

Abstract
Objective:
We undertook a systems immunology approach of the adaptive immune system in multiple sclerosis (MS), overcoming tradeoffs between scale and level of detail, in order to identify the immunologic signature of MS and the changes wrought by current immunomodulatory treatments.
Methods:
We developed a comprehensive flow cytometry platform measuring 38 immunologic cell types in the peripheral blood of 245 individuals in a routine clinical setting. These include patients with MS, untreated or receiving any of 4 current immunomodulatory treatments (interferon-β, glatiramer acetate, natalizumab, or fingolimod), patients with autoimmune thyroid disease, and healthy controls.
Results:
An increase in memory CD8+ T cells and B cells was observed in untreated patients with MS. Interferon-β and fingolimod induce significant changes upon multiple aspects of the peripheral immune system, with an unexpectedly prominent alteration of B cells. Overall, both treatments push the immune system in different directions, with only 2 significant effects shared across these treatments—an increase in transitional B cells and a decrease in class-switched B cells. We further identified heightened B cell-activating factor (BAFF) levels as regulating this shared B cell pathway.
Conclusions:
A systems immunology approach established different immunologic profiles induced by current immunomodulatory MS treatments, offering perspectives for personalized medicine. Pathways shared between the immunologic architecture of existing efficacious treatments identify targets for future treatment design.
The emergence of high-throughput genetics technology led to the identification of >100 risk variants for multiple sclerosis (MS), reinforcing the key role for adaptive immunity.1,2 The basis for understanding the immunology of MS is, by contrast, still largely founded upon animal models where intrinsic limitations in the fidelity of models to disease processes, interspecies barriers, and the highly complex nature of MS have hampered translational potential.3 Despite progress in the treatment of MS, the predictive capacity for successful treatments remains poor, with a trial and error approach being required, and the precise mechanism of action often remains unclear.3,4 These observations have spurred the call for characterization of the immune system in MS and upon treatment.3,5
In-depth analysis of variation in the adaptive immune system in humans has proven to be difficult due to trade-offs between scale and level of detail. Large-scale flow cytometry screens have now been able to characterize immune cells in the peripheral blood of healthy controls.6–8 We have here applied this systems immunology approach to the study of MS and, for comparison, autoimmune thyroid disease (AITD), characterized by familial clustering and shared genetic risk factors with MS.9,10 In an unbiased analysis, we revealed the immunologic signatures of MS and 4 current immunomodulatory treatments. The prominent alterations in B cell subsets, most importantly the increase in B cell-activating factor (BAFF) and transitional B cells induced by both interferon-β and fingolimod, indicate the importance of the early B cell pathway in treating MS.
METHODS
Study population.
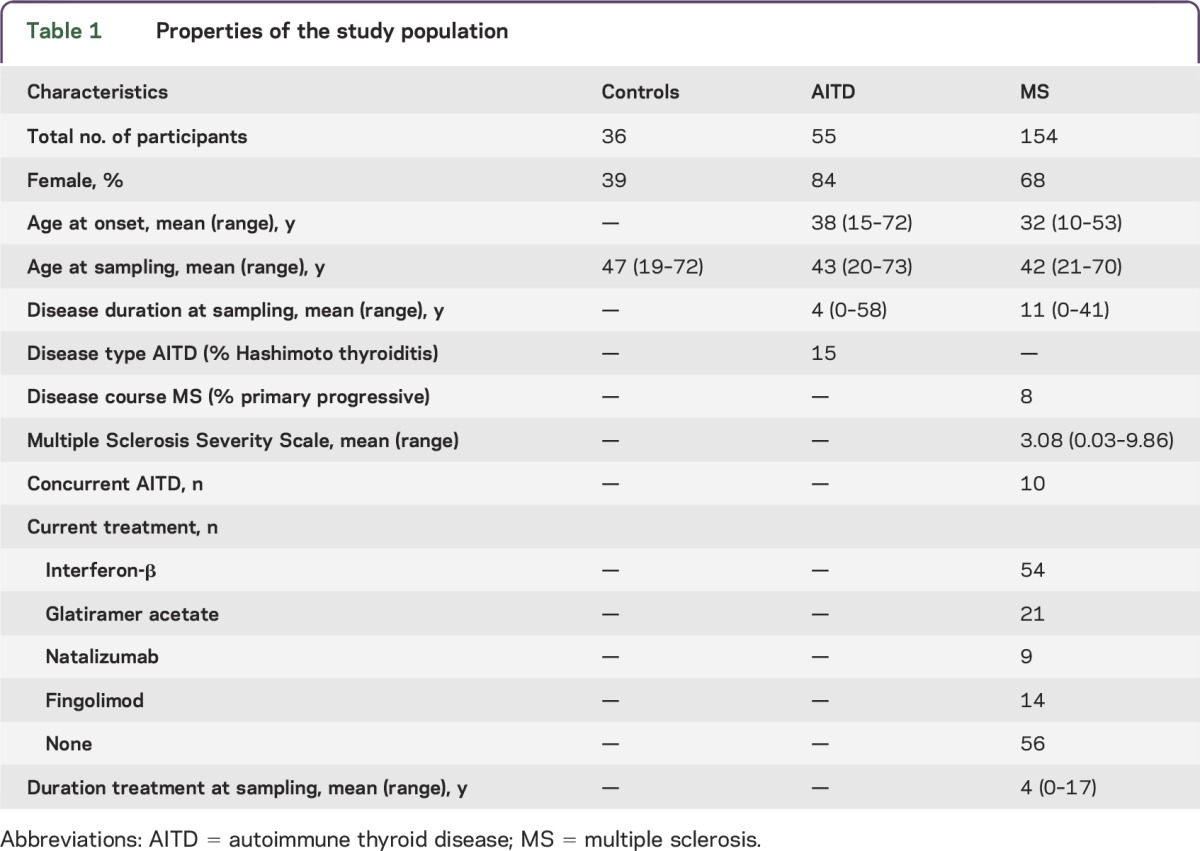
Unrelated patients of Caucasian descent fulfilling McDonald criteria for MS or criteria for clinical AITD, and spouses as healthy controls, were included in the study over a 22-month inclusion period (table 1). Extensive demographic and clinical data were collected through a questionnaire and medical records. Exclusion criteria were cancer, immunosuppressive treatments, antibiotics, anti-allergy or anti-inflammatory treatment in the week prior to sampling, and, for healthy controls, presence of known clinical autoimmune disease.
