. 5A), which indicates a failure to undergo HVD. Hypoxic ventilation response of HoxA4 RESCUE animals was identical to WT controls (Fig. 5A). In contrast, HoxA4 CKO animals showed abnormal HVD similar to NR animals (Fig. 5B). Thus, the MeCP2 function within the HoxA4 domain is both necessary and sufficient for normal HVD.
Figure 5.
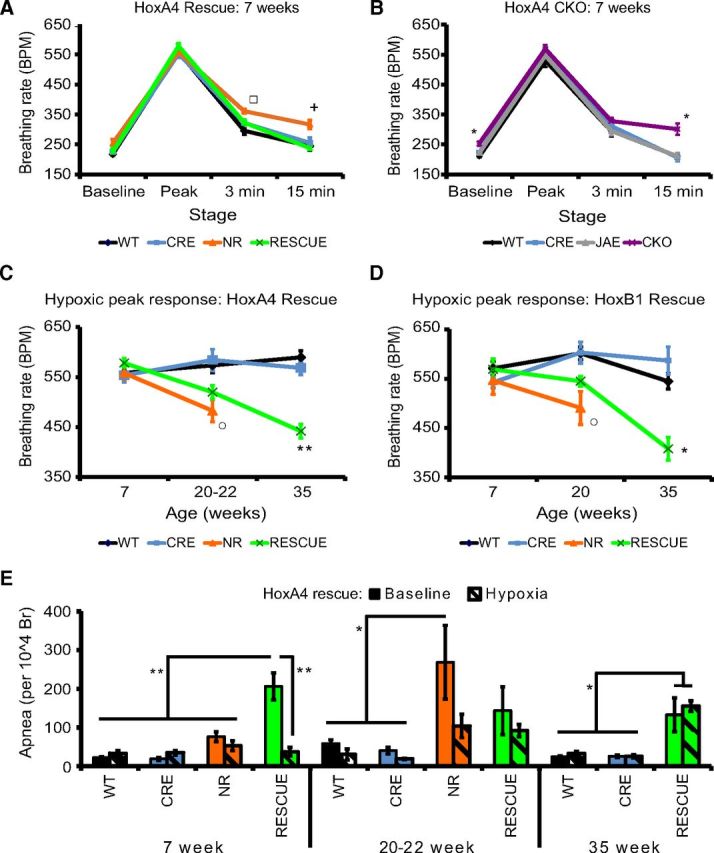
MeCP2 expression in pontine respiratory group and the HoxA4 domain is important for different aspects of the hypoxic ventilation response. NR animals (orange) showed normal peak response but had abnormal HVD (A) at 7 weeks of age. Restoring MeCP2 expression in HoxA4 domain (RESCUE, green) rescued the abnormal hypoxic decline (A). In contrast, removal of MeCP2 from HoxA4 domain (CKO) recapitulated HVD phenotype (B). Performing the hypoxic challenge on animals at different ages showed that the level of peak response was decreased in 20- to 22-week-old NR mice (C). HoxA4 RESCUE animals also showed a decline in peak response and reached the same level at 35 weeks of age (C). HoxB1 RESCUE animals showed similar decrease of the peak response at 35 weeks (D). Apnea of 7-week-old HoxA4 RESCUE animals was decreased to WT level during the first 5 min of hypoxic challenge (E). Hypoxic challenge also inhibited the apnea of 20- to 22-week-old NR animals, but not apnea of 35-week-old HoxA4 RESCUE animals (E). In hypoxic challenge experiments, the numbers of animals are as follows at 3 different time points (7, 20–22, 35 weeks): HoxA4 rescue experiment: WT: n = 11, 6, 11. CRE: n = 12, 6, 12. NR: n = 12, 8, 0. RESCUE: n = 13, 8, 12. HoxA4 CKO experiment: WT: n = 10. CRE: n = 10. JAE: n = 10. CKO: n = 12. HoxB1 rescue experiment (7, 20, 35 weeks): WT: n = 8, 8, 6. CRE: n = 5, 5, 4. NR: n = 8, 4, 0. RESCUE: n = 12, 9, 3. *p < 0.05 (one-way ANOVA with genotype as factor). **p < 0.001 (one-way ANOVA with genotype as factor). *p < 0.05 (Tukey post hoc pairwise comparisons). *NR and RESCUE versus WT and CRE, or CKO versus WT, CRE, and JAE. +NR versus WT, CRE, and RESCUE. ○ NR versus WT and CRE. □ represents NR versus WT. Error bars indicate SEM.
Although the peak response to hypoxia was normal at 7 weeks in all animals tested, NR animals showed decreased peak responses at 20 weeks of age (Fig. 5C), whereas HoxA4 RESCUE animals showed an intermediate level of decrease at 20 weeks (Fig. 5C), and the peak response of 35-week-old HoxA4 RESCUE animals was decreased to the level similar to the response of 20-week-old NR animals (Fig. 5C). These results indicate that the maintenance of normal peak response to hypoxia depends on MeCP2 function outside the HoxA4 domain. To address which component is likely responsible for maintenance of the peak response to hypoxia, we characterized the peak response in HoxB1 RESCUE animals because the HoxB1 domain is known to be sufficient for normal hypoxic ventilation response and excludes the PRG and the suprapontine structures (Ward et al., 2011). Similar to the decline we observed in HoxA4 RESCUE animals (Fig. 5C), the peak responses declined over time in HoxB1 RESCUE animals (Fig. 5D). From this, we conclude that maintenance of the peak response to hypoxia depends on MeCP2 function in components within the region rostral to the HoxB1 domain; and without MeCP2 in that region, the overall breathing control circuitry becomes progressively abnormal and unable to correctly respond to a hypoxic challenge by increasing the breathing rate.
Hypoxia is also able to suppress apneic events (Fig. 5E). At 7 weeks old, HoxA4 RESCUE animals had increased apneic events at baseline (Figs. 4B, 5E). At this age, hypoxic challenge significantly reduced the amount of apnea to the level similar to WT (Fig. 5E). At 20 weeks of age, when NR animals showed increased baseline apneas (Figs. 4B, 5E), exposure to hypoxic gas also inhibited apnea in NR animals (Fig. 5E). However, the amount of apnea of 35-week-old HoxA4 RESCUE animals during hypoxic challenge remained at the same elevated level as during baseline (Fig. 5E).
