

Abstract
LncRNAs represent a large class of noncoding RNA molecules that have important functions and play key roles in a variety of human diseases. There is an urgent need to develop bioinformatics tools as to gain insight into lncRNAs. This study developed a sequence-based bioinformatics method, LncDisease, to predict the lncRNA-disease associations based on the crosstalk between lncRNAs and miRNAs. Using LncDisease, we predicted the lncRNAs associated with breast cancer and hypertension. The breast-cancer-associated lncRNAs were studied in two breast tumor cell lines, MCF-7 and MDA-MB-231. The qRT-PCR results showed that 11 (91.7%) of the 12 predicted lncRNAs could be validated in both breast cancer cell lines. The hypertension-associated lncRNAs were further evaluated in human vascular smooth muscle cells (VSMCs) stimulated with angiotensin II (Ang II). The qRT-PCR results showed that 3 (75.0%) of the 4 predicted lncRNAs could be validated in Ang II-treated human VSMCs. In addition, we predicted 6 diseases associated with the lncRNA GAS5 and validated 4 (66.7%) of them by literature mining. These results greatly support the specificity and efficacy of LncDisease in the study of lncRNAs in human diseases. The LncDisease software is freely available on the Software Page: http://www.cuilab.cn/.
INTRODUCTION
Recently, analyses of human transcriptome revealed that protein-coding transcripts only account for a small portion of the whole-genome (1). Surprisingly, many of the transcripts in human transcriptome are long noncoding RNAs (lncRNAs) with lengths of more than 200 nucleotides (2). For a long time, the functionality of lncRNAs was frequently disputed (3) because of their low cross-species conservation, low expression levels and high tissue specificity. However, due to the fast development of lncRNAs, accumulating studies have reported that lncRNAs have important and diverse functions (4); thus, the dysfunction of lncRNAs is associated with some diseases, such as cardiovascular disease (5) and cancer (6). We previously built the long non-coding RNA disease database (LncRNADisease, http://www.cuilab.cn/lncrnadisease) (7), which shows that more than 200 diseases are associated with lncRNAs and more than 250 lncRNAs have roles in at least one disease. Thus, one emerging opinion is that lncRNAs could be novel molecules for disease diagnosis and therapy (8). Currently, a large number of lncRNAs have been identified. For example, the NONCODE database (9) and the MiTranscriptome database (10) have collected more than 90 000, and 60 000 human lncRNAs, respectively. Given the importance and the large number of lncRNAs, there is an increasing need to identify which lncRNAs are associated with which diseases on a genome-wide scale. However, at present, the relationship between most of the lncRNAs and most of the human diseases remains unknown. Therefore, it becomes important to develop bioinformatics methods to predict lncRNA-disease associations.
Hence, we built the lncRNA disease database (LncRNADisease) and presented a method to predict lncRNA-disease associations based on the genomic locus of lncRNAs (7,11). Recently, several studies presented bioinformatics methods based on co-expression of lncRNA and protein-coding genes for predicting the lncRNAs involved in lung cancer (12), human disease (13) and esophageal squamous cell carcinoma (14). In addition, a Laplacian Regularized Least Squares based method which integrated lncRNA-disease association data from the LncRNADisease database and lncRNA expression profile was developed (15). More recently, several network-based methods have been developed (16–20). The above presented methods collectively provide valuable help in dissecting the associations between lncRNAs and human diseases. However, limitations exist in the above methods. For example, regarding the genomic locus based methods, it is difficult to set a suitable genomic distance threshold. Also, lncRNAs and their neighbor genes may not always be functionally related. Moreover, only a small fraction of lncRNAs have neighbor protein-coding genes. In the case of the co-expression based methods, there are three major limitations. Firstly, only a small number of lncRNAs have matched tissue expression data with protein-coding genes. Secondly, some lncRNAs do not have co-expressed genes. Thirdly, co-expression does not always mean co-function. For the network-based methods, the major limitation is that they only focus on a limited number of lncRNAs (∼260) with known disease associations and cannot be applied to most of the human lncRNAs. Therefore, novel methods that are efficient and applicable in a large-scale are needed.