Table 1.
Properties of the study population
Standard protocol approvals, registrations, and patient consents.
Participants gave written informed consent, and the study was approved by the Ethics Committee of the University Hospitals Leuven.
Immunophenotyping and cytokine measurements.
Peripheral blood mononuclear cells (PBMCs) and plasma were isolated from heparinized blood and stored at −80°C prior to analysis. A multiplex flow cytometry platform, previously validated in healthy controls,8 was applied (table e-1 and figure e-1 at Neurology.org/nn). Some variables were measured in subsets of the samples only. Measurements are expressed as percentage of parental or grandparental cells as indicated. BAFF plasma levels were measured using a human BAFF Quantikine ELISA (R&D Systems, Minneapolis, MN). Averages were taken over 2–3 replicates after standardization using samples present on multiple plates. Sensitivity was 6.44 pg/mL and average standard deviation 15%.
Gene expression.
RNA was extracted from total PBMCs and reverse transcribed using a high-capacity cDNA reverse transcription kit (Life Technologies, Carlsbad, CA). Droplet digital PCR (Bio-Rad, Hercules, CA) with gene expression assays (Life Technologies) for the BAFF gene TNFSF13B (Hs00198106_m1) and housekeeping gene POLR2A (Hs00172187_m1) was performed using 4 ng/μL cDNA. Relative quantity of TNFSF13B vs POLR2A was measured with QuantaSoft v1.4 (Bio-Rad). Correlation between separate experiments (n = 68) was 0.75.
Statistical analysis.
For ratios of variables, the natural logarithm was taken. Spearman correlation for pairs of variables and significance in correlation coefficient between groups were calculated and plotted with heatmap.2 and cocor packages. Linear regression models in R 2.14.1 included the immunologic variable as function of disease or treatment and covariates sex, age, and, for disease effects, disease duration at sampling. Multiple testing correction was applied for 38 variables across 4 treatments (p = 0.00033). Nonmetric multidimensional scaling was used to visualize dissimilarities of samples between treatments. The ordination was created in R 3.1.0 using R package vegan with custom scripts. Distance matrix was calculated with Bray-Curtis dissimilarity index on original data. To test for the contribution of each variable to the resulting distance matrix, we applied Mantel test with 999 permutations comparing the original distance matrix to one with one variable removed at a time.
RESULTS
The adaptive immune profile of MS.
