

Abstract
A novel mechanism is unveiled to explain why a pro-mutagenic nucleotide lesion (oxidized guanine, 8-oxoG) causes the mammalian DNA repair polymerase-β (pol-β) to rapidly transition to an inactive open conformation. The mechanism involves unexpected features revealed recently in time-lapse crystallography. Specifically, a delicate water network associated with a lesion-stabilizing auxilliary product ion Mg(p) triggers a cascade of events that leads to poor active site geometry and the rupture of crucial molecular interactions between key residues in both the anti(8-oxoG:C) and syn(8-oxoG:A) systems. Once the base pairs in these lesioned systems are broken, dislocation of both Asp192 (a metal coordinating ligand) and the oxoG phosphate group (PO4) interfere with the hydrogen bonding between Asp192 and Arg258, whose rotation toward Asp192 is crucial to the closed-to-open enzyme transition. Energetically, the lesioned open states are similar in energy to those of the corresponding closed complexes after chemistry, in marked contrast to the unlesioned pol-β anti(G:C) system, whose open state is energetically higher than the closed state. The delicate surveillance system offers a fundamental protective mechanism in the cell that triggers DNA repair events which help deter insertion of oxidized lesions.
INTRODUCTION
DNA polymerase β (pol-β), a member of the X-family of DNA polymerases, is the smallest eukaryotic cellular DNA polymerase (1). Pol-β plays a crucial role in DNA repair synthesis during base excision repair (BER) (2–4) that is essential for the maintenance of the genome of living organisms. Malfunction of pol-β has been suggested to trigger premature aging, neurological diseases and various cancers (5–12).
Many aspects of the nucleotide insertion pathway by pol-β have been investigated by kinetic (13–14), structural (15–16) and computational (17–18) studies. The catalytic cycle of this enzyme involves well known steps (19). Pol-β binds DNA to form an open binary substrate complex and to the 2′-deoxyribonucleoside 5′ -triphosphate (dNTP) to form the ternary complex. This open ternary complex undergoes a conformational change to form a closed ternary complex that aligns catalytic groups for the chemical reaction. In the closed ternary complex, the nucleotide binding metal ion (Mg(n)) coordinates Asp190, Asp192 and non-bridging oxygens on the phosphate groups of the incoming dNTP, while the catalytic metal ion (Mg(c)) coordinates Asp190, Asp192 and Asp256, and a non-bridging oxygen of Pα (dNTP). This highly organized active site facilitates the nucleotidyl transfer reaction where the 3′-oxyanion of the primer strand attacks Pα of the incoming dNTP to extend the primer strand and form the ternary product complex (20–21). After chemistry, pol-β transitions to the open enzyme form and releases pyrophosphate (PPi) and ions.
DNA polymerase substrate discrimination is thought to involve conformational adjustments that include open and closed subdomain transitions and is related to a hybrid induced-fit/conformational sampling mechanism (22–31). The recruitment of complementary nucleotides for insertion opposite the template base is about 250000 times more accurate than incorrect nucleotide insertion (e.g., dATP opposite G), which is believed to misalign catalytic components and thereby hamper insertion (32).
One of the most common sources of DNA damage comes from reactive oxygen species generated from aerobic respiration and other environmental assaults (33–35). A common form of oxidized guanine is 8-oxo-7, 8-dihydro-2′-deoxyguanosine (8-oxoG), found in DNA, and also as a modified nucleotide (8-oxodGTP) (36) in the dNTP pool. In general, 8-oxoG in DNA and 8-oxodGTP are potentially dangerous and can lead to human diseases due to polymerase replication or repair errors (37–38). Errors stem from the dual coding potential dictated by the anti- or syn-glycosidic conformation of the oxidized guanine base (39). The fidelity of dATP insertion opposite 8-oxoG for pol-β is ∼2 (40), while the fidelity of 8-oxodGTP incorporation opposite A is <2 (41).
Both anti- and syn-conformations of 8-oxoG are observed both in DNA (i.e., as the templating base) and as a modified incoming nucleotide in the confines of the pol-β active site (40–45). Before dNTP binds to pol-β, an 8-oxoG template of DNA in the binary open pol-β complex has both anti- and syn-conformations (42). As pol-β forms the ternary structure, dCTP forms a Watson–Crick interaction with the 8-oxoG(anti), while dATP forms a Hoogsteen base pair with 8-oxoG(syn) (42). When 8-oxoG is in an anti-conformation, its O8 atom can clash with the negatively charged phosphate group. However, a local change of the phosphate backbone relieves this clash to maintain proper Watson–Crick interactions between dCTP and 8-oxoG(anti) (40). Kinetic and computational studies have shown that dCTP insertion opposite an 8-oxoG(anti) template is more favorable than that of dATP(anti) opposite 8-oxoG(syn) (43–45). When 8-oxodGTP binds to pol-β, the formation of 8-oxoG(anti) opposite template C shows significantly lower insertion efficiency than that of 8-oxodGTP(syn) opposite template A (42). The low insertion efficiency of the 8-oxodGTP(anti) is due to distortion of the active site through a clash between the phosphate group (i.e., Pα) and the O8 atom. Recently, X-ray crystallography and computational studies have revealed that a metal ion located near Pα of the incoming 8-oxodGTP(anti) can stabilize the active site (41). The syn-conformation of 8-oxoG in the template strand results in G to T transversions (46), while that of 8-oxoG in the primer strand generates A to C transversions (47–48) during subsequent rounds of DNA replication. Because alterations in DNA sequence can lead to human disease and cell death (37), it is important to understand how polymerases like the repair enzyme pol-β replicates DNA with oxidized substrates (damaged DNA and nucleotide pools).
