

 This work is licensed under a
This work is licensed under a Summary
We present a case of a young female patient with a rare cause of relapsing and remitting Cushing’s syndrome due to ectopic ACTH secretion from a thymic neuroendocrine tumour. A 34-year-old female presented with a constellation of symptoms of Cushing’s syndrome, including facial swelling, muscle weakness and cognitive impairment. We use the terms ‘relapsing and remitting’ in this case report, given the unpredictable time course of symptoms, which led to a delay of 2 years before the correct diagnosis of hypercortisolaemia. Diagnostic workup confirmed ectopic ACTH secretion, and a thymic mass was seen on mediastinal imaging. The patient subsequently underwent thymectomy with complete resolution of her symptoms. Several case series have documented the association of Cushing’s syndrome with thymic neuroendocrine tumours (NETs), although to our knowledge there are a few published cases of patients with relapsing and remitting symptoms. This case is also notable for the absence of features of the MEN-1 syndrome, along with the female gender of our patient and her history of non-smoking.
Learning points
Ectopic corticotrophin (ACTH) secretion should always be considered in the diagnostic workup of young patients with Cushing’s syndrome
There is a small but growing body of literature describing the correlation between ectopic ACTH secretion and thymic neuroendocrine tumours (NETs)
The possibility of a MEN-1 syndrome should be considered in all patients with thymic NETs, and we note the observational association with male gender and cigarette smoking in this cohort
An exception to these associations is the finding of relatively high incidence of thymic NETs among female non-smoking MEN-1 patients in the Japanese compared with Western populations
The relapsing and remitting course of our patient’s symptoms is noteworthy, given the paucity of this finding among other published cases
Background
Cushing’s syndrome results from hypercortisolaemia, can present with a broad constellation of symptoms, and is relatively uncommon with an estimated incidence of between 0.2 and 5 per million persons annually (1). Early diagnosis and initiation of treatment for Cushing’s syndrome is critical, given the morbidity and mortality associated with prolonged glucocorticoid exposure, including cardiovascular disease, thromboembolic complications, immunosuppression and neuro-cognitive disorders (1).
Cushing’s syndrome is most often due to a corticotrophin (ACTH)-producing pituitary tumour, or is secondary to cortisol secretion by an adrenal adenoma or carcinoma. A smaller proportion of Cushing’s syndrome is the result of ectopic ACTH secretion from a non-pituitary source, estimated to account for 5–10% of all cases (1). Ectopic ACTH secretion has been described in case series, most frequently arising from pulmonary carcinoids and pancreatic NETs, with very small numbers of thymic NETs, medullary thyroid cancer, small cell colon or small cell lung cancer, NETs of unidentified source, or phaeochromocytomas accounting for the remaining published cases (2, 3).
There is a modest body of literature describing Cushing’s syndrome resulting from ACTH secretion from thymic tumours. The relationship between a thymic mass and ectopic ACTH secretion was first proposed in 1931 by Leyton et al. (4, cited in 1). There have subsequently emerged case reports of 92 patients and a case series of 12 patients describing thymic NETs as the cause of Cushing’s syndrome (5). Features of this rare group include a young age at diagnosis (median age 35years), male preponderance, and the absence of cyclical features. Although cyclical Cushing’s syndrome due to a pituitary adenoma is a well-recognised phenomenon, within the published literature to date, there exist very few cases of relapsing and remitting Cushing’s syndrome due to ectopic ACTH secretion from any source, and only four identified cases due to thymic NETs (2, 3, 5, 6). Although the relapsing and remitting symptoms experienced by our patient are akin to those seen in cyclical Cushing’s syndrome of pituitary origin, the timing of the periods of her symptomatic hypercortisolaemia remained highly variable, a feature not yet widely described in this context.
Case presentation
A 34-year-old female patient of Japanese origin presented to her general practitioner (GP) with non-specific symptoms of 2-year duration. She described a multitude of symptoms including swelling of the face and peripheries, lethargy, muscle weakness, sleep disturbance and cognitive blunting, all of which caused significant detriment to her quality of life. She reported that since their onset in December 2012, her symptoms had worsened over a 5-month period and recurred in subsequent winter seasons over two consecutive years, during which time she kept meticulous personal records. Notably, her symptoms improved in the summer months and resolved completely between episodes.