In order to directly study the adaptive immune system across conditions, we applied a systems immunology approach in 154 patients with MS, 36 controls, and, for comparison, 55 patients with AITD (table 1). For each individual, the frequency of 38 leukocyte populations or ratios was assessed in the peripheral blood utilizing multiparameter flow cytometry, with a focus on T- and B cell subsets (table e-1 and figure e-1). Phenotype–phenotype correlations were assessed across each group (figure 1), identifying coregulated leukocyte populations that validate the systems immunology approach used here, as demonstrated previously.8 Treatment of MS resulted in substantial immunologic deviation (described below), so initial analysis was performed on untreated patients with MS. Comparison of the immunologic status of patients with AITD currently untreated or treated with the thyroperoxidase inhibitor methimazole revealed no consistent differences (data not shown), allowing merged analysis. Of the robust immunologic changes observed to occur in autoimmune disease patients compared to healthy individuals, there were, surprisingly, no overlaps between AITD and MS (table e-2). MS was correlated with perturbation in the CD8+ T-cell compartment compared to healthy controls, with a shift towards effector memory CD8+ T cells (CD4−CD8+CD45RA−CCR7−) as a percentage of all CD8+ T cells (p = 0.012), and increased the proportion of B cells, resulting in a decreased T/B cell ratio. The increase in B cells was mainly due to an increase in memory B cells (CD4−CD19+IgM+CD27+) as a percentage of total lymphocytes (p = 0.0042), and was especially prominent early on in disease (figure 2, A and B, and table e-2). Disease course (bout onset or primary progressive), time since last relapse, and multiple sclerosis severity scores did not confound these observations (data not shown). The relationship between different immunologic populations is largely left intact in patients with MS but the positive coregulation between B cells and the naive CD4+ T-cell cluster disappeared (r controls = 0.37, r MS = −0.36, p = 0.00064) (figure 1A). Four immunologic variables were associated with nominal significance with AITD but not MS: increased CD4+ recent thymic emigrant (RTE) (p = 0.047) and Th17 (p = 0.046) cells as percentage of CD4+ T cells and increased transitional (p = 0.046) with decreased switched B cells as percentage of B cells (p = 0.0019) (figure 2, C–F). A Th17-positive coregulation with effector memory CD4+ T cells (r controls = −0.16, r AITD = 0.51, p = 0.0092) and negative coregulation with the CD4+-naive cluster (r controls = 0.093, r AITD = −0.483, p = 0.027) appeared in patients with AITD that was not present in healthy individuals or patients with MS (figure 1B), indicating greater influence of Th17 cells over the immunologic landscape in AITD.
Figure 1. The human adaptive immune profile in multiple sclerosis (MS).

In A and B, the right upper half, above the diagonal, indicates coregulation between pairs of cell types in healthy controls (n = 36) (red: positive correlation coefficient, blue: negative correlation coefficient, light gray: no data available). Unbiased clustering of coefficients was performed to group coregulated cell types. Upon clustering, a main distinct naive cluster was observed, consisting of coregulated CD4+ T cells, CD4+/CD8+ ratio, and naive T cells, together with recent thymic emigrants (RTE) of both CD4+ and CD8+ lineages. This naive cluster was negatively correlated with activated and antigen-experienced lymphocyte subsets. Several microclusters were additionally observed, with coregulation of regulatory T cells (Tregs) with central memory CD4+ and CD8+ T cells (the antigen-experienced microcluster), coregulation of Th1 cells and interferon-γ-producing CD8+ cells (the Th1 microcluster), coregulation of Th2 and memory B cells (the Th2 microcluster), and so forth, revealing the emergence of known immunologic interactions through this systems immunology approach. In the lower left half, below the diagonal, dark gray indicates coregulation between pairs of cell types in (A) patients with untreated MS (MS_UNT, n = 56) and (B) patients with autoimmune thyroid disease (AITD, n = 55) that does not differ significantly from healthy controls. Coregulation between pairs of cell types that is significantly altered by disease (p < 0.05, and boxed if p < 0.01) in (A) untreated patients with MS vs controls and (B) patients with AITD vs controls is colored (red: positive correlation coefficient, blue: negative correlation coefficient, light gray: no data available).
Figure 2. Distinct changes to immunoprofile in autoimmune thyroid disease (AITD) and multiple sclerosis (MS).

A linear regression with covariates age, sex, and disease duration was applied to the immune profiling results of patients with AITD and untreated patients with MS vs healthy controls, and—within patients—untreated MS vs AITD. Significant differences in the peripheral immune system of untreated patients with MS compared to controls were restricted to (A) effector memory CD8+ cells and (B) memory B cells. Significant differences compared to controls observed for patients with AITD but not patients with MS include altered proportions of (C) recent thymic emigrant CD4+, (D) Th17, (E) transitional B cells, and (F) class-switched B cells. Median with boxes indicate 25th and 75th percentile and whiskers indicate 1.5 × interquartile range.
The immunologic profile induced by MS treatments.
After establishing the immunologic changes induced by MS in untreated patients, we investigated the immunologic signature induced by 4 common immunomodulatory treatments for MS (table e-3). Glatiramer acetate (Copaxone; Teva Pharmaceuticals, Castleford, UK; n = 21) and natalizumab (Tysabri; Biogen Idec, Maidenhead, UK; n = 9) induced several immunologic changes reaching nominal significance, including an increase in the frequency of RTE CD4+ T cells (p = 0.045) with glatiramer acetate and of switched B cells (p = 0.023) upon treatment with natalizumab; however, none of these changes survived multiple testing (152 tests corresponding to p = 0.00033).