Recent structural snapshots of pol-β undergoing chemistry using time-lapse X-ray crystallography for four systems have revealed surprising structural features. These systems include insertion of dCMP opposite G and misinsertion of dAMP opposite G (16), as well as 8-oxodGMP(anti) insertion opposite C and 8-oxodGMP(syn) insertion opposite A (41). We refer to these systems as G:C[In our computational study, we modified the identity of the correct base pair insertion as outlined in Materials and Methods to examine G or modified G incorporation.], A:G, oxoG(anti):C and oxoG(syn):A, respectively (16,41). The four complexes reveal different behavior and raise intriguing questions regarding the role of an adjunct metal ion as well as the mechanism of oxoG discrimination. Whereas the G:C system exhibits a stable active site geometry with a third product metal ion, the A:G system exhibits poor geometry, no product metal and rapid reopening (16). The two lesioned systems exhibit mixed features of both the G:C and A:G systems (41).
For the G:C system, the novel product-associated metal ion (product ion, Mg(p)) includes the extended primer terminus (dGMP), PPi, Mg(n) and a monovalent ion (Nac) in the catalytic metal binding site (PDB index: 4KLJ, Supplementary Figure S1A, upper panel) (16). When this product complex transitions to the open conformation, the PPi, Mg(n), Mg(p) and NaC dissociate from the complex, and the nascent Watson–Crick base pairing between the inserted nucleotide and template base remains intact (PDB index: 1BPZ, Supplementary Figure S1A lower panel).
In contrast, the product complex of the A:G system exhibits poor active site geometry and opens rapidly (PDB index: 4KLU, Supplementary Figure S1D lower panel) (16). The corresponding closed product complex contains the nucleotide metal ion and PPi, but not the product ion (PDB index: 4KLT, Supplementary Figure S1D, upper panel).
Both lesioned product complexes in the closed conformations contain the Mg(n), a sodium ion in the catalytic metal site, the third metal Mg(p) and PPi like the unlesioned G:C system (PDB index: 4UB3 and 4UAY, upper panel of Supplementary Figure S1B and C) (41). The oxoG(anti) nucleotide forms a Watson–Crick base pair with C, while the oxoG(syn) forms a Hoogsteen base pair with A. However, unlike the G:C system, both lesioned systems rapidly reopen and release the metal ions and PPi (PDB index: 4UB2 and 4UB1, lower panel of Supplementary Figure S1B and C). In addition, the base pairs between oxoG(anti) and C and oxoG(syn) and A are lost. Therefore, oxoG opposite C or A causes instability within the active site and induces rapid opening (41).
Why does the oxoG(syn- and anti-) insertion exhibit characteristics of correct nucleotide insertion on one hand (ion binding and stable base pairing in the closed conformation) but incorrect insertion on the other hand (lack of base pairing and rapid reopening in the open conformation)? How does oxoG(anti) insertion opposite C trigger the rapid reopening even as it forms a Watson–Crick base pair in the closed product complex? What molecular events and corresponding energy landscapes are associated with these opening conformational pathways?
To answer these questions, we apply transition path sampling (TPS) simulations to study the post-chemistry conformational changes of three systems based on three crystallographic reference structures—G:C, oxoG(anti):C and oxoG(syn):A—and report their molecular events, pathways and energy landscapes.
MATERIALS AND METHODS
Initial systems
We use three sets of pol-β systems for the closed and open conformations of G:C (dGMP:C, PDB index: 4KLJ (closed) and 1BPZ (open)), oxoG:C (PDB index: 4UB3 (closed) and 4UB2 (open)) and oxoG:A (PDB index: 4UAY (closed) and 4UB1 (open)). For comparison with oxoG systems, the initial dCMP:G base pair after chemistry is replaced by dGMP:C. All systems are solvated with TIP3P water molecules. The smallest image distance between the solute and the faces of the periodic cubic cell is set to 18 Å. Neutralizing ions (Na+) and 150 mM NaCl are added to all three systems.