In the 2years preceding referral to our centre, the patient was investigated by her GP in the United Kingdom and a physician in Japan. Diagnoses of vitamin D deficiency, hypothyroidism and polycythaemia were made, and treated with vitamin D and thyroxine replacement.
Between December 2014 and March 2015, the patient reported worsening of these symptoms, in addition to new dryness of the skin and areas of pigmentation. Preliminary investigations performed by the GP demonstrated hypokalaemia of 3.2mmol/L (normal range (NR) 3.5–5.5mmol/L), an elevated random serum cortisol of 1469nmol/L, and incomplete suppression to 158nmol/L after an overnight 1mg dexamethasone suppression test (NR<50nmol/L). Oral potassium supplementation was commenced, and the patient was referred to our Endocrinology service for further inpatient investigation of Cushing’s syndrome.
Physical examination revealed a slightly round facial appearance (not present in photographs taken 2years before), skin pigmentation over the dorsal aspects of the hands, and proximal muscle weakness. The patient was noted to be a non-smoker.
There was no family history of MEN-1.
Investigation
Investigations included: midnight serum cortisol concentration 331nmol/L (NR:<50nmol/L), 24h urine cortisol concentration 363nmol/L (NR: 10–150nmol/L), potassium 3.2mmol/L, and glycated haemoglobin 6.8% (NR: 4–6%). Low-dose dexamethasone suppression testing (2mg daily for 48h) revealed serum cortisol concentrations of 484, 648 and 1536nmol/L at 0, 24 and 48h respectively. A human sequence corticotrophin-releasing hormone (CRH) stimulation test demonstrated failure of ACTH or cortisol stimulation (Table 1).
Table 1.
CRH stimulation test demonstrating serum ACTH and cortisol values following administration of 100μg hCRH via intravenous route.
| Time (min) | CRH stimulation test | ||
|---|---|---|---|
| Time taken (h) | ACTH (ng/L) | Cortisol (nmol/L) | |
| −15 | 09:25 | 755 | 2369 |
| 0 | 09:40 | 686 | 2204 |
| CRH 100 μg administered via the intravenous route | |||
| 15 | 09:55 | 762 | 2122 |
| 30 | 10:10 | 751 | 2103 |
| 45 | 10:25 | 736 | 2210 |
| 60 | 10:40 | 693 | 2181 |
| 90 | 11:10 | 713 | 2380 |
| 120 | 11:40 | 686 | 2231 |
All other routine blood parameters, urea and other electrolytes were within normal limits.
Magnetic resonance imaging (MRI) of the pituitary demonstrated an asymmetrical gland, with a possible 8mm round nodule in the right side of the sella. Computed tomography (CT) scanning of the chest, abdomen and pelvis demonstrated a 24mm anterior mediastinal nodule (Fig. 1), felt to represent a thymoma, in addition to bulky appearances of the adrenal glands.
Figure 1.
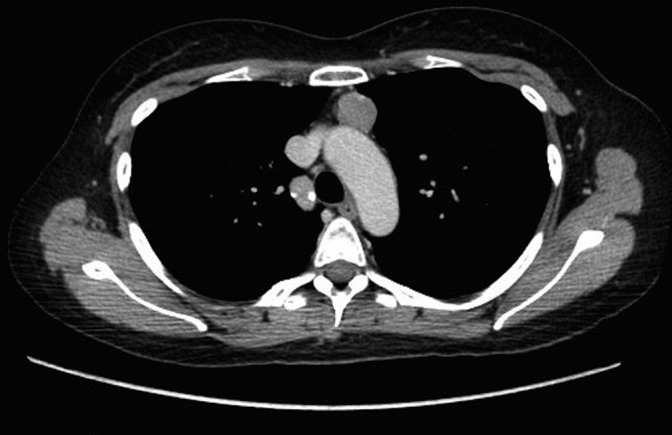
CT slice demonstrating anterior mediastinal nodule.
The findings of an elevated midnight cortisol and failure to suppress on low-dose dexamethasone testing confirmed the diagnosis of hypercortisolaemia and therefore Cushing’s syndrome. The elevated ACTH levels were suggestive of ACTH-dependent hypercortisolaemia, and the results of the CRH stimulation test pointed towards an ectopic source of ACTH. However, radiological investigations were suggestive of two possible sources of ACTH secretion, either the possible pituitary abnormality on pituitary MRI or the thymoma seen on CT of the chest.
