

Summary
Tumor proliferation is concomitant with autophagy, limited apoptosis, and resultant necrosis. Necrosis is associated with the release of damage-associated molecular pattern molecules (DAMPs), which act as ‘danger signals’, recruiting inflammatory cells, inducing immune responses, and promoting wound healing. Most of the current treatment strategies for cancer (chemotherapy, radiation therapy, hormonal therapy) promote DAMP release following therapy-induced tumor death by necroptosis and necrosis. Myeloid cells (monocytes, dendritic cells (DCs), and granulocytes), as well as mesenchymal stromal cells (MSCs) belong to the early immigrants in response to unscheduled cell death, initiating and modulating the subsequent inflammatory response. Responding to DAMPs, MSCs, and DCs promote an immunosuppressive milieu, while eosinophils induce oxidative conditions limiting the biologic activity of DAMPs over time and distance. Regulatory T cells are strongly affected by pattern recognition receptor signaling in the tumor microenvironment and limit immune reactivity coordinately with myeloid-derived suppressor cells. Means to ‘aerobically’ oxidize DAMPs provide a novel strategy for limiting tumor progression. The present article summarizes our current understanding of the impact of necrosis on the tumor microenvironment and the influence of oxidative conditions found within this setting.
I take thee to my weddyd wyf, to have and to hold fro thys day forwarde, for better for wors, for richer for porer, in sikenesse and in helthe, tyl deth us departe, yf holy Chyrche wyl it ordeyne; and thereto I plyght the my trouthe.' The Sarum Rite Saint Osmund, Bishop of Salisbury, England, 11th century
Until Death Do Us Part (Japanese Manga: 死がふたりを分かつまで Hepburn: Shi ga Futari wo Wakatsu made
Inflammation, Necrosis and Cancer
In sickness and in health, both scheduled (apoptotic) and unscheduled (necrotic, necroptotic, ferroptotic etc.) cell death, arising naturally or subsequent to disease, release factors which can promote chronic inflammation. Cancer is just as fundamentally a disorder of cell death as much as it is a disorder associated with proliferation, in almost half of individuals causing their death. Chronic infection and inflammation contributes to at least 25% of all cancer cases worldwide [1]. Autoimmunity, persistence of pathogens, and cancer are associated with chronic inflammation. This is the pathologically conducive microenvironment favoring both the initiation and progression of cancer [1,2]. Chronic inflammation promotes acquisition of genomic instability by enabling survival during prolonged exposure of tissue to endogenous mutagens, initially heightened apoptosis coupled with higher cellular turnover, prolonging autophagic survival, emergence of necrotic death, and promotion of neoplastic transformation.
Indeed, an inflammatory response may not necessarily impute by itself a good prognosis. Both T effector cells and T regulatory cells are found within the tumor microenvironment, providing complementary roles, as well as bone marrow-derived myeloid progenitors [2]. These include tumor-associated macrophages and CD103+ dendritic cells (DCs) promoting an immune response to tumor [3,4].
The production and consumption of released factors, including chemokines and cytokines, within stressed tissues as well as its chronicity may promote tumor death or progression.
The mammalian immune system is reciprocally composed of dynamic networks of immune cells and non-immune cells, enabling metabolic homeostasis, timely eradication of effete cells, and protection against pathogens. Simultaneous tolerance towards self-antigens and, conversely, reactivity to new or occult antigens occurs in settings of tissue damage and wound healing. When tissue homeostasis is perturbed, mediators such as cytokines, chemokines, matrix remodeling proteases, reactive oxygen species (ROS), and bioactive mediators such as histamine induce mobilization and infiltration of leukocytes and mesenchymal stromal cells (MSCs) into damaged tissue. Indeed, oxidative stress, in addition to its ability to recruit inflammatory cells, can limit metastasis of tumors [5,6]. Subsequently, the complementary processes of inflammation and wound healing begin. Inflammatory immune response focuses on the elimination of pathogens responsible for cell damage and induces a rather cytotoxic environment while wound healing is characterized by phagocytosis of cell debris and apoptotic cells, immune suppression, enabling re-epithelialization and synthesis of extracellular matrix (ECM), resolving inflammation and restoring tissue homeostasis.
The development of tumors over many years typically leads to reciprocal alterations in the host and the tumor, enabling tumor growth, paradoxically in the setting of substantial necrosis and (chronic) inflammation. In these circumstances, genomically unstable/neoplastic cells harness collaborative capabilities (see below) of immune cells and local non-mutated but injured tissues to favor their own survival and proliferation, in part by releasing immune suppressive factors such as transforming growth factor β (TGF-β), IL-10, and kynurenine. Tumor cells escaping eradication induce release of tissue healing factors promoting neo-vascularization and resultant nutrient supply.
Wound healing and tumor stroma formation share many important properties, including accumulation and activation of leukocytes and MSCs in response to released endogenous ‘danger signals’ from injured/necrotic tissue. Damage-associated molecule pattern molecules (DAMPs) are factors found in all stressed cells, including S100 proteins, uric acid, ATP, hyaluronan, heat shock proteins (HSPs), heparan, syndecan, versican, cold-inducible RNA-binding protein (CIRP) [7,8,9,10], and high-mobility group box 1 (HMGB1) [11,12,13,14,15,16].
Necrosis Is a Poor Prognostic Factor in the Tumor Microenvironment
Regardless of the origin and site of neoplastic cells, necrotic cell death with subsequent release of DAMPs is a characteristic feature of advanced solid tumors mainly due to three conditions:
– inadequate nutrient supply to tumor cells as a consequence of the imbalance between tumor growth and angiogenesis,
– the host's cytotoxic immune response to the tumor, and
– downregulation of programmed (apoptotic) cell death by the tumor itself.
Released DAMPs can critically impact the tumor microenvironment by enhancing wound healing processes such as angiogenesis and stroma formation or by influencing the immune response [17,18,19].
Tumors ‘addicted to death’ [20], stressed ‘internally’ with genomic stress, release DAMPs sustaining conditions associated with chronic inflammation and cell proliferation.
When evaluating a tumor, it is important to assess three elements within its microenvironment consisting of:
- – Factors released by
- • tumor cells themselves (including DAMPs) and/or
- • tumor-associated cells consisting of MSCs and fibroblasts, epithelial and endothelial cells, as well as infiltrating leukocytes.
– The quantity and quality of tumor-associated leukocytes.
– The state of activation of tumor and associated cells.
Eosinophilic granulocytes (Eos) and MSCs belong to the subset of cells often found in the setting of wound healing, chronic inflammation, and within the tumor microenvironment. Their presence is associated with the prognosis of cancer patients (see below). DAMPs including HMGB1 and S100A4 from necrotic cells act as chemoattractants and survival/proliferation factors for both Eos and MSCs [21,22]. Additionally, MSCs and DCs pulsed with S100A4 induce regulatory T cells (Tregs) [19,23]. Tregs also respond to and are promoted by DAMPs (see below)
Mesenchymal Stromal Cells
Biology of MSCs
Multipotent MSCs, unlike hematopoietic stem cells, originate within the stroma of the bone marrow. They lack typical hematopoietic cell surface markers, including CD34 and CD45, and have the potential to differentiate into several types of tissue cells, such as adipocytes, osteoblasts, and chondrocytes [24].
MSCs were first identified in the 1960s and were regarded as colony-forming fibroblasts. Later, they were shown to have the ability to be induced to differentiate according to external stimuli. Although MSCs reside predominantly in the bone marrow, they are also distributed throughout many other tissues, where they are thought to serve as local sources of dormant (quiescent) stem cells [25] contributing to tissue healing. The contributions of MSCs to tissue formation become apparent in cases of tissue remodeling following injury or chronic inflammation. These conditions are typically accompanied by mobilization of multipotent MSCs and their subsequent recruitment to the site of damage [25]. MSCs contribute to the formation of fibrous scars following injury [26] and, in the setting of wound healing, can act as ‘nurse-like cells’ [27]. This is accomplished by:
– scavenging ROS [28] which are provided by inflammatory cells migrating into sites of damage [29,30],
– paracrine mechanisms promoting cell proliferation and inducing angiogenesis, and
– induction of local immunosuppression thus protecting regenerating tissue from cytotoxic collateral damage caused by activated local leukocytes ‘sensing damage and danger’ [29,30].
In the setting of wound healing, MSCs can act as immune regulatory cells by releasing IL-10 or by producing the enzyme indoleamine-dioxygenase (IDO) associated with induction of tolerance and a shift from Th1 to Th2 immune response [31]. Because of their immunosuppressive capacities, MSCs have been applied in clinical trials to treat:
– autoimmune diseases including ulcerative colitis,
– graft-versus-host disease (GvHD) following transplantation of hematopoietic stem cells,
– nonhealing wounds due to local aseptic chronic inflammation.
Immunosuppressive MSCs Are Recruited to Necrotic Sites within Tumors
MSCs within tumor tissue influence the biological behavior of tumors as well as the host's immune response to them. Systemically transferred MSCs can migrate into colon carcinomas [32]. In breast cancer, MSCs promote tumor metastasis [33]. We could show that active DAMPs from necrotic cells are capable of enhancing proliferation and chemotaxis of MSCs in vitro [22] while oxidized DAMPs lose their stimulatory capacities on MSCs [22,34]. Uric acid, HMGB1, and S100A4 act as chemoattractants and proliferation-promoting factor to MSCs [22]. Moreover, in the presence of S100A4, MSCs induce CD4+ CD25+ FoxP3+ Tregs expressing IL-10 and IDO [23]. Interestingly, HMGB1 and S100A4 are both sensitive to oxidation and need reducing conditions for preserving their biologic activities [35,36]. Consistently, uric acid (acting as an antioxidant) protects S100A4 from oxidation (thus inactivation) showing a synergistic chemotactic effect when combined with S100A4 [23]. Considering the clinical data demonstrating that the presence of MSCs within the tumor microenvironment is rather disadvantageous for cancer patients [33], induction of oxidative conditions within tumor tissue could significantly impact the biology and response of MSCs to DAMPs and thus provide a therapeutic option.
Stranger and Danger Models (Signal 0s in the Immune Response)
Three decades ago, Charles Janeway [37] proposed a conceptual working frame model of the immune system: it protected against infectious pathogens rather than other innocuous foreign antigens, and this discrimination between infectious and innocuous antigens was achieved by antigen-presenting cells (APCs) through recognition of pathogen-associated molecular patterns (PAMPs) by their interaction with pattern recognition receptors. The significance of this ‘stranger model’ concept has been well appreciated with Toll-like receptors (TLRs) identified successively, which collectively have been termed pattern recognition receptors (PRRs). Janeway's model failed to explain initiation of sterile inflammatory responses to tumors, tissue damage or injury including myocardial infarction and cerebrovascular accidents, tissue transplants, allergy and in autoimmune diseases (although microorganisms might contribute to the course of responses in some cases). This inconformity led Polly Matzinger [29] to ponder and advance the ‘danger hypothesis’ in 1994. She postulated that the immune system not only responds to pathogens but also to endogenous signals arising from nonphysiological cell stress, damage, and death. In this model, dying cells release endogenous danger signals (DAMPs). This so-called ‘sterile inflammation’ event initiated by DAMPs (Signal 0) facilitates an early innate and adaptive immune response (Signal 1-4) through activation of APCs in response to injury, trauma, ischemia, cancer, and other tissue damage, regardless of the presence of overt pathogenic infection [38]. It is now well established that necrosis, pyroptosis (inflammatory cell death), necroptosis, ferroptosis, late apoptosis, and autophagy would lead to DAMP release [39], at least under certain circumstances if not all. Notably, many receptors are shared between DAMPs and PAMPs, including members of TLRs, NOD-like receptors (NLRs), AIM2-like receptors (ALRs), RIG-I-like receptors (RLRs) and others, providing colloquy between nonmicrobial and microbial inflammatory responses.
In response to DAMPs and PAMPs, APCs are central to initiating the immune responses and modifying the magnitude of the ongoing response. The most efficient APCs are professional DCs that are pivotal for recognition of antigens/immunogens and manipulation of a series of responses. With such capacities, DCs are indispensable for how immunity is directed and enabled [40]. As professional sentinels, DCs continuously sense and translate the surrounding cues, arising from infection, tissue damage or injury and cytokines, orchestrating a repertoire of immune responses in the settings of various physiological and pathological conditions. Hence, improved understanding of how DCs respond to DAMPs may allow for detailed pathways for immune optimization and new approaches to numerous diseases, including cancer.
Dendritic Cells
DC Maturation
The immune system is classically separated into innate and adaptive (acquired) immunity. The distinctive feature of innate immunity is its rapid response to a limited repertoire of substrates, including PAMPs and DAMPs, which is promoted and realized by innate myeloid and lymphoid cells. In contrast, adaptive immunity engages two primary types of lymphocytes (T and B cells) encountering antigen and clonally expressing antigen-specific receptors, promoted by VDJC somatic recombination. The transition from innate to adaptive immunity involves DC capture and presentation of antigens to naive T and B cells to promote their effector function.
Schuler and Steinman [41] first identified DC function in vitro in 1985, with evidence that cultured epidermal Langerhans cells (LCs) could drive T-cell clonal expansion more efficiently than freshly isolated LCs. DCs are capable of transiting from immature, antigen-capturing and preserving cells to mature, antigen-presenting cells. ‘Maturation’ is preferential to ‘activation’ because the former represents a process of large-scale differentiation with gradual gain of function, whereas the latter typically refers to on-off or limited arrays of events [40].