In summary, a stable, normal hypoxic breathing response requires MeCP2 function within multiple components of the neural circuitry that control breathing. Restoring MeCP2 function solely within the HoxA4 domain initially normalizes the breathing response to hypoxia, with a normal peak response (Fig. 5A), HVD (Fig. 5A), and hypoxia suppression of apneas (Fig. 5E). In contrast, removing MeCP2 only from the HoxA4 domain disrupts the normal HVD (Fig. 5B). Although the response to hypoxia is normal in HoxA4 RESCUE animals at 7 weeks of age, the overall breathing circuitry is unstable over time with a progressive loss of normal peak response (Fig. 5C) and a loss of the ability of hypoxia to suppress apneas (Fig. 5E).
In vitro abnormal respiratory rhythm in animals lacking MeCP2 expression rostral to the HoxA4 domain
Within the respiratory network, the preBötC contains neurons with population rhythmic properties that have been associated with the breathing cycle, and this activity can be observed in isolated in vitro brainstem slices or in situ working heart-brainstem preparations (Smith et al., 1991, 2007; Koshiya and Smith, 1999; Johnson et al., 2001). In vitro brainstem slice recording can detect the population rhythmic activity of preBötC, and a previous study showed that preBötC of postnatal day 14 (P14) NULL mice generates irregular rhythmic activity (Viemari et al., 2005).
In HoxA4 RESCUE animals, MeCP2 expression is partially restored in approximately one-fourth of neuron cells in the major part of preBötC (Fig. 2, bottom). Our previous result from HoxB1 RESCUE animals showed that restoring MeCP2 protein to 50% of the cells within the hindbrain is sufficient to rescue the life span and other autonomic and breathing abnormalities (Ward et al., 2011). To determine whether the partial rescue of MeCP2 function in the preBötC restores its normal rhythmic activity, we measured the rhythmic properties of the preBötC in brain slices from WT, NR, HoxA4 RESCUE, and HoxA4 CKO animals. Representative raw recordings in 1 min, integrated activities of similar recordings, and interburst intervals in 10 min are presented (Fig. 6A–L). As previously demonstrated (Viemari et al., 2005), WT animals showed regular extracellular population discharges (Fig. 6A,E,I). Slices from NR animals showed very irregular patterns of discharges (Fig. 6B,F,J), which is consistent with recordings from NULL animals (Viemari et al., 2005). HoxA4 RESCUE animals show more irregular duration of interburst interval than WT animals, but not as disrupted as in NR animals (Fig. 6C,G,K). Slices from HoxA4 CKO animals show a pattern of interburst intervals similar to that seen in WT slices (Fig. 6D,H,L), albeit with a slightly lower interburst interval.
Figure 6.

Irregularity of pre-Bötzinger rhythmic activity was observed in NR and HoxA4 RESCUE mice. preBötC extracellular rhythmic bursts recorded in brainstem medullary slices from WT, MecP2 non-rescue (NR), HoxA4 RESCUE (RESCUE), and HoxA4 CKO animals. Top (A–D) and middle (E–H) portions show representative 1 min traces (raw and integrated). Bottom, Interburst intervals measured over representative 10 min segment (I–L). Regular spontaneous bursts were generated in preBötC of WT animals (A, E, I). Removal of MeCP2 from the whole body (NR) caused irregular burst generation in the preBötC (B, F, J). Null animals with MeCP2 reexpression in HoxA4 domain (RESCUE) also generated irregular bursts (C, G, K). However, removal of MeCP2 from HoxA4 domain (CKO) did not affect the regularity of bursts in the preBötC (D, H, L). Cumulative distribution plot shows variability of interburst intervals among WT, NR, rescue, and CKO mice (232–685 burst cycles per slice) (M). Distribution of interburst intervals of NR mice is significantly different from WT and CKO (M). Distribution of HoxA4 RESCUE mice is intermediate and significantly different from that in WT/CKO and MR animals. Interburst intervals of NR animals show increased irregularity compared with other groups (N). Number of animals: WT = 8, NR = 5, HoxA4 RESCUE = 5, and CKO = 4. *Significantly different from other groups (panel M, two-sample Kolmogorov–Smirnov test; panel N, ANOVA with Holm Sidak multiple-comparison test).
WT and HoxA4 CKO animals showed very similar distributions of interburst intervals (Fig. 6M); however, NR animals showed a markedly wider distribution and an increased median interburst interval compared with either WT or CKO animals (Fig. 6M). Interestingly, HoxA4 RESCUE animals showed an intermediate shape of variation that is significantly different from either WT/CKO or NR animals (Fig. 6M), with an increase in the interburst interval duration at both tails of the distribution. In addition, NR animals showed increased short-term irregularity scores compared with the other three groups (Fig. 6N). Results from these in vitro experiments show that partial rescue of MeCP2 expression in the preBötC of HoxA4 RESCUE animals reduces the variation of interburst interval but does not fully restore the normal pattern of rhythmic activity. In contrast, removing MeCP2 function by HoxA4-Cre did not impede the rhythmic activity of the preBötC in HoxA4 CKO animals. Combined with our in vivo breathing recording, our results suggest a connection between the abnormal rhythmic activity of the preBötC and the development of the apnea and breathing irregularity.
MeCP2 expression in HoxA4 domain is necessary and sufficient for normal life span
Previously, we determined that MeCP2 expression in HoxB1-derived tissues is both necessary and sufficient for normal life span and heart rate (Ward et al., 2011), and here we show that MeCP2 function within HoxB1-derived tissues is also necessary and sufficient for normal breathing patterns aside from basal hyperventilation and the maintenance of peak response during hypoxic challenge (Figs. 3A–D, 5D). In contrast, restoring MeCP2 function in the more restricted HoxA4 domain does not rescue the abnormal breathing rate (Fig. 3E–H) and increased apnea (Fig. 4B). An important issue is whether these breathing abnormalities also contribute to early lethality. To explore this, we compared the survival of NR, HoxA4 RESCUE, and HoxA4 CKO animals.