It is known that lncRNAs could exert their functions by interacting with miRNAs (21,22). The human miRNA-disease associations have been collected and annotated in a large scale in our Human microRNA disease database (HMDD) database since 2007 (23,24). Therefore, it seems possible to predict lncRNA-disease associations by enrichment analysis of the miRNAs interacting with the given lncRNAs in specific disease-associated miRNA sets. We previously presented a miRNA enrichment analysis tool (25), TAM and confirmed its usefulness in mining miRNA-related knowledge (26). In a recent study, Chen revealed that integrating the HMDD data (human miRNA-disease associations) and the starBase data (miRNA–lncRNA interactions) can indeed infer lncRNA-disease associations (27). However, two major limitations exist in the study. Firstly, the lncRNA–miRNA interaction data the author used is from the starBase database (21), which included only the experimentally supported data set. This makes Chen's method only feasible in a very small fraction of lncRNAs and human diseases. The reason is that the lncRNA–miRNA interactions in starBase only contain 1114 lncRNAs, which only cover ∼1.2% (∼1114/90000) of the total human lncRNAs. Therefore, Chen's method cannot be applied to ∼98.8% of the total human lncRNAs. Moreover, the lncRNA–miRNA interactions in starBase only contain 132 miRNAs, which only cover ∼23.1% (132/572) of the total miRNAs in the HMDD database. This will also largely restrict the application of Chen's method. Secondly, Chen's study did not provide web-based or standalone software for users, which also greatly limit the application of the method. Based on the above observations, our study developed a standalone tool, LncDisease, to predict lncRNA-disease associations based on disease enrichment analysis of the miRNAs interacting with the given lncRNA. For a given lncRNA sequence, LncDisease first predicted the potential miRNAs interacting with the given lncRNA. Next, LncDisease performed enrichment analysis for the predicted miRNAs on the disease-associated miRNA sets, which are derived from our HMDD database (23,24). Afterward, LncDisease predicted the significant diseases as the potential diseases associated with the given lncRNA. Finally, to validate the accuracy of LncDisease, we selected the 12 most significant lncRNAs predicted to be associated with breast cancer for further biological experiments in two breast cancer cell lines, MCF7 and MDA-MB-231, respectively. Moreover, four lncRNAs predicted to be associated with hypertension were also validated in human vascular smooth muscle cells (VSMCs) stimulated with angiotensin II (Ang II). The results showed that 11 (91.7%) of the 12 predicted-breast-cancer-associated lncRNAs were significantly deregulated in both breast cancer cell lines. Also, 3 (75.0%) of the 4 predicted hypertension-associated lncRNAs were significantly deregulated in Ang II-treated human VSMCs. In addition, we predicted the diseases associated with the lncRNA GAS5 and literature mining showed that 66.7% of the predicted diseases have literature evidence. These results suggest that LncDisease could be a useful tool in predicting the lncRNA-disease associations.
MATERIALS AND METHODS
The data used in this study
We downloaded the sequence data of human lncRNAs from the lncipedia database (28) (http://www.lncipedia.org/) and the miRNA sequence data from the miRBase database. Also, we downloaded the human miRNA-disease association data set from the HMDD database (23,24).
Predicting lncRNA–miRNA interactions
Currently, there is no computational tool or pipeline developed for the prediction of lncRNA–miRNA interactions. According to current knowledge, miRNAs regulate lncRNAs through the same mechanism of regulating mRNAs, which is mainly based on the base pairing of target RNA sequences and the seed regions of miRNAs (29). Given that TargetScan and miRanda represent two of the most popular and efficient computational tools for miRNA target prediction (30), in our study, LncDisease used TargetScan (31) and miRanda (32) to predict the lncRNA–miRNA interactions. In addition, the users can use the union or intersection of the predictions by the two tools. For TargetScan, the criteria for target prediction and ranking include stringent seed pairing, site number, site type and site context; whereas for miRanda, the criteria include moderately stringent seed pairing and site number (30). From this, we can conclude that TargetScan has more stringent criteria than miRanda. Therefore, under normal circumstances, miRanda will predict more miRNA targets and more binding sites than TargetScan.
Predicting lncRNA-associated disease
Given that the accuracy of the current miRNA target predictions tools (including TargetScan) is not high (30), we cannot predict that the diseases associated with the lncRNA-binding miRNAs are the potential associated diseases of the given lncRNA. Because the predicted targets could be true targets with higher probability than random predictions (30), statistical enrichment analysis becomes a useful solution to predict associated diseases of a given lncRNA. This study used the TAM method (25), which was previously developed to discover novel knowledge for a group of inputted miRNAs by performing hypergeometric test based enrichment analysis of the inputted miRNAs in a number of miRNA sets. miRNA sets were defined as a group of miRNAs with similar or the same biological meaning (24). For example, the miRNAs associated with breast cancer are grouped into the breast cancer miRNA set. Assuming that the number of miRNAs included in all miRNA sets is P; the number of miRNAs in miRNA set A is S; the number of input miRNAs included in the P total miRNAs is HP; and the number of input miRNAs included in the S miRNAs of miRNA set A is HS, we then calculated the probability of HS miRNAs of interest in the miRNA set A using Equation (1):
 |
(1) |
where the symbol ‘C’ is the combination operation. Thus, we calculated the statistical significance of the enrichment of the input miRNAs in miRNA set A using Equation (2):
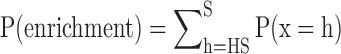 |
(2) |
Finally, the P-values for all miRNA sets were adjusted by Bonferroni correction. The flowchart of LncDisease is shown in Figure 1. In total, LncDisease collected 372 disease-associated miRNA sets based on the miRNA-disease association data set in the HMDD database. One disease-associated miRNA set is defined as a group of miRNAs that is associated with one disease. For example, the total miRNAs that are associated with breast cancer are assigned to the breast cancer miRNA set. LncDisease works according to the following flow. For a given lncRNA sequence, LncDisease first predicts the miRNAs interacting with the given lncRNA. Next, LncDisease performs enrichment analysis of the predicted miRNAs in the 372 disease-associated miRNA sets based on the TAM method. Moreover, LncDisease outputs the enrichment significance of the predicted miRNAs in each disease-associated miRNA set. Finally, we may take the significant results as the potential diseases associated with the inputted lncRNA.