Despite serum cortisol levels in excess of 1000nmol/L at presentation, following the period of initial investigation, this level of hypercortisolaemia was not seen again on repeated biochemical testing. The patient continued to run weekly serum cortisols at 09:00h of approximately 250–350nmol/L, suggesting that the period of hypercortisolaemia had remitted. The impression that the ACTH source was behaving in a periodic secretory pattern was therefore derived from the detailed presenting complaint obtained from the patient and her records, and the demonstrable change in the degree of hypercortisolaemia in the short time frame between her presentation to our service and further monitoring of serum cortisol only 3–4weeks later.
Bilateral inferior petrosal sinus sampling (BIPSS) was performed in order to evaluate the significance of the pituitary lesion seen on MRI. Despite following basal serum cortisol levels for 2months following initial presentation, further biochemical evidence of hypercortisolaemia could not be demonstrated. As such, a decision was taken to perform BIPSS irrespective of the normal serum cortisol of 289nmol/L, given concern regarding delay in further evaluation of the thymic tumour.
BIPSS revealed no significant gradient between sinus ACTH and peripheral ACTH, and was therefore not suggestive of a pituitary source of ACTH. As such, the pituitary lesion was felt to be an incidental finding as can be detected in a significant minority of the general population undergoing MRI.
Given the possibility of a potentially malignant thymic tumour, a decision was made to refer directly for surgical resection rather than preliminary biopsy. Octreotide radionuclide scanning demonstrated no uptake by the thymic lesion.
Treatment
During admission, the patient was continued on potassium replacement for hypokalaemia, which proved difficult to treat and required maximum dosing with oral potassium tablets as well as a potassium-sparing diuretic (amiloride). She received thromboembolic prophylaxis with low-molecular-weight heparin and subsequently proceeded to thymectomy under the Cardiothoracic Surgery service. There was no suggestion of macroscopic extension or persistent disease at operation. Histopathology confirmed a well-differentiated grade 1 neuroendocrine tumour (Fig. 2) with focal microscopic involvement at the surgical margin. Immunostaining demonstrated ACTH staining within the tumour, thus confirming the source of ectopic ACTH secretion.
Figure 2.
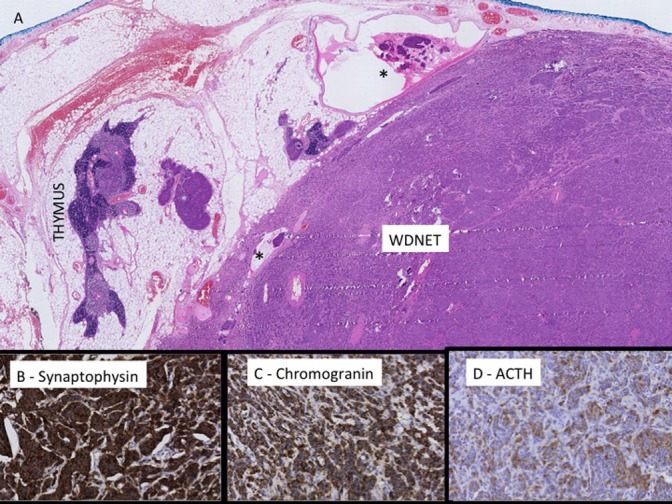
(A) H&E of residual thymic tissue and well-differentiated neuroendocrine tumour (WDNET) with foci of lymphatic space invasion (*). The mitotic count is 1 per 10 HPFs and there is no necrosis. Immunohistochemical studies show positive immunoreactivity with synaptophysin (B), chromogranin (C) and ACTH (D).
Outcome and follow-up
Monitoring during the perioperative period demonstrated marked reduction in plasma ACTH concentrations (Table 2).
Table 2.