Interferon-β (Avonex; Biogen Idec, Maidenhead, UK; Betaferon; Bayer Pharma AG, Berlin, Germany; Rebif; Merck Serono, London, UK; n = 54) and fingolimod (Gilenya; Novartis Pharma AG, Horsham, UK; n = 14), on the other hand, were observed to alter multiple aspects of the peripheral immune system, both in a multidimensional analysis including all 38 variables (figure 3) and for individual variables (table e-3). A total of 4 variables regulated by treatment with interferon-β survived multiple testing (p < 0.00033) (figure 4, A–D). These include 2 changes in frequency of the B cell population, with an increase in transitional B cells (p = 0.00030) and decrease in switched B cells (p = 8.0E-05). The final significant changes were a decrease in frequency of myeloid dendritic cells (mDCs) (p = 9.22E-06) and interferon-γ-producing CD8+ cells (p = 0.00032), with an additional 12 variables reaching nominal significance (p < 0.05) (table e-3), including an increase in total (p = 0.0064) and naive (p = 0.00037) B cells, and a decrease in Th1 (p = 0.0014). The decrease in mDCs and in Th1 cells is correlated with the increase in B cells (p = 0.023 and p = 0.033, respectively), suggesting the B cell changes as primary drivers in the interferon-β-driven immune deviation.
Figure 3. Multiple sclerosis (MS) immunomodulatory treatments interferon-β (IFNB) and fingolimod (FTY720) result in global perturbation of the immune system.

Patients with MS being treated with one of 4 immunomodulatory treatments (GA = glatiramer acetate; NAT = natalizumab) were plotted together with controls (CON) and untreated (UNT) patients with MS using nonparametric multidimensional scaling over 38 immunologic variables. Individual patients and distance from the average of each condition are shown. Variation explained by each axis is indicated in the parentheses. Pairwise distance of samples was calculated based on Bray-Curtis dissimilarity index.
Figure 4. Shared and unique immune changes induced by multiple sclerosis (MS) immunomodulatory treatments.

The effect of 4 immunomodulatory treatments (interferon-β [IFNB], glatiramer acetate [GA], natalizumab [NAT], fingolimod [FTY720]) was compared with combined controls (CON) and untreated (UNT) patients with MS, after establishing no significant difference between the latter 2 groups (except for panels E and F). A linear regression with covariates age and sex was applied. Significant differences (after multiple testing correction) in the peripheral immune system of patients with MS following treatment compared to controls and untreated patients with MS included altered proportions of (A) transitional B cells, (B) switched B cells, (C) myeloid dendritic cells (mDC), (D) interferon-γ-producing CD8+ T cells, (E) B cells, (F) T cell/B cell ratio, (G) naive B cells, (H) plasmablasts, (I) CD4+/CD8+ T cell ratio, (J) Th2 cells, (K) interleukin-2 (IL-2)–producing CD4+ T cells, (L) CD8+ terminally differentiated effector memory T cells (TEMRA), (M) IL-2-secreting CD8+ T cells, and (N) natural killer T cell (NKT) cells. Median with boxes indicating 25th and 75th percentile and whiskers indicating 1.5 × interquartile range.
Fingolimod induced the strongest changes in the peripheral immune system of the 4 routine MS treatments (figure 3), with changes in the frequency of 13/38 of the measured immunologic variables surviving multiple testing (p < 0.00033) (figure 4), and another 14 reaching nominal significance (p < 0.05) (table e-3). Within the T-cell compartment, the observed reductions in the frequency of C-chemokine receptor type 7–expressing naive and central memory populations were consistent with the biological function for fingolimod,11 but within the effector population, Th2 cells proved relatively refractory to fingolimod compared to Th1, Th17, and Tfh cells (table e-3, figure e-2). Compared to the decrease in the T-cell compartment, the observed decrease in B cell frequency (p = 1.16E-10) (figure 4E) was, however, more profound. These alterations were driven by reduced switched (p = 0.00014) and naive (p = 6.44E-09) B cells (figure 4, B and G), with transitional B cells (p = 0.0012) and plasmablasts (p = 3.50E-09) (figure 4, A and H) increased as a proportion of B cells but remaining constant as a proportion of total lymphocytes (figure e-3).
Early B cell changes as shared immunologic alterations across treatments.
At a global level, the 2 strongest immunomodulatory treatments pushed the immune system in different directions (figure 3), with only 2 significant effects shared across treatments—a decrease in switched B cells and an increase in transitional B cells as percentage of total B cells upon treatment with both interferon-β and fingolimod (figure 4, A and B). As BAFF drives the differentiation of transitional B cells,12,13 we additionally measured levels of BAFF in the same individuals. We observed upregulated BAFF plasma levels for the AITD disease process (p = 0.0011). In MS, levels were unaltered in untreated patients but were upregulated during treatment with interferon-β (high-dose p = 2.69E-09) and fingolimod (p = 2.17E-10), in parallel with changes in the B cell compartment (figure 5, A–C). Treatment-induced changes followed a dose-dependent model (for interferon-β) (figure 5, A–C) and decreased by increased treatment duration (data not shown). Changes in BAFF plasma levels were correlated with the observed changes in B cell immunophenotype, i.e., the increase in the frequency of transitional B cells (r2 = 0.30, p = 0.0047) and decrease in switched B cells (r2 = 0.072, p = 0.022) as percentage of B cells, after accounting for disease/treatment group, sex, and age (figure 5, D–E). In line with protein levels, transcription levels of the BAFF gene (TNFSF13B) in total PBMCs were upregulated in patients with AITD (p = 0.046) as well as upon treatment of patients with MS with high-dose interferon-β (p = 0.035) or fingolimod (p = 0.00087) (figure 5F).
