

Abstract
Visualization of chromosomal dynamics is important for understanding many fundamental intra-nuclear processes. Efficient and reliable live-cell multicolor labeling of chromosomal loci can realize this goal. However, the current methods are constrained mainly by insufficient labeling throughput, efficiency, flexibility as well as photostability. Here we have developed a new approach to realize dual-color chromosomal loci imaging based on a modified single-guide RNA (sgRNA) of the CRISPR/Cas9 system. The modification of sgRNA was optimized by structure-guided engineering of the original sgRNA, consisting of RNA aptamer insertions that bind fluorescent protein-tagged effectors. By labeling and tracking telomeres, centromeres and genomic loci, we demonstrate that the new approach is easy to implement and enables robust dual-color imaging of genomic elements. Importantly, our data also indicate that the fast exchange rate of RNA aptamer binding effectors makes our sgRNA-based labeling method much more tolerant to photobleaching than the Cas9-based labeling method. This is crucial for continuous, long-term tracking of chromosomal dynamics. Lastly, as our method is complementary to other live-cell genomic labeling systems, it is therefore possible to combine them into a plentiful palette for the study of native chromatin organization and genome ultrastructure dynamics in living cells.
INTRODUCTION
Accumulating evidence has suggested that mammalian genomes have hierarchical structures (1–6). Fundamental transitions during cell differentiation and between developmental stages are often accompanied by changes in chromatin architecture and gene relocalization (7–10). Moreover, defects in nuclear reorganization, such as the spatiotemporal mislocalization of genomic loci, can lead to severe human diseases (11). However, a causal relationship between a gene's spatial position and its level of expression has not been established. It remains unclear whether the translocation of a gene is the cause or the consequence of the changing expression level (12–15). Addressing these questions will require live cell multicolor fluorescent labeling and long-term tracking of chromosomal loci.
To visualize the spatial distribution of genomic elements and probe changes therein under different conditions and differentiation states, DNA fluorescence in situ hybridization (DNA–FISH) and fluorescent repressor and operator system (FROS), such as artificial LacO arrays, have been developed as traditional genomic loci labeling methods in the last two decades (16–21). However, while the robust and widely-used FISH method can provide high specificity and signal-to-noise ratio (SNR), it must be performed on fixed samples, making it incompatible with living cells. For the FROS labeling method, although it allows long term dynamic tracking of chromosomal loci in living cells, the intrusive insertion of long LacO repeats into the locus of interest is labor-intensive and may perturb the native chromosomal structure and dynamics.
In recent years, several new endogenous genomic labeling approaches in the living cell have been developed based on gene targeting techniques including the zinc-finger nucleases, the transcription activator-like effector (TALE) and the clustered regulatory interspaced short palindromic repeats (CRISPR/Cas9) system (22–28). These non-intrusive techniques are composed of programmable, sequence-specific DNA-binding modules fused to fluorescent proteins by various linkers (29–33). The repetitive building block assembly of ZFs and TALEs for each target is labor-intensive and costly to implement (24). In contrast, the CRISPR/Cas9 system recognizes target DNA by a short guide RNA via Watson–Crick base pairing, making it easier to perform gene targeting in a high-throughput manner (34). Recently, the CRISPR/Cas9 system was adapted for imaging, providing a robust method to visualize and track the dynamics of both repetitive and non-repetitive genomic loci in living human cells (25).
Labeling chromosomal loci using multiple colors allows spatial resolution of individual alleles (33), as well as observation of fundamental processes such as double strand break-induced translocations (18,21) and promoter-enhancer looping (35–37). Because the targeting specificity of the commonly used Streptococcus pyogenes (SP) Cas9 system is solely determined by sgRNA base pairing instead of the Cas9 protein, resolving different chromosomal loci with multiple fluorophores has remained challenging. Until recently, dual-color CRISPR/Cas9 imaging was accomplished based on an orthogonal Cas9 system to visualize inter- and intra-chromosomal repetitive sequences (38). However, unlike the SP Cas9, the efficiency of the NM (Neisseria meningitidis) and ST1 (Streptococcus thermophilus) Cas9 still requires optimization (39). Additionally, the required protospacer adjacent motifs (PAM) sequences of the NM and ST1 Cas9 systems are more complicated than that of the SP Cas9 system, making it difficult to design a pool of sgRNAs for the target regions.
Here we describe a new approach to realize multicolor labeling of multiple chromosomal loci using modified sgRNA scaffolds that only require an SP dCas9. The multicolor imaging system consists of an endonuclease-deactivated Cas9 protein (dCas9) (25) and structurally modified sgRNAs with RNA aptamer MS2/PP7 insertions that bind fluorescent protein-tagged tdMCP and tdPCP (40,41). We demonstrate that the new multicolor imaging approach enables robust imaging of repetitive elements of both telomeres and centromeres simultaneously for a long term. Compared with the approach based on the orthogonal Cas9 systems, the modified sgRNA approach offers multicolor imaging capacity with higher specificity and flexibility as the PAM sequence of the SP CRISPR/Cas9 system is more abundant in the human genome. In addition, because the exchange rates for tdMCP and tdPCP are much faster than that of the dCas9 protein, our approach is particularly suitable for long term tracking by compensating for photobleaching at the target locus. Lastly, our modified sgRNA approach is complementary to the orthogonal Cas9 imaging system. Therefore, these two multicolor genomic labeling methods can be combined into an expansive toolkit for the study of native chromatin organization and genomic higher-order structural dynamics in living human cells.
MATERIALS AND METHODS
Construction of dCas9, modified sgRNA and aptamer coating protein expression plasmids
The NLSSV40-dCas9-NLSSV40 fragment was amplified by polymerase chain reaction (PCR) from pSLQ1645-dCas9-GFP (a gift from Bo Huang, University of California, San Francisco) and then ligated into plasmid pcDNA3.1(+)-TRE3G by Gibson Assembly (NEB). The dCas9-mCherry expression plasmid was constructed by ligation of NLSSV40-dCas9-NLSSV40 into pcDNA3.1(+)-TRE3G-mCherry by Gibson Assembly (NEB). The tdMCP fragment was amplified by PCR from phage-ubc-nls-ha-tdMCP-gfp (a gift from Robert H. Singer, Albert Einstein College of Medicine) and then ligated into plasmid pHR-TRE3G-mCherry (a gift from Ping Wei, Peking University) by standard ligation-dependent cloning using MluI-HF (NEB) and BamHI-HF (NEB) enzymes. In order to construct a stable cell line, NLSSV40-dCas9-NLSSV40 was ligated into PiggyBac plasmid pB-TRE3G-BsmBI-EF1α-HygroR-P2A-rtTA by golden gate cloning. NLSSV40-tdMCP-mCherry and NLSSV40-tdPCP-EGFP were amplified by PCR and then ligated into PiggyBac plasmid pB-TRE3G-BsmBI-EF1α-PuroR-P2A-rtTA by golden gate cloning.