As observed previously (Ward et al., 2011), NR animals had a median survival of 119 d (Fig. 7A). In contrast, when MeCP2 expression was restored within the HoxA4 domain, HoxA4 RESCUE animals showed a substantial improvement in survival (Fig. 7A). Of 13 HoxA4 RESCUE animals, one animal died at 189 d old, and another was killed because of penile prolapse and severe self-induced penile injury at 323 d old. However, all the remaining animals survived beyond 1 year, at which time they were killed for tissue collection. MeCP2 function within HoxA4-defined tissues is thus clearly sufficient for improved survival. HoxA4 CKO animals showed early lethality similar to NR animals, with a median survival of 101 d (Fig. 3A). The results from HoxA4 RESCUE and CKO animals indicate that, although MeCP2 expression in the HoxA4 lineage is not sufficient for the normal breathing regulation, it is both necessary and sufficient for the survival.
Figure 7.
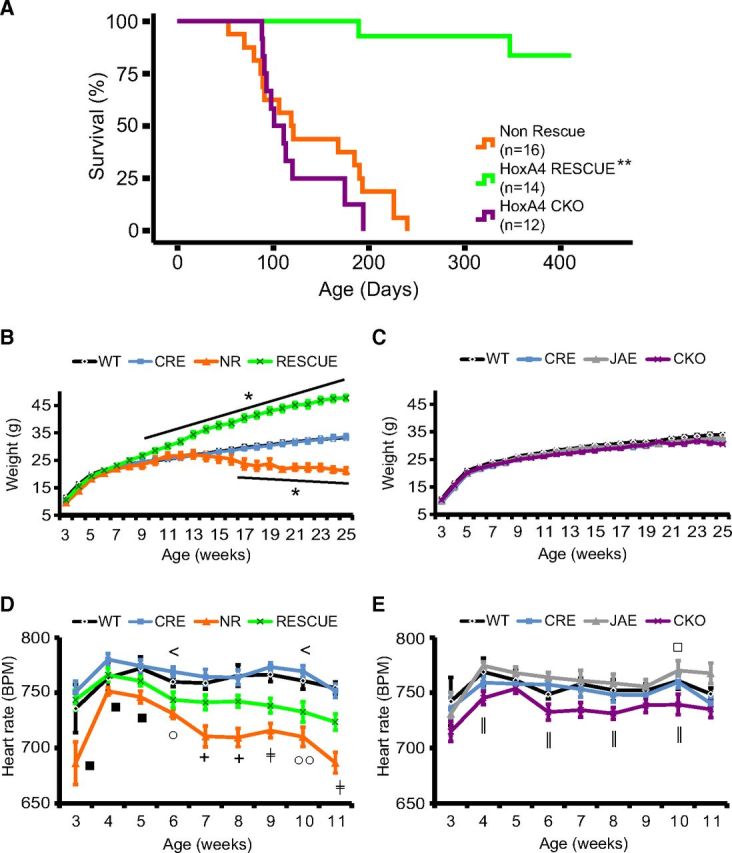
MeCP2 expression in the HoxA4 domain is necessary and sufficient for the survival. Non-rescue mice (Non-Rescue, orange, n = 16) showed early lethality with a median survival of 119 d (A). HoxA4 CKO mice (CKO, purple, n = 12) had early lethality with a median survival of 101 d. HoxA4 rescue mice (A, RESCUE, green, n = 13, 2 deaths) showed increased survival, with the majority surviving > 1 year (p < 0.0001 comparing RESCUE with Non-Rescue or CKO). HoxA4 RESCUE animals developed obesity compared with the control siblings (B), but non-rescue mice (NR) lost weight when they were old and close to death (B). Although HoxA4 CKO animals showed early lethality similar to NR animals, they did not lose weight like NR animals (C). Heart rates of animals were recorded by pulse oximetry in restrained mice weekly at indicated ages (D, E). Loss of MeCP2 in the whole body (NR, orange) causes a progressive decrease of heart rates compared with control animals (WT, black; CRE, blue) in young adults (D). HoxA4 RESCUE animals showed an intermediate heart rate between control siblings and NR mice (D). Removal of MeCP2 expression from the HoxA4 domain (CKO, purple) resulted in a slight decrease of the heart rate (E), and the heart rate of CKO animals was significantly lower compared with JAE control siblings in several time points (WT, black; CRE, blue; JAE, gray). *p < 0.05 (one-way ANOVA with genotype as factor). **p < 0.001 (one-way ANOVA with genotype as factor). *p < 0.05 (Tukey post hoc pairwise comparisons). *NR and RESCUE versus WT and CRE, or CKO versus WT, CRE, and JAE. +NR versus WT, CRE, and RESCUE. □ represents NR versus WT or CKO versus WT. ■ represents NR versus CRE or CKO versus CRE. < represents RESCUE versus CRE. ○ represents NR versus WT and CRE. ‡RESCUE versus NR versus WT and CRE. ‖CKO versus JAE. Error bars indicate SEM.
Boys with congenital encephalopathy due to RTT-causing mutations in MECP2 are often found to have respiratory arrest (Kankirawatana et al., 2006) at death. However, our results suggested that severe breathing disorders do not directly cause early lethality in MeCP2-deficient animals. To find the possible physiological changes that may correlate to the early lethality, we compared the body weight and the heart rate of different groups of animals at different ages. Weekly body weight measurement revealed that, in addition to premature lethality, NR animals lost body weight significantly from 17 weeks of age (Fig. 7B), about the time they reached their median survival. In contrast, restoring MeCP2 in HoxA4 domain reversed this weight loss, and the HoxA4 RESCUE animals became obese (Fig. 7B), similar to animals with MeCP2 rescue within HoxB1 domain (Ward et al., 2011). Although HoxA4 CKO animals showed early lethality, their body weight was not significantly different from WT animals (Fig. 7C). This result is consistent with the previous results indicating that normal weight homeostasis requires MeCP2 function within the hypothalamus (Fyffe et al., 2008; Torres-Andrade et al., 2014). The normal body weight observed in HoxA4 CKO animals before death suggested that the mechanism underlying premature lethality can be dissociated from abnormal growth.