DC maturation plays an essential role in dictating the fate of T cells and the subsequent adaptive immunity. Encountering environmental changes, DCs respond quickly and differentiate to mature cells capable of eliciting T-cell acquired immunity. Hence, maturation indicates a comprehensive differentiation and alteration, involving redistribution of major histocompatibility complex (MHC) complex to the surface, acquisition of antigen presentation/cross-presentation capability, up-regulation of surface co-stimulatory molecules, secretion of cytokines and chemokines, increased capacity to migrate into lymph nodes, and morphological changes/cytoskeleton reconstitution.
Characteristics of Tumor-Associated-DCs (TA-DCs)
DCs are found in the microenvironment of several types of cancer and confer a better prognosis [42,43,44,45,46,47]. DAMPs and tumor-associated cytokines including VEGF, IL-10, and prostaglandin E2 (PGE2) can profoundly limit the ability of DCs to promote a TH1 response [19,48]. TA-DCs are immunosuppressive and can also induce regulatory T-cell expansion [19,46,49]. Besides their immunosuppressive nature, TA-DCs are able to produce angiogenic factors promoting tumor survival, progression, and metastasis [42,47,50]. Additionally, DCs cultured in the presence of tumor factors can lose CD14/CD45 and express endothelial markers including CD31, CD34, von Willebrand factor, vascular endothelial growth factor receptor 2 (VEGFR-2), and VE-cadherin [51,52,53,54,55].
The Tumor Microenvironment Limits the Efficacy of DC Vaccination
TA-DCs are usually described as immature cells with low expression of co-stimulatory molecules and incapable of inducing robust antitumor immune responses [55,56,57,58,59]. Ex vivo matured DCs loaded with tumor antigens are trapped within the tumor microenvironment and thus incapable of migrating into regional lymph nodes [60]. Current strategies to circumvent the immunosuppressive influence of the tumor microenvironment target either local bioactive factors such as cytokines and chemokines, or tumor-associated cells including myeloid-derived suppressor cells and Tregs. Indeed, depletion of Tregs before vaccination with carcinoembryonic antigen (CEA) pulsed DCs can enhance specific T-cell response to CEA [61]. Similar effects of Treg depletion could be observed in patients with metastatic renal cell carcinomas [62].
Interestingly, depletion of TA-DCs from the tumor microenvironment of ovarian cancer boost antitumor immune responses in a murine model [63]. Thus, depletion of these cells prior to DC-based vaccination may also induce effective antitumor immune responses. The group from the University of Pennsylvania and University of Lausanne found that oxidation of antigen was critically important for delivery of immunogenic antigen from tumors to immunogenic DCs [64]. This is consistent with earlier observations that oxidation of HMGB1 was critical for conferring tolerance to antigens delivered by tolerogenic DCs [36].
DAMPs critically impact the tumor microenvironment by enhancing angiogenesis or influencing the immune response [20,65]. Current studies and therapeutic strategies addressing the immunosuppressive nature of the tumor microenvironment focus on cytokines and chemokines released from tumor or tumor-associated cells. We have recently shown that necrotic material (DAMPs) enhance the chemotaxis and proliferation of MSCs [22] with potent immune regulatory capacity. DAMPs, including S100A4, induce regulatory DCs [19]. S100A4 promotes metastasis, stimulates angiogenesis, induces cell motility, and increases expression of matrix metalloproteinases [66,67,68,69] and is associated with poor prognosis of cancer patients [70,71,72,73]. S100A4 loses its stimulatory capacity following oxidation. Given the hypoxic and reducing conditions prevalent within the tumor microenvironment, tumor-associated S100A4 is protected from oxidation. The stimulatory effect of DAMPs on MSCs was abrogated following oxidation of these factors [22]. Providing oxidizing conditions could abrogate the immunosuppressive activity of DAMPs on tumor-associated MSCs and DCs.
Eosinophilic Granulocytes
Biology of Eosinophils
Compartments with abundant resident populations of eosinophilic granulocytes (Eos) include tissues with substantial cellular turnover and regenerative capacity such as the bone marrow, the primary and secondary lymphoid tissues (e.g., spleen, lymph nodes, and thymus) [74], the uterus [75], and nearly the entire gastrointestinal tract (with the exception of the esophagus) [74,76]. This linkage with cell turnover and tissue repair also may explain the presence of Eos at sites of wound repair [77] and the commonality of an Eos infiltrate among solid tumors [78].
Eos contain and can release several cationic proteins, which in addition to their cytotoxic character are also potentially important for tissue remodeling and clearance of cellular debris [79]. Eos are falsely thought to be responsible for the cell death and tissue damage commonly observed in disease states associated with increased numbers and tissue-specific recruitment [80].
Eos mediate their effects via at least three independent mechanisms in addition to the release of cytotoxic granule proteins. These mechanisms enable Eos to modulate the intensity of inflammation: i) Eos are potent regulators of local inflammatory responses [81]; ii) recruited Eos are a source of ROS [82] and established small molecule lipid mediators of inflammation. In particular, Eos generate cysteinyl leukotrienes (i.e., LTC4, LTD4, LTE4, and LTB4) [83,84], 5-HETE [85], PGE2[86], and platelet-activating factor (PAF) [87]. The capability of cysteinyl leukotrienes to mediate primary inflammatory responses such as edema [88], the recruitment of other pro-inflammatory leukocytes [89], and the induction of tissue histopathology [90] uniquely positions these molecules as mediators of inflammation. iii) Eos are a prodigious source of cytokines associated with tissue repair and remodeling (see table 2 in [17]).
A growing body of literature suggests that both Eos-mediated immune regulation and tissue repair/remodeling in particular may represent important non-overlapping Eos effector functions [79].
In addition to their capacity to synthesize and release a variety of immune regulatory molecules [17], some studies have suggested that Eos may function as APCs [91,92,93,94,95,96]. In the presence of exogenous CpG-DNA, a PAMP, human eosinophils enhance maturation of autologous DCs [81]. While Eos-derived neurotoxin did not contribute to the described effect, DCs internalized MBP, released from CpG-stimulated Eos [81]. Thus, induction of DC maturation by Eos may be due to released MBP, in response to necrosis.
Eos may also affect local T-cell responses by modulating the balance between Th1 and Th2 immune responses (e.g., through Eos-derived IDO production of kynurenine [31]). Importantly, IDO is essential for the induction of tolerance by tissue-recruited T cells [31,97,98]. Thus, similar to other Eos-mediated immune suppressive activities (e.g., the potential induction of Tregs through TGF-β production [99]), Eos-derived IDO may also play a crucial role in immune suppression. Eos are capable of both synthesizing and releasing cytokines characterized as either Th1 (e.g., IFN-γ), Th2 (e.g., IL-4, IL-5, and IL-13), Th3 (TGF-β, IL-10), and possibly Th4 (IL-17 [100,101,102] also known as Th17) cytokines, or acute phase responses (e.g., TNF-α, IL-1, IL-6, and VEGF) (see tables 1 and 2 in [17])
Eosinophils Are Recruited to Necrotic Sites in Cancer
Neoplastic tissues undergoing necrosis activate Eos and induce their migration in vitro [21,103] and in vivo [103,104]. A quantitative assessment of Eos recruitment/accumulation in solid tumors showed that the tissue eosinophilia is apparently mediated by one or more factors released directly from necrotic tissues within the tumor [79,105]. Studies linking Eos recruitment and activations with cell death and necrosis abound. In particular, Stenfeldt and colleagues [103] discovered that damaged epithelial cell lines (e.g., genital (HeLa), respiratory (HEp-2), and intestinal (HT29) cells) induce Eos migration, the release of putative tissue-damaging factors such as Eos secondary granule proteins, and secretion of Eos-derived pro-fibrotic factors such as fibroblast growth factors (FGF-1 and −2) and TGF-β1. In a B16-F10 melanoma murine model, eosinophilic infiltration of tumors occurred from the earliest palpable stages with significant accumulation only within the necrotic center and the outer capsular regions [105]. Tumor-associated tissue eosinophilia or blood eosinophilia of patients with gastrointestinal cancers is a highly significant and favorable prognostic factor [17,106,107,108,109,110].
The two most dominant effector functions ascribed to Eos results from the production of ROS (due to their oxidative burst) and the release of cationic granule proteins (following degranulation). Indeed, DAMPs not only promote Eos' survival but also induce their degranulation (release of MBP and EPO) and their oxidative burst in a dose-dependent manner [21]. Interestingly the oxidative burst of Eos is the most sensitive detectable response to necrosis-associated factors (DAMPs) regardless of the origin/neoplastic transformation of necrotic cells [21].
Compared with other leukocytes, Eos have the highest capacity to produce ROS with their capacity being 10- to 100-fold greater than neutrophils [111,112,113]. The stimulatory effect of necrotic material is abolished following oxidation [21,22] suggesting a possible role for Eos capable of oxidizing necrotic cell debris. Dose-dependent release of MBP (degranulation) follows stimulation of Eos with necrotic material [21]. MBP also induces neutrophil O2 production [114,115] which may also contribute to oxidation of necrotic material. MBP serves as a factor amplifying [79] local ‘danger signals’ released from stressed cells. Additionally, we assessed the dose-dependent release of EPO following stimulation with DAMPs [21]. EPO acts as an enzyme catalyzing the oxidation of chloride, bromide, and thiocyanate to their respective hypohalous acids with even higher oxidative capacity than peroxide itself, which is consistent with the oxidative role of Eos within stressed/necrotic tissues.
We propose that the response to necrotic material is a fundamental mechanism that promotes eosinophil trafficking into stressed/necrotic tissues such as that found in cancer and in chronic inflammatory diseases including asthma or ulcerative colitis. Production of ROS by Eos encountering necrotic/stressed cells induces oxidation and thus inactivation of DAMPs.
Eos are not only able to eliminate effete and damaged tissue but are also attracted and activated by stressed and damaged cells. It is likely that stressed cells attract and activate Eos by expression of molecules such as MHC class I chain-related A (MIC-A), MIC-B, Letal [116] as well as UL16 binding proteins (ULBP). These stress-associated molecules serve as ligands for NKG2D, described first on NK cells [117] and subsequently on eotaxin-activated Eos [118] and T cells [119]. Thus, tumor-associated Eos appear to have at least two dominant non-overlapping activities: i) destructive effector functions which may limit tumor growth and cause recruitment and activation of other leukocytes and ii) immune regulative and remodeling activities which suppress immune response and release of cytokines, promoting wound healing and resultant tumor proliferation.
Consistent with the hypothesis that DAMPs initiate innate immune cell activation when encountering microbes or parasites [30], Eos are often first responders to tissue damage and likely mediate some aspects of tissue remodeling and repair. Interestingly, Eos infiltration is used by some groups as a poor prognostic factor associated with transplant rejection [120,121].
Eosinophils Are Sentinels of Effective Tumor Immunotherapy
Eos are frequently observed within regressing tumors in the setting of immunotherapy with IL-2 [122,123], IL-4 [124,125], GM-CSF [126], and antibodies to CTLA-4, but their appearance has been an interesting sidenote the significance of which has remained largely unknown. In particular, the anti-tumor effects of successful cytokine therapy of cancer with IL-2 had been associated with the identification of degranulating Eos within the tumor [122,123], suggesting that Eos effector functions (e.g., direct [122] or antibody-dependent [127] tumor cell lysis or the immune regulative capacity of Eos modulating the local tumor microenvironment) may play a role in the anti-cancer activities mediated by systemic IL-2 administration. However, despite the promises of these potential Eos-mediated anti-tumor activities, the presence of Eos has not been found to be prognostically important for high-dose IL-2-treated patients.
Murine studies suggesting a link between Eos and the therapeutic value of the anti-tumor responses associated with IL-4 administration [125,128] have also led to clinical trials evaluating these responses in cancer patients. In a phase I clinical trial of IL-4 administered to cancer patients, Sosman and colleagues [124] showed that IL-4 therapy induced systemic Eos degranulation with increases in serum and urine MBP levels. The increase in serum MBP was IL-4 dose-dependent. Unfortunately, the linkage of anti-tumor activities in these patients to Eos is only correlative, and - similar to the observations in patients following IL-2 administration - no definitive conclusions as to if and how Eos modulate tumor growth could be made so far.
Efforts to demonstrate experimentally a role for Eos in tumor immunity have also been fraught with complicating variables that yielded qualified interpretations. Most notably, considerable excitement was generated by data from the studies of Tepper and colleagues [125,129,130] which demonstrated in athymic nude mice that malignant cell lines transfected for constitutive expression of IL-4 elicited a tumor-associated macrophage and Eos infiltrate that led to the attenuation of tumor growth. This provoked a series of studies, all done with transplantable tumors (see for example [131,132]), in an attempt to define the cellular and molecular mechanisms of this apparent IL-4-mediated anti-tumor effect. Although these studies have shown that even spontaneous tumors evidenced tumor regression associated with tumor infiltrating Eos [132], none of these studies has resolved the role(s) of Eos in tumor rejection reactions.