Figure 5. Increased B cell-activating factor (BAFF) levels are shared between immunomodulatory treatments.

(A) Plasma BAFF levels, (B) transitional B cells, (C) switched B cells, and (F) total peripheral blood mononuclear cells (PBMC) BAFF gene expression levels (RQ = relative quantity compared to housekeeping gene, with addition of n = 13 additional controls not part of the immunophenotyping cohort) were quantified in controls (CON), patients with autoimmune thyroid disease (AITD), and patients with multiple sclerosis (MS) either untreated (MS_UNT) or treated with 4 immunomodulatory treatments (interferon [IFN]–β low and IFN-β high = IFN-β low or high dose; GA = glatiramer acetate; NAT = natalizumab; FTY720 = fingolimod). Median with boxes indicating 25th and 75th percentile and whiskers indicating 1.5 × interquartile range. A linear regression with covariates age and sex was applied. Correlation between plasma BAFF levels and (D) transitional B cells and (E) switched B cells in the entire cohort after accounting for disease/treatment group, age, and sex.
DISCUSSION
In this study, we have undertaken a comprehensive and unbiased analysis of the adaptive immune system measuring 38 immunologic variables in 245 individuals. Our approach allows us to directly compare, on the same large-scale platform, MS and AITD, 2 distinct autoimmune diseases with elements of shared genetic risk9,10 as well as 4 common immunomodulatory treatments for MS (interferon-β, glatiramer acetate, natalizumab, fingolimod), of which the mechanism of action is not fully understood.3,4
Patients with MS displayed systematic immune deviations, with an overall increase in effector memory CD8+ T cells and memory B cells. Both cell types have recently been reported as part of the inflammatory response in MS14,15 and as populating the CNS in MS,16–18 providing a biological parallel to these systems–immunology associations. Despite the shared genetic risk factors, the immunologic deviations observed for MS were distinct from AITD. This result opposes a model (implied from genome-wide association studies) where common immunologic processes underlie different autoimmune diseases, with the unique clinical manifestations being based on the specificity of the escaping forbidden clones. By contrast, the results here support a more complex gene–immune relationship, where largely overlapping genetic polymorphisms nevertheless synergize to form immunologic deviations that are distinct at the systems level.
In addition to the different efficacy and safety profile of the 4 compounds in routine MS treatment, each induced unique constellations of immune deviations, which offers perspectives to the challenge of personalized medicine.5 Alterations in B cells were unexpectedly prominent upon treatment; indeed, the only shared treatment effects were notably B cell-related. Our observations on the crucial role of B cells in immunomodulatory treatment of MS hence add to emerging evidence emphasizing the importance of B cells, which have long stood in the shadow of T cells, in both MS pathogenesis and treatment.19
Our immunophenotyping platform suggests a janus role for BAFF in autoimmune disease and treatment. The pathogenic face of BAFF is exposed in AITD, where BAFF is upregulated and may contribute to Th17-mediated disease. By contrast, the protective face of BAFF is revealed as the convergence point of effective immunomodulatory treatment in MS. Here we confirm previously reported changes in BAFF protein level upon interferon-β treatment20 and establish it as a unique shared mechanism with fingolimod. This pathway is furthermore shared with promising emerging lymphocyte and B cell depletion therapies for MS.21,22 We demonstrate that these protein level changes are not merely due to reduced consumption by BAFF receptors on B cells but involve active upregulation of transcription. Both in AITD and upon immunomodulatory treatment of MS, we now correlate BAFF plasma changes to an increase in transitional and decrease in switched B cells as percentage of B cells and propose the BAFF pathway as the mechanism of action driving these B cell alterations. Transitional B cells may be enriched for autoreactive B cells and have been implicated in the pathogenesis of other autoimmune diseases such as Sjögren syndrome and systemic lupus erythematosus.23 At the same time, they may also have regulatory properties supporting a beneficial effect as part of the therapeutic mechanisms of MS immunomodulatory treatments.24
BAFF antagonists have been proposed as a promising strategy based on animal models for MS.25,26 However, 2 clinical trials in MS with atacicept, neutralizing all forms of BAFF and the related a proliferation-inducing ligand (APRIL), had to be stopped prematurely because of increased inflammatory activity in patients with MS, and a 2-fold higher rate of conversion from clinically isolated syndrome to clinically definite MS in the treated compared to placebo group.27,28 A third trial with a monoclonal antibody against BAFF (tabalumab) was completed but no results are available.29 Of note, animal models leading to these clinical trials had a Th17-mediated component,26 which in our study we observe in human patients with AITD but not MS. The immunomodulatory treatments for which we observe an increase in BAFF, interferon-β and fingolimod, have notably distinct effects between MS, where they are beneficial, and the neuroinflammatory disease neuromyelitis optica (NMO), where these treatments are not only ineffective but may be harmful and result in excessive inflammatory activity and increased relapse rate and disability.30,31 This is in line with our observation that the unique shared signature of both treatments is an increase in BAFF coupled to already increased levels in BAFF seen in NMO but not MS.32 We propose that the distinct and shared immunologic architecture of disease and of existing efficacious treatments may inform further design of novel treatment strategies. In particular, our data suggest that antagonism of BAFF may be counterproductive to MS treatment but may be further pursued in NMO, where current immunosuppressive or immunomodulatory treatments are only partially effective.33