Marked bradycardia is also found in boys with congenital encephalopathy due to RTT-causing mutations in MECP2 at death (Schule et al., 2008). Decreased heart rate was observed in NULL and HoxB1 CKO animals but not HoxB1 RESCUE animals (Ward et al., 2011). Therefore, we compared the heart rate of MeCP2-deficient animals at different ages to determine whether MeCP2 expression in the HoxA4 domain is required for normal heart rate. NR mice showed age-dependent decrease of heart rate and had significantly decreased heart rate compared with control siblings from 6- to 11-week-olds (Fig. 7D). Restoring MeCP2 function in the HoxA4 domain partially rescued the decreasing heart rate (Fig. 7D). Removing MeCP2 from the HoxA4 domain (HoxA4 CKO) caused a decrease in heart rate compared with JAE control animals (Fig. 7E). Thus, MeCP2 function within the HoxA4 domain contributes to normal heart rate, and the disruption or restoration of this function likely is a contributing factor to normal survival.
Discussion
Previously, we demonstrated that MeCP2 function within specific anatomic regions within the hindbrain is important for specific breathing phenotypes (Ward et al., 2011). In this work, we further explored the role of MeCP2 in the caudal brainstem in relationship to breathing problems seen in RTT. Our current results demonstrate that MeCP2 expression within the caudal, HoxA4-derived hindbrain is both necessary and sufficient for survival, but neither necessary nor sufficient for normal basal breathing rate, prevention of apneas, or regular breathing pattern. This was unexpected and separates the role of abnormal breathing from survival in these animals. The survival effect appears to correlate with partial rescue of heart rate when MeCP2 expression is restricted to the HoxA4 domain. Interestingly, we found evidence that the requirement for MeCP2 function for normal breathing is within a highly distributed network. The various breathing phenotypes and a summary of the experimental findings are presented in Table 1.
Table 1.
Comparison of respiratory phenotypes in MeCP2 conditional knock-out and rescue animalsa
| Non-rescue | HoxB1 RESCUE | HoxA4 RESCUE | HoxA4 CKO | |
|---|---|---|---|---|
| Breathing circuitry components expressing MeCP2 | None | RTN, BötC, preBötC, rVRG, cVRG, NTS, raphe | NTS, rVRG, cVRG, raphe | PRG, RTN, BötC, preBötC |
| Breathing circuitry components lacking MeCP2 | All | PRG | PRG, RTN, BötC, preBötC | NTS, rVRG, cVRG, raphé |
| Basal breathing rate | ↑ | ↑ | ↑ (age) | ↑ |
| Apnea | ↑ (age) | Rescued | ↑ (age) | Normal |
| Hypoxic peak response | ↓ ↓ (age) | ↓ (age) | ↓ (age) | Normal |
| Hypoxic decline | ↓ | Normal | Normal | ↓ |
| Life span | ↓ | Rescued | Rescued | ↓ |
a↑, Increased level of breathing rate, response, or decline; ↓, decreased level of breathing rate, response, or decline; age, phenotype is age-dependent. Two arrows indicate an increased change compared with other genotypes. The phenotypes that are rescued (i.e., indistinguishable from WT) in the regional rescue experiment are listed as “rescued.”
Basal breathing regulation and irregularity
Restoring MeCP2 expression in the HoxB1 domain did not rescue basal hyperventilation, suggesting that MeCP2 function within the PRG or suprapontine structures is required for normal breathing rate. Surprisingly, either removing MeCP2 from the HoxA4 domain (HoxA4 CKO) or restoring MeCP2 function in the HoxA4 domain (HoxA4 RESCUE) caused basal hyperventilation. This was completely unexpected because, in HoxA4 CKO animals, MeCP2 function is present within the previously implicated PRG or suprapontine structures. These seemingly contradictory pieces of data implicate that disrupting MeCP2 function in a number of components of the breathing control circuitry can lead to hyperventilation. Other experiments support the idea of a distributed and interconnected network required for maintenance of normal breathing rates. For example, both the PRG and the rVRG have been proposed to regulate breathing rhythmogenesis (Dobbins and Feldman, 1994; Dutschmann and Herbert, 2006). Increased PRG GABA inhibitory signaling resulted in decreased breathing rate (Damasceno et al., 2014), whereas acute increase of glutamate excitatory signaling in the anterior rVRG causes bradypnea (Monnier et al., 2003). MeCP2 deficiency causes decreases in multiple neurotransmitters, notably Glu and GABA (Viemari et al., 2005; Medrihan et al., 2008; Samaco et al., 2009; Chao et al., 2010; El-Khoury et al., 2014); thus, the basal hyperventilation observed may result from an imbalance between PRG GABAergic tone and glutamatergic tone in the rVRG. Future analysis of changes in neurotransmitter levels in these brain regions will be needed to test this hypothesis.
One of the most interesting results is the age-dependent changes seen in breathing rate in HoxA4 RESCUE animals, which start at a lower rate than WT animals, increase to hyperventilation, and then declines again later in life. Age-dependent changes in HoxA4 RESCUE animals were also observed in other breathing parameters (irregularity and apnea); however, in the middle-age points (20–35 weeks), the hyperventilation is most abnormal, whereas irregularity and apnea are “normalized.” One possible explanation for the “normalization” of the irregularity and apnea at these middle-age points is that the abnormally increased breathing rate during this time is masking or completely inhibiting the breathing irregularity and apneas. This masking cannot be maintained over time; and eventually, the entire circuit degenerates to the point that it can no longer maintain the elevated breathing rate, maintain a regular rate, or prevent apneas. Differences between breathing abnormalities at early or late stage were observed in previous studies (Voituron et al., 2010a, 2010b; Ren et al., 2012), and our findings suggest that losing MeCP2 function causes progressive deficits in multiple respiratory components; and their dynamic interactions result in the age-dependent phenotypes. In the future, manipulation of MeCP2 expression in specific components, such as in the preBötC, via viral injection, could help further dissect the MeCP2 requirement.