Regulatory T Cells
Biology of Tregs
Tregs represent 4-5% of the CD4+ T cells and have a potent immune regulatory capacity. They were first described in 1995 by Sakaguchi and colleagues [133]. Tregs comprise several subsets of phenotypically similar cells able to inhibit immune response to self- and non-self antigens through distinct mechanisms [134]. To date at least two Treg subsets have been recognized in humans. Naturally occurring Tregs (nTregs), which originate in the thymus, mediate suppression via cell contact-dependent mechanisms involving the granzyme B/perforin or Fas/FasL pathways and constitute a major Treg subset for maintaining peripheral tolerance [135]. The phenotypic characteristics are still a matter of debate. nTregs were commonly characterized by expression IL-2 receptor alpha chain (CD25), which is expressed on the cell surface. However, only CD4+ CD25high T cells are considered immunosuppressive Tregs [136], because this marker is also expressed on activated CD4+ T cells (tumor effector T cells; Teffs) at a medium expression level. But defining the cut-off level of CD25 expression density on Tregs based on the mean fluorescence intensity has not been entirely reliable. Other approaches to identify and isolate nTregs are the absence of Treg markers such as the IL-7 receptor, CD127, and an integrin-αsubunit, CD49d, that are expressed on conventional T cells, which have been utilized for nTregs isolation from PBMC by negative selection [137,138]. The most prominent marker for identifying Tregs is the intracellular marker FOXP3, the transcription factor forkhead box P3, which belongs to the forkhead/winged-helix family [139]. The development of nTregs depends on FOXP3, and its expression is required for the ability of CD4+ T-cell populations to mediate immune suppression by inhibiting proliferation and IL-2 production in Teffs. The absence of FOXP3+ nTregs results in autoimmune disease [140]. FOXP3 is expressed in other immune cells including activated CD4+ lymphocytes and even in cancer cells [141]. Stable expression of FOXP3 in nTregs depends on the demethylation status of the foxp3 gene. The relevant sequence of the foxp3 gene is called ‘Treg-specific demethylated region’ (TSDR), which allows discriminating Tregs from activated T cells by sequencing [142,143]. Nevertheless, intracellular localization of FOXP3 in nTregs makes it unsuitable for their viable isolation. Other surface molecules expressed on nTregs have been considered as cell markers able to reliably identify Tregs and allow for their isolation. The cytotoxic T lymphocyte antigen-4 (CTLA-4, CD152), the glucocorticoid-induced tumor necrosis factor receptor(GITR)-related protein [144]) and ICOS (CD278, inducible T-cell co-stimulator, CD28 superfamily co-stimulatory molecule) as well as Programmed-death-1 (PD-1) are all expressed on human Tregs, but neither is specific for nTregs [145,146,147].
The other major subtype of Tregs is the group of inducible or adaptive Tregs (iTreg), also referred to as type 1 Tregs (Tr1), which are induced in the periphery in response to environmental signals, including tumor-derived antigens, cytokines or other soluble factors, and mediate powerful suppression of Teff functions by a variety of mechanisms [148]. Tr1 mediate suppression by contact-independent mechanisms through the production of TGF-β, IL-10 and other immunosuppressive factors. Further, iTregs can assume functions that either favor tumor growth by down-regulating activity of anti-tumor immune cells or inhibit tumor progression by suppressing inflammation, which is thought to contribute to cancer development [149]. Therefore, the consequences of iTreg presence and activity in cancer and precancerous inflammatory lesions may be profound. Their depletion may or may not be beneficial to cancer patients, depending on the environmental context [96].
Necrosis Promotes Treg Persistence in Cancer
The tumor orchestrates its surrounding microenvironment by secreting several soluble factors, including adenosine [150,151], TGF-β [152], prostaglandins [153,154], kynurenine [155,156] and HMGB1 as well as other DAMPs. Tumor-induced immune tolerance in this environment is sustainably mediated by Tregs, which are strongly affected by DAMPs [157], Tumor-induced dysfunction extends to tumor antigen-specific T cells as well as innate immunity components such as natural killer (NK) cells and macrophages [158]. The profile and severity of immune dysfunction in immune cells of different patients with cancer can vary and appears to be dependent on the ability of each tumor to create a unique immune inhibitory environment and to engineer an escape from immune control [159]. Furthermore, H2O2 at low concentrations of about 20 µmol/l within the tumor microenvironment is considered part of the immunosuppressive tumor microenvironment with lymphocytes exhibiting aberrant function upon exposure [160].
Tregs also strongly interact with other immune cells present in the tumor microenvironment and mediate inhibition of the respective immune cell functions [161]. In particular, several soluble factors released by Tregs (e.g., galectin-1, PGE2) may directly suppress [162,163] or induce cell death (e.g., perforin, granzyme) of Teffs and NK cells [164]. Ectoenzymes located on the cell membrane of Tregs (e.g., CD39, CD73) mediate the metabolization of ATP to adenosine, a potential suppressant of T cells [165]. APCs are functionally modulated (e.g., by IL-10, soluble TGF-β) contributing to a tolerogenic tumor milieu. TGF-β is activated by oxidation and inactivated by free thiols, demonstrating the need for the appropriate redox microenvironment for its function [166,167]. Cell-to-cell contact between Tregs and immune cells is obligatory for certain direct and indirect suppressive pathways. CTLA-4 and LAG-3 surface molecules constitutively expressed on nTregs contribute to the cell-to-cell-dependent suppressive mechanisms via interactions with CD80 and CD86 on APCs [168]. Tregs limit Teff and NK cell responses by membrane-bound TGF-β as well as cAMP release [169]. Close interaction with APCs (e.g. via LAG-3, galectin) reduces their immune stimulatory capacity through attenuation of co-stimulation and antigen presentation, while increases their tolerizing potential [170,171], especially by a CTLA-4-mediated up-regulation of the enzyme IDO [172]. IDO activity leads to a depletion of tryptophan accompanied by an accumulation of kynurenine, both with a negative impact on T cells. Tregs induce IDO in DCs, which catalyzes the oxidative catabolism of tryptophan [173]. They require IL-2 for proper function, which is produced by conventional T cells. Accordingly, nTregs express increased levels of the high-affinity heterotrimeric receptor for IL-2 composed of CD25, CD122, and CD132. Competitive depletion of available IL-2 by Tregs and the resultant starvation of activated, dividing T cells has been proposed as a minor suppressive mechanism at minimum within the tumor microenvironment [174]. Lastly, Tregs alter the redox balance of T cells by inhibition of their supply of thiols provided by APCs mainly in form of cysteines, which are obligatory for sustained activation. The proliferating response of activated T cells require glutathione (GSH), an abundant intracellular antioxidant [175]. The synthesis of GSH is limited by the availability of cysteine, and Teffs are inefficient at transporting cysteine [176], the predominate form of this amino acid in the extracellular milieu. Therefore, the cysteine/cystine redox couple is a quantitative significant determinant of the extracellular redox potential, which plays a critical regulatory role in cell proliferation, differentiation, and apoptosis. Exhaustion of free thiol groups by a process similar to cytokine depletion can also produce a negative effect on activated T cells [177]. DCs provide molecular cysteine to the T cells [178], enabling a reducing microenvironment within the immune synapse. Tregs interfere with this process with one very likely mechanism being competitive consumption of thiols including cysteine, as Tregs exhibit increased levels of intra- and extracellular thiols [179].
Cancer is associated with oxidative stress mediated through enhanced ROS production. Malignant cells and, more importantly, cells recruited or induced by tumor cells within the microenvironment, such as tumor-associated macrophages, activated granulocytes, and myeloid-derived suppressor cells, produce significant amounts of ROS [180,181]. The detrimental effect of ROS such as H2O2 and reactive nitrogen species such as NO on NK and T cells is well established and described in malignant and chronic inflammatory diseases [160,182,183,184]. Further immature myeloid cells (ImC) from tumor-bearing mice are able to inhibit the Ag-specific response of CD8+ T cells. Inhibition of ROS in ImC completed abrogated the inhibitory effect of these cells on T cells, indicating a ROS-dependent mechanism of suppression of cytotoxic T cells. Paradoxically, Treg levels can be increased in this hostile (for lymphocytes) milieu, indicating that Tregs can persist in this environment of oxidative stress leading to a selective enrichment of Tregs in the tumor microenvironment. nTregs are more resistant to oxidative stress-mediated cell death compared to conventional CD4+ T cells from healthy individuals [185]. Tregs maintain their suppressive activity even at H2O2 levels that are lethal for half of the conventional effector CD4+ CD25low/- T-cell population. Greater expression levels of surface thiols and a stronger intracellular antioxidative capacity observed in Tregs may contribute to their reduced sensitivity to oxidative stress.
The prototypic DAMP, HMGB1, is passively released from stressed and necrotic cells and is actively secreted by inflammatory cells. Released HMGB1 mediates the response to infection and injury by binding with high affinity to several receptors, including the receptor for advanced glycation end products (RAGE) and TLR-2, TLR-4 and TLR-9, thereby promoting inflammation. The pathogenic role of HMGB1 has been identified during inflammation-associated cell necrosis [186] or as a means by which acute immune responses are initiated against tumor cells undergoing chemotherapy-induced necrosis [187]. HMGB1 is a critical regulator of autophagy in cancer cells. Autophagy allows tumor cells to survive bioenergetic stress by clearing damaged cell organelles or mutant or unfolded proteins and generating glycolic substrates [188]. We have shown that HMGB1 release is associated with sustained autophagy. Moreover, the redox state of HMGB1 is critical, as exogenous delivery of reduced HMGB1 protein promotes autophagy while oxidized HMGB1 promotes apoptosis in cancer cells [189]. Oxidative stress occurs when the generation of ROS in a system exceeds its ability to neutralize and eliminate them. Compared with normal cells, both ROS and autophagy are altered in cancer cells. On the one hand, ROS can induce autophagy through several distinct mechanisms involving ATG4, catalase and the mitochondrial electron transport chain. On the other hand, defective autophagy can increase oxidative stress in tumor cells. This suggests that autophagy defects may increase oxidized HMGB1. Tregs, especially in cancer patients, express both HMGB1-recognizing receptors TLR4 and RAGE [190]. HMGB1 increased secretion of IL-10 and the suppressive capacity in Tregs in a RAGE-dependent manner. Furthermore, our studies revealed that HMGB1 inhibited IFN-γ secretion in Teff. In summary, HMGB1 directly enhanced immune regulatory functions of Tregs and impairs effector function of Teffs [191]. Further, we have demonstrated that HMGB1 is greatly expressed in cancer cells in patients with head and neck cancer and with elevated HMGB1 serum levels [192]. HMGB1 is integral to oxidative stress and downstream apoptosis or survival within the tumor microenvironment. There is a clear association between the release of oxidized HMGB1 and increased ROS production in tumor cells [36] with a subsequent activation of autophagy. Thus, ROS production in tumors not only puts them in a state of apoptotic resistance/autophagy but also sustains tolerance promoting release of HMGB1 and Tregs.
Summary and Perspective
The inflammatory response mediated by innate effectors are quite sensitive to ‘internalized’ stress and unscheduled cell death, characteristic of cancer. In some instances, indeed the adaptive immune response to tumor can enhance tumor progression by promoting non-apoptotic, non-autophagic (i.e. necrotic) cell death and subsequent release of DAMPs. In case of dysplastic transformation of cells, it could be reasonable to modify the local microenvironment that would limit inflammation using immunosuppression, anti-angiogenic agents and apoptosis-inducing drugs, since phagocytosis of apoptotic cells triggers powerful anti-inflammatory signals [193,194]. The hypothesis of necrosis-associated tumorigenesis and tumor proliferation is based on the capacity of released DAMPs to induce tissue proliferation, and most recently to limit apoptosis [20]. Eosinophils may be capable of breaking this vicious circle by limiting tumor cells with release of highly cytotoxic granules and - at the same time - inducing an oxidative milieu inactivating factors released from necrotic cells. DAP10- or DAP12-associated receptors as well as ST2, the receptor for IL-33 are candidate molecules regulating recruitment of inflammatory cells such as eosinophils and the wound healing phenotype within tissues [195,196]. Even in the setting of metastasis, the ability of tumor-derived factors such as DAMPs to promote ‘metastatic’ inflammation has been overlooked [195,196,197]. Inflammation associated with chronic allergic respiratory provocation and substantial local eosinophil accumulation is also capable of significantly enhancing metastasis to the lung in a murine tumor model [198]. The tumor microenvironment is characterized by reducing and hypoxic conditions protecting DAMPs from oxidation. Hypoxia-inducible factor-1 alpha (HIF-1α), upregulation and stabilization, normally degraded by proteasomal ubiquitination under normoxic conditions is observed in tumors. Under hypoxic conditions, the ubiquitination system for HIF-1α is inhibited. Hypoxia and accumulation of HIF-1α in solid tumor tissues is associated with resistance to chemotherapy, radiotherapy, and immunotherapy and poor prognosis. An increase in VEGF levels induces HIF-1α accumulation and promotes tumor metastasis by enhancing angiogenesis [199]. The impact of H2O2 within inflammatory tissue (including tumor tissue) is still controversial. H2O2 at low levels acts as an important ‘second messenger’ during lymphocyte activation [200]. It has both proliferative and apoptotic effects on T cells through various signalling pathways [201,202,203]. Human NK cells can be subdivided into two subsets based on their level of CD56 expression. CD56dim NK cells are potent cytotoxic cells, expressing high levels of granzymes and perforin, and can mediate Ab-dependent cellular cytotoxicity through the FcR CD16. CD56bright NK cells, on the other hand, represent about 5-10% of the NK cells in peripheral blood and, following stimulation with IL-12, IL-15, and IL-18, produce high levels of inflammatory cytokines like IFNγ [204,205]. Interestingly, CD56bright NK cells are very resistant to oxidative stress whereas their counterparts CD56dim NK cells undergo cell death when exposed to ROS.