Perioperative ACTH monitoring.
| Day pre- or post-operation | ACTH (ng/L) |
|---|---|
| −14 | 93.5 |
| −7 | 94.2 |
| +1 | <5.0 |
| +28 | 28.9 |
At a 3-month review, no clinical or biochemical evidence of persisting disease was elicited. The patient reported complete resolution of her presenting symptoms. Serum cortisol was 193nmol/L and plasma ACTH was 29.8ng/L (NR 0–46ng/L). Repeat CT of the chest at both 3 and 6months demonstrated no recurrence of the thymic mass.
Genetic testing for MEN-1 mutations including testing for multiplex ligation-dependent probe amplification was negative.
The patient has remained clinically well following thymectomy, and is now approximately 1year into follow-up.
In view of the finding of the pituitary lesion, it is anticipated that the patient will continue annual MRI surveillance for 1–2years.
Discussion
This patient presented with a constellation of relapsing and remitting symptoms over a 2-year period, before correct identification of the hypercortisolaemia due to a thymic NET. Although her symptoms were intermittent and seasonal, biochemical evidence of a constant and predictable pattern of hypercortisolaemia could not be demonstrated. As such, the term ‘cyclic’ Cushing’s syndrome is not used in reference to our case, and we use the terms ‘relapsing and remitting’ instead. The difficulty of diagnosis of periodic hypercortisolaemia in the context of intermittent symptoms is evident in this case, given the critical timing of stimulatory tests in relation to the patient cycling in or out of the symptomatic phase.
Ectopic secretion of ACTH is reported to be the cause of cyclic Cushing’s syndrome in up to 25% of cases (7). Although the initial CRH test in our patient was indeed suggestive of an ectopic source, the radiographic findings of a pituitary abnormality in addition to the thymoma provided a significant differential diagnosis for consideration. Furthermore, the finding of a normal serum cortisol level at the time of her BIPSS was a significant consideration. Nonetheless, the BIPSS findings supported an ectopic source of ACTH; in conjunction with the finding of a thymic mass on CT imaging, this was concluded to be the culprit lesion. When a pituitary or adrenal cause for Cushing’s syndrome is not evident, early workup and timely recognition of an ectopic source of ACTH is extremely important, allowing prompt correction of hypercortisolaemia by curative surgery or adjunctive treatment. Despite the resolution of hypercortisolaemia and clinical symptoms with resection of the thymic tumour in this case, the importance of ongoing surveillance of any incidentally noted pituitary tumour cannot be overlooked.
In general, published cases of thymic NET associated with MEN-1 syndrome document a strong correlation between both male gender and cigarette smoking, as well as the absence of Cushing’s syndrome (5, 7, 8). This case is, however, notable for the gender and ethnicity of our patient. While Western case series report an incidence of thymic NETs in female patients with MEN-1 of between 0 and 9% (8, 9), a Japanese series documented a significantly higher incidence of 36% among female patients (10). Although the authors note that prevalence among females remained significantly lower than in male patients (3.2% vs 7.6%), this has significant implications for surveillance among female MEN-1 patients who may previously have been considered to be at very low risk of thymic NET. Furthermore, this finding was not limited to smokers. The precise mechanism behind such a discrepancy between ethnic groups remains unknown and requires further investigation; however, clinicians should be aware of the need to consider a thymic NET as a possible diagnosis even in female non-smoking patients with ectopic ACTH.
The periodic nature of the clinical symptoms and highly variable degree of hypercortisolaemia in this case is also noteworthy, as to our knowledge there are only four published cases of relapsing and remitting Cushing’s syndrome due to thymic NETs (2, 3, 5, 6). These features may be considered more suggestive of a pituitary source; however, this was excluded in this case by the normal BIPSS and normalisation of clinical and biochemical features following resection of the thymic tumour.
We do note that the young age of our patient is consistent with other published case series – to which end we conclude that ectopic ACTH is an infrequent but important differential for exclusion in patients of this age group presenting with Cushing’s syndrome. In broader terms, given the implications of MEN-1 diagnosis on morbidity and life expectancy, genetic screening of any patients presenting with a thymic mass remains imperative.
Finally, we cannot account for persistent hypokalaemia in the presence of normalised cortisol prior to thymectomy in this patient. In general, the hypokalaemia of Cushing’s syndrome is directly related to hypercortisolaemia; however, in our case, serum potassium remained persistently low despite resolution of hypercortisolaemia. In this case the discordance is unexpected; although highly speculative, it is possible that another proopiomelanocortin-related peptide is responsible.