Apnea
The increased apneas and breathing irregularity observed in animals completely lacking MeCP2 were not observed in HoxB1 RESCUE animals, indicating that the critical region requiring MeCP2 function to prevent these breathing abnormalities lies within the HoxB1 domain. Interestingly, removing MeCP2 from the caudal-most region of the HoxB1 domain in the HoxA4 CKO animals also did not result in increased apnea or irregular breathing, pointing to a critical requirement of MeCP2 function in the HoxB1 domain rostral to the HoxA4 domain. The HoxB1 domain rostral to the HoxA4 domain includes a number of known components of the breathing circuitry (RTN, BötC, majority of the preBötC, rostral NTS). The BötC and preBötC form the core rhythm-generating circuitry (Smith et al., 2013), and it is likely that MeCP2 function within this core rhythm generating circuitry is critical to prevent the development of apneas and irregularity. Our results from the isolated medullary slices containing the preBötC are consistent with this notion. Restoring MeCP2 expression within the HoxA4 domain (HoxA4 RESCUE) caused a partial rescue of the prolonged interburst intervals in the isolated medullary slices compared with slices from NR animals, which suggests that restoring MeCP2 function in a portion of cells (∼25%) within the preBötC improves, but does not fully restore, the rhythmic property. Additionally, increased breathing irregularity and apnea were observed in HoxA4 RESCUE animals at 7 weeks of age as well. These findings suggests that the small percentage of MeCP2-positive cells in the preBötC of HoxA4 RESCUE animal are not sufficient for restoring normal breathing regulation.
We observed some unusual and unexpected pattern of results with regards to irregularity and apnea. First, the irregularity and apnea in HoxA4 RESCUE animals at 7 week of age were more dramatically increased than the abnormalities observed in medullary slice bursting rhythms at 2 weeks of age (P14). This discrepancy may be due to the timing of onset of apneas in the whole animal. Unfortunately, it is not technically possible to perform medullary slice recordings at the older time point (7 weeks), and it is possible that the rhythm generator in the preBötC at P14 is not as disrupted as seen at later ages. As mentioned previously, the initial partial rescue of bursting pattern seen in medullary slices at P14 may be due to the small portion of MeCP2-positive cells (∼25%) within the preBötC. By 7 weeks of age, the contribution of limited MeCP2 expression in this region is not sufficient to maintain normal breathing rhythm and results in apneas and increased irregularity.
The second surprising finding is that, at 7 weeks of age, the HoxA4 RESCUE animals show more irregularity and apneas than the NR animals, which again points to an initial instability of the circuitry when some components of the breathing circuitry (VRG, NTS) have normal amount of MeCP2 expression, whereas other components (BötC, RTN, PRG, suprapontine structures) either have no or significantly reduced amount of MeCP2. Unexpectedly, there is a dynamic pattern in the middle ages in HoxA4 RESCUE animals with a “normalization” of apneas and irregularity, followed by increases in these phenotypes at older ages. Interestingly, the qualitative differences in the type of apneas observed in young versus old HoxA4 and NR animals support the idea that there are distinct circuit-level changes occurring over time in these animals. As discussed above in the hyperventilation section, we suspect that there are circuit-level changes that normalize breathing in these animals, but these adaptations cannot be maintained; and eventually, circuit function degenerates and results in the observed phenotypes at old ages. Ultimately, understanding the circuit-level changes that occur during the adaptation and the ensuing degradation may provide insight into the regression of skills over time seen in people with RTT.
Hypoxic breathing response
We previously found that NULL mice also show increased hypoxic ventilation response (Ward et al., 2011), and that MeCP2 expression in the HoxB1 domain is both necessary and sufficient for this response. This current work demonstrated that increased hypoxic ventilation response in Mecp2-deficient animals results from a failed transition into proper HVD, but not from an increase of hypoxic peak response (HPR; Fig. 5A,B). Furthermore, we show that MeCP2 function in the HoxA4 domain is both necessary and sufficient for the normal HVD. The HoxA4 domain contains both the NTS and the VRG, and previous work suggests that the NTS is a key region for the proper response to hypoxia (Ward et al., 2011; Ramirez et al., 2013). Furthermore, we found that HPR of NR mice decreases with age, and restoring MeCP2 expression in either the HoxA4 or HoxB1 domain only slows, but does not prevent, the decline of HPR. Hypoxia-excited neurons in NTS send axonal projections to the PRG (Song et al., 2011), and a reasonable hypothesis is that MeCP2 expression in the PRG is required for normal HPR.
Survival and cardiac function
Although MeCP2 function in components that are considered the central rhythm generator (Smith et al., 2007) is dispensable for survival, MeCP2 function in the breathing circuits within the HoxB1-specific domain is critical for maintaining proper breathing. MeCP2 expression in different breathing circuitry components is necessary for specific manners of breathing regulation, but restoring MeCP2 expression in only parts of the respiratory network may result in unbalanced outputs and induce more severe breathing disorders as observed in young HoxA4 RESCUE animals. Future experiments to measure the changes of signal inputs and the responses of the rhythm generator of Mecp2-deficient animals at different ages may help us discover the complex interactions underlying breathing regulation and RTT breathing disorders. On the other hand, because severe breathing irregularity does not directly correlate to the early lethality in Mecp2-deficient mice, cardiac dysfunction becomes the major candidate underlying early lethality (McCauley et al., 2011). Future work is needed to explore whether restoring MeCP2 function within the HoxA4 domain also corrects the QTc prolongation and susceptibility to ventricular tachycardia observed in NULL animals (McCauley et al., 2011). Together, our work supports a model of a functionally diverse respiratory network and suggests that RTT breathing disorders are the results of interactions within the entire respiratory network instead of the malfunction of a single, critical breathing component.