Figure 1.

Flowchart of LncDisease for the prediction of lncRNA-disease associations.
Breast cancer cell culture and reagents
The human breast cancer cell lines MCF7 and MDA-MB-231 were purchased from ATCC (Manassas, VA, USA) and both were cultured using Dulbecco's modified eagle medium (DMEM) (Invitrogen, Carlsbad, CA, USA) with 10% fetal bovine serum (FBS) (Atlanta Biologicals, Flowery Branch, GA, USA). The human mammary epithelial cell line HMEC was purchased from Lonza (Allendale, NJ, USA), and cultured in Mammary Epithelial Cell Growth Medium MEGM (Lonza) containing BulletKit (Lonza) with serum-free.
RNA isolation
Total RNA was extracted by Trizol reagent (Invitrogen). Cells were harvested and dissolved in 1 ml of Trizol reagent and then 200 μl of 1-bromo-3-chloropropane solution was added (Molecular Research Center, Inc. Cincinnati, OH, USA) and mixed thoroughly by inverting the tube until well-blended. After the centrifugation, 14 000 rpm for 15 min at 4°C, the upper aqueous phase was removed to a new 1.5 ml tube, an equal volume of isopropanol (Sigma-Aldrich, St Louis, MO, USA) for precipitation subsequently. Extracted RNA was dissolved in nuclease-free water after washing the pellets using 75% ethanol twice, and then the RNA concentration was determined by Nanodrop (Thermo, Worcester, MA, USA).
Quantitative real-time PCR
The total RNA was used to synthesize cDNA by a high capacity cDNA reverse transcriptase kit (Applied Biosystems, Foster City, CA, USA). The relative mRNAs expression of lncRNAs was determined by quantitative real-time PCR (qRT-PCR), and the reaction mixtures was consisted of 10 μl 2x SYBR master mix (Roche, Indianapolis, IN, USA), 2 μl synthesized forward primer and reverse primer mixture, 1 μl cDNA and 7 μl nuclease-free water. The RT-PCR was performed on a Stratagene Mx3000p with the following cycling conditions: 95°C for 5 min followed by 40 cycles of 95°C for 30 s, 59°C for 30 s, 72°C for 30 s. After finishing 40 cycles, a final extension at 72°C for 7 min was performed. Quantitative values were obtained as threshold PCR cycle number (Ct) when the increase in the fluorescent signal of PCR product showed exponential amplification. Target lncRNA level was normalized to that of housekeeping gene in the same sample. In brief, the relative expression level of the target gene compared with that of a housekeeping gene was calculated as 2−ΔCt, where ΔCt = Cttarget lncRNA − Cthousekeeping gene. The ratio of the relative expression of the target gene in treated cells or cancer cells to that of untreated cells or normal cells was then calculated as 2−ΔΔCt, where ΔΔCt = ΔCttreated cell – Ctcontrol cell. Each sample was measured in duplicate or triplicate for each experiment. Moreover, melting and amplification curves for each PCR product were analyzed to ensure the specificity of the amplification product (33,34).
Parameters were: 95°C for 10 min, and then 40 cycles of 95°C for 10 s and 58°C for 35 s. GAPDH was taken as the endogenous control. The comparative cycle threshold (CT) method was used to compute the relative quantification of lncRNAs in human breast cancer cells by comparing them with the normal cells. The primer sequences used for qRT-PCR in the study are listed in Tables 1 and 2.