Minimization and dynamics
All systems are minimized with fixed positions for all heavy atoms of pol-β and DNA for 10 000 steps. The equilibration process is started with a 200 ps simulation at 300 K using Langevin dynamics, while keeping all heavy atoms of pol-β and DNA fixed (49). This is followed by unconstrained minimization consisting of 20000 steps. The systems are then equilibrated for 500 ps at constant pressure and temperature. Pressure is maintained at 1 atm using Langevin piston method and the temperature is maintained at 300 K using weakly coupled Langevin dynamics of non-hydrogen atoms with a damping coefficient of 10 ps−1. The systems are simulated in periodic boundary conditions with full electrostatics computed using the particle mesh Ewald (PME) method (50). The equilibrated systems are subjected to targeted molecular dynamics (TMD) simulations (see below for details). All MD simulations for this study including TPS simulations are performed by the NAMD (51) simulation package with the CHARMM36 force field (52–58). The treatment of divalent ions is indeed less accurate than monovalent ions, but force fields need not be perfect to be valuable (59). The energetics of divalent ions are described by the phenomenological Lennard–Jones potential and Coulombic interactions (60–63). Thus, while data generated for divalent ions with these force fields are generally useful and informative, ligand/ion distances may be shorter than those observed in high resolution X-ray crystal structures. Because our study compares the behavior associated with divalent ions on different systems, we believe that those trends among closely-related systems are meaningful.
TPS
The initial trajectories that connect the closed to open conformations during the opening transition are obtained by TMD simulations. During the TMD simulations, as the closed pol-β in each test set is changed to open conformation, Mg(n), Mg(p), NaC and PPi are removed from the active site and displaced 16 Å away from the pol-β surface so that the ions and PPi are out of the interaction range from the pol-β when pol is completely open. From the initial TMD trajectories, each transition region is selected, and unconstrained dynamics simulations are applied to generate new physical and unbiased trajectories to connect the closed and open states. The sole purpose of the TMD is to generate initial points for the TPS simulations. Since the TPS simulations use these initial conformations with various configurational and momenta perturbations in a process similar to a Monte Carlo process in MD trajectory space, the choice of starting points does not affect the final pathway. Because of the way TPS is formulated, any initial path can be used to ‘harvest’ trajectories. The choice of initial point affects the computational time to generate a trajectory that captures a transition state, but not the nature of the transition state per se (64). The theoretical grounding of TPS is described in detail in ref. (65).
The appropriate order parameters for TPS simulations are chosen from the crystallographic data (16,41), molecular dynamics (17,18) and prior TPS studies (45,65–68). These works have shown that key active site residues (Asp190, Asp192, Arg258 and Phe272) and metal ions motions serve as measures of pol-β closing pathway before chemistry. In this work, we assess the values associated with these residues, base pair breaking, and the two metal ion motions (Mg(p) and Mg(n)). The complete set of order parameters is listed in Table 1.
Table 1. Transition states properties for the opening conformational profile of the G:C and oxoG Pol-β systems.
| Event | χ-order parameter | χmax state A | χmin state B |
|---|---|---|---|
| Arg258 initial flip | Dihedral angle Cγ—Cδ—Nϵ—Cζ | 280° (G:C, oxoG:C, oxoG:A) | 200° (G:C, oxoG:C, oxoG:A) |
| Phe272 | Dihedral angle Cα—Cβ—Cγ—Cδ2 | 190° (G:C) | 130° (G:C) |
| 270° (oxoG:C) | 210° (oxoG:C) | ||
| 210° (oxoG:A) | 110° (oxoG:A) | ||
| Asp190 | Dihedral angle N—Cα—Cβ—Cγ | 290° (G:C) | 210° (G:C) |
| Asp192 | Dihedral angle Cγ—Cβ—Cα—C | 170° (oxoG:C) | 100° (oxoG:C) |
| 160° (oxoG:A) | 90° (oxoG:A) | ||
| Arg258 second flip | Dihedral angle Cγ—Cδ—Nϵ—Cζ | 180° (G:C, oxoG:C) | 100° (G:C, oxoG:C) |
| 210° (oxoG:A) | 110° (oxoG:A) | ||
| Mg(p) dissociation | Distance of Mg(p) to P atom at dGMP and oxoG | 1.9 Å (G:C, oxoG:C, oxoG:A) | 4.2 Å (G:C) |
| 4.0 Å (oxoG:C, oxoG:A) | |||
| Mg(n) dissociation | Distance of Mg(n) to OD2 atom at Asp192 | 1.9 Å (G:C, oxoG:C, oxoG:A) | 4.5 Å (G:C) |
| 3.3 Å (oxoG:C) | |||
| 4.4 Å (oxoG:A) | |||
| Base pair breaking | Distance of H1 (oxoG(anti)) to N3 of CYT6 | 2.0 Å (oxoG:C, oxoG:A) | 3.0 Å (oxoG:C) |
| Distance of H7 (oxoG(syn)) to N1 of ADE6 | 3.4 Å (oxoG:A) |
The atomic momenta of the selected region are perturbed and integrated by the Newtonian equations of motion forward and backward over short trajectories of length 5–20 ps. Using the newly generated physical trajectory as starting trajectory, we apply transition path sampling to each individual conformational change with the shooting and shifting algorithm and a Monte Carlo protocol (65). The entire process is performed using a PERL script that interfaces with NAMD. The Verlet integrator in NAMD with a time step of 1 fs is used to generate the individual MD trajectories in TPS (64,69). All other parameters are the same as those in the equilibration process. To obtain an acceptance rate of 30–45% in TPS, the momentum perturbation magnitudes (dP) of each transition state are varied from 0.001 to 0.005 amu·Å/fs. To identify the transition states, 100 accepted trajectories for each transition state are collected. In our study, TPS simulations are applied to only the major transition events, such as the rotation of key residues in the active site, base pair breaking and the dissociations of metal ions (Table 1).