The DNA fragments of each modified sgRNA scaffold were synthesized by Shanghai GeneArray biotech (Shanghai, China) with about a 15-bp homologous arm flanking each end and ligated into the original sgRNA expression plasmid by Gibson Assembly, which was PCR amplified into a linear form to remove the native sgRNA scaffold. The telomere, centromere and MUC4-E2 targeting sgRNA expression plasmids were made by replacing the lethal gene ccdB with a 20-bp annealed oligo using golden gate cloning with enzyme BsmBI and T4 ligase (NEB). For each modified sgRNA design and cloning, the sequence and primer of each construct, see Supplementary Materials.
Cell culture, transfection, stable cell line construction and immunofluorescence
Human cell line MDA-MB-231 and HeLa cells were maintained in Dulbecco's modified Eagle medium with high glucose (Lifetech), 10% Fetal bovine serum (FBS) (Lifetech), 1× penicillin/streptomycin (Lifetech). All cells were maintained at 37°C and 5% CO2 in a humidified incubator. All plasmids were transfected with Lipofectamine 2000 (Lifetech) in accordance with the manufacturer's protocol. To construct a stable cell line, MDA-MB-231 cells were spread onto a 6-well plate one day before transfection. On the next day, the cells were transfected with 500 ng pB-TRE3G-dCas9-EF1α-HygroR-P2A-rtTA, 500 ng pB-TRE3G-tdPCP-EGFP-EF1α-PuroR-P2A-rtTA, 500 ng pB-TRE3G-tdMCP-mCherry-EF1α-PuroR-P2A-rtTA and 200 ng pCAG-hyPBase using Lipo 2000. After 48 h, cells were transferred onto a 35 mm dish, incubated for another 48 h and then subjected to hygromycin (200 μg/ml) and puromycin (5 μg/ml) selection. After incubation for 2 weeks, cells with appropriate expression level of tdPCP-EGFP and tdMCP-mCherry were selected using FACS. For immunofluorescence, cells were washed with phosphate buffered saline (PBS) briefly once and incubated with extraction buffer (0.1 M PIPS, 1 mM EGTA, 1 mM MgCl2, 0.2% TritonX-100) for 1 min to extract cytoplasm diffusive proteins. Cells were washed once with PBS, fixed with 4% PFA for 15 min and then incubated in 0.5% Triton followed by 30 min 5% BSA blocking. The cells were then stained with human anti-CREST antibodies (Antibodies Incorporated, #15–235-F) in blocking buffer for 60 min, washed with PBS and then stained with Cy5-labeled secondary antibodies in blocking buffer for 1 h. The labeled cells were washed with PBS, post-fixed with 4% PFA for 10 min at room temperature.
Optical setup and image acquisition
All images were taken on an UltraVIEW VoX spinning disc microscope (PerkinElmer). For imaging, cells were grown on 35-mm glass-bottom dishes. The microscope stage incubation chamber was maintained at 37°C and 5% CO2.
Fluorescence recovery after photobleaching (FRAP) experiments were performed using 10 iterations with the acousto-optic tunable filter (AOTF) of the 488 nm laser line or 561 nm laser line set to 100% transmission. To reduce the phototoxic effect, for acquisition of post-bleach images, the 488 nm laser line was set to 20% transmission and the 561 nm laser line was set to 30% transmission. In one cell, two rectangle bleach regions, each covering one centromere, were chosen to be photobleached. For sgRNA2.02-MS2-EGFP, after 10 pre-bleach frames with a time interval of 200 ms, 150 post-bleach frames were recorded with a time interval of 200 ms followed by 150 post-bleach frames with a time interval of 5 s. For dCas9-mCherry, after 10 pre-bleach frames with a time interval of 10 s, 236 post-bleach frames were recorded with a time interval of 10 s.
In the long-term time-lapse tracking of cell division, a z-stack of 31 layers of both channels with a step size of 0.4 μm were acquired every 10 min for about 3 h. In the longer time scale imaging, one layer of both channels were acquired every 5 min for about 26 h. In both cases, to reduce the photo-damage effect to the cells, both 488 and 561 nm lasers were set to 10% transmission.
Image analysis
Dual-color single particle tracking analysis
Dual-color telomere and centromere image stacks were analyzed by MATLAB tracking package ‘u-track’ (42). Fluorescent puncta were identified in each frame with Gaussian fitting after Fourier low pass filtering. The coordinates of the fluorescent puncta were determined. Trajectories were created by linking identified puncta to their nearest neighbors within a maximum distance range of 5 camera pixels (715 nm) in the previous frame. Trajectory gap larger than 10 consecutive frames were treated as two particles.
For each trajectory, the mean square displacement (MSD) as a function of time delay was calculated by the following equation:
 |
where δt is the time interval between two successive frames, x(t) and y(t) are the coordinates at time t, N is the total number of frames and n is the number of time intervals. To maximize the precision in the long-range MSD, the intervals smaller than N/10 were used for the calculation.
The analysis of MSD curves was carried out using custom MATLAB scripts. The averaged MSD curves were fitted by least-squares regression to a model for confined diffusion, macroscopic diffusion and active transport as previously described (25,43):
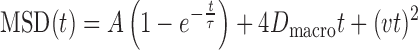 |
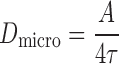 |
FRAP analysis
Each centromere region was cropped into a discrete image file. The selected fluorescent centromere was converted into a binary image and then the intensity of selected region was analyzed. The measurements and calculations were performed with ImageJ and MATLAB scripts. The normalized intensity was calculated by,
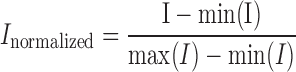 |
To quantify the bleaching effect, the whole cell fluorescence was used to calculate the total intensity as a control,
 |
The resulting intensity was then fitted by an exponential recovery function,
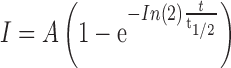 |
where, A is the intensity in t→∞, when the recovery reaches to a steady state. t1/2 is the time when the intensity reaches to half of the steady state.
Colocalization analysis
The red channel and green channel images of a z stack were projected into a 2D image by maximum intensity projection and then converted into 8 bit images. Colocalization analysis was carried out using the Image J plugin ‘Image Correlator plus’ with crosshair size = 3 pixels. The Pearson's correlation Rr and overlap coefficient R were calculated to estimate the colocalization efficiency of the two channels. The colocalized pixels were used to render the colocalization images.