Table 1. The 12 candidate breast-cancer-associated lncRNAs and their primers for qRT-PCR validation experiments.
| Primers | Primer sequence (5′ to 3′) | |
|---|---|---|
| lnc-CEP170–1:2 | Sense | TGCGAGATTGAGATGATGA |
| Antisense | GCTGAGAACTTACCAGAGT | |
| lnc-CHAD-1:1 | Sense | GTGATGGAGCAAGACTGT |
| Antisense | GGCTGAAGTGTTGAAGGA | |
| lnc-GGCT-1:4 | Sense | TGACAAGAGGAGGAAGGAA |
| Antisense | TGCTGAGATTATAGGTGTGAG | |
| lnc-IL5RA-4:1 | Sense | AAGAGGAGCCAGCACTTC |
| Antisense | AGCCACTGTCCTGATGAAT | |
| lnc-PARN-7:2 | Sense | AGGTGCTGGAGTCAAGAA |
| Antisense | GGTGTGGTTGGTAGGAAG | |
| lnc-PPHLN1–1:1 | Sense | CACACCAAGACGGACTATC |
| Antisense | GAATACTGAAGATGCTGACTG | |
| lnc-PSCA-1:1 | Sense | AGACGAGGCTAATCACTGT |
| Antisense | GGCGGTTGTAAGAGGATG | |
| lnc-SCN5A-1:1 | Sense | CCAGGTCAGGTATCATAATAAG |
| Antisense | CAGTTGTCAAGTAAGCAGTT | |
| lnc-SMARCA5–3:1 | Sense | AGTTAGACCATAATGCCTCT |
| Antisense | GTGTCAATGTGTTACCTTCA | |
| lnc-USP8–2:3 | Sense | GGTAGCAGGTAGGTGTGA |
| Antisense | GTGAAGACATTACTATCCTCCT | |
| lnc-PPHLN1–1:2 | Sense | CACACCAAGACGGACTATC |
| Antisense | GAATACTGAAGATGCTGACTG | |
| lnc-WBSCR16–1:1 | Sense | TCAGTACACCATCCATCCA |
| Antisense | AAGAGTTGAGCAGAGTTCC | |
| GAPDH | Sense | TTGGCTACAGCAACAGGGTG |
| Antisense | GGTCTACATGGCAACTGTGAG |
Table 2. The 4 candidate hypertension-associated lncRNAs and their primers for qRT-PCR validation experiments.
| Primers | Primer sequence (5′ to 3′) | |
|---|---|---|
| lnc-C16orf95–1:5 | Sense | ACATCCAGAACAGGCAAAGC |
| Antisense | AATGTTAGGTCTCCCAGCCC | |
| lnc-RASA1–3:9 | Sense | GGGACGAACAGCGTGACAAT |
| Antisense | TGCAGTCACCTCATGTCCAAAA | |
| lnc-SLC17A9–1:1 | Sense | GATCACTTGAGCCCAGGAGT |
| Antisense | GACAGGGTCTTTCTCCGTCA | |
| lnc-SPATA9–1:2 | Sense | ATTTGACCCATGTAACGCGG |
| Antisense | CAGTGCGTTTGGGAATGTCA | |
| β-actin | Sense | GGTGGGAATGGGTCAGAAGG |
| Antisense | GTACATGGCTGGGGTGTTGA |
VSMC stimulated with Ang II
One popular way of mimicking hypertension in cells is by treating VSMCs with Ang II, a key mediator of hypertension (35); hence, we treated human VSMC cell line T/G HA-VSMC with 0.5 M Ang II for 24 h. Then, the expression levels of the four predicted hypertension-associated lncRNAs, lnc-C16orF95–1:5, lnc-RASA1–3:9, lnc-SLC17A9–1:1 and lnc-SPATA9–1:2, were determined using qRT-PCR analysis. The primer sequences are listed in Table 2.
Statistical analysis
The qRT-PCR result data were presented as mean ± S.E.M. The statistical significance of differences between groups was analyzed by t-test.
RESULTS AND DISCUSSION
The LncDisease standalone software
As shown in Figure 2, the users can copy and paste the sequence(s) of one or multiple lncRNAs in FASTA format into the ‘LncRNAs Input’ panel. An alternative solution for inputting the lncRNA sequences is to load the file containing the sequences in FASTA format by clicking the ‘Import’ button. After inputting the lncRNA sequences, the users can then click the ‘Predict’ button to run LncDisease. During the running process, the status of LncDisease will be shown in the ‘Log’ panel. When LncDisease finishes the task, the prediction results will be shown in the ‘Results’ panel. The users can rank the predicted lncRNA-disease entries by using several items, such as lncRNA name, disease name and the significance (P-value). In addition, the users can output the prediction results into a file by clicking the ‘Save All’ button.
Figure 2.

The user interface of the LncDisease software.