Free energy computation by BOLAS
The free energy barriers for transition states are evaluated using the ‘BOLAS’ protocol (70), an efficient procedure for computing free energies with relatively low error bars using the TPS trajectory harvesting idea. To calculate the free energy for each subpath, 600–1000 trajectories for each transition state (6–10 overlapping windows, 100 trajectories per window) are collected. The potential of mean force plots obtained for each window are combined by adding/subtracting a constant to match the free energy values of the overlapping region. The free energy barriers for the conformational transitions are then calculated from the free energy plots. The error bar for the free energy calculations is determined by repeating umbrella sampling on one window of a transition for 10 times with the same initial trajectory but different starting pseudorandom numbers. From these calculations, the standard deviation for each barrier (2−3 kBT) is obtained and listed as the error bar in Table 2. Our BOLAS protocol is currently implemented in the NAMD.
Table 2. Free energy barrier and rate kTST estimated by transition state theory.
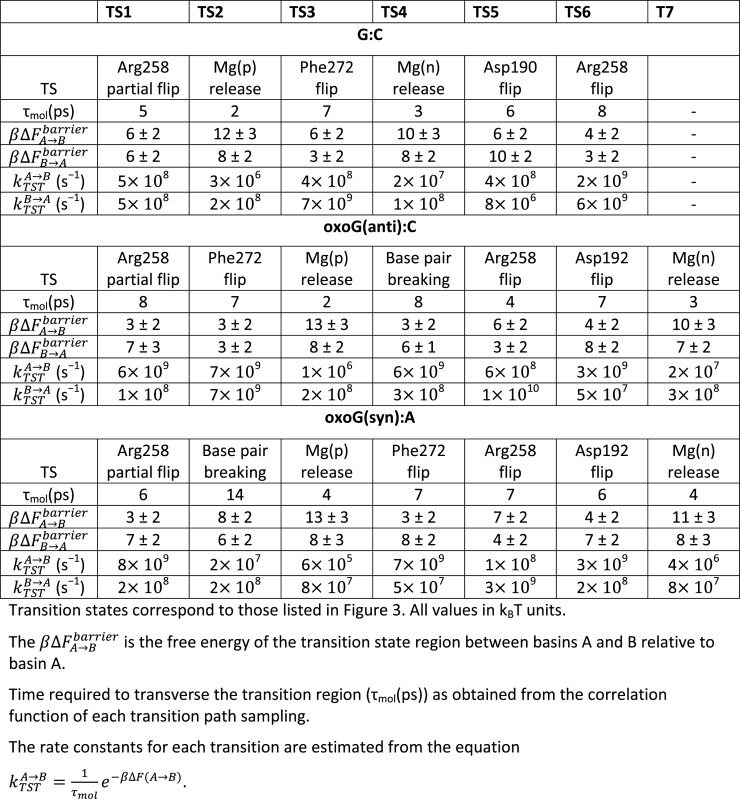
|
RESULTS
TPS simulations allow us to follow the conformational pathway between two experimental endpoints. As shown for the closing conformational pathway of pol-β before chemistry (65), conformational closing involves a directed sequence of local checkpoints or gates (30,31) including, in turn, N-subdomain closing, Arg192 flip, Arg258 rotation, Phe272 flip and ion rearrangement. Moreover, while the closed and open states before chemistry are energetically equal for correct base pair insertion (G:C), the closed state is higher in energy for the incorrect, mismatched system (G:A), thereby hampering chemistry (66). See Supplementary Figure S2 here. This energetic trend also explains behavior for the pol-β E295K mutant (67) and 8-oxoG template pol-β systems (dCTP:8-oxoG and dATP:8-oxoG) before chemistry (45).
Here, we show that the opening conformational change after chemistry for the G:C system essentially reverses the local motions found in the closing before chemistry (Figure 1A). For the lesioned systems, release of the product ion triggers base pair rupturing via a network of water interactions (Figure 2) and does not follow an uphill energy path like the G:C system (Figure 3).
Figure 1.



Snapshots of transition states during the opening conformational pathway of the (A) G:C, (B) oxoG(anti):C and (C) oxoG(syn):A pol-β systems. The active site geometries before and after each transition are aligned and superimposed with each other. The residues before each transition are shown in light red and those of after are indicated with a multi-color code based on atom type. Two metal ions, Mg(n) and Mg(p) are indicated by green and blue, respectively.
Figure 2.