Tregs also are resistant to oxidative stress and insensitive to ROS, thiols and cystine, enabling immune selection within the tumor microenvironment which eventually results in increased numbers of these cells with high capacity of mediating immune tolerance against the tumor. IL-2 administration at high doses was perhaps the first means to enable overcoming Treg effects [206].
Oxidizing conditions should be considered for therapeutic approaches targeting the tumor microenvironment. In particular, adoptive transfer of eosinophils or induction of eosinophilia in patients with tumor may be helpful considering that these cells are attracted into necrotic areas of tumor and respond to DAMPs by oxidizing them (fig. 1).
Fig. 1.
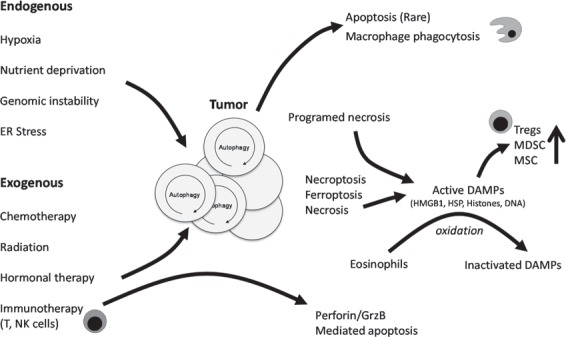
The tumor microenvironment is disordered and characterized by necrosis, redox stress, and DAMPs. Autophagy within the tumor can be promoted by several endogenous (hypoxia, nutrient deprivation, genomic instability, ER stress) and exogenous (chemotherapy, radiation, hormonal therapy) stressors. Most of the current treatment strategies for cancer promote (DAMP molecule release (HMGB1, HSP, histones, DNA etc.) following therapy-induced unscheduled tumor death (often by necroptosis and necrosis). Solely apoptotic cell death in the tumor, intrinsic and driven by p53 pathways, extrinsic, promoted by tissue macrophages and other immune cells expressing TNF family members, is a rare finding. Immunotherapy promoting NK and Teff function can also lead to cytolytic (Perforin/GrzB) mediated apoptosis in the tumor. Therapy promoted DAMP release leads to recruitment of myeloid cells (monocytes, DCs, and granulocytes), as well as MSCs. They belong to the early immigrants in response to unscheduled cell death, initiating and modulating the subsequent inflammatory response. MSCs as well as DCs respond to DAMPs by promoting an immunosuppressive milieu, while eosinophils induce oxidative conditions limiting the biologic activity of DAMPs over time and distance. Tregs are strongly affected by pattern recognition receptor signaling in the tumor microenvironment and limit immune reactivity coordinately with myeloid-derived suppressor cells.
Disclosure Statement
The authors have no conflicting financial interests related to this manuscript.
Acknowledgments/ Financial Support
The study was funded by the University of Ulm; and by 3P30CA047904-22S1 (Davidson); R01 CA160417-01 (Tang); and R01 CA181450-01 (Lotze/Zeh).
References
- 1.Perwez Hussain S, Harris CC. Inflammation and cancer: an ancient link with novel potentials. Int J Cancer. 2007;121:2373–2380. doi: 10.1002/ijc.23173. [DOI] [PubMed] [Google Scholar]
- 2.Kim S, Takahashi H, Lin WW, Descargues P, Grivennikov S, Kim Y, Luo JL, Karin M. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature. 2009;457:102–106. doi: 10.1038/nature07623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJ, Barczak A, Rosenblum MD, Daud A, Barber DL, Amigorena S, Van't Veer LJ, Sperling AI, Wolf DM, Krummel MF. Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell. 2014;26:638–652. doi: 10.1016/j.ccell.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ruffell B, Chang-Strachan D, Chan V, Rosenbusch A, Ho CM, Pryer N, Daniel D, Hwang ES, Rugo HS, Coussens LM. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell. 2014;26:623–637. doi: 10.1016/j.ccell.2014.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Piskounova E, Agathocleous M, Murphy MM, Hu Z, Huddlestun SE, Zhao Z, Leitch AM, Johnson TM, DeBerardinis RJ, Morrison SJ. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature. 2015;527:186–191. doi: 10.1038/nature15726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harris IS, Brugge JS. Cancer: The enemy of my enemy is my friend. Nature. 2015;527:170–171. doi: 10.1038/nature15644. [DOI] [PubMed] [Google Scholar]
- 7.Idrovo JP, Jacob A, Yang WL, Wang Z, Yen HT, Nicastro J, Coppa GF, Wang P. A deficiency in cold-inducible RNA-binding protein accelerates the inflammation phase and improves wound healing. Int J Mol Med. 2016;10 doi: 10.3892/ijmm.2016.2451. [DOI] [PubMed] [Google Scholar]
- 8.Zhou Y, Dong H, Zhong Y, Huang J, Lv J, Li J. The Cold-Inducible RNA-Binding Protein (CIRP) Level in peripheral blood predicts sepsis outcome. PLoS One. 2015;10:e0137721. doi: 10.1371/journal.pone.0137721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Godwin A, Yang WL, Sharma A, Khader A, Wang Z, Zhang F, Nicastro J, Coppa GF, Wang P. Blocking cold-inducible RNA-binding protein protects liver from ischemia-reperfusion injury. Shock. 2015;43:24–30. doi: 10.1097/SHK.0000000000000251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qiang X, Yang WL, Wu R, Zhou M, Jacob A, Dong W, Kuncewitch M, Ji Y, Yang H, Wang H, Fujita J, Nicastro J, Coppa GF, Tracey KJ, Wang P. Cold-inducible RNA-binding protein (CIRP) triggers inflammatory responses in hemorrhagic shock and sepsis. Nat Med. 2013;19:1489–1495. doi: 10.1038/nm.3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vogel S, Bodenstein R, Chen Q, Feil S, Feil R, Rheinlaender J, Schaffer TE, Bohn E, Frick JS, Borst O, Munzer P, Walker B, Markel J, Csanyi G, Pagano PJ, Loughran P, Jessup ME, Watkins SC, Bullock GC, Sperry JL, Zuckerbraun BS, Billiar TR, Lotze MT, Gawaz M, Neal MD. Platelet-derived HMGB1 is a critical mediator of thrombosis. J Clin Invest. 2015;125:4638–4654. doi: 10.1172/JCI81660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen M, Liu Y, Varley P, Chang Y, He XX, Huang H, Tang D, Lotze MT, Lin J, Tsung A. High-mobility group box 1 promotes hepatocellular carcinoma progression through miR-21-mediated matrix metalloproteinase activity. Cancer Res. 2015;75:1645–1656. doi: 10.1158/0008-5472.CAN-14-2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu Y, Yan W, Tohme S, Chen M, Fu Y, Tian D, Lotze M, Tang D, Tsung A. Hypoxia induced HMGB1 and mitochondrial DNA interactions mediate tumor growth in hepatocellular carcinoma through Toll-like receptor 9. J Hepatol. 2015;63:114–121. doi: 10.1016/j.jhep.2015.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu X, Messer JS, Wang Y, Lin F, Cham CM, Chang J, Billiar TR, Lotze MT, Boone DL, Chang EB. Cytosolic HMGB1 controls the cellular autophagy/apoptosis checkpoint during inflammation. J Clin Invest. 2015;125:1098–1110. doi: 10.1172/JCI76344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kang R, Chen R, Zhang Q, Hou W, Wu S, Cao L, Huang J, Yu Y, Fan XG, Yan Z, Sun X, Wang H, Wang Q, Tsung A, Billiar TR, Zeh HJ, III, Lotze MT, Tang D. HMGB1 in health and disease. Mol Aspects Med. 2014;40:1–116. doi: 10.1016/j.mam.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang L, Xie M, Yang M, Yu Y, Zhu S, Hou W, Kang R, Lotze MT, Billiar TR, Wang H, Cao L, Tang D. PKM2 regulates the Warburg effect and promotes HMGB1 release in sepsis. Nat Commun. 2014;5:4436. doi: 10.1038/ncomms5436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lotfi R, Lee JJ, Lotze MT. Eosinophilic granulocytes and damage-associated molecular pattern molecules (DAMPs): role in the inflammatory response within tumors. J Immunother. 2007;30:16–28. doi: 10.1097/01.cji.0000211324.53396.f6. [DOI] [PubMed] [Google Scholar]
- 18.Lotfi R, Schrezenmeier H, Lotze MT. Immunotherapy for cancer: promoting innate immunity. Front Biosci. 2009;14:818–832. doi: 10.2741/3280. [DOI] [PubMed] [Google Scholar]
- 19.Lotfi R, Wiegmann DS, Asseck L, Erle A, Yildiz T, Jahrsdörfer B, Schrezenmeier H. Necrosis-associated factors (DAMPs) including S100A4 used to pulse dendritic cells (DCs) induce regulatory T cells. J Cell Sci Ther. 2012;134:2. [Google Scholar]
- 20.Zeh HJ, III, Lotze MT. Addicted to death: invasive cancer and the immune response to unscheduled cell death. J Immunother. 2005;28:1–9. doi: 10.1097/00002371-200501000-00001. [DOI] [PubMed] [Google Scholar]
- 21.Lotfi R, Herzog GI, DeMarco RA, Beer-Stolz D, Lee JJ, Rubartelli A, Schrezenmeier H, Lotze MT. Eosinophils oxidize damage-associated molecular pattern molecules derived from stressed cells. J Immunol. 2009;183:5023–5031. doi: 10.4049/jimmunol.0900504. [DOI] [PubMed] [Google Scholar]
- 22.Lotfi R, Eisenbacher J, Solgi G, Fuchs K, Yildiz T, Nienhaus C, Rojewski MT, Schrezenmeier H. Human mesenchymal stem cells respond to native but not oxidized damage associated molecular pattern molecules from necrotic (tumor) material. Eur J Immunol. 2011;41:2021–2028. doi: 10.1002/eji.201041324. [DOI] [PubMed] [Google Scholar]
- 23.Eisenbacher JL, Schrezenmeier H, Jahrsdorfer B, Kaltenmeier C, Rojewski MT, Yildiz T, Beyer T, Erle A, Wiegmann DS, Grassl S, Hang R, Korper S, Wiesneth M, Lotze MT, Lotfi R. S100A4 and uric acid promote mesenchymal stromal cell induction of IL-10+/IDO+ lymphocytes. J Immunol. 2014;192:6102–6110. doi: 10.4049/jimmunol.1303144. [DOI] [PubMed] [Google Scholar]
- 24.Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, Moorman MA, Simonetti DW, Craig S, Marshak DR. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284:143–147. doi: 10.1126/science.284.5411.143. [DOI] [PubMed] [Google Scholar]
- 25.Young HE, Steele TA, Bray RA, Hudson J, Floyd JA, Hawkins K, Thomas K, Austin T, Edwards C, Cuzzourt J, Duenzl M, Lucas PA, Black AC., Jr Human reserve pluripotent mesenchymal stem cells are present in the connective tissues of skeletal muscle and dermis derived from fetal, adult, and geriatric donors. Anat Rec. 2001;264:51–62. doi: 10.1002/ar.1128. [DOI] [PubMed] [Google Scholar]
- 26.Gregory CA, Prockop DJ, Spees JL. Non-hematopoietic bone marrow stem cells: molecular control of expansion and differentiation. Exp Cell Res. 2005;306:330–335. doi: 10.1016/j.yexcr.2005.03.018. [DOI] [PubMed] [Google Scholar]
- 27.Horwitz EM, Prockop DJ, Fitzpatrick LA, Koo WW, Gordon PL, Neel M, Sussman M, Orchard P, Marx JC, Pyeritz RE, Brenner MK. Transplantability and therapeutic effects of bone marrow-derived mesenchymal cells in children with osteogenesis imperfecta. Nat Med. 1999;5:309–313. doi: 10.1038/6529. [DOI] [PubMed] [Google Scholar]
- 28.Taniguchi IE, Gonzalez-Nieto D, Ghiaur G, Dunn SK, Ficker AM, Murali B, Madhu M, Gutstein DE, Fishman GI, Barrio LC, Cancelas JA. Connexin-43 prevents hematopoietic stem cell senescence through transfer of reactive oxygen species to bone marrow stromal cells. Proc Natl Acad Sci U S A. 2012;109:9071–9076. doi: 10.1073/pnas.1120358109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994;12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015. [DOI] [PubMed] [Google Scholar]
- 30.Matzinger P. An innate sense of danger. Ann N Y Acad Sci. 2002;961:341–342. doi: 10.1111/j.1749-6632.2002.tb03118.x. [DOI] [PubMed] [Google Scholar]
- 31.Odemuyiwa SO, Ghahary A, Li Y, Puttagunta L, Lee JE, Musat-Marcu S, Moqbel R. Cutting edge: human eosinophils regulate T cell subset selection through indoleamine 2,3-dioxygenase. J Immunol. 2004;173:5909–5913. doi: 10.4049/jimmunol.173.10.5909. [DOI] [PubMed] [Google Scholar]
- 32.Hung SC, Deng WP, Yang WK, Liu RS, Lee CC, Su TC, Lin RJ, Yang DM, Chang CW, Chen WH, Wei HJ, Gelovani JG. Mesenchymal stem cell targeting of microscopic tumors and tumor stroma development monitored by noninvasive in vivo positron emission tomography imaging. Clin Cancer Res. 2005;11:7749–7756. doi: 10.1158/1078-0432.CCR-05-0876. [DOI] [PubMed] [Google Scholar]