Footnotes
This work was supported by National Institutes of Health/National Institute of Child Health and Human Development R01HD062553 to J.L.N., U54 HD083092, sub 6924 to B.R.A., International Rett Syndrome Foundation research funds to J.L.N., Anthony and Cynthia Petrello Scholar Fund at the Jan and Dan Duncan Neurological Research Institute at Texas Children's Hospital to J.L.N., and the Baylor College of Medicine Mouse Neurobehavioral Core funded by the Baylor College of Medicine-IDDRC P30HD024064 and U54 HD083092. The content is solely the responsibility of the authors and does not necessarily represent the official views of the Eunice Kennedy Shriver National Institute of Child Health and Development of the National Institutes of Health. We thank Drs. Roy V. Sillitoe and Joshua White for help with bursts analysis, and thank Dr. Jan Marino Ramirez with instruction and guidance in medullary slice electrophysiology.
The authors declare no competing financial interests.
References
- Alheid GF, McCrimmon DR. The chemical neuroanatomy of breathing. Respir Physiol Neurobiol. 2008;164:3–11. doi: 10.1016/j.resp.2008.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arenkiel BR, Gaufo GO, Capecchi MR. Hoxb1 neural crest preferentially form glia of the PNS. Dev Dyn. 2003;227:379–386. doi: 10.1002/dvdy.10323. [DOI] [PubMed] [Google Scholar]
- Behringer RR, Crotty DA, Tennyson VM, Brinster RL, Palmiter RD, Wolgemuth DJ. Sequences 5′ of the homeobox of the Hox-1.4 gene direct tissue-specific expression of lacZ during mouse development. Development. 1993;117:823–833. doi: 10.1242/dev.117.3.823. [DOI] [PubMed] [Google Scholar]
- Bianchi AL, Denavit-Saubié M, Champagnat J. Central control of breathing in mammals: neuronal circuitry, membrane properties, and neurotransmitters. Physiol Rev. 1995;75:1–45. doi: 10.1152/physrev.1995.75.1.1. [DOI] [PubMed] [Google Scholar]
- Bissonnette JM, Schaevitz LR, Knopp SJ, Zhou Z. Respiratory phenotypes are distinctly affected in mice with common Rett syndrome mutations MeCP2 T158A and R168X. Neuroscience. 2014;267:166–176. doi: 10.1016/j.neuroscience.2014.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao HT, Chen H, Samaco RC, Xue M, Chahrour M, Yoo J, Neul JL, Gong S, Lu HC, Heintz N, Ekker M, Rubenstein JL, Noebels JL, Rosenmund C, Zoghbi HY. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature. 2010;468:263–269. doi: 10.1038/nature09582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen RZ, Akbarian S, Tudor M, Jaenisch R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet. 2001;27:327–331. doi: 10.1038/85906. [DOI] [PubMed] [Google Scholar]
- Cohen MI. Neurogenesis of respiratory rhythm in the mammal. Physiol Rev. 1979;59:1105–1173. doi: 10.1152/physrev.1979.59.4.1105. [DOI] [PubMed] [Google Scholar]
- Damasceno RS, Takakura AC, Moreira TS. Regulation of the chemosensory control of breathing by Kolliker-Fuse neurons. Am J Physiol Regul Integr Comp Physiol. 2014;307:R57–R67. doi: 10.1152/ajpregu.00024.2014. [DOI] [PubMed] [Google Scholar]
- Depuy SD, Kanbar R, Coates MB, Stornetta RL, Guyenet PG. Control of breathing by raphe obscurus serotonergic neurons in mice. J Neurosci. 2011;31:1981–1990. doi: 10.1523/JNEUROSCI.4639-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobbins EG, Feldman JL. Brainstem network controlling descending drive to phrenic motoneurons in rat. J Comp Neurol. 1994;347:64–86. doi: 10.1002/cne.903470106. [DOI] [PubMed] [Google Scholar]
- Dutschmann M, Herbert H. The Kolliker-Fuse nucleus gates the postinspiratory phase of the respiratory cycle to control inspiratory off-switch and upper airway resistance in rat. Eur J Neurosci. 2006;24:1071–1084. doi: 10.1111/j.1460-9568.2006.04981.x. [DOI] [PubMed] [Google Scholar]
- Easton PA, Slykerman LJ, Anthonisen NR. Ventilatory response to sustained hypoxia in normal adults. J Appl Physiol. 1986;61:906–911. doi: 10.1152/jappl.1986.61.3.906. [DOI] [PubMed] [Google Scholar]
- El-Khoury R, Panayotis N, Matagne V, Ghata A, Villard L, Roux JC. GABA and glutamate pathways are spatially and developmentally affected in the brain of Mecp2-deficient mice. PLoS One. 2014;9:e92169. doi: 10.1371/journal.pone.0092169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman JL, Smith JC. Neural control of respiratory pattern in mammals: an overview. In: Dempsey JA, Pack AI, editors. Regulation of breathing. New York: Marcel Dekker; 1995. pp. 39–69. [Google Scholar]
- Fyffe SL, Neul JL, Samaco RC, Chao HT, Ben-Shachar S, Moretti P, McGill BE, Goulding EH, Sullivan E, Tecott LH, Zoghbi HY. Deletion of Mecp2 in Sim1-expressing neurons reveals a critical role for MeCP2 in feeding behavior, aggression, and the response to stress. Neuron. 2008;59:947–958. doi: 10.1016/j.neuron.2008.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia AJ, 3rd, Zanella S, Koch H, Doi A, Ramirez JM. Chapter 3-networks within networks: the neuronal control of breathing. Prog Brain Res. 2011;188:31–50. doi: 10.1016/B978-0-444-53825-3.00008-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy J, Hendrich B, Holmes M, Martin JE, Bird A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat Genet. 2001;27:322–326. doi: 10.1038/85899. [DOI] [PubMed] [Google Scholar]