The base pair breaking mechanism of oxoG(anti):C (left) and oxoG(syn):A (right) pol-β systems. The Mg(p) metal ion is shown as a blue sphere. The red dashed line and yellow circle indicate hydrogen bonding interactions. The black dashed lines are distance measurements between atoms. (A) In the oxoG(anti):C system, the Mg(p) ion stabilizes the base pair interaction by reducing electrostatic repulsion between O8 of the guanine residue and the phosphate group and strengthening the hydrogen bonding interaction between a water molecule and O8 (top image). After the Mg(p) ion dissociates, both the distance between O8 and the phosphate group and the glycosyl angle χ increase so that the base pair interaction is destabilized (middle image). The base pair then breaks and the χ angle adjusts slightly (bottom image). (B) In the oxoG(syn):A system, the syn-conformation is stabilized by the hydrogen bonding interaction between N2 of the guanine residue and O2P of the phosphate group (top image). Because the water molecules that coordinate Mg(p) are also located between the phosphate group and base, as water movement occurs, the hydrogen bonding interaction between N2 and O2P is broken and the Hoogsteen base pair interaction is also broken (middle image). After the Mg(p) ion dissociates, the hydrogen bonding interaction between N2 and O2P cannot be recovered, and the Hoogsteen base pair remains broken.
Figure 3.

The free energy profiles of the G:C (black line), oxoG(anti):C (red line) and oxoG(syn):A (blue line) pol-β systems. The transition T2 and T4 in oxoG(anti):C and oxoG(syn):A systems are indicated with red and blue, respectively, while all other transition states, which are identical between two oxoG systems are indicated with violet.
The opening conformational pathway for the G:C system
The conformational opening pathway of the pol-β system after chemistry is shown in Figure 1A. From the bimodality of the probability distribution (Supplementary Figure S3), we see that the sequence of transition states (TS) from closed to open state is: Arg258 partial flip (TS1), Mg(p) ion dissociation (TS2), Phe272 rotation (TS3), Mg(n) ion dissociation (TS4), Asp190 flip (TS5) and Arg258 full flip (TS6). This sequence for the opening conformational change after chemistry (rearrangements for Arg258, Phe272 and Mg(n)) occurs in a reverse order of transition states compared to the closing conformational pathway before chemistry (65).
For each transition state, snapshots before and after transitions are shown in Figure 1A. The first step is Arg258's partial flip (TS1). As Mg(p) dissociates from the dGMP phosphate group, we see that the water shell, which is coordinated with Mg(p), also separates from dGMP (TS2 in Figure 1A). When the Mg(p)-water shell is released from the binding site, no major interaction between the water shell and the dGMP base is disrupted; the Watson–Crick base pair interaction between dGMP and C is well maintained. Following the Mg(p) ion dissociation, Phe272 rotates (TS3 in Figure 1A) and creates space for the Arg258 flip. As Mg(n) dissociates from the binding site, Asp192 approaches Arg258 (TS4 in Figure 1A). The short distance between Asp192 and Arg258 initiates a hydrogen bond interaction between OD1 (Asp192) and HH12-NH1 (Arg258), which triggers the flipping of Arg258 (TS6 in Figure 1A) and formation of an additional hydrogen bond interaction with Asp256 following flipping of Asp190 (TS5). Multiple hydrogen bond interactions between Arg258 and Asp192/Asp256 combined with the stable Watson–Crick base pair interaction between dGMP and C present a well-maintained active site geometry in the pol-β system during the entire opening conformational pathway.
The opening conformational pathway for the oxoG(anti):C system
From the probability distributions of the order parameters for the transition states in the oxoG(anti):C system (Supplementary Figure S4), we observe a different sequence of transition states after chemistry compared to the G:C system: Arg258 partial flip (TS1), Phe272 rotation (TS2), Mg(p) ion dissociation (TS3), Watson–Crick base pair breaking between oxoG(anti) and C (TS4), Arg258 full flip (TS5), Asp192 flip (TS6) and Mg(n) ion dissociation (TS7). Compared to the G:C unlesioned system, the major differences are nascent base pair breaking in the middle of the conformational change and Mg(n) ion dissociation at the last step. These combined factors trigger rapid opening in the oxoG(anti):C system.
In more detail, the Arg258 partial flip (TS1) is followed by the Phe272 rotation (TS2). The Mg(p) ion bound to the oxoG phosphate group remains coordinated with water molecules and forms a water shell. One water molecule in the water shell forms a hydrogen bond interaction with the O8 atom in oxoG and stabilizes the anti-conformation (Figure 2A, top image). The Mg(p) ion and hydrogen bonding with the water shell also reduce the electrostatic clash between O5’ and O8, so that the two oxygen atoms (O5’ and O8) of oxoG maintain a relatively short distance (∼3.0 Å) (TS2 in Supplementary Figure S5A). As the Mg(p) ion dissociates from the binding site, the associated water shell is disrupted (TS3 in Figures 1B and 2A). This dissociation of the Mg(p)-water shell combined with the electrostatic clash between the phosphate group and O8 leads to a rotation of the oxoG base so that its χ angle increases (TS4 in Supplementary Figure S5B). The distance between O5’ and O8 subsequently increases to ∼4 Å. This unstable configuration of the oxoG system causes base pair breaking between oxoG(anti) and C (TS4, Figures 1B and 2A, bottom image). Once broken, the χ angle decreases and the distance between O5’ and O8 remains large (∼4.5 Å) (TS5 in Supplementary Figure S5). Therefore, the dissociation of the Mg(p) ion triggers an electrostatic clash between the phosphate group and O8 and causes the destabilization and rupture of the base pair between oxoG(anti) and C. Once broken, Arg258 flips (TS5 in Figure 1B). However, because Arg258 is too far from Asp192 and Asp256, and Asp192 cannot approach Arg258 because of the bound Mg(n) ion, Arg258 and Asp192 do not form a hydrogen bond. Interestingly, the overall characteristics of the active site geometry at TS5 are very similar to those of the closed product complex in the mismatched pol-β system (Supplementary Figure S1D), in terms of the configurations of Arg258, Phe272 and Asp192, broken base pair, and the bound Mg(n) ion at Asp192. Thus, beyond TS5, the opening conformational pathway in the oxoG(anti) pol-β system resembles that of the mismatched system, which opens rapidly. After Arg258 flips, Asp192 rotates (TS6 in Figure 1B) and Mg(n) dissociates (TS7 in Figure 1B); the slight shift of Asp192 toward Arg258 forms a hydrogen bond interaction with Arg258.