Our study was based on a routine clinical setting in a tertiary outpatient clinic. While we corrected for heterogeneity in age and sex, additional variables affecting treatment decisions, patient compliance, and treatment response are potential confounders. Preexisting variation among the immune profiles of patients with MS is, however, unlikely to be responsible for the immunologic responses to treatment. While patients with MS were assigned to treatment groups based on standard clinical criteria (driven by disease factors), assignment between low-dose interferon-β, high-dose interferon-β, and glatiramer acetate was determined by patient-preferred regimen, a research design that would not produce the dose-dependent effects observed with interferon-β or the distinct immunologic changes between interferon-β and glatiramer acetate–treated groups. Likewise, of the 14 fingolimod patients, 10 were included as part of a randomized clinical trial determined by patient preference for an oral treatment, a design structure that avoids segregation based on preexisting clinical variation. Furthermore, our recapitulation of previously reported findings in studies of single treatments typically limited by sample size and number of cell types investigated34–38 validates the methodology and thus the novel associations also observed in this systems immunology approach. While we were only able to assess changes in the peripheral blood, and not the target tissue, there is active exchange between the CNS and peripheral blood,18,39 and changes in the peripheral blood have previously been shown to be able to capture genetic variation important for MS.40 Moreover, immunomodulatory treatments are not administered directly into the CNS, and thus the likely location of activity is in the periphery, where our analysis took place.
In response to the challenge put forward for large-scale evaluation of the adaptive immune system in patients with MS,3 we demonstrate that a comprehensive common immunophenotyping platform is able to dissect the unique immunologic signatures of MS and immunomodulatory treatments. The different effects of immunomodulatory treatments suggest that the immunologic profile may in the future assist the neurologist in taking treatment decisions and optimizing the efficacy and safety for subgroups of patients, hence offering perspectives to the challenge of personalized medicine in MS.5 We propose that further design of novel treatment strategies takes into account the shared immunologic architecture of existing efficacious treatments, such as the early B cell changes with increase in BAFF and transitional B cells.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the patients and their relatives for participating in this study; Cindy Thys, Katleen Clysters, and Klara Mallants for assistance in sample collection and management; and Dr. V. Lagou, Dr. S. Humblet-Baron, and Prof. P. Dupont for discussions.
GLOSSARY
- AITD
autoimmune thyroid disease
- BAFF
B cell-activating factor
- mDC
myeloid dendritic cell
- MS
multiple sclerosis
- NMO
neuromyelitis optica
- PBMC
peripheral blood mononuclear cell
- RTE
recent thymic emigrant
Footnotes
Supplemental data at Neurology.org/nn
AUTHOR CONTRIBUTIONS
J.D., I.P., D.F., I.S., J.E.G.-P., K.H., D.D.-A., and L.D.M. performed experiments. I.S., J.T., A.T.L.N., and A.G. performed statistical analyses. B. Decallonne and B. Dubois provided samples and clinical data. J.D., I.P., B. Decallonne, B. Dubois, A.L., and A.G. designed the experiment. A.L. and A.G. supervised the experiment and wrote the first draft of the manuscript. All authors contributed to data interpretation and writing of the manuscript.
STUDY FUNDING
B. Decallonne and B. Dubois are Clinical Investigators of the Research Foundation Flanders (FWO-Vlaanderen). This project was supported by the Research Fund KU Leuven (OT/11/087, to B. Dubois and A.G.), Research Foundation Flanders (G073415N, to A.G. and B. Dubois), MS-Liga Vlaanderen (to A.L. and A.G.), Belgian Charcot Foundation (to A.G. and I.S.), and ERC Start Grant IMMUNO (to A.L.).
DISCLOSURE
J. Dooley reports no disclosures. I. Pauwels is employed by Ismar Healthcare. D. Franckaert received research support from Becton-Dickinson and his partner is an ex-employee of Boehringer-Ingelheim and current employee of Becton-Dickinson. I. Smets received travel funding from Biogen Idec and Boehringer and received research support from the Belgian Charcot Foundation. J. Garcia-Perez, K. Hilven, and D. Danso-Abeam report no disclosures. J. Terbeek's spouse is employed by and received travel funding from Janssen Pharmaceuticals and received a fellowship from Research Foundation Flanders (FWO). A.T.L. Nguyen reports no disclosures. L. De Muynck is employed by Janssen R&D. B. Decallonne reports no disclosures. B. Dubois served on the advisory boards for Biogen Idec and Merck, received travel funding from Novartis, Merck Serono, BSP, Sanofi-Aventis, and Biogen Idec, and received research support from Serono, Biogen Idec, Teva, and Novartis. A. Liston is deputy editor for Immunology and Cell Biology and Clinical and Translational Immunology, is on the editorial board for Frontiers in Immune Tolerance, his spouse is an ex-employee of UCB, and he received research support from JDRF, European Research Council, Research Foundation Flanders (FWO), KU Leuven, VIB, Interuniversity Attraction Poles, Jeffrey Modell Foundation, and Juvenile Diabetes Research Foundation. A. Goris received research support from Research Foundation Flanders (FWO), Belgian Charcot Foundation, Belgian Neurological Society, and Wetenschappelijk Onderzoek Multiple Sclerose. Go to Neurology.org/nn for full disclosure forms.