- Guy J, Gan J, Selfridge J, Cobb S, Bird A. Reversal of neurological defects in a mouse model of Rett syndrome. Science. 2007;315:1143–1147. doi: 10.1126/science.1138389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera JA, Ward CS, Pitcher MR, Percy AK, Skinner S, Kaufmann WE, Glaze DG, Wehrens XH, Neul JL. Treatment of cardiac arrhythmias in Rett Syndrome with sodium channel blocking antiepileptic drugs. Dis Model Mech. 2015;8:363–371. doi: 10.1242/dmm.020131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill AA, Garcia AJ, 3rd, Zanella S, Upadhyaya R, Ramirez JM. Graded reductions in oxygenation evoke graded reconfiguration of the isolated respiratory network. J Neurophysiol. 2011;105:625–639. doi: 10.1152/jn.00237.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang WH, Tupal S, Huang TW, Ward CS, Neul JL, Klisch TJ, Gray PA, Zoghbi HY. Atoh1 governs the migration of postmitotic neurons that shape respiratory effectiveness at birth and chemoresponsiveness in adulthood. Neuron. 2012;75:799–809. doi: 10.1016/j.neuron.2012.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SM, Koshiya N, Smith JC. Isolation of the kernel for respiratory rhythm generation in a novel preparation: the pre-Botzinger complex “island.”. J Neurophysiol. 2001;85:1772–1776. doi: 10.1152/jn.2001.85.4.1772. [DOI] [PubMed] [Google Scholar]
- Julu PO, Kerr AM, Apartopoulos F, Al-Rawas S, Engerström IW, Engerström L, Jamal GA, Hansen S. Characterisation of breathing and associated central autonomic dysfunction in the Rett disorder. Arch Dis Child. 2001;85:29–37. doi: 10.1136/adc.85.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kankirawatana P, Leonard H, Ellaway C, Scurlock J, Mansour A, Makris CM, Dure LS, 4th, Friez M, Lane J, Kiraly-Borri C, Fabian V, Davis M, Jackson J, Christodoulou J, Kaufmann WE, Ravine D, Percy AK. Early progressive encephalopathy in boys and MECP2 mutations. Neurology. 2006;67:164–166. doi: 10.1212/01.wnl.0000223318.28938.45. [DOI] [PubMed] [Google Scholar]
- Kerr AM, Armstrong DD, Prescott RJ, Doyle D, Kearney DL. Rett syndrome: analysis of deaths in the British survey. Eur Child Adolesc Psychiatry. 1997;6(Suppl 1):71–74. [PubMed] [Google Scholar]
- Koshiya N, Smith JC. Neuronal pacemaker for breathing visualized in vitro. Nature. 1999;400:360–363. doi: 10.1038/22540. [DOI] [PubMed] [Google Scholar]
- Laurvick CL, de Klerk N, Bower C, Christodoulou J, Ravine D, Ellaway C, Williamson S, Leonard H. Rett syndrome in Australia: a review of the epidemiology. J Pediatr. 2006;148:347–352. doi: 10.1016/j.jpeds.2005.10.037. [DOI] [PubMed] [Google Scholar]
- McCauley MD, Wang T, Mike E, Herrera J, Beavers DL, Huang TW, Ward CS, Skinner S, Percy AK, Glaze DG, Wehrens XH, Neul JL. Pathogenesis of lethal cardiac arrhythmias in Mecp2 mutant mice: implication for therapy in Rett syndrome. Sci Transl Med. 2011;3:113ra125. doi: 10.1126/scitranslmed.3002982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medrihan L, Tantalaki E, Aramuni G, Sargsyan V, Dudanova I, Missler M, Zhang W. Early defects of GABAergic synapses in the brain stem of a MeCP2 mouse model of Rett syndrome. J Neurophysiol. 2008;99:112–121. doi: 10.1152/jn.00826.2007. [DOI] [PubMed] [Google Scholar]
- Monnier A, Alheid GF, McCrimmon DR. Defining ventral medullary respiratory compartments with a glutamate receptor agonist in the rat. J Physiol. 2003;548:859–874. doi: 10.1113/jphysiol.2002.038141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulkey DK, Stornetta RL, Weston MC, Simmons JR, Parker A, Bayliss DA, Guyenet PG. Respiratory control by ventral surface chemoreceptor neurons in rats. Nat Neurosci. 2004;7:1360–1369. doi: 10.1038/nn1357. [DOI] [PubMed] [Google Scholar]
- Neul JL, Kaufmann WE, Glaze DG, Christodoulou J, Clarke AJ, Bahi-Buisson N, Leonard H, Bailey ME, Schanen NC, Zappella M, Renieri A, Huppke P, Percy AK. Rett syndrome: revised diagnostic criteria and nomenclature. Ann Neurol. 2010;68:944–950. doi: 10.1002/ana.22124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogier M, Wang H, Hong E, Wang Q, Greenberg ME, Katz DM. Brain-derived neurotrophic factor expression and respiratory function improve after ampakine treatment in a mouse model of Rett syndrome. J Neurosci. 2007;27:10912–10917. doi: 10.1523/JNEUROSCI.1869-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Franklin K. The mouse brain in stereotaxic coordinates. Ed 3. Amsterdam: Elsevier; 2008. [Google Scholar]
- Pitcher MR, Ward CS, Arvide EM, Chapleau CA, Pozzo-Miller L, Hoeflich A, Sivaramakrishnan M, Saenger S, Metzger F, Neul JL. Insulinotropic treatments exacerbate metabolic syndrome in mice lacking MeCP2 function. Hum Mol Genet. 2013;22:2626–2633. doi: 10.1093/hmg/ddt111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitcher MR, Herrera JA, Buffington SA, Kochukov MY, Merritt JK, Fisher AR, Schanen NC, Costa-Mattioli M, Neul JL. Rett syndrome like phenotypes in the R255X Mecp2 mutant mouse are rescued by MECP2 transgene. Hum Mol Genet. 2015;24:2662–2672. doi: 10.1093/hmg/ddv030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez JM, Quellmalz UJ, Richter DW. Postnatal changes in the mammalian respiratory network as revealed by the transverse brainstem slice of mice. J Physiol. 1996;491:799–812. doi: 10.1113/jphysiol.1996.sp021258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez JM, Ward CS, Neul JL. Breathing challenges in Rett syndrome: lessons learned from humans and animal models. Respir Physiol Neurobiol. 2013;189:280–287. doi: 10.1016/j.resp.2013.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J, Ding X, Funk GD, Greer JJ. Anxiety-related mechanisms of respiratory dysfunction in a mouse model of Rett syndrome. J Neurosci. 2012;32:17230–17240. doi: 10.1523/JNEUROSCI.2951-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter DW. Neural regulation of respiration: rhythmogenesis and afferent control. In: Gregor R, Windhorst U, editors. Comprehensive human physiology, Vol II. Berlin: Springer; 1996. pp. 2079–2095. [Google Scholar]