The opening conformational pathway of the oxoG(syn):A system
For the lesioned oxoG(syn):A system, the base pair is broken earlier in the opening conformational changes, so that the sequence of transition states is (Figure 1C; see Supplementary Figure S6 for the probability distributions): Arg258 partial flip (TS1), Hoogsteen base pair breaking between oxoG(syn) and A (TS2), Mg(p) ion dissociation (TS3), Phe272 rotation (TS4), Arg258 full flip (TS5), Asp192 flip (TS6) and Mg(n) ion release (TS7). Thus, a different role of the Mg(p)-water shell complex in this Hoogsteen base paired system causes the base pair to break (TS2).
As shown in the X-ray structure of the oxoG:A, a relatively weak Hoogsteen base pair interaction can be stabilized by a hydrogen bond interaction between O1P at the phosphate group and H22-N2 atoms in the oxoG(syn)-conformation (TS2 in Figures 1C and 2B, top image). However, water molecules that coordinate with the Mg(p) ion interfere and break the hydrogen bond interaction between O1P and N2. As a result, the Hoogsteen base pair is destabilized and broken as the Mg(p)-water shell forms between the O1P and N2 atoms (TS2 in Figures 1C and 2B, middle image). Once the oxoG(syn):A base pair is broken, the Mg(p)-water shell dissociates from the binding site (TS3, Figure 1C). The rotation of Phe272 at TS4 vacates space for the Arg258 flip (TS5, Figure 1C). However, as observed in the oxoG(anti):C pol-β system, Arg258 is too far to form hydrogen bond interactions with Asp192 and Asp256. Thus, the characteristics of the active site geometry in the oxoG(syn):A system also resemble those of the mismatched A:G system (Supplementary Figure S1D) in terms of the geometry of Asp256, Asp 192 and Arg258, broken base pair, and the bound Mg(n) ion at Asp192. After Arg258 flips, Asp192 rotates while still bound to the Mg(n) ion (TS6 in Figure 1C). As the Mg(n) ion dissociates from the binding site of Asp192, Asp192 rotates toward Arg258 and forms a weak hydrogen bond interaction with Asp258. Therefore, the oxoG(syn):A pol-β system also exhibits poor active site geometry during the opening conformational change.
Free energy profiles of the G:C and two oxoG systems
The free energy profiles of the G:C and two oxoG pol-β systems in Figure 3 are calculated by BOLAS protocol. The free energy values of each transition in the three systems are listed in Table 2. The free energy analysis is performed for each system separately with respect to its starting state. The initial energies for the closed state after chemistry for each lesioned system are shown (zero value references) in Figure 3.
In the beginning of opening pathway for the G:C system, the partial flip of Arg258 (TS1) has a larger energy barrier (∼3 kBT,) than that in the two oxoG systems. Although the events of metal ion dissociations from each system occur in different transition order, the dissociations of metal ions in all three systems have consistent high free energy characteristics. The free energy barriers of Mg(p) and Mg(n) dissociations from each system are ∼12–13 kBT and ∼10–11 kBT, respectively (Table 2). These free energy patterns of metal ion dissociations during the opening pathway after chemistry are opposite those in the closing pathway of the pol-β before chemistry (68).
Other barriers for the G:C substrate pol-β system are 6 kBT, 6 kBT and 4 kBT for Phe272, Asp190 and Arg258 second flips, respectively. Following this last transition, the free energy of the G:C pol-β remains 13 kBT higher than the beginning of the opening pathway (Figure 3). Therefore, overall, the free energy of the G:C system continues uphill as the transitions evolve.
In the oxoG(anti) system, the free energy remains at the same level following the Phe272 flip (TS2), while base pair breaking in the oxoG(syn) system (TS2) increases the free energy of the system by ∼2 kBT. As the Mg(p) ion dissociates from both systems (TS3), the difference in free energy between oxoG(anti) and oxoG(syn) remains at the same level (2 kBT). As the base pair is broken in oxoG(anti) system (TS4), the free energy of the system decreases by 3 kBT, while the Phe272 flip in the oxoG(syn) system decreases free energy by 5 kBT. Thus, while both oxoG systems evolve differently between TS2 and TS4, the two systems’ free energy characteristics after TS4 are very similar (see Figure 3). The Hoogsteen base pair breaking in oxoG(syn) system has a higher free energy barrier (8 kBT) than the Watson–Crick base pair breaking in oxoG(anti) system (3 kBT). The electrostatic repulsion between O8 and the phosphate in the anti- system after Mg(p) dissociation causes the Watson–Crick base pair to break and reduces the free energy barrier. In addition, the free energy of the oxoG(anti) system after base pair breaking decreases by 5 kBT, probably due to the relaxation of the electrostatic repulsion and base stacking, while that of the oxoG(syn) system increases 2 kBT. The Arg258 flips in the two oxoG systems are energetically less favorable than for the G:C system which is energetically more favorable and stable due to multiple hydrogen bond interactions.