Patient consent
The authors confirm that written informed consent was obtained from the patient for publication of the submitted article and accompanying images before submission.
Author contribution statement
Drs M Trott, G Farah and V Stokes attended the patient during inpatient stay. Dr M Trott wrote the first draft. Professor A Grossman supervised the care of the patient as the named Consultant. Dr L M Wang provided the histopathology slides included as figures in this report.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This research did not receive any specific grant from any funding agency in the public, commercial or not-for-profit sector.
References
- 1. Lacroix A, Feelders RA, Stratakis CA & Nieman LK 2015. Cushing’s syndrome. Lancet 386 913–927. 10.1016/S0140-6736(14)61375-1 [DOI] [PubMed] [Google Scholar]
- 2. Ilias I, Torpy DJ, Pacak K, Mullen N, Wesley RA & Nieman LK 2005. Cushing’s syndrome due to ectopic corticotropin secretion: twenty year’s experience at the National Institutes of Health . Journal of Clinical Endocrinology and Metabolism 90 4955–4962. 10.1210/jc.2004-2527 [DOI] [PubMed] [Google Scholar]
- 3. Isidori AM, Kaltsas GA, Pozza C, Frajese V, Newel-Price J, Reznek DH, Jenkins PJ, Monson JP, Grossman AB & Besser GM 2006. The ectopic adrenocorticotropin clinical features, diagnosis, management, and long-term follow up. Journal of Clinical Endocrinology and Metabolism 91 371–377. 10.1210/jc.2005-1542 [DOI] [PubMed] [Google Scholar]
- 4. Leyton O, Turnbull HM & Bratton AB 1931. Primary cancer of the thymus with pluriglandular disturbance. Journal of Pathology and Bacteriology 34 635–660. 10.1002/(ISSN)1555-2039 [DOI] [Google Scholar]
- 5. Neary NM, Lopez-Chavez A, Abel BS, Boyce AM, Schaub N, Kwong K, Stratakis CA, Moran CA, Giaccone G & Nieman LK 2012. Neuroendocrine ACTH-producing tumour of the thymus – experience with 12 patients over 25 years. Journal of Clinical Endocrinology and Metabolism 97 2223–2230. 10.1210/jc.2011-3355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Estopinan V, Varela C, Riobo P, Dominguez JR & Sancho J 1987. Ectopic Cushing’s syndrome with periodic hormonogenesis – a case suggesting a pathogenetic mechanism. Postgraduate Medical Journal 63 887–889. 10.1136/pgmj.63.744.887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Meinardi JR, Wolffenbuttel BHR & Dullaart RPF 2007. Cyclic Cushing’s a clinical challenge. European Journal of Endocrinology 157 245–254. 10.1530/EJE-07-0262 [DOI] [PubMed] [Google Scholar]
- 8. de Laat JM, Pieterman CR, van den Broek MD, Twisk JW, Hermus AR, Dekkers OM, de Herder WW, van der Horst-Schrivers AN, Drent ML & Bisschop PH et al. 2014. Natural course and survival of neuroendocrine tumors of thymus and lung in MEN-1 patients. Journal of Clinical Endocrinology and Metabolism 99 3325–3333. 10.1210/jc.2014-1560 [DOI] [PubMed] [Google Scholar]
- 9. Ferolla P, Falchetti A, Filosso P, Tomassetti P, Tamburrano G, Avenia N, Daddi G, Puma F, Ribacchi R & Santeusanio F et al. 2005. Thymic neuroendocrine carcinoma (carcinoid) in multiple endocrine neoplasia type 1 syndrome: the Italian series. Journal of Clinical Endocrinology and Metabolism 90 2603–2609. 10.1210/jc.2004-1155 [DOI] [PubMed] [Google Scholar]
- 10. Sakurai A, Imai T, Kikumori T, Horiuchi K, Okamoto T, Uchino S, Kosugi S, Suzuki S, Suyama K & Yamazaki M et al. 2013. Thymic neuroendocrine tumour in multiple endocrine neoplasia type 1: female patients are not rare exceptions. Clinical Endocrinology 78 248–254. 10.1111/j.1365-2265.2012.04467.x [DOI] [PubMed] [Google Scholar]