Validation of putative lncRNAs associated with breast cancer
To evaluate the accuracy of the predictions of LncDisease, we selected the 12 most significant predicted lncRNAs associated with breast cancer to explore the differential expression by biological experiments. qRT-PCR analysis was used to determine the level of lncRNAs in breast cancer cells (MCF7 and MDA-MB-231) and HMEC cells. For each lncRNA, we compared its expression level in MCF7 with that in the control cell (HMEC) using t-test. In addition, we also compared the expression level of each lncRNA in MDA-MB-231 with that in HMEC. But we did not compare the expression level of each lncRNA in MCF7 with that in MDA-MB-231. We found that 11 (91.7%) of the 12 selected lncRNAs had a significantly elevated level in both breast cancer cell lines compared with HMEC cells (Figure 3). The results suggest that these putative lncRNAs could indeed be involved in breast cancer, which further suggests that the presented method has a reliable accuracy.
Figure 3.

Validation results of the 12 candidate lncRNAs associated with breast cancer. * P < 0.05.
Validation of putative lncRNAs associated with hypertension
To evaluate the accuracy of the predictions of LncDisease, four predicted lncRNAs associated with hypertension were selected to further explore the differential expression by biological experiments. qRT-PCR analysis was used to determine the level of lncRNAs in Ang II-treated VSMCs and we found that 3 (75.0%) of the 4 selected lncRNAs exhibited a significantly decreased expression after treatment with Ang II (Figure 4). The results suggest that these putative lncRNAs could indeed be involved in hypertension, which further suggests that the presented method has a reliable accuracy.
Figure 4.
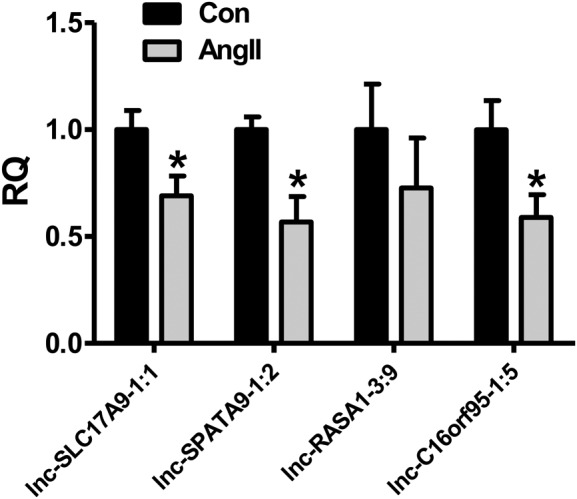
Validation results of the four candidate lncRNAs associated with hypertension. * P < 0.05.
Literature mining for diseases predicted to be associated with the lncRNA GAS5
We predicted six diseases (liver cirrhosis, hepatocellular carcinoma, fibrosis, kidney diseases, Duchenne muscular dystrophy and lung cancer) which are significantly associated with the lncRNA GAS5 (P < 0.01). By literature mining, we found that literature supports four of these diseases, hepatocellular carcinoma (P = 1.11e-3) (36–39), fibrosis (P = 1.17e-3) (40), kidney diseases (P = 3.16e-3) (41,42) and lung cancer (P = 9.76e-3) (43,44). For liver cirrhosis, we did not find any evidence linking it to GAS5. Given that GAS5 is significantly associated with hepatocellular carcinoma and liver fibrosis, two diseases highly associated with liver cirrhosis, it is reasonable to suggest that GAS5 may also be associated with liver cirrhosis. The results collectively suggest that the presented method is valuable in predicting lncRNA-disease associations.
Current limitations and future perspectives
LncRNAs are one class of crucial non-coding molecules, which play critical roles in some biological processes. Therefore, their dysfunctions are associated with a variety of human diseases. LncRNAs represent potential new molecules for disease diagnosis and therapy. Recently, a large number of human lncRNAs have been identified. For example, the NONCODE database and the MiTranscriptome have collected more than 90 000 human lncRNAs and 60 000 human lncRNAs, respectively. However, for most of the human lncRNAs, their relations with human diseases remain unknown. Given the large number of lncRNAs and their important functions, it becomes critically important to identify the relations between lncRNAs and diseases. For this purpose, we presented a sequence-based method, LncDisease, to predict associations between lncRNAs and human diseases through disease enrichment analysis of the miRNAs interacting with the given lncRNAs. Moreover, a biological experiment confirmed that the presented method has a reliable accuracy. However, several limitations exist in the current method. Firstly, the exact mechanism by which miRNAs regulate lncRNAs is not clear. This makes the prediction of lncRNA targets of miRNAs to have high false positives and high false negatives. Secondly, the miRNA-disease association data set is far from completeness. For example, the disease number is 372, which is greatly less than the total number of human diseases. As more precise knowledge of miRNA–lncRNA interaction and more miRNA-disease association data become available, LncDisease will be improved continuously. Thirdly, miRNA target prediction currently has high false positives. When better miRNA target prediction tools become available in the future, LncDisease will be improved. In addition, it is interesting that the 11 predicted breast cancer lncRNAs are all up-regulated in breast cancer while the 3 predicted hypertension lncRNAs are all down-regulated in hypertension. The reasons why these lncRNAs predicted by LncDisease show the same change in direction remains unknown. Further explorations are needed to find out whether these lncRNAs have special relations with disease-associated miRNAs. Finally, although the above limitations exist, we believe that LncDisease is a convenient tool for researchers to dissect the relations between lncRNAs and diseases in a large-scale, which could be helpful in identifying potential lncRNAs for disease diagnosis and therapy.