Besides the Mg(p) and Mg(n) dissociations, most transition events in the two oxoG systems do not increase the overall free energy while those of the G:C proceed uphill. This trend is evident from Table 2, which shows that the reverse barrier 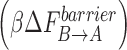 is higher than the forward barrier
is higher than the forward barrier 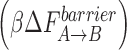 for the key active site residues in the two oxoG systems, while the pattern is reversed in the G:C system. In addition, compared to the G:C system, the free energy landscapes of the two oxoG systems have a lower maximum barrier (18 kBT versus 10 kBT) as well as a 8 kBT lower final state than the G:C system. These trends explain the more rapid opening of both oxoG systems compared to G:C. These energy trends corroborate our earlier studies and predictions regarding uphill/downhill energy trends as related to the conformational change (66,67). Thus, the important conclusion from the free energy calculations is that the free energies of the last transition in both oxoG systems are almost the same or slightly lower than the corresponding first transition, while the free energy of the G:C system increases as transitions progress. Therefore, the free energy profile of G:C indicates that the system energetically prefers the closed state while the two oxoG systems prefer the open state.
for the key active site residues in the two oxoG systems, while the pattern is reversed in the G:C system. In addition, compared to the G:C system, the free energy landscapes of the two oxoG systems have a lower maximum barrier (18 kBT versus 10 kBT) as well as a 8 kBT lower final state than the G:C system. These trends explain the more rapid opening of both oxoG systems compared to G:C. These energy trends corroborate our earlier studies and predictions regarding uphill/downhill energy trends as related to the conformational change (66,67). Thus, the important conclusion from the free energy calculations is that the free energies of the last transition in both oxoG systems are almost the same or slightly lower than the corresponding first transition, while the free energy of the G:C system increases as transitions progress. Therefore, the free energy profile of G:C indicates that the system energetically prefers the closed state while the two oxoG systems prefer the open state.
DISCUSSION
We can understand the opening conformational pathway after chemistry by examining the closing conformational pathway before chemistry. In both cases, the sequence of side-chain and ion transitions presents a cascade of checkpoints to direct the pathway. In the open state of pol-β before chemistry, Arg258 interacts with Asp192, before the nucleotide and catalytic metal ions bind to Asp192. As the conformation of pol-β changes to the closed form, Asp192 flips and Arg258 loses the interaction with Asp192. The rotation of Phe272 blocks the interaction between Arg258 and Asp192. The flipped Arg258 forms hydrogen bond interactions with Glu295/Tyr296, while Asp192 remains bound to the nucleotide and catalytic metal ions for the chemical reaction (71). During the opening conformational pathway after chemistry with the G:C system, those events eventually reverse. Here, the dissociation of the Mg(p)-water shell does not affect the Watson–Crick base pair interactions between dGMP and C for the G:C system, since there is no major interaction between the dGMP base and the water shell (TS2, Figure 1A). Phe272 rotates and permits interactions between Arg258 and Asp192 (TS3, Figure 1A). The Mg(n) ion then dissociates from the binding site of Asp192 (TS4, Figure 1A), Asp192 moves toward Arg258, and forms multiple hydrogen bond interactions with it. As Arg258 flips, stable hydrogen bond interactions with Asp192 and Asp256 (TS6, Figure 1A) result. Therefore, the precise behavior of key residues (Asp192, Arg258 and Phe272) and proper order of metal ion dissociations in the G:C system are key elements that optimize substrate discrimination before and after chemistry. The significance of the key residues in the G:C system before the opening conformation pathway and after the Arg258 flip (TS5) is illustrated in Figure 4A.
Figure 4.

The configuration of key residues in the active sites of (A) the G:C, (B) oxoG(anti):C and (C) oxoG(syn):A pol-β systems. Structures on the left and right correspond to before opening conformational pathway, and the after Arg258 flip, respectively. The red dashed lines and yellow regions indicate hydrogen bond interactions between atoms. The black dashed lines indicate long distances which do not allow hydrogen bond interactions.