- 33.Karnoub AE, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW, Richardson AL, Polyak K, Tubo R, Weinberg RA. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 2007;449:557–563. doi: 10.1038/nature06188. [DOI] [PubMed] [Google Scholar]
- 34.Meng E, Guo Z, Wang H, Jin J, Wang J, Wang H, Wu C, Wang L. High mobility group box 1 protein inhibits the proliferation of human mesenchymal stem cells and promotes their migration and differentiation along osteoblastic pathway. Stem Cells Dev. 2008;17:805–813. doi: 10.1089/scd.2007.0276. [DOI] [PubMed] [Google Scholar]
- 35.Lotze MT, Zeh HJ, Rubartelli A, Sparvero LJ, Amoscato AA, Washburn NR, Devera ME, Liang X, Tor M, Billiar T. The grateful dead: damage-associated molecular pattern molecules and reduction/oxidation regulate immunity. Immunol Rev. 2007;220:60–81. doi: 10.1111/j.1600-065X.2007.00579.x. [DOI] [PubMed] [Google Scholar]
- 36.Kazama H, Ricci JE, Herndon JM, Hoppe G, Green DR, Ferguson TA. Induction of immunological tolerance by apoptotic cells requires caspase-dependent oxidation of high-mobility group box-1 protein. Immunity. 2008;29:21–32. doi: 10.1016/j.immuni.2008.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Janeway CA., Jr. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol. 1989;54:1–13. doi: 10.1101/sqb.1989.054.01.003. [DOI] [PubMed] [Google Scholar]
- 38.Tang D, Kang R, Coyne CB, Zeh HJ, Lotze MT. PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol Rev. 2012;249:158–175. doi: 10.1111/j.1600-065X.2012.01146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kang R, Zhang Q, Zeh HJ, III, Lotze MT, Tang D. HMGB1 in cancer: good, bad, or both? Clin Cancer Res. 2013;19:4046–4057. doi: 10.1158/1078-0432.CCR-13-0495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Steinman RM. Decisions about dendritic cells: past, present, and future. Annu Rev Immunol. 2012;30:1–22. doi: 10.1146/annurev-immunol-100311-102839. [DOI] [PubMed] [Google Scholar]
- 41.Schuler G, Steinman RM. Murine epidermal Langerhans cells mature into potent immunostimulatory dendritic cells in vitro. J Exp Med. 1985;161:526–546. doi: 10.1084/jem.161.3.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Curiel TJ, Cheng P, Mottram P, Alvarez X, Moons L, Evdemon-Hogan M, Wei S, Zou L, Kryczek I, Hoyle G, Lackner A, Carmeliet P, Zou W. Dendritic cell subsets differentially regulate angiogenesis in human ovarian cancer. Cancer Res. 2004;64:5535–5538. doi: 10.1158/0008-5472.CAN-04-1272. [DOI] [PubMed] [Google Scholar]
- 43.Baleeiro RB, Anselmo LB, Soares FA, Pinto CA, Ramos O, Gross JL, Haddad F, Younes RN, Tomiyoshi MY, Bergami-Santos PC, Barbuto JA. High frequency of immature dendritic cells and altered in situ production of interleukin-4 and tumor necrosis factor-alpha in lung cancer. Cancer Immunol Immunother. 2008;57:1335–1345. doi: 10.1007/s00262-008-0468-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shurin MR, Shurin GV, Lokshin A, Yurkovetsky ZR, Gutkin DW, Chatta G, Zhong H, Han B, Ferris RL. Intratumoral cytokines/chemokines/growth factors and tumor infiltrating dendritic cells: friends or enemies? Cancer Metastasis Rev. 2006;25:333–356. doi: 10.1007/s10555-006-9010-6. [DOI] [PubMed] [Google Scholar]
- 45.Whiteside TL. The role of immune cells in the tumor microenvironment. Cancer Treat Res. 2006;130:103–124. doi: 10.1007/0-387-26283-0_5. [DOI] [PubMed] [Google Scholar]
- 46.Whiteside TL. The tumor microenvironment and its role in promoting tumor growth. Oncogene. 2008;27:5904–5912. doi: 10.1038/onc.2008.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mantovani A, Sozzani S, Locati M, Schioppa T, Saccani A, Allavena P, Sica A. Infiltration of tumours by macrophages and dendritic cells: tumour-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Novartis Found Symp. 2004;256:137–145. [PubMed] [Google Scholar]
- 48.Liu Q, Zhang C, Sun A, Zheng Y, Wang L, Cao X. Tumor-educated CD11bhighIalow regulatory dendritic cells suppress T cell response through arginase I. J Immunol. 2009;182:6207–6216. doi: 10.4049/jimmunol.0803926. [DOI] [PubMed] [Google Scholar]
- 49.Benencia F, Sprague L, McGinty J, Pate M, Muccioli M. Dendritic cells the tumor microenvironment and the challenges for an effective antitumor vaccination. J Biomed Biotechnol. 2012;2012:425476. doi: 10.1155/2012/425476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Markiewski MM, DeAngelis RA, Benencia F, Ricklin-Lichtsteiner SK, Koutoulaki A, Gerard C, Coukos G, Lambris JD. Modulation of the antitumor immune response by complement. Nat Immunol. 2008;9:1225–1235. doi: 10.1038/ni.1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Conejo-Garcia JR, Benencia F, Courreges MC, Kang E, Mohamed-Hadley A, Buckanovich RJ, Holtz DO, Jenkins A, Na H, Zhang L, Wagner DS, Katsaros D, Caroll R, Coukos G. Tumor-infiltrating dendritic cell precursors recruited by a beta-defensin contribute to vasculogenesis under the influence of Vegf-A. Nat Med. 2004;10:950–958. doi: 10.1038/nm1097. [DOI] [PubMed] [Google Scholar]
- 52.Gottfried E, Kreutz M, Haffner S, Holler E, Iacobelli M, Andreesen R, Eissner G. Differentiation of human tumour-associated dendritic cells into endothelial-like cells: an alternative pathway of tumour angiogenesis. Scand J Immunol. 2007;65:329–335. doi: 10.1111/j.1365-3083.2007.01903.x. [DOI] [PubMed] [Google Scholar]
- 53.Fernandez PB, Lucibello FC, Zuzarte M, Lutjens P, Muller R, Havemann K. Dendritic cells derived from peripheral monocytes express endothelial markers and in the presence of angiogenic growth factors differentiate into endothelial-like cells. Eur J Cell Biol. 2001;80:99–110. doi: 10.1078/0171-9335-00136. [DOI] [PubMed] [Google Scholar]
- 54.Lu J, Liu K, Zhao J, Zhao J, Ma J, Yang H, Huang Y, Qin Z, Bai R, Jiang L, Lv F, Li P, Yan W, Zhao M, Dong Z. VEGF-A not Ang2 mediates endothelial-like differentiation of immature DCs by ERK1/2 signaling in the microenvironment of human colon adenocarcinoma. Int J Oncol. 2011;38:1579–1588. doi: 10.3892/ijo.2011.989. [DOI] [PubMed] [Google Scholar]
- 55.Lu J, Zhao J, Liu K, Zhao J, Yang H, Huang Y, Qin Z, Bai R, Li P, Ma J, Yan W, Zhao M, Dong Z. MAPK/ERK1/2 signaling mediates endothelial-like differentiation of immature DCs in the microenvironment of esophageal squamous cell carcinoma. Cell Mol Life Sci. 2010;67:2091–2106. doi: 10.1007/s00018-010-0316-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Almand B, Resser JR, Lindman B, Nadaf S, Clark JI, Kwon ED, Carbone DP, Gabrilovich DI. Clinical significance of defective dendritic cell differentiation in cancer. Clin Cancer Res. 2000;6:1755–1766. [PubMed] [Google Scholar]
- 57.Vicari AP, Chiodoni C, Vaure C, Ait-Yahia S, Dercamp C, Matsos F, Reynard O, Taverne C, Merle P, Colombo MP, O'Garra A, Trinchieri G, Caux C. Reversal of tumor-induced dendritic cell paralysis by CpG immunostimulatory oligonucleotide and anti-interleukin 10 receptor antibody. J Exp Med. 2002;196:541–549. doi: 10.1084/jem.20020732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vermi W, Bonecchi R, Facchetti F, Bianchi D, Sozzani S, Festa S, Berenzi A, Cella M, Colonna M. Recruitment of immature plasmacytoid dendritic cells (plasmacytoid monocytes) and myeloid dendritic cells in primary cutaneous melanomas. J Pathol. 2003;200:255–268. doi: 10.1002/path.1344. [DOI] [PubMed] [Google Scholar]
- 59.Ugolini C, Basolo F, Proietti A, Vitti P, Elisei R, Miccoli P, Toniolo A. Lymphocyte and immature dendritic cell infiltrates in differentiated, poorly differentiated, and undifferentiated thyroid carcinoma. Thyroid. 2007;17:389–393. doi: 10.1089/thy.2006.0306. [DOI] [PubMed] [Google Scholar]
- 60.Feijoo E, Alfaro C, Mazzolini G, Serra P, Penuelas I, Arina A, Huarte E, Tirapu I, Palencia B, Murillo O, Ruiz J, Sangro B, Richter JA, Prieto J, Melero I. Dendritic cells delivered inside human carcinomas are sequestered by interleukin-8. Int J Cancer. 2005;116:275–281. doi: 10.1002/ijc.21046. [DOI] [PubMed] [Google Scholar]
- 61.Morse MA, Hobeika AC, Osada T, Serra D, Niedzwiecki D, Lyerly HK, Clay TM. Depletion of human regulatory T cells specifically enhances antigen-specific immune responses to cancer vaccines. Blood. 2008;112:610–618. doi: 10.1182/blood-2008-01-135319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dannull J, Nair S, Su Z, Boczkowski D, DeBeck C, Yang B, Gilboa E, Vieweg J. Enhancing the immunostimulatory function of dendritic cells by transfection with mRNA encoding OX40 ligand. Blood. 2005;105:3206–3213. doi: 10.1182/blood-2004-10-3944. [DOI] [PubMed] [Google Scholar]
- 63.Huarte E, Cubillos-Ruiz JR, Nesbeth YC, Scarlett UK, Martinez DG, Buckanovich RJ, Benencia F, Stan RV, Keler T, Sarobe P, Sentman CL, Conejo-Garcia JR. Depletion of dendritic cells delays ovarian cancer progression by boosting antitumor immunity. Cancer Res. 2008;68:7684–7691. doi: 10.1158/0008-5472.CAN-08-1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chiang CL, Kandalaft LE, Tanyi J, Hagemann AR, Motz GT, Svoronos N, Montone K, Mantia-Smaldone GM, Smith L, Nisenbaum HL, Levine BL, Kalos M, Czerniecki BJ, Torigian DA, Powell DJ, Jr, Mick R, Coukos G. A dendritic cell vaccine pulsed with autologous hypochlorous acid-oxidized ovarian cancer lysate primes effective broad antitumor immunity: from bench to bedside. Clin Cancer Res. 2013;19:4801–4815. doi: 10.1158/1078-0432.CCR-13-1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vakkila J, Lotze MT. Inflammation and necrosis promote tumour growth. Nat Rev Immunol. 2004;4:641–648. doi: 10.1038/nri1415. [DOI] [PubMed] [Google Scholar]
- 66.Schmidt-Hansen B, Ornas D, Grigorian M, Klingelhofer J, Tulchinsky E, Lukanidin E, Ambartsumian N. Extracellular S100A4(mts1) stimulates invasive growth of mouse endothelial cells and modulates MMP-13 matrix metalloproteinase activity. Oncogene. 2004;23:5487–5495. doi: 10.1038/sj.onc.1207720. [DOI] [PubMed] [Google Scholar]
- 67.Belot N, Pochet R, Heizmann CW, Kiss R, Decaestecker C. Extracellular S100A4 stimulates the migration rate of astrocytic tumor cells by modifying the organization of their actin cytoskeleton. Biochim Biophys Acta. 2002;1600:74–83. doi: 10.1016/s1570-9639(02)00447-8. [DOI] [PubMed] [Google Scholar]
- 68.Semov A, Moreno MJ, Onichtchenko A, Abulrob A, Ball M, Ekiel I, Pietrzynski G, Stanimirovic D, Alakhov V. Metastasis-associated protein S100A4 induces angiogenesis through interaction with annexin II and accelerated plasmin formation. J Biol Chem. 2005;280:20833–20841. doi: 10.1074/jbc.M412653200. [DOI] [PubMed] [Google Scholar]
- 69.Schmidt-Hansen B, Klingelhofer J, Grum-Schwensen B, Christensen A, Andresen S, Kruse C, Hansen T, Ambartsumian N, Lukanidin E, Grigorian M. Functional significance of metastasis-inducing S100A4(Mts1) in tumor-stroma interplay. J Biol Chem. 2004;279:24498–24504. doi: 10.1074/jbc.M400441200. [DOI] [PubMed] [Google Scholar]
- 70.Boye K, Maelandsmo GM. S100A4 and metastasis: a small actor playing many roles. Am J Pathol. 2010;176:528–535. doi: 10.2353/ajpath.2010.090526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mencia N, Selga E, Rico I, de Almagro MC, Villalobos X, Ramirez S, Adan J, Hernandez JL, Noe V, Ciudad CJ. Overexpression of S100A4 in human cancer cell lines resistant to methotrexate. BMC Cancer. 2010;10:250–210. doi: 10.1186/1471-2407-10-250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang HY, Zhang JY, Cui JT, Tan XH, Li WM, Gu J, Lu YY. Expression status of S100A14 and S100A4 correlates with metastatic potential and clinical outcome in colorectal cancer after surgery. Oncol Rep. 2010;23:45–52. [PubMed] [Google Scholar]