- Rohdin M, Fernell E, Eriksson M, Albåge M, Lagercrantz H, Katz-Salamon M. Disturbances in cardiorespiratory function during day and night in Rett syndrome. Pediatr Neurol. 2007;37:338–344. doi: 10.1016/j.pediatrneurol.2007.06.009. [DOI] [PubMed] [Google Scholar]
- Samaco RC, Mandel-Brehm C, Chao HT, Ward CS, Fyffe-Maricich SL, Ren J, Hyland K, Thaller C, Maricich SM, Humphreys P, Greer JJ, Percy A, Glaze DG, Zoghbi HY, Neul JL. Loss of MeCP2 in aminergic neurons causes cell-autonomous defects in neurotransmitter synthesis and specific behavioral abnormalities. Proc Natl Acad Sci U S A. 2009;106:21966–21971. doi: 10.1073/pnas.0912257106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schüle B, Armstrong DD, Vogel H, Oviedo A, Francke U. Severe congenital encephalopathy caused by MECP2 null mutations in males: central hypoxia and reduced neuronal dendritic structure. Clin Genet. 2008;74:116–126. doi: 10.1111/j.1399-0004.2008.01005.x. [DOI] [PubMed] [Google Scholar]
- Smith CS, Tisak J, Bauman T, Green E. Psychometric equivalence of a translated circadian rhythm questionnaire: implications for between- and within-population assessments. J Appl Psychol. 1991;76:628–636. doi: 10.1037/0021-9010.76.5.628. [DOI] [PubMed] [Google Scholar]
- Smith JC, Abdala AP, Koizumi H, Rybak IA, Paton JF. Spatial and functional architecture of the mammalian brain stem respiratory network: a hierarchy of three oscillatory mechanisms. J Neurophysiol. 2007;98:3370–3387. doi: 10.1152/jn.00985.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JC, Abdala AP, Borgmann A, Rybak IA, Paton JF. Brainstem respiratory networks: building blocks and microcircuits. Trends Neurosci. 2013;36:152–162. doi: 10.1016/j.tins.2012.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song G, Xu H, Wang H, Macdonald SM, Poon CS. Hypoxia-excited neurons in NTS send axonal projections to Kolliker-Fuse/parabrachial complex in dorsolateral pons. Neuroscience. 2011;175:145–153. doi: 10.1016/j.neuroscience.2010.11.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stettner GM, Huppke P, Brendel C, Richter DW, Gärtner J, Dutschmann M. Breathing dysfunctions associated with impaired control of postinspiratory activity in Mecp2-/y knockout mice. J Physiol. 2007;579:863–876. doi: 10.1113/jphysiol.2006.119966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teppema LJ, Dahan A. The ventilatory response to hypoxia in mammals: mechanisms, measurement, and analysis. Physiol Rev. 2010;90:675–754. doi: 10.1152/physrev.00012.2009. [DOI] [PubMed] [Google Scholar]
- Torres-Andrade R, Moldenhauer R, Gutierrez-Bertín N, Soto-Covasich J, Mancilla-Medina C, Ehrenfeld C, Kerr B. The increase in body weight induced by lack of methyl CpG binding protein-2 is associated with altered leptin signalling in the hypothalamus. Exp Physiol. 2014;99:1229–1240. doi: 10.1113/expphysiol.2014.079798. [DOI] [PubMed] [Google Scholar]
- Viemari JC, Roux JC, Tryba AK, Saywell V, Burnet H, Peña F, Zanella S, Bévengut M, Barthelemy-Requin M, Herzing LB, Moncla A, Mancini J, Ramirez JM, Villard L, Hilaire G. Mecp2 deficiency disrupts norepinephrine and respiratory systems in mice. J Neurosci. 2005;25:11521–11530. doi: 10.1523/JNEUROSCI.4373-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voituron N, Zanella S, Menuet C, Dutschmann M, Hilaire G. Early breathing defects after moderate hypoxia or hypercapnia in a mouse model of Rett syndrome. Respir Physiol Neurobiol. 2009;168:109–118. doi: 10.1016/j.resp.2009.05.013. [DOI] [PubMed] [Google Scholar]
- Voituron N, Menuet C, Dutschmann M, Hilaire G. Physiological definition of upper airway obstructions in mouse model for Rett syndrome. Respir Physiol Neurobiol. 2010a;173:146–156. doi: 10.1016/j.resp.2010.07.006. [DOI] [PubMed] [Google Scholar]
- Voituron N, Zanella S, Menuet C, Lajard AM, Dutschmann M, Hilaire G. Early abnormalities of post-sigh breathing in a mouse model of Rett syndrome. Respir Physiol Neurobiol. 2010b;170:173–182. doi: 10.1016/j.resp.2009.12.009. [DOI] [PubMed] [Google Scholar]
- Ward CS, Arvide EM, Huang TW, Yoo J, Noebels JL, Neul JL. MeCP2 is critical within HoxB1-derived tissues of mice for normal life span. J Neurosci. 2011;31:10359–10370. doi: 10.1523/JNEUROSCI.0057-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weese-Mayer DE, Lieske SP, Boothby CM, Kenny AS, Bennett HL, Silvestri JM, Ramirez JM. Autonomic nervous system dysregulation: breathing and heart rate perturbation during wakefulness in young girls with Rett syndrome. Pediatr Res. 2006;60:443–449. doi: 10.1203/01.pdr.0000238302.84552.d0. [DOI] [PubMed] [Google Scholar]