RESULTS
Design and optimization of modified sgRNAs
The structure-guided engineering of the Cas9–sgRNA complex has recently led to several robust tools for gene regulation (44–46). To engineer sgRNAs for the endogenous chromosomal loci labeling, we modified the sgRNA guided by the structural information (47–49) so that the perturbation to the Cas9–sgRNA complex may be minimized (Figure 1A). Specifically, by inserting the MS2/PP7 aptamer to the tail, loop 2 or tetraloop, seven types of modified sgRNAs, named as sgRNA1.0, 1.1, 1.2, 2.0, 1.12, 1.22 and 2.02, were generated for screening (Figure 1B and Supplementary Figure S1). Among them, sgRNA1.12, 1.22 and 2.02 adopt an A-U base pair flip as well as an extended hairpin structure, which were suggested to prevent the premature termination of U6 Pol-III transcription and increase the assembly stability of the dCas9 and sgRNA (25). In order to increase the nuclear importing efficiency of the dCas9 protein, two nuclear localization sequences (NLS) were added to both the N- and C- termini (Figure 1C). The MS2 binding protein MCP and the PP7 binding protein PCP were expressed in a tandem dimer to enhance the binding affinity to their respective aptamer stem loop (50). Fluorescent proteins EGFP and mCherry were fused to tdPCP and tdMCP, respectively (Figure 1C).
Figure 1.

Schematic diagram of the modified sgRNA scaffolds for dual-color imaging of genomic sequences in living cells. (A) Overview of the dual-color imaging by modified sgRNA scaffolds. sgRNA molecules are extended with additional domains (MS2/PP7) to recruit RNA binding proteins that are fused with fluorescent pro3teins (tdMCP-mCherry, tdPCP-EGFP). The modified sgRNA can be classified into several motifs including the DNA targeting motif, the dCas9 binding motif and the effector recruitment motif. Enrichment of the fluorescence signal by multiple fluorescently labeled modified sgRNAs allows imaging of different genomic elements in living cells. The color annotations in the nucleus stand for different chromosomes. (B) Motif annotations of the modified sgRNA scaffolds. The original sgRNA can be classified into eight motifs. The RNA aptamers (MS2/PP7) can be inserted into the tail (named as sgRNA1.0), the tetraloop (sgRNA1.1), loop 2 (sgRNA1.2), or both tetraloop and loop2 (sgRNA2.0). sgRNAs with additional optimizations are named as sgRNA1.12, 1.22 and 2.02 (see Supplementary Figure S1). (C) Three components of the modified dual-color CRISPR imaging system: a doxycycline-inducible dCas9 expression plasmid, two doxycycline-inducible fluorescent protein-tagged tdMCP/tdPCP plasmids and target-specific modified sgRNAs expressed from a murine U6 promoter. NLS sequences were added to dCas9 as well as tdMCP and tdPCP to increase their nuclear localization.
To evaluate the genomic labeling performance of the modified sgRNAs, we designed modified sgRNAs that targeted the repetitive sequences of the human centromeric α satellites and telomeric repeats. For the validation of labeling specificity, SP dCas9-mCherry, modified sgRNA-MS2/PP7 and tdMCP/tdPCP-EGFP were co-expressed to co-label the target sequences in human MDA-MB-231 and HeLa cells. Direct labeling by dCas9-mCherry would demonstrate that the modified sgRNAs have no influences on the integrity of the targeting mechanism, while ‘bridge’ labeling by sgRNAs-tdMCP/tdPCP-EGFP shows that the inserted aptamers can recruit fluorescent protein-tagged aptamer binding proteins to the designed target locus successfully. The colocalization level of the two fluorescence channels was used to evaluate the labeling performance of the modified sgRNAs.
For sgRNA1.0, in which MS2 or PP7 was inserted at the end of the sgRNA scaffold, no distinct puncta of telomeres were observed. In contrast, the dCas9-mCherry was able to label the telomeres in about 60% of the cells imaged (Supplementary Figure S2). Additionally, dCas9-mCherry and sgRNA1.0-MS2/PP7-EGFP were both found to largely localize to the nucleoli (Supplementary Figure S2). The poor labeling efficiency and the non-specific spatial distribution imply that the changes in the sgRNA structure may cause a certain level of competition between dCas9 and tdMCP/tdPCP in sgRNA binding. Since the terminal insertion in sgRNA1.0 was not suitable for labeling, we tried to insert the aptamer to the loops outside of the dCas9–sgRNA complex (47–49) and created sgRNA1.1, 1.2, 2.0, 1.12, 1.22 and 2.02 (Figure 1B). Dual-color co-labeling of the centromeres by the modified sgRNA-MS2/PP7-EGFP and dCas9-mCherry were used to quantify the performance of modified sgRNAs based on two parameters, i.e. the SNR and the total number of labeled centromeres per cell, in addition to the colocalization level (Figure 2). The number of labeled centromeres was counted using the binary analysis (Supplementary Figure S3). Figure 2A and B show that sgRNA1.1-MS2-EGFP puncta colocalized well with dCas9-mCherry puncta, suggesting that insertions in the middle of the sgRNA can reduce the competition between dCas9 and tdMCP/tdPCP in the sgRNA binding and thus increase the labeling efficiency of both dCas9 and modified sgRNA (Supplementary Movies S1 and 2). Nevertheless, although the SNRs of the centromeres in both channels were similar, dCas9-mCherry was found to more severely localize in the nucleoli, resulting in a poor SNR between the centromere signal and the nucleolus signal (Figure 2C). In contrast, sgRNA1.1-MS2 showed minimal nucleoli-localized signal and a good SNR (Figure 2C). Interestingly, the normalized line intensity profiles show that both dCas9-mCherry and sgRNA1.1-MS2-EGFP formed two peaks, which correspond to the valleys of DAPI signal (Figure 2D, Supplementary Figure S4). These results may be due to denaturing of dsDNA by dCas9/sgRNA binding (47), which causes a drop of DAPI staining efficiency on the target site. The above analysis was repeated on telomere labeling, which reaches similar conclusions (Supplementary Figure S5, Movies S3 and S4). In addition, the modified sgRNA was also found to be able to label the centromeres during mitosis (Supplementary Figure S6).
Figure 2.