- 73.Kang YG, Jung CK, Lee A, Kang WK, Oh ST, Kang CS. Prognostic significance of S100A4 mRNA and protein expression in colorectal cancer. J Surg Oncol. 2012;105:119–124. doi: 10.1002/jso.22070. [DOI] [PubMed] [Google Scholar]
- 74.Rothenberg ME, Mishra A, Brandt EB, Hogan SP. Gastrointestinal eosinophils. Immunol Rev. 2001;179:139–155. doi: 10.1034/j.1600-065x.2001.790114.x. [DOI] [PubMed] [Google Scholar]
- 75.Jones RL, Hannan NJ, Kaitu'u TJ, Zhang J, Salamonsen LA. Identification of chemokines important for leukocyte recruitment to the human endometrium at the times of embryo implantation and menstruation. J Clin Endocrinol Metab. 2004;89:6155–6167. doi: 10.1210/jc.2004-0507. [DOI] [PubMed] [Google Scholar]
- 76.Matthews AN, Friend DS, Zimmermann N, Sarafi MN, Luster AD, Pearlman E, Wert SE, Rothenberg ME. Eotaxin is required for the baseline level of tissue eosinophils. Proc of the Natl Acad Sci U S A. 1998;95:6273–6278. doi: 10.1073/pnas.95.11.6273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yang J, Torio A, Donoff RB, Gallagher GT, Egan R, Weller PF, Wong DT. Depletion of eosinophil infiltration by anti-IL-5 monoclonal antibody (TRFK-5) accelerates open skin wound epithelial closure. Am J Pathol. 1997;151:813–819. [PMC free article] [PubMed] [Google Scholar]
- 78.Samoszuk M. Eosinophils and human cancer. Histol Histopathol. 1997;12:807–812. [PubMed] [Google Scholar]
- 79.Lee JJ, Lee NA. Eosinophil degranulation: an evolutionary vestige or a universally destructive effector function? Clin Exp Allergy. 2005;35:986–994. doi: 10.1111/j.1365-2222.2005.02302.x. [DOI] [PubMed] [Google Scholar]
- 80.Afshar K, Vucinic V, Sharma OP. Eosinophil cell: pray tell us what you do! Curr Opin Pulm Med. 2007;13:414–421. doi: 10.1097/MCP.0b013e328224b90b. [DOI] [PubMed] [Google Scholar]
- 81.Lotfi R, Lotze MT. Eosinophils induce DC maturation, regulating immunity. J Leukoc Biol. 2008;83:456–460. doi: 10.1189/jlb.0607366. [DOI] [PubMed] [Google Scholar]
- 82.Nagata M, Sedgwick JB, Bates ME, Kita H, Busse WW. Eosinophil adhesion to vascular cell adhesion molecule-1 activates superoxide anion generation. J Immunol. 1995;155:2194–2202. [PubMed] [Google Scholar]
- 83.Bandeira-Melo C, Woods LJ, Phoofolo M, Weller PF. Intracrine cysteinyl leukotriene receptor-mediated signaling of eosinophil vesicular transport-mediated interleukin-4 secretion. J Exp Med. 2002;196:841–850. doi: 10.1084/jem.20020516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Henderson WR, Harley JB, Fauci AS. Arachidonic acid metabolism in normal and hypereosinophilic syndrome human eosinophils: generation of leukotrienes B4, C4, D4 and 15-lipoxygenase products. Immunology. 1984;51:679–686. [PMC free article] [PubMed] [Google Scholar]
- 85.Turk J, Maas RL, Brash AR, Roberts LJ, Oates JA. Arachidonic acid 15-lipoxygenase products from human eosinophils. J Biol Chem. 1982;257:7068–7076. [PubMed] [Google Scholar]
- 86.Bruijnzeel P, Kok P, Kijne G, Verhagen J. Leukotriene synthesis by isolated granulocytes from intrinsic and extrinsic asthmatics and age-matched controls. Agents Actions Suppl. 1989;28:191–194. [PubMed] [Google Scholar]
- 87.Lee T, Lenihan DJ, Malone B, Roddy LL, Wasserman SI. Increased biosynthesis of platelet-activating factor in activated human eosinophils. J Biol Chem. 1984;259:5526–5530. [PubMed] [Google Scholar]
- 88.Hay DW, Torphy TJ, Undem BJ. Cysteinyl leukotrienes in asthma: old mediators up to new tricks. Trends Pharmacol Sci. 1995;16:304–309. doi: 10.1016/s0165-6147(00)89059-8. [DOI] [PubMed] [Google Scholar]
- 89.Ford-Hutchinson AW, Bray MA, Doig MV, Shipley ME, Smith MJ. Leukotriene B, a potent chemokinetic and aggregating substance released from polymorphonuclear leukocytes. Nature. 1980;286:264–265. doi: 10.1038/286264a0. [DOI] [PubMed] [Google Scholar]
- 90.Busse WW. Leukotrienes and inflammation. Am J Respir Crit Care Med. 1998;157:S210–S213. discussion S247-248. [PubMed] [Google Scholar]
- 91.Akuthota P, Wang H, Weller PF. Eosinophils as antigen-presenting cells in allergic upper airway disease. Curr Opin Allergy Clin Immunol. 2010;10:14–19. doi: 10.1097/ACI.0b013e328334f693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lucey DR, Dorsky DI, Nicholson-Weller A, Weller PF. Human eosinophils express CD4 protein and bind human immunodeficiency virus 1 gp120. J Exp Med. 1989;169:327–332. doi: 10.1084/jem.169.1.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mattes J, Yang M, Mahalingam S, Kuehr J, Webb DC, Simson L, Hogan SP, Koskinen A, McKenzie AN, Dent LA, Rothenberg ME, Matthaei KI, Young IG, Foster PS. Intrinsic defect in T cell production of interleukin (IL)-13 in the absence of both IL-5 and eotaxin precludes the development of eosinophilia and airways hyperreactivity in experimental asthma. J Exp Med. 2002;195:1433–1444. doi: 10.1084/jem.20020009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Padigel UM, Lee JJ, Nolan TJ, Schad GA, Abraham D. Eosinophils can function as antigen-presenting cells to induce primary and secondary immune responses to Strongyloides stercoralis. Infect Immun. 2006;74:3232–3238. doi: 10.1128/IAI.02067-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Shi HZ, Humbles A, Gerard C, Jin Z, Weller PF. Lymph node trafficking and antigen presentation by endobronchial eosinophils. J Clin Invest. 2000;105:945–953. doi: 10.1172/JCI8945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Weller PF, Rand TH, Barrett T, Elovic A, Wong DT, Finberg RW. Accessory cell function of human eosinophils. HLA-DR-dependent, MHC-restricted antigen-presentation and IL-1 alpha expression. J Immunol. 1993;150:2554–2562. [PubMed] [Google Scholar]
- 97.Astigiano S, Morandi B, Costa R, Mastracci L, D'Agostino A, Ratto GB, Melioli G, Frumento G. Eosinophil granulocytes account for indoleamine 2,3-dioxygenase-mediated immune escape in human non-small cell lung cancer. Neoplasia. 2005;7:390–396. doi: 10.1593/neo.04658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Braun D, Longman RS, Albert ML. A two-step induction of indoleamine 2,3 dioxygenase (IDO) activity during dendritic-cell maturation. Blood. 2005;106:2375–2381. doi: 10.1182/blood-2005-03-0979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Schramm C, Huber S, Protschka M, Czochra P, Burg J, Schmitt E, Lohse AW, Galle PR, Blessing M. TGFbeta regulates the CD4+CD25+ T-cell pool and the expression of Foxp3 in vivo. Int Immunol. 2004;16:1241–1249. doi: 10.1093/intimm/dxh126. [DOI] [PubMed] [Google Scholar]
- 100.Iwakura Y, Ishigame H. The IL-23/IL-17 axis in inflammation. J Clin Invest. 2006;116:1218–1222. doi: 10.1172/JCI28508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kim MR, Manoukian R, Yeh R, Silbiger SM, Danilenko DM, Scully S, Sun J, DeRose ML, Stolina M, Chang D, Van GY, Clarkin K, Nguyen HQ, Yu YB, Jing S, Senaldi G, Elliott G, Medlock ES. Transgenic overexpression of human IL-17E results in eosinophilia, B-lymphocyte hyperplasia, and altered antibody production. Blood. 2002;100:2330–2340. doi: 10.1182/blood-2002-01-0012. [DOI] [PubMed] [Google Scholar]
- 102.Molet S, Hamid Q, Davoine F, Nutku E, Taha R, Page N, Olivenstein R, Elias J, Chakir J. IL-17 is increased in asthmatic airways and induces human bronchial fibroblasts to produce cytokines. J Allergy Clin Immunol. 2001;108:430–438. doi: 10.1067/mai.2001.117929. [DOI] [PubMed] [Google Scholar]
- 103.Stenfeldt AL, Wenneras C. Danger signals derived from stressed and necrotic epithelial cells activate human eosinophils. Immunology. 2004;112:605–614. doi: 10.1111/j.1365-2567.2004.01906.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cormier SA, Larson KA, Yuan S, Mitchell TL, Lindenberger K, Carrigan P, Lee NA, Lee JJ. Mouse eosinophil-associated ribonucleases: a unique subfamily expressed during hematopoiesis. Mamm Genome. 2001;12:352–361. doi: 10.1007/s003350020007. [DOI] [PubMed] [Google Scholar]
- 105.Cormier SA, Taranova AG, Bedient C, Nguyen T, Protheroe C, Pero R, Dimina D, Ochkur SI, O'Neill K, Colbert D, Lombari TR, Constant S, McGarry MP, Lee JJ, Lee NA. Pivotal Advance: eosinophil infiltration of solid tumors is an early and persistent inflammatory host response. J Leukoc Biol. 2006;79:1131–1139. doi: 10.1189/jlb.0106027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Cuschieri A, Talbot IC, Weeden S. Influence of pathological tumour variables on long-term survival in resectable gastric cancer. Br J Cancer. 2002;86:674–679. doi: 10.1038/sj.bjc.6600161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Fernandez-Acenero MJ, Galindo-Gallego M, Sanz J, Aljama A. Prognostic influence of tumor-associated eosinophilic infiltrate in colorectal carcinoma. Cancer. 2000;88:1544–1548. [PubMed] [Google Scholar]
- 108.Nagtegaal ID, Marijnen CA, Kranenbarg EK, Mulder-Stapel A, Hermans J, van de Velde CJ, van Krieken JH. Local and distant recurrences in rectal cancer patients are predicted by the nonspecific immune response; specific immune response has only a systemic effect - a histopathological and immunohistochemical study. BMC Cancer. 2001;1:7. doi: 10.1186/1471-2407-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Nielsen HJ, Hansen U, Christensen IJ, Reimert CM, Brunner N, Moesgaard F. Independent prognostic value of eosinophil and mast cell infiltration in colorectal cancer tissue. J Pathol. 1999;189:487–495. doi: 10.1002/(SICI)1096-9896(199912)189:4<487::AID-PATH484>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 110.Pretlow TP, Keith EF, Cryar AK, Bartolucci AA, Pitts AM, Pretlow TG, Kimball PM, Boohaker EA. Eosinophil infiltration of human colonic carcinomas as a prognostic indicator. Cancer Res. 1983;43:2997–3000. [PubMed] [Google Scholar]
- 111.Petreccia DC, Nauseef WM, Clark RA. Respiratory burst of normal human eosinophils. J Leukoc Biol. 1987;41:283–288. doi: 10.1002/jlb.41.4.283. [DOI] [PubMed] [Google Scholar]
- 112.Someya A, Nishijima K, Nunoi H, Irie S, Nagaoka I. Study on the superoxide-producing enzyme of eosinophils and neutrophils - comparison of the NADPH oxidase components. Arch Biochem Biophys. 1997;345:207–213. doi: 10.1006/abbi.1997.0252. [DOI] [PubMed] [Google Scholar]
- 113.Yagisawa M, Yuo A, Yonemaru M, Imajoh-Ohmi S, Kanegasaki S, Yazaki Y, Takaku F. Superoxide release and NADPH oxidase components in mature human phagocytes: correlation between functional capacity and amount of functional proteins. Biochem Biophys Res Commun. 1996;228:510–516. doi: 10.1006/bbrc.1996.1691. [DOI] [PubMed] [Google Scholar]
- 114.Haskell MD, Moy JN, Gleich GJ, Thomas LL. Analysis of signaling events associated with activation of neutrophil superoxide anion production by eosinophil granule major basic protein. Blood. 1995;86:4627–4637. [PubMed] [Google Scholar]
- 115.Moy JN, Gleich GJ, Thomas LL. Noncytotoxic activation of neutrophils by eosinophil granule major basic protein. Effect on superoxide anion generation and lysosomal enzyme release. J Immunol. 1990;145:2626–2632. [PubMed] [Google Scholar]
- 116.Conejo-Garcia JR, Benencia F, Courreges MC, Gimotty PA, Khang E, Buckanovich RJ, Frauwirth KA, Zhang L, Katsaros D, Thompson CB, Levine B, Coukos G. Ovarian carcinoma expresses the NKG2D ligand Letal and promotes the survival and expansion of CD28- antitumor T cells. Cancer Res. 2004;64:2175–2182. doi: 10.1158/0008-5472.can-03-2194. [DOI] [PubMed] [Google Scholar]