FUNDING
National Basic Research program of China [2012CB517506 to Q.C.]; National High Technology Research and Development Program of China [2014AA021102 to Q.C.]; National Natural Science Foundation of China [91339106 to Q.C., 81422006 to Q.C.]; NIH/NCI R01 [1R01CA192395 to Y.X.]; American Cancer Society Research Scholar [RSG-13–265–01-RMC to Y.X.]; NIH/NCI R21 [1R21CA160280 and 1R21CA182754 to Y.X.]. Funding for open access charge: National High Technology Research and Development Program of China [2014AA021102].
Conflict of interest statement. None declared.
REFERENCES
- 1.Bertone P., Stolc V., Royce T.E., Rozowsky J.S., Urban A.E., Zhu X., Rinn J.L., Tongprasit W., Samanta M., Weissman S., et al. Global identification of human transcribed sequences with genome tiling arrays. Science. 2004;306:2242–2246. doi: 10.1126/science.1103388. [DOI] [PubMed] [Google Scholar]
- 2.Kapranov P., Cheng J., Dike S., Nix D.A., Duttagupta R., Willingham A.T., Stadler P.F., Hertel J., Hackermuller J., Hofacker I.L., et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science. 2007;316:1484–1488. doi: 10.1126/science.1138341. [DOI] [PubMed] [Google Scholar]
- 3.Ponting C.P., Oliver P.L., Reik W. Evolution and functions of long noncoding RNAs. Cell. 2009;136:629–641. doi: 10.1016/j.cell.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 4.Chu C., Spitale R.C., Chang H.Y. Technologies to probe functions and mechanisms of long noncoding RNAs. Nat. Struct. Mol. Biol. 2015;22:29–35. doi: 10.1038/nsmb.2921. [DOI] [PubMed] [Google Scholar]
- 5.Kataoka M., Wang D.Z. Non-coding RNAs including miRNAs and lncRNAs in cardiovascular biology and disease. Cells. 2014;3:883–898. doi: 10.3390/cells3030883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chakravarty D., Sboner A., Nair S.S., Giannopoulou E., Li R., Hennig S., Mosquera J.M., Pauwels J., Park K., Kossai M., et al. The oestrogen receptor alpha-regulated lncRNA NEAT1 is a critical modulator of prostate cancer. Nat. Commun. 2014;5:5383. doi: 10.1038/ncomms6383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen G., Wang Z., Wang D., Qiu C., Liu M., Chen X., Zhang Q., Yan G., Cui Q. LncRNADisease: a database for long-non-coding RNA-associated diseases. Nucleic Acids Res. 2013;41:D983–D986. doi: 10.1093/nar/gks1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wapinski O., Chang H.Y. Long noncoding RNAs and human disease. Trends Cell Biol. 2011;21:354–361. doi: 10.1016/j.tcb.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 9.Bu D., Yu K., Sun S., Xie C., Skogerbo G., Miao R., Xiao H., Liao Q., Luo H., Zhao G., et al. NONCODE v3.0: integrative annotation of long noncoding RNAs. Nucleic Acids Res. 2012;40:D210–D215. doi: 10.1093/nar/gkr1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iyer M.K., Niknafs Y.S., Malik R., Singhal U., Sahu A., Hosono Y., Barrette T.R., Prensner J.R., Evans J.R., Zhao S., et al. The landscape of long noncoding RNAs in the human transcriptome. Nat. Genet. 2015;47:199–208. doi: 10.1038/ng.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li J., Gao C., Wang Y., Ma W., Tu J., Wang J., Chen Z., Kong W., Cui Q. A bioinformatics method for predicting long noncoding RNAs associated with vascular disease. Sci. China. Life Sci. 2014;57:852–857. doi: 10.1007/s11427-014-4692-4. [DOI] [PubMed] [Google Scholar]
- 12.Sun L., Luo H., Liao Q., Bu D., Zhao G., Liu C., Liu Y., Zhao Y. Systematic study of human long intergenic non-coding RNAs and their impact on cancer. Sci. China. Life Sci. 2013;56:324–334. doi: 10.1007/s11427-013-4460-x. [DOI] [PubMed] [Google Scholar]
- 13.Liu M.X., Chen X., Chen G., Cui Q.H., Yan G.Y. A computational framework to infer human disease-associated long noncoding RNAs. PloS One. 2014;9:e84408. doi: 10.1371/journal.pone.0084408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hao Y., Wu W., Shi F., Dalmolin R.J., Yan M., Tian F., Chen X., Chen G., Cao W. Prediction of long noncoding RNA functions with co-expression network in esophageal squamous cell carcinoma. BMC Cancer. 2015;15:168. doi: 10.1186/s12885-015-1179-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen X., Yan G.Y. Novel human lncRNA-disease association inference based on lncRNA expression profiles. Bioinformatics. 2013;29:2617–2624. doi: 10.1093/bioinformatics/btt426. [DOI] [PubMed] [Google Scholar]