Comparison of the modified sgRNA method and the fluorescent dCas9 method by co-labeling human centromeric α-satellites. (A) Co-labeling of human centromeric α-satellites using sgRNA1.1 (green) and dCas9-mCherry (red). The nucleus was stained by DAPI (blue). The merged image shows the two types of labeling are well co-localized, but dCas9-mCherry concentrates in the nucleoli, compared with sgRNA1.1. The inset shows the magnified image of the boxed region. (B) Upper panel: fluorescence colocalization analysis of the two channels. The scatter plot shows the intensity correlation of the two channels for each individual pixel. Pearson's correlation Rr = 0.759, Overlap coefficient R = 0.997; Lower panel: a colocalization image reconstructed from the correlated pixels. It indicates that the large fraction of uncorrelated red pixels are from the nucleoli signal. The color bar denotes the colocalization percentage. (C) Signal-to-noise ratios (SNRs) of the two channels. The SNRs of the centromere signal relative to the nuclear background is slightly higher for the sgRNA1.1 (G/B) than that of the dCas9 labeling (R/B). The SNRs referred to the nucleoli regions is about 3-fold higher for the sgRNA1.1 (G/N) than that of the dCas9 labeling (R/N). N = 10 cells. (D) Normalized line intensity profiles along the white line in A inset. Both GFP and mCherry channels form two intensity peaks, where the DAPI channel forms valleys. (E) Representative images of all 12 modified sgRNAs that target centromeres. Among them, sgRNA1.12, 1.22 and 2.02 show notably less nucleoli localizations than sgRNA1.1, 1.2 and 2.0. (F) Representative images of a MDA-MB-231 cell line stably expressing dCas9, tdPCP-EGFP and tdMCP-mCherry. (G and H) Total number of centromeres detected per cell and SNRs for all 12 modified sgRNAs. Cyan color represents the MS2 insertion, blue color represents the PP7 insertion and gray color represents the stable cell line. Error bars are standard deviations. N = 10 cells. All scale bars are 5 μm.
We next set out to compare the six modified sgRNAs. Even though all modified sgRNAs were able to target the centromeres with similar numbers of detected centromeres per cell (Figure 2E and G), sgRNA2.02 exhibited a better SNR than other modified sgRNAs, likely due to the double aptamer insertion in sgRNA2.02 (Figure 2H). As the nucleolar localization of dCas9, tdPCP-EGFP and tdMCP-mCherry highly depended on their expression levels (25,38), we also established a cell line stably expressing these three proteins at a minimal level, which indeed exhibited lower nucleolar localization than that in transiently transfected cells (Figure 2F, Supplementary Figure S7). For labeling of centromeres and telomeres, sgRNA2.02 in both transfected cells and the stable cell line showed similar SNRs (Figure 2H).
In order to quantitatively determine the targeting specificity, we co-labeled centromeres with sgRNA2.02-PP7-EGFP and anti-CREST-Cy5 (51) (Figure 3A). Data analysis indicates that the sgRNA-labeled fluorescent puncta showed about 90% colocalization with the anti-CREST-Cy5 labeled fluorescent puncta (Figure 3B and C). Considering the un-consensus nature of human α satellite DNA sequences (Supplementary Figure S8), the high colocalization ratio suggests that there is no obvious undercounting by the modified sgRNA labeling method. For telomeres that were co-labeled with sgRNA2.02-PP7-EGFP and mCherry-TRF2 (52), the sgRNA-labeled fluorescent puncta showed more than 95% colocalization with the mCherry-TRF2 labeled fluorescent puncta (Figure 3D–F). Note that for both centromeres and telomeres, a tiny fraction (<5%) of the sgRNA-labeled fluorescent puncta showed no apparent colocalization with the puncta labeled by the conventional labeling methods (Figure 3C and F). This kind of inconsistence was also observed in the dCas9-based labeling method reported previously (25). On all accounts, the minor uncolocalized fractions might origin from either insufficient labeling efficiency or competition between the two labeling methods.
Figure 3.

Targeting specificity of the modified sgRNA labeling method. (A) Co-labeling of centromeres using sgRNA2.02-PP7-EGFP (green) and anti-CREST-Cy5 (red). The nucleus was stained by DAPI (blue). Scale bar: 5 μm. (B) Three boxed regions in the lower right panel of A are magnified to display three typical colocalization scenarios. Scenario 1: apparent colocalization between sgRNA-labeled punctum and anti-CREST-Cy5 labeled punctum; Scenario 2: anti-CREST-Cy5 labeled punctum without apparent sgRNA-labeled punctum; Scenario 3: sgRNA-labeled punctum without apparent anti-CREST-Cy5 labeled punctum. Scale bar: 1 μm. (C) Quantification of centromere targeting specificity based on the three colocalization scenarios in 10 cells with Scenario 1 marked in yellow, Scenario 2 marked in red and Scenario 3 marked in green. (D) Co-labeling of telomeres using sgRNA2.02-PP7-EGFP (green) and mCherry-TRF2 (red). The nucleus was stained by DAPI (blue). Scale bar: 5 μm. (E) Three boxed regions in the lower right panel of D are magnified to display three typical colocalization scenarios. Scenario 1: apparent colocalization between sgRNA-labeled punctum and mCherry-TRF2 labeled punctum; Scenario 2: mCherry-TRF2 labeled punctum without apparent sgRNA-labeled punctum; Scenario 3: sgRNA-labeled punctum without apparent mCherry-TRF2 labeled punctum. Scale bar: 0.3 μm. (F) Quantification of telomere targeting specificity based on the three colocalization scenarios in 12 cells with Scenario 1 marked in yellow, Scenario 2 marked in red and Scenario 3 marked in green.
We then used sgRNA2.02-MS2-EGFP to label a single repetitive genomic locus. The second exon of MUC4 gene of human MDA-MB-231 cells, which contains about 400 tandem repeats, were targeted by the modified sgRNAs. The same region was co-labeled with dCas9-mCherry for comparison. Both channels presented three allelic loci in the nucleus (Supplementary Figures S9 and S10A). Interestingly, the direct visualization of sgRNA via tdMCP-EGFP revealed that sgRNAs were able to enter the cytoplasm and form large particles, colocalizing with DCP1A, a marker protein of P-bodies. P-bodies are known for RNA degradation (Supplementary Figure S10). Therefore, this result suggests that the excrescent expression of sgRNAs would be transported to P-bodies for degradation.
Taken together, these results suggest that sgRNA2.02 with the double insertions in the tetraloop and loop 2 as well as the A-U flip and stem loop extension is the optimal genomic labeling probe among the seven modified sgRNAs.
Fast exchange rates of tdMCP/tdPCP make the modified sgRNA suitable for continuous, long-term tracking
The DAPI staining results in fixed cells suggest that the dCas9–sgRNA complex binds to the target DNA sequence with high affinity (Figure 2D, Supplementary Figures S4A–F and S5C). Although the high affinity would enhance the specificity of dCas9-based labeling, it may limit the time length of observation that is constrained by photobleaching at the labeled locus. In order to make a quantitative evaluation, we compared the on-off kinetics of sgRNA2.02-MS2-EGFP and dCas9-mCherry on the centromeres using the FRAP assay. In each cell, two centromeres were selectively photobleached for the recovery kinetics measurements of dCas9-mCherry (Figure 4A and Supplementary Movie S5) and sgRNA2.02-MS2-EGFP (Figure 4B and Supplementary Movie S6), respectively. The dCas9-mCherry channel recovered about 40 times slower than the sgRNA2.02-MS2-EGFP channel (t1/2 = 12 min versus t1/2 = 19 s). In addition, the immobile fraction (Fi = 0.55) of the dCas9-mCherry channel was larger than that of the sgRNA2.02-MS2-EGFP channel (Fi = 0.22) (Figure 4C and D). Overall, the fast recovery rate of the sgRNA2.02-MS2-EGFP, similar to the previously reported rate of exchange for tdMCP on the MS2 aptamer (53), makes our sgRNA-based labeling approach particularly suitable for long-term tracking by buffering the effects of photobleaching at the target site. This point was confirmed by continuous, long-term imaging of telomeres co-labeled with sgRNA1.12-MS2-EGFP and dCas9-mCherry (Supplementary Movie S7). Direct comparison of photobleaching tolerance was conducted by measuring photobleaching rates of telomeres labeled separately by dCas9-EGFP and sgRNA1.12-PP7-EGFP under identical imaging conditions for cells with similar EGFP expression levels (Figure 5A and Supplementary Movies S8 and S9). The results indicate that our sgRNA-based labeling method is more than 2-fold tolerant to photobleaching than the Cas9-based labeling method (Figure 5B).