- 117.Costello RT, Fauriat C, Sivori S, Marcenaro E, Olive D. NK cells: innate immunity against hematological malignancies? Trends Immunol. 2004;25:328–333. doi: 10.1016/j.it.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 118.Kataoka S, Konishi Y, Nishio Y, Fujikawa-Adachi K, Tominaga A. Antitumor activity of eosinophils activated by IL-5 and eotaxin against hepatocellular carcinoma. DNA Cell Biol. 2004;23:549–560. doi: 10.1089/dna.2004.23.549. [DOI] [PubMed] [Google Scholar]
- 119.Burgess SJ, Marusina AI, Pathmanathan I, Borrego F, Coligan JE. IL-21 down-regulates NKG2D/DAP10 expression on human NK and CD8+ T cells. J Immunol. 2006;176:1490–1497. doi: 10.4049/jimmunol.176.3.1490. [DOI] [PubMed] [Google Scholar]
- 120.Goldman M, Le MA, Braun M, Flamand V, Abramowicz D. A role for eosinophils in transplant rejection. Trends Immunol. 2001;22:247–251. doi: 10.1016/s1471-4906(01)01893-2. [DOI] [PubMed] [Google Scholar]
- 121.Le MA, Surquin M, Demoor FX, Noel JC, Nahori MA, Pretolani M, Flamand V, Braun MY, Goldman M, Abramowicz D. IL-5 mediates eosinophilic rejection of MHC class II-disparate skin allografts in mice. J Immunol. 1999;163:3778–3784. [PubMed] [Google Scholar]
- 122.Huland E, Huland H. Tumor-associated eosinophilia in interleukin-2-treated patients: evidence of toxic eosinophil degranulation on bladder cancer cells. J Cancer Res Clin Oncol. 1992;118:463–467. doi: 10.1007/BF01629431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Simon HU, Plotz S, Simon D, Seitzer U, Braathen LR, Menz G, Straumann A, Dummer R, Levi-Schaffer F. Interleukin-2 primes eosinophil degranulation in hypereosinophilia and Wells' syndrome. Eur J Immunol. 2003;33:834–839. doi: 10.1002/eji.200323727. [DOI] [PubMed] [Google Scholar]
- 124.Sosman JA, Bartemes K, Offord KP, Kita H, Fisher SG, Kefer C, Ellis TA, Fisher RI, Higgins TJ, Gleich GJ. Evidence for eosinophil activation in cancer patients receiving recombinant interleukin-4: effects of interleukin-4 alone and following interleukin-2 administration. Clin Cancer Res. 1995;1:805–812. [PubMed] [Google Scholar]
- 125.Tepper RI, Coffman RL, Leder P. An eosinophil-dependent mechanism for the antitumor effect of interleukin-4. Science. 1992;257:548–551. doi: 10.1126/science.1636093. [DOI] [PubMed] [Google Scholar]
- 126.Bristol JA, Zhu M, Ji H, Mina M, Xie Y, Clarke L, Forry-Schaudies S, Ennist DL. In vitro and in vivo activities of an oncolytic adenoviral vector designed to express GM-CSF. Mol Ther. 2003;7:755–764. doi: 10.1016/s1525-0016(03)00103-5. [DOI] [PubMed] [Google Scholar]
- 127.Rivoltini L, Viggiano V, Spinazze S, Santoro A, Colombo MP, Takatsu K, Parmiani G. In vitro anti-tumor activity of eosinophils from cancer patients treated with subcutaneous administration of interleukin 2. Role of interleukin 5. Int J Cancer. 1993;54:8–15. doi: 10.1002/ijc.2910540103. [DOI] [PubMed] [Google Scholar]
- 128.Wong HL, Costa GL, Lotze MT, Wahl SM. Interleukin (IL) 4 differentially regulates monocyte IL-1 family gene expression and synthesis in vitro and in vivo. J Exp Med. 1993;177:775–781. doi: 10.1084/jem.177.3.775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Tepper RI, Pattengale PK, Leder P. Murine interleukin-4 displays potent anti-tumor activity in vivo. Cell. 1989;57:503–512. doi: 10.1016/0092-8674(89)90925-2. [DOI] [PubMed] [Google Scholar]
- 130.Tepper RI. The eosinophil-mediated antitumor activity of interleukin-4. J Allergy Clin Immunol. 1994;94:1225–1231. doi: 10.1016/0091-6749(94)90336-0. [DOI] [PubMed] [Google Scholar]
- 131.Pericle F, Giovarelli M, Colombo MP, Ferrari G, Musiani P, Modesti A, Cavallo F, Di PF, Novelli F, Forni G. An efficient Th2-type memory follows CD8+ lymphocyte-driven and eosinophil-mediated rejection of a spontaneous mouse mammary adenocarcinoma engineered to release IL-4. J Immunol. 1994;153:5659–5673. [PubMed] [Google Scholar]
- 132.Sacco M, Benedetti S, Cato EM, Caniatti M, Ceruti R, Scanziani E, Pirola B, Villa A, Finocchiaro G, Vezzoni P. Retrovirus-mediated IL-4 gene therapy in spontaneous adenocarcinomas from MMTV-neu transgenic mice. Gene Ther. 1999;6:1893–1897. doi: 10.1038/sj.gt.3301017. [DOI] [PubMed] [Google Scholar]
- 133.Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155:1151–1164. [PubMed] [Google Scholar]
- 134.Shevach EM. Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity. 2009;30:636–645. doi: 10.1016/j.immuni.2009.04.010. [DOI] [PubMed] [Google Scholar]
- 135.Raimondi G, Turner MS, Thomson AW, Morel PA. Naturally occurring regulatory T cells: recent insights in health and disease. Crit Rev Immunol. 2007;27:61–95. doi: 10.1615/critrevimmunol.v27.i1.50. [DOI] [PubMed] [Google Scholar]
- 136.Strauss L, Bergmann C, Whiteside TL. Functional and phenotypic characteristics of CD4+CD25highFoxp3+ Treg clones obtained from peripheral blood of patients with cancer. Int J Cancer. 2007;121:2473–2483. doi: 10.1002/ijc.23001. [DOI] [PubMed] [Google Scholar]
- 137.Liu W, Putnam AL, Xu-Yu Z, Szot GL, Lee MR, Zhu S, Gottlieb PA, Kapranov P, Gingeras TR, Fazekas de St GB, Clayberger C, Soper DM, Ziegler SF, Bluestone JA. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med. 2006;203:1701–1711. doi: 10.1084/jem.20060772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kleinewietfeld M, Starke M, Di MD, Borsellino G, Battistini L, Rotzschke O, Falk K. CD49d provides access to ‘untouched' human Foxp3+ Treg free of contaminating effector cells. Blood. 2009;113:827–836. doi: 10.1182/blood-2008-04-150524. [DOI] [PubMed] [Google Scholar]
- 139.Gavin MA, Rasmussen JP, Fontenot JD, Vasta V, Manganiello VC, Beavo JA, Rudensky AY. Foxp3-dependent programme of regulatory T-cell differentiation. Nature. 2007;445:771–775. doi: 10.1038/nature05543. [DOI] [PubMed] [Google Scholar]
- 140.Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133:775–787. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 141.Roncarolo MG, Gregori S. Is FOXP3 a bona fide marker for human regulatory T cells? Eur J Immunol. 2008;38:925–927. doi: 10.1002/eji.200838168. [DOI] [PubMed] [Google Scholar]
- 142.Polansky JK, Schreiber L, Thelemann C, Ludwig L, Kruger M, Baumgrass R, Cording S, Floess S, Hamann A, Huehn J. Methylation matters: binding of Ets-1 to the demethylated Foxp3 gene contributes to the stabilization of Foxp3 expression in regulatory T cells. J Mol Med (Berl) 2010;88:1029–1040. doi: 10.1007/s00109-010-0642-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ohkura N, Kitagawa Y, Sakaguchi S. Development and maintenance of regulatory T cells. Immunity. 2013;38:414–423. doi: 10.1016/j.immuni.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 144.McHugh RS, Whitters MJ, Piccirillo CA, Young DA, Shevach EM, Collins M, Byrne MC. CD4(+)CD25(+) immunoregulatory T cells: gene expression analysis reveals a functional role for the glucocorticoid-induced TNF receptor. Immunity. 2002;16:311–323. doi: 10.1016/s1074-7613(02)00280-7. [DOI] [PubMed] [Google Scholar]
- 145.Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol. 2012;30:531–564. doi: 10.1146/annurev.immunol.25.022106.141623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Fife BT, Bluestone JA. Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunol Rev. 2008;224:166–182. doi: 10.1111/j.1600-065X.2008.00662.x. [DOI] [PubMed] [Google Scholar]
- 147.Strauss L, Bergmann C, Szczepanski MJ, Lang S, Kirkwood JM, Whiteside TL. Expression of ICOS on human melanoma-infiltrating CD4+CD25highFoxp3+ T regulatory cells: implications and impact on tumor-mediated immune suppression. J Immunol. 2008;180:2967–2980. doi: 10.4049/jimmunol.180.5.2967. [DOI] [PubMed] [Google Scholar]
- 148.Bilate AM, Lafaille JJ. Induced CD4+Foxp3+ regulatory T cells in immune tolerance. Annu Rev Immunol. 2012;30:733–758. doi: 10.1146/annurev-immunol-020711-075043. [DOI] [PubMed] [Google Scholar]
- 149.Lin WW, Karin M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J Clin Invest. 2007;117:1175–1183. doi: 10.1172/JCI31537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Hoskin DW, Mader JS, Furlong SJ, Conrad DM, Blay J. Inhibition of T cell and natural killer cell function by adenosine and its contribution to immune evasion by tumor cells (Review) Int J Oncol. 2008;32:527–535. [PubMed] [Google Scholar]
- 151.Blay J, White TD, Hoskin DW. The extracellular fluid of solid carcinomas contains immunosuppressive concentrations of adenosine. Cancer Res. 1997;57:2602–2605. [PubMed] [Google Scholar]
- 152.Chen W, Konkel JE. TGF-beta and ‘adaptive' Foxp3(+) regulatory T cells. J Mol Cell Biol. 2010;2:30–36. doi: 10.1093/jmcb/mjp004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kalinski P. Regulation of immune responses by prostaglandin E2. J Immunol. 2012;188:21–28. doi: 10.4049/jimmunol.1101029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Wang D, Dubois RN. Eicosanoids and cancer. Nat Rev Cancer. 2010;10:181–193. doi: 10.1038/nrc2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Munn DH, Mellor AL. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J Clin Invest. 2007;117:1147–1154. doi: 10.1172/JCI31178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, Parmentier N, Boon T, Van den Eynde BJ. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med. 2003;9:1269–1274. doi: 10.1038/nm934. [DOI] [PubMed] [Google Scholar]
- 157.Mandapathil M, Szczepanski MJ, Szajnik M, Ren J, Jackson EK, Johnson JT, Gorelik E, Lang S, Whiteside TL. Adenosine and prostaglandin E2 cooperate in the suppression of immune responses mediated by adaptive regulatory T cells. J Biol Chem. 2010;285:27571–27580. doi: 10.1074/jbc.M110.127100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Whiteside TL. Immune responses to malignancies. J Allergy Clin Immunol. 2010;125(suppl 2):S272–S283. doi: 10.1016/j.jaci.2009.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Ferrone S, Whiteside TL. Tumor microenvironment and immune escape. Surg Oncol Clin N Am. 2007;16:755–774. doi: 10.1016/j.soc.2007.08.004. viii. [DOI] [PubMed] [Google Scholar]
- 160.Malmberg KJ, Arulampalam V, Ichihara F, Petersson M, Seki K, Andersson T, Lenkei R, Masucci G, Pettersson S, Kiessling R. Inhibition of activated/memory (CD45RO(+)) T cells by oxidative stress associated with block of NF-kappaB activation. J Immunol. 2001;167:2595–2601. doi: 10.4049/jimmunol.167.5.2595. [DOI] [PubMed] [Google Scholar]
- 161.Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol. 2008;8:523–532. doi: 10.1038/nri2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Mahic M, Yaqub S, Johansson CC, Tasken K, Aandahl EM. FOXP3+CD4+CD25+ adaptive regulatory T cells express cyclooxygenase-2 and suppress effector T cells by a prostaglandin E2-dependent mechanism. J Immunol. 2006;177:246–254. doi: 10.4049/jimmunol.177.1.246. [DOI] [PubMed] [Google Scholar]
- 163.Bergmann C, Strauss L, Zeidler R, Lang S, Whiteside TL. Expansion of human T regulatory type 1 cells in the microenvironment of cyclooxygenase 2 overexpressing head and neck squamous cell carcinoma. Cancer Res. 2007;67(18):8865–8873. doi: 10.1158/0008-5472.CAN-07-0767. [DOI] [PubMed] [Google Scholar]
- 164.Cao X, Cai SF, Fehniger TA, Song J, Collins LI, Piwnica-Worms DR, Ley TJ. Granzyme B and perforin are important for regulatory T cell-mediated suppression of tumor clearance. Immunity. 2007;27:635–646. doi: 10.1016/j.immuni.2007.08.014. [DOI] [PubMed] [Google Scholar]