REFERENCES
- 1.The International Multiple Sclerosis Genetics Consortium. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat Genet 2013;45:1353–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.The International Multiple Sclerosis Genetics Consortium and The Wellcome Trust Case Control Consortium 2. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 2011;476:214–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roep BO, Buckner J, Sawcer S, Toes R, Zipp F. The problems and promises of research into human immunology and autoimmune disease. Nat Med 2012;18:48–53. [DOI] [PubMed] [Google Scholar]
- 4.Linker RA, Kieseier BC, Gold R. Identification and development of new therapeutics for multiple sclerosis. Trends Pharmacol Sci 2008;29:558–565. [DOI] [PubMed] [Google Scholar]
- 5.Hauser SL, Chan JR, Oksenberg JR. Multiple sclerosis: prospects and promise. Ann Neurol 2013;74:317–327. [DOI] [PubMed] [Google Scholar]
- 6.Orru V, Steri M, Sole G, et al. Genetic variants regulating immune cell levels in health and disease. Cell 2013;155:242–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brodin P, Jojic V, Gao T, et al. Variation in the human immune system is largely driven by non-heritable influences. Cell 2015;160:37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carr EJ, Dooley J, Garcia-Perez JE, et al. The cellular composition of the human immune system is maintained in multiple stable equilibriums shaped by age and cohabitation. Nat Immunol 2016;17:461–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Broadley SA, Deans J, Sawcer SJ, Clayton D, Compston DA. Autoimmune disease in first-degree relatives of patients with multiple sclerosis. a UK survey. Brain 2000;123:1102–1111. [DOI] [PubMed] [Google Scholar]
- 10.Cooper JD, Simmonds MJ, Walker NM, et al. Seven newly identified loci for autoimmune thyroid disease. Hum Mol Genet 2012;21:5202–5208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ricklin ME, Lorscheider J, Waschbisch A, et al. T-cell response against varicella-zoster virus in fingolimod-treated MS patients. Neurology 2013;81:174–181. [DOI] [PubMed] [Google Scholar]
- 12.Rowland SL, Leahy KF, Halverson R, Torres RM, Pelanda R. BAFF receptor signaling aids the differentiation of immature B cells into transitional B cells following tonic BCR signaling. J Immunol 2010;185:4570–4581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jacobi AM, Huang W, Wang T, et al. Effect of long-term belimumab treatment on B cells in systemic lupus erythematosus: extension of a phase II, double-blind, placebo-controlled, dose-ranging study. Arthritis Rheum 2009;62:201–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haegele KF, Stueckle CA, Malin JP, Sindern E. Increase of CD8+ T-effector memory cells in peripheral blood of patients with relapsing-remitting multiple sclerosis compared to healthy controls. J Neuroimmunol 2007;183:168–174. [DOI] [PubMed] [Google Scholar]
- 15.Seifert M, Przekopowitz M, Taudien S, et al. Functional capacities of human IgM memory B cells in early inflammatory responses and secondary germinal center reactions. Proc Natl Acad Sci USA 2015;112:E546–E555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Battistini L, Piccio L, Rossi B, et al. CD8+ T cells from patients with acute multiple sclerosis display selective increase of adhesiveness in brain venules: a critical role for P-selectin glycoprotein ligand-1. Blood 2003;101:4775–4782. [DOI] [PubMed] [Google Scholar]
- 17.Ifergan I, Kebir H, Alvarez JI, et al. Central nervous system recruitment of effector memory CD8+ T lymphocytes during neuroinflammation is dependent on alpha4 integrin. Brain 2011;134:3560–3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stern JN, Yaari G, Vander Heiden JA, et al. B cells populating the multiple sclerosis brain mature in the draining cervical lymph nodes. Sci Transl Med 2014;6:248ra107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krumbholz M, Derfuss T, Hohlfeld R, Meinl E. B cells and antibodies in multiple sclerosis pathogenesis and therapy. Nat Rev Neurol 2012;8:613–623. [DOI] [PubMed] [Google Scholar]
- 20.Krumbholz M, Faber H, Steinmeyer F, et al. Interferon-beta increases BAFF levels in multiple sclerosis: implications for B cell autoimmunity. Brain 2008;131:1455–1463. [DOI] [PubMed] [Google Scholar]