Figure 4.

FRAP analysis of the dCas9 channel and the modified sgRNA scaffold channel. (A and B) Snapshots of the FRAP image series of dCas9-mCherry (A) and sgRNA2.02-MS2-EGFP (B). Localized photobleaching was applied to the centromeres marked by the black and purple circles. The centromere marked by the red circle was used as the control. The time lapse snapshots are shown in the right panel. All scale bars are 5 μm. The lookup tables on the lower right corners indicate the fluorescence intensity. (C and D) Quantifications of the FRAP experiments (11 centromeres for the dCas9-mCherry channel and 26 centromeres for the sgRNA2.02-MS2-EGFP channel). The data are background-subtracted and photobleach-corrected by the whole cell fluorescence. The dCas9-mCherry recovers at a much slower speed (t1/2 = 12 min) than that of sgRNA2.02-MS2-EGFP channel (t1/2 = 19 s) and the immobile fraction (Fi = 0.55) is larger than that of sgRNA2.02-MS2-EGFP channel (Fi = 0.22). Error bars are standard deviations.
Figure 5.

The modified sgRNA labeling system allows continuous, long-term imaging and tracking of chromosomal loci. (A) Selected frames from a time-lapse series of telomeres labeled with dCas9-EGFP or sgRNA1.12-PP7-EGFP. Scale bars are 1 μm. (B) The fluorescence intensity of telomeres in A was normalized and averaged. The data are displayed as mean ± sem. N = 24 puncta (3 cells) for dCas9-EGFP and N = 26 puncta (3 cells) for sgRNA1.12-PP7-EGFP. (C) Time-lapse snapshots from live imaging of a MDA-MB-231 cell through mitosis. Telomeres and centromeres were dual-color labeled with sgRNA2.02-MS2-mCherry and sgRNA2.02-PP7-EGFP, respectively. In this case, dCas9 was transfected to bring sgRNAs to the target sites and all fluorescence signal was from the sgRNA channels. Scale bar is 5 μm.
With the capability of long-term imaging of the sgRNA labeling, we tracked telomeres and centromeres in live cells every 5 min for about 26 h (Supplementary Movie S10). Note that the cell did not divide after 26 h, likely due to accumulated phototoxicity during the long-term imaging, which is less dependent on the labeling methods but more dependent on the light dose. We then used the sgRNA labeling method to study the dynamics of centromeres and telomeres through mitosis (Figure 5C and Supplementary Movie S11). During metaphase, centromeres and telomeres were aligned at the metaphase plate in an approximately-mirrored distribution. As the sister chromatids were separating during anaphase, centromeres and telomeres continued to condense. Upon entering telophase and during cytokinesis, centromeres and telomeres started to decondense. These results indicate that the chromatin condensation during mitosis has negligible effects on the binding of the dCas9–sgRNA complex to its target sequences, in contrast to the cell cycle-dependent labeling by fluorescent TALEs (54). Besides the local chromatin structure, whether and how other local chromatin epigenetic environments such as histone modifications may influence the dCas9–sgRNA binding activity require further study.
Dual-color continuous tracking of centromeres and telomeres in living cells
We first used sgRNA2.02-MS2-EGFP to track the dynamics of individual telomeres in living human MDA-MB-231 cells. The telomeres were co-labeled with dCas9-mCherry for comparison. Both channels showed high SNRs to allow long-term single particle tracking (Figure 6A). Regardless of the differential expression levels and the chromatic aberration, the trajectories of two channels mostly overlapped with each other during a 5-min tracking duration (Figure 6B and Supplementary Movie S12). All telomeres exhibited confined diffusive motion with intermittent hopping between consecutive confined diffusion areas (Figure 6C). This type of diffusion trajectories can be described by the walking confined diffusion model. The model uses Dmicro (the microscopic diffusion coefficient) to characterize the confined movement within a domain and Dmacro (the macroscopic diffusion coefficient) to measure the diffusion of the domain itself (43). The MSD analysis gives similar diffusion coefficients for both channels (Figure 6D). Interestingly, dCas9-mCherry presented a slightly larger Dmicro than sgRNA2.02-MS2-EGFP. This could be due to that in the dCas9–sgRNA complex, dCas9 is located relatively distal from the DNA binding site compared with the inserted aptamers in the sgRNA. This observation is consistent with a previous finding that transcriptional activation by dCas9 is less robust than that of the modified sgRNA (45).
Figure 6.

Dual-color continuous tracking of centromeres and telomeres in living cells. (A) A confocal section image of telomeres labeled with the sgRNA2.02-MS2-EGFP (green) and dCas9-mCherry (red), exhibiting a good level of colocalization with Pearson's correlation coefficient Rr = 0.741, overlap coefficient R = 1.0. Tracking of individual telomeres is performed by Gaussian fitting and marked by cyan circles (sgRNA) and red circles (dCas9), respectively. Scale bar: 5 μm. (B) The tracking trajectories of labeled telomeres. Red trajectories represent the dCas9-mCherry channel and green trajectories represent the sgRNA-MS2-EGFP channel. Scale bar: 5 μm. (C) Three telomere trajectories in B are magnified to display by the color-coded time points with green for the starting, blue for the intermediate and red for the ending frames. Scale bar: 0.5 μm. (D) The averaging MSD curves of telomere trajectories in both channels. The colored shade area represents the 95% fitting confidence interval. (E) Dual-color labeling of telomeres (sgRNA2.02-PP7-EGFP, green) and centromeres (sgRNA2.02-MS2-mCherry, red) simultaneously in living HeLa cells using modified sgRNAs. In this case, dCas9 was transfected to bring sgRNAs to the target sites and all fluorescence signal was from the sgRNA channels. Scale bar: 5 μm. (F) The averaging MSD curves of both telomeres (green) and centromeres (red). The colored shade area represents the 95% fitting confidence interval.