- 165.Whiteside TL, Mandapathil M, Szczepanski M, Szajnik M. Mechanisms of tumor escape from the immune system: adenosine-producing Treg, exosomes and tumor-associated TLRs. Bull Cancer. 2011;98:E25–E31. doi: 10.1684/bdc.2010.1294. [DOI] [PubMed] [Google Scholar]
- 166.Nakamura K, Kitani A, Strober W. Cell contact-dependent immunosuppression by CD4(+)CD25(+) regulatory T cells is mediated by cell surface-bound transforming growth factor beta. J Exp Med. 2001;194:629–644. doi: 10.1084/jem.194.5.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Blakytny R, Erkell LJ, Brunner G. Inactivation of active and latent transforming growth factor beta by free thiols: potential redox regulation of biological action. Int J Biochem Cell Biol. 2006;38:1363–1373. doi: 10.1016/j.biocel.2006.01.017. [DOI] [PubMed] [Google Scholar]
- 168.Sakaguchi S, Setoguchi R, Yagi H, Nomura T. Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in self-tolerance and autoimmune disease. Curr Top Microbiol Immunol. 2006;305:51–66. doi: 10.1007/3-540-29714-6_3. [DOI] [PubMed] [Google Scholar]
- 169.Ghiringhelli F, Menard C, Terme M, Flament C, Taieb J, Chaput N, Puig PE, Novault S, Escudier B, Vivier E, Lecesne A, Robert C, Blay JY, Bernard J, Caillat-Zucman S, Freitas A, Tursz T, Wagner-Ballon O, Capron C, Vainchencker W, Martin F, Zitvogel L. CD4+CD25+ regulatory T cells inhibit natural killer cell functions in a transforming growth factor-beta-dependent manner. J Exp Med. 2005;202:1075–1085. doi: 10.1084/jem.20051511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Huang CT, Workman CJ, Flies D, Pan X, Marson AL, Zhou G, Hipkiss EL, Ravi S, Kowalski J, Levitsky HI, Powell JD, Pardoll DM, Drake CG, Vignali DA. Role of LAG-3 in regulatory T cells. Immunity. 2004;21:503–513. doi: 10.1016/j.immuni.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 171.Huang CT, Workman CJ, Flies D, Pan X, Marson AL, Zhou G, Hipkiss EL, Ravi S, Kowalski J, Levitsky HI, Powell JD, Pardoll DM, Drake CG, Vignali DA. Role of LAG-3 in regulatory T cells. Immunity. 2004;21:503–513. doi: 10.1016/j.immuni.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 172.Joosten SA, van Meijgaarden KE, Savage ND, de BT, Triebel F, van der Wal A, de HE, Klein MR, Geluk A, Ottenhoff TH. Identification of a human CD8+ regulatory T cell subset that mediates suppression through the chemokine CC chemokine ligand 4. Proc Natl Acad Sci U S A. 2007;104:8029–8034. doi: 10.1073/pnas.0702257104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Fallarino F, Grohmann U, Hwang KW, Orabona C, Vacca C, Bianchi R, Belladonna ML, Fioretti MC, Alegre ML, Puccetti P. Modulation of tryptophan catabolism by regulatory T cells. Nat Immunol. 2003;4:1206–1212. doi: 10.1038/ni1003. [DOI] [PubMed] [Google Scholar]
- 174.von Boehmer H. Mechanisms of suppression by suppressor T cells. Nat Immunol. 2005;6:338–344. doi: 10.1038/ni1180. [DOI] [PubMed] [Google Scholar]
- 175.Suthanthiran M, Anderson ME, Sharma VK, Meister A. Glutathione regulates activation-dependent DNA synthesis in highly purified normal human T lymphocytes stimulated via the CD2 and CD3 antigens. Proc Natl Acad Sci U S A. 1990;87:3343–3347. doi: 10.1073/pnas.87.9.3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Ishii T, Sugita Y, Bannai S. Regulation of glutathione levels in mouse spleen lymphocytes by transport of cysteine. J Cell Physiol. 1987;133:330–336. doi: 10.1002/jcp.1041330217. [DOI] [PubMed] [Google Scholar]
- 177.Banjac A, Perisic T, Sato H, Seiler A, Bannai S, Weiss N, Kolle P, Tschoep K, Issels RD, Daniel PT, Conrad M, Bornkamm GW. The cystine/cysteine cycle: a redox cycle regulating susceptibility versus resistance to cell death. Oncogene. 2008;27:1618–1628. doi: 10.1038/sj.onc.1210796. [DOI] [PubMed] [Google Scholar]
- 178.Angelini G, Gardella S, Ardy M, Ciriolo MR, Filomeni G, Di TG, Clarke F, Sitia R, Rubartelli A. Antigen-presenting dendritic cells provide the reducing extracellular microenvironment required for T lymphocyte activation. Proc Natl Acad Sci U S A. 2002;99:1491–1496. doi: 10.1073/pnas.022630299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Yan Z, Garg SK, Kipnis J, Banerjee R. Extracellular redox modulation by regulatory T cells. Nat Chem Biol. 2009;5:721–723. doi: 10.1038/nchembio.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Szatrowski TP, Nathan CF. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991;51:794–798. [PubMed] [Google Scholar]
- 181.Kusmartsev S, Nefedova Y, Yoder D, Gabrilovich DI. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J Immunol. 2004;172:989–999. doi: 10.4049/jimmunol.172.2.989. [DOI] [PubMed] [Google Scholar]
- 182.Gringhuis SI, Leow A, Papendrecht-Van Der Voort EA, Remans PH, Breedveld FC, Verweij CL. Displacement of linker for activation of T cells from the plasma membrane due to redox balance alterations results in hyporesponsiveness of synovial fluid T lymphocytes in rheumatoid arthritis. J Immunol. 2000;164:2170–2179. doi: 10.4049/jimmunol.164.4.2170. [DOI] [PubMed] [Google Scholar]
- 183.Schmielau J, Finn OJ. Activated granulocytes and granulocyte-derived hydrogen peroxide are the underlying mechanism of suppression of t-cell function in advanced cancer patients. Cancer Res. 2001;61:4756–4760. [PubMed] [Google Scholar]
- 184.Li W, Lidebjer C, Yuan XM, Szymanowski A, Backteman K, Ernerudh J, Leanderson P, Nilsson L, Swahn E, Jonasson L. NK cell apoptosis in coronary artery disease: relation to oxidative stress. Atherosclerosis. 2008;199:65–72. doi: 10.1016/j.atherosclerosis.2007.10.031. [DOI] [PubMed] [Google Scholar]
- 185.Mougiakakos D, Johansson CC, Kiessling R. Naturally occurring regulatory T cells show reduced sensitivity toward oxidative stress-induced cell death. Blood. 2009;113:3542–3545. doi: 10.1182/blood-2008-09-181040. [DOI] [PubMed] [Google Scholar]
- 186.Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–195. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- 187.Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, Mignot G, Maiuri MC, Ullrich E, Saulnier P, Yang H, Amigorena S, Ryffel B, Barrat FJ, Saftig P, Levi F, Lidereau R, Nogues C, Mira JP, Chompret A, Joulin V, Clavel-Chapelon F, Bourhis J, Andre F, Delaloge S, Tursz T, Kroemer G, Zitvogel L. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13:1050–1059. doi: 10.1038/nm1622. [DOI] [PubMed] [Google Scholar]
- 188.Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, Mukherjee C, Shi Y, Gelinas C, Fan Y, Nelson DA, Jin S, White E. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10:51–64. doi: 10.1016/j.ccr.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Tang D, Kang R, Cheh CW, Livesey KM, Liang X, Schapiro NE, Benschop R, Sparvero LJ, Amoscato AA, Tracey KJ, Zeh HJ, Lotze MT. HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene. 2010;29:5299–5310. doi: 10.1038/onc.2010.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Wild CA, Brandau S, Lindemann M, Lotfi R, Hoffmann TK, Lang S, Bergmann C. Toll-like receptors in regulatory T cells of patients with head and neck cancer. Arch Otolaryngol Head Neck Surg. 2010;136:1253–1259. doi: 10.1001/archoto.2010.195. [DOI] [PubMed] [Google Scholar]
- 191.Wild CA, Bergmann C, Fritz G, Schuler P, Hoffmann TK, Lotfi R, Westendorf A, Brandau S, Lang S. HMGB1 conveys immunosuppressive characteristics on regulatory and conventional T cells. Int Immunol. 2012;24:485–494. doi: 10.1093/intimm/dxs051. [DOI] [PubMed] [Google Scholar]
- 192.Wild CA, Brandau S, Lotfi R, Mattheis S, Gu X, Lang S, Bergmann C. HMGB1 is overexpressed in tumor cells and promotes activity of regulatory T cells in patients with head and neck cancer. Oral Oncol. 2012;48:409–416. doi: 10.1016/j.oraloncology.2011.12.009. [DOI] [PubMed] [Google Scholar]
- 193.Savill J, Fadok V. Corpse clearance defines the meaning of cell death. Nature. 2000;407:784–788. doi: 10.1038/35037722. [DOI] [PubMed] [Google Scholar]
- 194.Stuart LM, Lucas M, Simpson C, Lamb J, Savill J, Lacy-Hulbert A. Inhibitory effects of apoptotic cell ingestion upon endotoxin-driven myeloid dendritic cell maturation. J Immunol. 2002;168:1627–1635. doi: 10.4049/jimmunol.168.4.1627. [DOI] [PubMed] [Google Scholar]
- 195.Kuchler AM, Pollheimer J, Balogh J, Sponheim J, Manley L, Sorensen DR, De Angelis PM, Scott H, Haraldsen G. Nuclear interleukin-33 is generally expressed in resting endothelium but rapidly lost upon angiogenic or proinflammatory activation. Am J Pathol. 2008;173:1229–1242. doi: 10.2353/ajpath.2008.080014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Lanier LL. DAP10- and DAP12-associated receptors in innate immunity. Immunol Rev. 2009;227:150–160. doi: 10.1111/j.1600-065X.2008.00720.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Chiang AC, Massague J. Molecular basis of metastasis. N Engl J Med. 2008;359:2814–2823. doi: 10.1056/NEJMra0805239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Lee JJ, Lotze MT. Molecular basis of metastasis. N Engl J Med. 2009;360:1679–1680. [PubMed] [Google Scholar]
- 199.Yasuda H. Solid tumor physiology and hypoxia-induced chemo/radio-resistance: novel strategy for cancer therapy: nitric oxide donor as a therapeutic enhancer. Nitric Oxide. 2008;19:205–216. doi: 10.1016/j.niox.2008.04.026. [DOI] [PubMed] [Google Scholar]
- 200.Reth M. Hydrogen peroxide as second messenger in lymphocyte activation. Nat Immunol. 2002;3:1129–1134. doi: 10.1038/ni1202-1129. [DOI] [PubMed] [Google Scholar]
- 201.Devadas S, Zaritskaya L, Rhee SG, Oberley L, Williams MS. Discrete generation of superoxide and hydrogen peroxide by T cell receptor stimulation: selective regulation of mitogen-activated protein kinase activation and fas ligand expression. J Exp Med. 2002;195:59–70. doi: 10.1084/jem.20010659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Gulow K, Kaminski M, Darvas K, Suss D, Li-Weber M, Krammer PH. HIV-1 trans-activator of transcription substitutes for oxidative signaling in activation-induced T cell death. J Immunol. 2005;174:5249–5260. doi: 10.4049/jimmunol.174.9.5249. [DOI] [PubMed] [Google Scholar]
- 203.Kwon J, Devadas S, Williams MS. T cell receptor-stimulated generation of hydrogen peroxide inhibits MEK-ERK activation and lck serine phosphorylation. Free Radic Biol Med. 2003;35:406–417. doi: 10.1016/s0891-5849(03)00318-6. [DOI] [PubMed] [Google Scholar]
- 204.Cooper MA, Fehniger TA, Turner SC, Chen KS, Ghaheri BA, Ghayur T, Carson WE, Caligiuri MA. Human natural killer cells: a unique innate immunoregulatory role for the CD56(bright) subset. Blood. 2001;97:3146–3151. doi: 10.1182/blood.v97.10.3146. [DOI] [PubMed] [Google Scholar]
- 205.Harlin H, Hanson M, Johansson CC, Sakurai D, Poschke I, Norell H, Malmberg KJ, Kiessling R. The CD16- CD56(bright) NK cell subset is resistant to reactive oxygen species produced by activated granulocytes and has higher antioxidative capacity than the CD16+ CD56(dim) subset. J Immunol. 2007;179:4513–4519. doi: 10.4049/jimmunol.179.7.4513. [DOI] [PubMed] [Google Scholar]
- 206.Lotze MT, Frana LW, Sharrow SO, Robb RJ, Rosenberg SA. In vivo administration of purified human interleukin 2. I. Half-life and immunologic effects of the Jurkat cell line-derived interleukin 2. J Immunol. 1985;134:157–166. [PubMed] [Google Scholar]