- 21.Lavie F, Miceli-Richard C, Ittah M, Sellam J, Gottenberg JE, Mariette X. Increase of B cell-activating factor of the TNF family (BAFF) after rituximab treatment: insights into a new regulating system of BAFF production. Ann Rheum Dis 2007;66:700–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thompson SA, Jones JL, Cox AL, Compston DA, Coles AJ. B-cell reconstitution and BAFF after alemtuzumab (Campath-1H) treatment of multiple sclerosis. J Clin Immunol 2010;30:99–105. [DOI] [PubMed] [Google Scholar]
- 23.Vossenkamper A, Lutalo PM, Spencer J. Translational Mini-Review Series on B cell subsets in disease: transitional B cells in systemic lupus erythematosus and Sjogren's syndrome: clinical implications and effects of B cell-targeted therapies. Clin Exp Immunol 2012;167:7–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bouaziz JD, Yanaba K, Tedder TF. Regulatory B cells as inhibitors of immune responses and inflammation. Immunol Rev 2008;224:201–214. [DOI] [PubMed] [Google Scholar]
- 25.Huntington ND, Tomioka R, Clavarino C, et al. A BAFF antagonist suppresses experimental autoimmune encephalomyelitis by targeting cell-mediated and humoral immune responses. Int Immunol 2006;18:1473–1485. [DOI] [PubMed] [Google Scholar]
- 26.Zhou X, Xia Z, Lan Q, et al. BAFF promotes Th17 cells and aggravates experimental autoimmune encephalomyelitis. PLoS One 2011;6:e23629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kappos L, Hartung HP, Freedman MS, et al. Atacicept in multiple sclerosis (ATAMS): a randomised, placebo-controlled, double-blind, phase 2 trial. Lancet Neurol 2014;13:353–363. [DOI] [PubMed] [Google Scholar]
- 28.Sergott RC, Bennett JL, Rieckmann P, et al. ATON: results from a phase II randomized trial of the B-cell-targeting agent atacicept in patients with optic neuritis. J Neurol Sci 2015;351:174–178. [DOI] [PubMed] [Google Scholar]
- 29.Vincent FB, Saulep-Easton D, Figgett WA, Fairfax KA, Mackay F. The BAFF/APRIL system: emerging functions beyond B cell biology and autoimmunity. Cytokine Growth Factor Rev 2013;24:203–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Warabi Y, Matsumoto Y, Hayashi H. Interferon beta-1b exacerbates multiple sclerosis with severe optic nerve and spinal cord demyelination. J Neurol Sci 2007;252:57–61. [DOI] [PubMed] [Google Scholar]
- 31.Min JH, Kim BJ, Lee KH. Development of extensive brain lesions following fingolimod (FTY720) treatment in a patient with neuromyelitis optica spectrum disorder. Mult Scler 2012;18:113–115. [DOI] [PubMed] [Google Scholar]
- 32.Vaknin-Dembinsky A, Brill L, Orpaz N, Abramsky O, Karussis D. Preferential increase of B-cell activating factor in the cerebrospinal fluid of neuromyelitis optica in a white population. Mult Scler 2010;16:1453–1457. [DOI] [PubMed] [Google Scholar]
- 33.Weinshenker BG, Barron G, Behne JM, et al. Challenges and opportunities in designing clinical trials for neuromyelitis optica. Neurology 2015;84:1805–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miyazaki Y, Niino M, Fukazawa T, et al. Suppressed pro-inflammatory properties of circulating B cells in patients with multiple sclerosis treated with fingolimod, based on altered proportions of B-cell subpopulations. Clin Immunol 2014;151:127–135. [DOI] [PubMed] [Google Scholar]
- 35.Kowarik MC, Pellkofer HL, Cepok S, et al. Differential effects of fingolimod (FTY720) on immune cells in the CSF and blood of patients with MS. Neurology 2011;76:1214–1221. [DOI] [PubMed] [Google Scholar]
- 36.Planas R, Jelcic I, Schippling S, Martin R, Sospedra M. Natalizumab treatment perturbs memory- and marginal zone-like B-cell homing in secondary lymphoid organs in multiple sclerosis. Eur J Immunol 2011;42:790–798. [DOI] [PubMed] [Google Scholar]
- 37.Schubert RD, Hu Y, Kumar G, et al. IFN-beta treatment requires B cells for efficacy in neuroautoimmunity. J Immunol 2015;194:2110–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Claes N, Dhaeze T, Fraussen J, et al. Compositional changes of B and T cell subtypes during fingolimod treatment in multiple sclerosis patients: a 12-month follow-up study. PLoS One 2014;9:e111115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Palanichamy A, Apeltsin L, Kuo TC, et al. Immunoglobulin class-switched B cells form an active immune axis between CNS and periphery in multiple sclerosis. Sci Transl Med 2014;6:248ra106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Achiron A, Gurevich M. Peripheral blood gene expression signature mirrors central nervous system disease: the model of multiple sclerosis. Autoimmun Rev 2006;5:517–522. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.