Next, we examined the dual-color imaging performance of two modified sgRNAs, i.e. sgRNA2.02-MS2-mCherry and sgRNA2.02-PP7-EGFP. Using a cross-talk assay, we proved that the sgRNA2.02-MS2 only interacted with its partner tdMCP-mCherry while the sgRNA2.02-PP7 only bound its partner tdPCP-EGFP (Supplementary Figure S11). The orthogonality of the modified imaging system guarantees that each targeting sgRNA can only recruit its binding protein and gather at the right site to amplify the fluorescence signal. Simultaneous tracking of the telomeres and centromeres in living human MDA-MB-231 cells shows that the sgRNA2.02 labeling seemed to be more efficient and specific for the centromeres than the telomeres (Figure 6E, Supplementary Movie S13), even though it worked equally well in the single color labeling of telomeres or centromeres (Figure 2A and Supplementary Figure S6). This differential labeling efficiency may be because the more abundant centromeric repeats outcompete telomeric sites for dCas9 recruitment.
The dual-color tracking also revealed interesting dynamics of the centromeres and telomeres. Specifically, even though both centromeres and telomeres showed similar micro-diffusive motion, their macro-diffusion coefficients Dmacro were different, implicating that centromeres may be tethered in the interphase (Figure 6F and Supplementary Movie S14). These data demonstrate that the modified sgRNA imaging system is suitable for dual-color labeling of chromosomal loci in living cells.
DISCUSSION
Visualization of chromatin dynamics in the nucleus is critical for unveiling the mechanisms of fundamental biological processes including transcription, DNA replication and DNA repair. Here we report a live-cell dual-color chromosomal loci imaging technique based on modified sgRNAs of the CRISPR/Cas9 system. To create a modular sgRNA-based system for locus-specific imaging, we modified the original sgRNA to include an MS2 or PP7 RNA aptamer that specifically recruits effector proteins tdMCP and tdPCP, respectively. In complex with dCas9, the structurally modified sgRNA can bind specifically to its target DNA sequence and the fluorescence labeling is realized by the fluorescent protein-tagged tdMCP or tdPCP effectors. Thus, the constructed modular sgRNA scaffold is able to encode two types of information. One is to execute the targeting function at user-defined locus and the other is to specify the labeling color by recruiting specific fluorescent protein-fused effector at the particular locus.
We found that a few structural and sequence modifications are essential for the optimal labeling performance of the modified sgRNAs. Firstly, the RNA aptamer insertion in the tetraloop or loop 2 has negligible interference with the binding affinity of the dCas9–sgRNA complex to the target site, while insertion in the tail largely impairs the affinity and causes a poor signal-to-background ratio in the fluorescence imaging. Secondly, the A-U flip and stem loop extension are found to further improve the labeling efficiency of the modified sgRNAs. Thirdly, double insertion in both the tetraloop and loop 2 is able to further increase the SNR compared with the single insertion in either loop. Besides MS2 and PP7, other RNA aptamers such as λN (55), Spinach (56) and com (44) can also be inserted into sgRNA tetraloop or loop 2 and engineered for multicolor imaging. What's more, the engineered multi-functional sgRNAs can be used to regulate gene expression and monitor its position and dynamics simultaneously.
We demonstrate the performance of the modified sgRNA in live-cell dual-color labeling by simultaneous imaging of telomeres and centromeres. Our new method offers several advantages over the previously reported dCas9 or TALE-based labeling methods in the following aspects. Firstly, in terms of multicolor imaging techniques, even though TALE-based labeling approach is by nature compatible with multicolor labeling, the laborious module constructions generally limit it to labeling of repetitive sequences. In contrast, labeling non-repetitive sequences would be in principle more feasible with the CRISPR/Cas9 system. Secondly, regarding the issue of multicolor labeling efficiency and specificity, the recently developed orthogonal Cas9 labeling system is constrained by the low targeting efficiency of the NM and ST1 Cas9 as well as the problem that their PAM sequences are remarkably less frequent than that of the SP Cas9. In contrast, our multicolor labeling method based on the modified sgRNA only uses the optimal SP Cas9. Thirdly, we also observed less pronounced off-target nucleolar fluorescence using the modified sgRNA system than the dCas9 labeling method. Lastly and importantly, compared with the dCas9-based labeling method, the fast exchange kinetics of the fluorescent effectors makes our method more suitable for long term tracking of the chromosomal dynamics, particularly for short repeats or non-repetitive sequences, which would be more prone to suffering from photobleach under continuous exposure. To this end, our new approach enables to study the chromosomal dynamics at both short and long time scales.
As all current multicolor chromosomal labeling methods are complementary to each other, one important future direction would be to combine these methods to achieve better SNRs or higher levels of multiplexing labeling, providing capabilities of simultaneous imaging of multiple chromosomal loci in living cells.
Supplementary Material
Acknowledgments
We thank Dr Bo Huang (Department of Pharmaceutical Chemistry, Department of Biochemistry and Biophysics, University of California, San Francisco) for providing us the dCas9 plasmid, Dr Robert H. Singer (Albert Einstein College of Medicine, the Yeshiva university) for providing MS2, PP7, tdMCP and tdPCP plasmids, Dr Ping Wei (The Center for Quantitative Biology, Peking University) for providing pHR-mCherry plasmid and Dr Wei Guo (Department of Biology, University of Pennsylvania) for providing the MDA-MB-231 cell line. We thank Dr Bo Huang and Dr Lei Stanley Qi (Department of Bioengineering, Stanford University) for the helpful discussion. We thank Robert Warneford-Thomson (University of Pennsylvania) and Lei Chang (Peking University) for polishing the manuscript. We also thank the core imaging facility of the School of Life Sciences, Peking University for imaging support.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Science Foundation of China [21573013, 21390412, 31271423, 31327901, 863 Program SS2015AA020406 to Y.S.]. Funding for open access charge: National Science Foundation of China [21390412, 31327901, 973 Program 2011CB809106, 863 Program SS2015AA020406, Thousand Youth Talents Program to Y.S.].
Conflict of interest statement. None declared.