- 16.Bialkowska-Hobrzanska H., Driman D.K., Fletcher R., Harry V., Razvi H. Expression of human telomerase reverse transcriptase, Survivin, DD3 and PCGEM1 messenger RNA in archival prostate carcinoma tissue. Can. J. Urol. 2006;13:2967–2974. [PubMed] [Google Scholar]
- 17.Ganegoda G.U., Li M., Wang W., Feng Q. Heterogeneous network model to infer human disease-long intergenic non-coding RNA associations. IEEE Trans. Nanobioscience. 2015;14:175–183. doi: 10.1109/TNB.2015.2391133. [DOI] [PubMed] [Google Scholar]
- 18.Sun J., Shi H., Wang Z., Zhang C., Liu L., Wang L., He W., Hao D., Liu S., Zhou M. Inferring novel lncRNA-disease associations based on a random walk model of a lncRNA functional similarity network. Mol. BioSyst. 2014;10:2074–2081. doi: 10.1039/c3mb70608g. [DOI] [PubMed] [Google Scholar]
- 19.Yang X., Gao L., Guo X., Shi X., Wu H., Song F., Wang B. A network based method for analysis of lncRNA-disease associations and prediction of lncRNAs implicated in diseases. PloS One. 2014;9:e87797. doi: 10.1371/journal.pone.0087797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou M., Wang X., Li J., Hao D., Wang Z., Shi H., Han L., Zhou H., Sun J. Prioritizing candidate disease-related long non-coding RNAs by walking on the heterogeneous lncRNA and disease network. Mol. BioSyst. 2015;11:760–769. doi: 10.1039/c4mb00511b. [DOI] [PubMed] [Google Scholar]
- 21.Li J.H., Liu S., Zhou H., Qu L.H., Yang J.H. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014;42:D92–D97. doi: 10.1093/nar/gkt1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paraskevopoulou M.D., Georgakilas G., Kostoulas N., Reczko M., Maragkakis M., Dalamagas T.M., Hatzigeorgiou A.G. DIANA-LncBase: experimentally verified and computationally predicted microRNA targets on long non-coding RNAs. Nucleic Acids Res. 2013;41:D239–D245. doi: 10.1093/nar/gks1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Y., Qiu C., Tu J., Geng B., Yang J., Jiang T., Cui Q. HMDD v2.0: a database for experimentally supported human microRNA and disease associations. Nucleic Acids Res. 2014;42:D1070–D1074. doi: 10.1093/nar/gkt1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu M., Zhang Q., Deng M., Miao J., Guo Y., Gao W., Cui Q. An analysis of human microRNA and disease associations. PloS One. 2008;3:e3420. doi: 10.1371/journal.pone.0003420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu M., Shi B., Wang J., Cao Q., Cui Q. TAM: a method for enrichment and depletion analysis of a microRNA category in a list of microRNAs. BMC Bioinformatics. 2010;11:419. doi: 10.1186/1471-2105-11-419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qiu C., Chen G., Cui Q. Towards the understanding of microRNA and environmental factor interactions and their relationships to human diseases. Sci. Rep. 2012;2:318. doi: 10.1038/srep00318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen X. Predicting lncRNA-disease associations and constructing lncRNA functional similarity network based on the information of miRNA. Sci. Rep. 2015;5:13186. doi: 10.1038/srep13186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Volders P.J., Helsens K., Wang X., Menten B., Martens L., Gevaert K., Vandesompele J., Mestdagh P. LNCipedia: a database for annotated human lncRNA transcript sequences and structures. Nucleic Acids Res. 2013;41:D246–D251. doi: 10.1093/nar/gks915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Poliseno L., Salmena L., Zhang J., Carver B., Haveman W.J., Pandolfi P.P. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature. 2010;465:1033–1038. doi: 10.1038/nature09144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bartel D.P. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Friedman R.C., Farh K.K., Burge C.B., Bartel D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Betel D., Wilson M., Gabow A., Marks D.S., Sander C. The microRNA.org resource: targets and expression. Nucleic Acids Res. 2008;36:D149–D153. doi: 10.1093/nar/gkm995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang C., Chen Z., Li S., Zhang Y., Jia S., Li J., Chi Y., Miao Y., Guan Y., Yang J. Hepatic overexpression of ATP synthase beta subunit activates PI3K/Akt pathway to ameliorate hyperglycemia of diabetic mice. Diabetes. 2014;63:947–959. doi: 10.2337/db13-1096. [DOI] [PubMed] [Google Scholar]
- 34.Wang C., Chi Y., Li J., Miao Y., Li S., Su W., Jia S., Chen Z., Du S., Zhang X., et al. FAM3A activates PI3K p110alpha/Akt signaling to ameliorate hepatic gluconeogenesis and lipogenesis. Hepatology. 2014;59:1779–1790. doi: 10.1002/hep.26945. [DOI] [PubMed] [Google Scholar]
- 35.Prasad A.M., Morgan D.A., Nuno D.W., Ketsawatsomkron P., Bair T.B., Venema A.N., Dibbern M.E., Kutschke W.J., Weiss R.M., Lamping K.G., et al. Calcium/calmodulin-dependent kinase II inhibition in smooth muscle reduces angiotensin II-induced hypertension by controlling aortic remodeling and baroreceptor function. J Am. Heart Assoc. 2015;4:e001949. doi: 10.1161/JAHA.115.001949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chang L., Li C., Lan T., Wu L., Yuan Y., Liu Q., Liu Z. Decreased expression of long non-coding RNA GAS5 indicates a poor prognosis and promotes cell proliferation and invasion in hepatocellular carcinoma by regulating vimentin. Mol. Med. Rep. 2015;13:1541–1550. doi: 10.3892/mmr.2015.4716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hu L., Ye H., Huang G., Luo F., Liu Y., Liu Y., Yang X., Shen J., Liu Q., Zhang J. Long noncoding RNA GAS5 suppresses the migration and invasion of hepatocellular carcinoma cells via miR-21. Tumour Biol. 2015 doi: 10.1007/s13277-015-4111-x. doi:10.1007/s13277-015-4111-x. [DOI] [PubMed] [Google Scholar]
- 38.Tao R., Hu S., Wang S., Zhou X., Zhang Q., Wang C., Zhao X., Zhou W., Zhang S., Li C., et al. Association between indel polymorphism in the promoter region of lncRNA GAS5 and the risk of hepatocellular carcinoma. Carcinogenesis. 2015;36:1136–1143. doi: 10.1093/carcin/bgv099. [DOI] [PubMed] [Google Scholar]
- 39.Tu Z.Q., Li R.J., Mei J.Z., Li X.H. Down-regulation of long non-coding RNA GAS5 is associated with the prognosis of hepatocellular carcinoma. Int. J. Clin. Exp. Pathol. 2014;7:4303–4309. [PMC free article] [PubMed] [Google Scholar]
- 40.Yu F., Zheng J., Mao Y., Dong P., Lu Z., Li G., Guo C., Liu Z., Fan X. Long non-coding RNA growth arrest-specific transcript 5 (GAS5) inhibits liver fibrogenesis through a mechanism of competing endogenous RNA. J. Biol. Chem. 2015;290:28286–28298. doi: 10.1074/jbc.M115.683813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Qiao H.P., Gao W.S., Huo J.X., Yang Z.S. Long non-coding RNA GAS5 functions as a tumor suppressor in renal cell carcinoma. Asian Pac. J. Cancer Prev. 2013;14:1077–1082. doi: 10.7314/apjcp.2013.14.2.1077. [DOI] [PubMed] [Google Scholar]
- 42.Zhou S., Wang J., Zhang Z. An emerging understanding of long noncoding RNAs in kidney cancer. J. Cancer Res. Clin. Oncol. 2014;140:1989–1995. doi: 10.1007/s00432-014-1699-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dong S., Qu X., Li W., Zhong X., Li P., Yang S., Chen X., Shao M., Zhang L. The long non-coding RNA, GAS5, enhances gefitinib-induced cell death in innate EGFR tyrosine kinase inhibitor-resistant lung adenocarcinoma cells with wide-type EGFR via downregulation of the IGF-1R expression. J. Hematol. Oncol. 2015;8:43. doi: 10.1186/s13045-015-0140-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou Y., Wu K., Jiang J., Huang J., Zhang P., Zhu Y., Hu G., Lang J., Shi Y., Hu L., et al. Integrative analysis reveals enhanced regulatory effects of human long intergenic non-coding RNAs in lung adenocarcinoma. J. Genet. Genomics. 2015;42:423–436. doi: 10.1016/j.jgg.2015.07.001. [DOI] [PubMed] [Google Scholar]