REFERENCES
- 1.Dixon J.R., Selvaraj S., Yue F., Kim A., Li Y., Shen Y., Hu M., Liu J.S., Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–380. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mateos-Langerak J., Bohn M., de Leeuw W., Giromus O., Manders E.M., Verschure P.J., Indemans M.H., Gierman H.J., Heermann D.W., van Driel R., et al. Spatially confined folding of chromatin in the interphase nucleus. Proc. Natl. Acad. Sci. U.S.A. 2009;106:3812–3817. doi: 10.1073/pnas.0809501106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sexton T., Cavalli G. The role of chromosome domains in shaping the functional genome. Cell. 2015;160:1049–1059. doi: 10.1016/j.cell.2015.02.040. [DOI] [PubMed] [Google Scholar]
- 4.Hirano T. Chromosome territories meet a condensin. PLoS Genet. 2012;8:e1002939. doi: 10.1371/journal.pgen.1002939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bolzer A., Kreth G., Solovei I., Koehler D., Saracoglu K., Fauth C., Muller S., Eils R., Cremer C., Speicher M.R., et al. Three-dimensional maps of all chromosomes in human male fibroblast nuclei and prometaphase rosettes. PLoS Biol. 2005;3:e157. doi: 10.1371/journal.pbio.0030157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cremer T., Cremer M. Chromosome territories. Cold Spring Harb. Perspect. Biol. 2010;2:a003889. doi: 10.1101/cshperspect.a003889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Doerks T., Copley R.R., Schultz J., Ponting C.P., Bork P. Systematic identification of novel protein domain families associated with nuclear functions. Genome Res. 2002;12:47–56. doi: 10.1101/gr.203201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hu Q., Kwon Y.S., Nunez E., Cardamone M.D., Hutt K.R., Ohgi K.A., Garcia-Bassets I., Rose D.W., Glass C.K., Rosenfeld M.G., et al. Enhancing nuclear receptor-induced transcription requires nuclear motor and LSD1-dependent gene networking in interchromatin granules. Proc. Natl. Acad. Sci. U.S.A. 2008;105:19199–19204. doi: 10.1073/pnas.0810634105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Essers J., van Cappellen W.A., Theil A.F., van Drunen E., Jaspers N.G., Hoeijmakers J.H., Wyman C., Vermeulen W., Kanaar R. Dynamics of relative chromosome position during the cell cycle. Mol. Biol. Cell. 2005;16:769–775. doi: 10.1091/mbc.E04-10-0876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kind J., Pagie L., Ortabozkoyun H., Boyle S., de Vries S.S., Janssen H., Amendola M., Nolen L.D., Bickmore W.A., van Steensel B. Single-cell dynamics of genome-nuclear lamina interactions. Cell. 2013;153:178–192. doi: 10.1016/j.cell.2013.02.028. [DOI] [PubMed] [Google Scholar]
- 11.Misteli T. Higher-order genome organization in human disease. Cold Spring Harb. Perspect. Biol. 2010;2:a000794. doi: 10.1101/cshperspect.a000794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Therizols P., Illingworth R.S., Courilleau C., Boyle S., Wood A.J., Bickmore W.A. Chromatin decondensation is sufficient to alter nuclear organization in embryonic stem cells. Science. 2014;346:1238–1242. doi: 10.1126/science.1259587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reddy K.L., Zullo J.M., Bertolino E., Singh H. Transcriptional repression mediated by repositioning of genes to the nuclear lamina. Nature. 2008;452:243–247. doi: 10.1038/nature06727. [DOI] [PubMed] [Google Scholar]
- 14.Kumaran R.I., Spector D.L. A genetic locus targeted to the nuclear periphery in living cells maintains its transcriptional competence. J. Cell Biol. 2008;180:51–65. doi: 10.1083/jcb.200706060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kumaran R.I., Thakar R., Spector D.L. Chromatin dynamics and gene positioning. Cell. 2008;132:929–934. doi: 10.1016/j.cell.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Langer-Safer P.R., Levine M., Ward D.C. Immunological method for mapping genes on Drosophila polytene chromosomes. Proc. Natl. Acad. Sci. U.S.A. 1982;79:4381–4385. doi: 10.1073/pnas.79.14.4381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Belmont A.S. Visualizing chromosome dynamics with GFP. Trends Cell Biol. 2001;11:250–257. doi: 10.1016/s0962-8924(01)02000-1. [DOI] [PubMed] [Google Scholar]
- 18.Soutoglou E., Dorn J.F., Sengupta K., Jasin M., Nussenzweig A., Ried T., Danuser G., Misteli T. Positional stability of single double-strand breaks in mammalian cells. Nat. Cell Biol. 2007;9:675–682. doi: 10.1038/ncb1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robinett C.C., Straight A., Li G., Willhelm C., Sudlow G., Murray A., Belmont A.S. In vivo localization of DNA sequences and visualization of large-scale chromatin organization using lac operator/repressor recognition. J. Cell Biol. 1996;135:1685–1700. doi: 10.1083/jcb.135.6.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beliveau B.J., Joyce E.F., Apostolopoulos N., Yilmaz F., Fonseka C.Y., McCole R.B., Chang Y., Li J.B., Senaratne T.N., Williams B.R., et al. Versatile design and synthesis platform for visualizing genomes with Oligopaint FISH probes. Proc. Natl. Acad. Sci. U.S.A. 2012;109:21301–21306. doi: 10.1073/pnas.1213818110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roukos V., Voss T.C., Schmidt C.K., Lee S., Wangsa D., Misteli T. Spatial dynamics of chromosome translocations in living cells. Science. 2013;341:660–664. doi: 10.1126/science.1237150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shalem O., Sanjana N.E., Zhang F. High-throughput functional genomics using CRISPR-Cas9. Nat. Rev. Genet. 2015;16:299–311. doi: 10.1038/nrg3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hsu P.D., Lander E.S., Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157:1262–1278. doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Badique F., Stamov D.R., Davidson P.M., Veuillet M., Reiter G., Freund J.N., Franz C.M., Anselme K. Directing nuclear deformation on micropillared surfaces by substrate geometry and cytoskeleton organization. Biomaterials. 2013;34:2991–3001. doi: 10.1016/j.biomaterials.2013.01.018. [DOI] [PubMed] [Google Scholar]
- 25.Chen B., Gilbert L.A., Cimini B.A., Schnitzbauer J., Zhang W., Li G.W., Park J., Blackburn E.H., Weissman J.S., Qi L.S., et al. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell. 2013;155:1479–1491. doi: 10.1016/j.cell.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li J., Zhang B.B., Ren Y.G., Gu S.Y., Xiang Y.H., Du J.L. Intron targeting-mediated and endogenous gene integrity-maintaining knockin in zebrafish using the CRISPR/Cas9 system. Cell Res. 2015;25:634–637. doi: 10.1038/cr.2015.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wan H., Feng C., Teng F., Yang S., Hu B., Niu Y., Xiang A.P., Fang W., Ji W., Li W., et al. One-step generation of p53 gene biallelic mutant Cynomolgus monkey via the CRISPR/Cas system. Cell Res. 2015;25:258–261. doi: 10.1038/cr.2014.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu Y., Zhou H., Fan X., Zhang Y., Zhang M., Wang Y., Xie Z., Bai M., Yin Q., Liang D., et al. Correction of a genetic disease by CRISPR-Cas9-mediated gene editing in mouse spermatogonial stem cells. Cell Res. 2015;25:67–79. doi: 10.1038/cr.2014.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma H., Reyes-Gutierrez P., Pederson T. Visualization of repetitive DNA sequences in human chromosomes with transcription activator-like effectors. Proc. Natl. Acad. Sci. U.S.A. 2013;110:21048–21053. doi: 10.1073/pnas.1319097110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pederson T. Repeated TALEs visualizing DNA sequence localization and chromosome dynamics in live cells. Nucleus. 2014;5:28–31. doi: 10.4161/nucl.28143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miyanari Y. TAL effector-mediated genome visualization (TGV) Methods. 2014;69:198–204. doi: 10.1016/j.ymeth.2014.03.030. [DOI] [PubMed] [Google Scholar]
- 32.Anton T., Bultmann S., Leonhardt H., Markaki Y. Visualization of specific DNA sequences in living mouse embryonic stem cells with a programmable fluorescent CRISPR/Cas system. Nucleus. 2014;5:163–172. doi: 10.4161/nucl.28488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miyanari Y., Ziegler-Birling C., Torres-Padilla M.E. Live visualization of chromatin dynamics with fluorescent TALEs. Nat. Struct. Mol. Biol. 2013;20:1321–1324. doi: 10.1038/nsmb.2680. [DOI] [PubMed] [Google Scholar]
- 34.Wang T., Wei J.J., Sabatini D.M., Lander E.S. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014;343:80–84. doi: 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lucas J.S., Zhang Y., Dudko O.K., Murre C. 3D trajectories adopted by coding and regulatory DNA elements: first-passage times for genomic interactions. Cell. 2014;158:339–352. doi: 10.1016/j.cell.2014.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Levine M. The contraction of time and space in remote chromosomal interactions. Cell. 2014;158:243–244. doi: 10.1016/j.cell.2014.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fanucchi S., Shibayama Y., Burd S., Weinberg M.S., Mhlanga M.M. Chromosomal contact permits transcription between coregulated genes. Cell. 2013;155:606–620. doi: 10.1016/j.cell.2013.09.051. [DOI] [PubMed] [Google Scholar]
- 38.Ma H., Naseri A., Reyes-Gutierrez P., Wolfe S.A., Zhang S., Pederson T. Multicolor CRISPR labeling of chromosomal loci in human cells. Proc. Natl. Acad. Sci. U.S.A. 2015;112:3002–3007. doi: 10.1073/pnas.1420024112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Esvelt K.M., Mali P., Braff J.L., Moosburner M., Yaung S.J., Church G.M. Orthogonal Cas9 proteins for RNA-guided gene regulation and editing. Nat. Methods. 2013;10:1116–1121. doi: 10.1038/nmeth.2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Larson D.R., Zenklusen D., Wu B., Chao J.A., Singer R.H. Real-time observation of transcription initiation and elongation on an endogenous yeast gene. Science. 2011;332:475–478. doi: 10.1126/science.1202142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bertrand E., Chartrand P., Schaefer M., Shenoy S.M., Singer R.H., Long R.M. Localization of ASH1 mRNA particles in living yeast. Mol. Cell. 1998;2:437–445. doi: 10.1016/s1097-2765(00)80143-4. [DOI] [PubMed] [Google Scholar]
- 42.Jaqaman K., Loerke D., Mettlen M., Kuwata H., Grinstein S., Schmid S.L., Danuser G. Robust single-particle tracking in live-cell time-lapse sequences. Nat. Methods. 2008;5:695–702. doi: 10.1038/nmeth.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Daumas F., Destainville N., Millot C., Lopez A., Dean D., Salome L. Confined diffusion without fences of a g-protein-coupled receptor as revealed by single particle tracking. Biophys. J. 2003;84:356–366. doi: 10.1016/S0006-3495(03)74856-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zalatan J.G., Lee M.E., Almeida R., Gilbert L.A., Whitehead E.H., La Russa M., Tsai J.C., Weissman J.S., Dueber J.E., Qi L.S., et al. Engineering complex synthetic transcriptional programs with CRISPR RNA scaffolds. Cell. 2015;160:339–350. doi: 10.1016/j.cell.2014.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Konermann S., Brigham M.D., Trevino A.E., Joung J., Abudayyeh O.O., Barcena C., Hsu P.D., Habib N., Gootenberg J.S., Nishimasu H., et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. 2014;517:583–588. doi: 10.1038/nature14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mali P., Aach J., Stranges P.B., Esvelt K.M., Moosburner M., Kosuri S., Yang L., Church G.M. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat. Biotechnol. 2013;31:833–838. doi: 10.1038/nbt.2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nishimasu H., Ran F.A., Hsu P.D., Konermann S., Shehata S.I., Dohmae N., Ishitani R., Zhang F., Nureki O. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell. 2014;156:935–949. doi: 10.1016/j.cell.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Anders C., Niewoehner O., Duerst A., Jinek M. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature. 2014;513:569–573. doi: 10.1038/nature13579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jinek M., Jiang F., Taylor D.W., Sternberg S.H., Kaya E., Ma E., Anders C., Hauer M., Zhou K., Lin S. Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science. 2014;343:1215–1236. doi: 10.1126/science.1247997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu B., Chao J.A., Singer R.H. Fluorescence fluctuation spectroscopy enables quantitative imaging of single mRNAs in living cells. Biophys. J. 2012;102:2936–2944. doi: 10.1016/j.bpj.2012.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shoji I., Takagi T., Kasukawa R. Anti-centromere antibody and CREST syndrome in patients with primary biliary cirrhosis. Intern. Med. 1992;31:1348–1355. doi: 10.2169/internalmedicine.31.1348. [DOI] [PubMed] [Google Scholar]
- 52.Griffith J.D., Comeau L., Rosenfield S., Stansel R.M., Bianchi A., Moss H., de Lange T. Mammalian telomeres end in a large duplex loop. Cell. 1999;97:503–514. doi: 10.1016/s0092-8674(00)80760-6. [DOI] [PubMed] [Google Scholar]
- 53.Ben-Ari Y., Brody Y., Kinor N., Mor A., Tsukamoto T., Spector D.L., Singer R.H., Shav-Tal Y. The life of an mRNA in space and time. J. Cell Sci. 2010;123:1761–1774. doi: 10.1242/jcs.062638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Thanisch K., Schneider K., Morbitzer R., Solovei I., Lahaye T., Bultmann S., Leonhardt H. Targeting and tracing of specific DNA sequences with dTALEs in living cells. Nucleic Acids Res. 2014;42:e38. doi: 10.1093/nar/gkt1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Daigle N., Ellenberg J. LambdaN-GFP: an RNA reporter system for live-cell imaging. Nat. Methods. 2007;4:633–636. doi: 10.1038/nmeth1065. [DOI] [PubMed] [Google Scholar]
- 56.Strack R.L., Disney M.D., Jaffrey S.R. A superfolding Spinach2 reveals the dynamic nature of trinucleotide repeat–containing RNA. Nat. Methods. 2013;10:1219–1224. doi: 10.1038/nmeth.2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.