The dissociation of the Mg(p) and Mg(n) ions performs a crucial role in the opening conformational pathways of both oxoG systems. As the Mg(p)-water shell dissociates from the binding site, the base pair between oxoG(anti) and C is destabilized and broken by the electrostatic repulsion between the phosphate group and O8 atoms (Figure 2A); for the syn-system, the bound Mg(p)-water shell destabilizes and breaks the Hoogsteen base pair of the oxoG(syn) and A by interfering instead with the hydrogen bond interaction between O1P and N2 atoms (Figure 2B). After the base pair is broken and Arg258 flips, the active site geometries of both oxoG systems are significantly distorted (Figure 4B and C). When Arg258 flips, the dislocated phosphate group due to the broken base pair influences the location of Asp192 via the bound Mg(n) ion. This geometry positions Asp192 out of interaction range from Arg258 (Figure 4B and C). In addition, for the oxoG(syn):A system, the interaction between Arg258 and Glu295 is maintained even after Arg258 flips. When the Mg(n) is released from the systems at the last transition event, Asp192 begins to establish an interaction with Arg258 (Figure 1B and C). Therefore, as the transition states evolve during the opening conformational pathway in the oxoG systems, the active site geometries of both oxoG systems are significantly distorted and the key molecular interactions between Asp192 and Arg258 cannot form. In addition, the configuration of the active site geometry when Arg258 flips resembles that of the mismatched A:G system, which opens rapidly. Therefore, the absence of crucial molecular interactions between key residues in both oxoG systems causes rapid opening, resembling that of the mismatched A:G system.
Before chemistry, the electrostatic clash between the O8 atom and the phosphate group of oxoG results in a the lower insertion efficiency of oxoG(anti) opposite C than oxoG(syn) insertion opposite A (72). The X-ray structures and MD simulations from our previous work (41) showed that the product ion Mg(p) and associated water shell compensate for the electrostatic clash and stabilize the anti-conformation during the closing pathway before chemistry. Here, we propose that the Mg(p) and associated water shell have a crucial role in destabilizing the oxoG base pair during the opening conformational change after chemistry and trigger rapid opening. In the oxoG(anti):C system, the Mg(p) ion stabilizes the anti-conformation by reducing electrostatic repulsion between O8 atom in the base and the phosphate group of oxoG(anti). As Mg(p) dissociates from the binding site, the electrostatic repulsion destabilizes Watson–Crick hydrogen bond interactions and causes base breaking (Figure 2A). In contrast, the Mg(p) ion in the oxoG(syn):A system destabilizes the Hoogsteen base pair by interfering with the hydrogen bond interaction between N2 of the base and its phosphate group, which stabilizes the Hoogsteen base pair (Figure 2B). These subtle interactions promote rapid reopening of the lesioned systems, while other side chains rearrange in an orchestrated cascade.
The free energy characteristics between the G:C and two oxoG systems are also illuminating. The free energy of the G:C system shows an overall uphill pattern so that the open conformation is 8 kBT higher than the closed state, while the closed and open states for both oxoG systems are about equal in energy, supporting the rapid reopening. In addition, despite different nucleotide insertions, the free energies for the oxoG(anti):C and oxoG(syn):A systems are overall similar. These free energy profiles indicate that the opening conformational change after chemistry is hampered for the unlesioned system by the relatively stable active site geometry of the closed conformation with respect to the open state. In contrast, the lesioned systems rapidly open due to the unstable active site and the rupture of crucial molecular interactions associated with the oxoG base pairs, Mg(p) ions and water shells. Thus, conformational changes following nucleotide insertion depend on the identity of the incoming nucleotide, as well as the Watson–Crick hydrogen bond interactions with the nascent base pair.
As a summary, Figure 5 compares the opening mechanism of the unlesioned G:C pol-β system (top image) to that of the lesioned oxoG(anti):C system (bottom). When the Mg(p)-water shell is released from the unlesioned pol-β system, no significant disruptions occur between the water shell and dGMP; the Watson-Crick base pair interaction between dGMP and C remains intact (Figure 5A and B). The location of the phosphate group maintained by the Watson-Crick base pair stabilizes the active site geometry as the transition states evolve. After the nucleotide metal ion (Mg(n)) dissociates from Asp192, Asp192 relocates and approaches Arg258. The short distance between Asp192 and Arg258 seals hydrogen bond interactions which trigger the rotation of Arg258 (Figure 5C).
Figure 5.

Summary of key structural and energetic aspects distinguishing the opening conformational change of the lesioned versus unlesioned pol-β systems. In particular, we highlight differences associated with Mg(p) binding and its associated water shell. See text for details.
In contrast, as the Mg(p)-water shell dissociates from the oxoG(anti):C system, the electrostatic repulsion between the phosphate group and the O8 atom in the oxoG causes the Watson–Crick base pair to break (Figure 5D and E). Once broken, the phosphate group is dislocated and distorts the active site. When Arg258 flips toward Asp192, Arg258 is too far from Asp192, so Arg258 and Asp192 do not form a hydrogen bond (Figure 5F). Therefore, the broken base pair, due to the Mg(p) ion dissociation, causes the cascade of events leading to poor active site geometry and the rupture of crucial molecular interactions among key residues. A similar cascade of distortions occur for the oxoG(syn):A system (Figure 4C). Alanine substitution of Arg258 has been shown to influence conformational steps before and after the chemical step for insertion of a correct dNTP (73–74).
Energetically, the lesioned open states are similar in energy to those of the corresponding closed complexes after chemistry, in marked contrast to the unlesioned system, whose open state is energetically higher than the closed state (Figure 5, middle image).
Conformational transitions subsequent to chemistry have important kinetic and biological implications. DNA polymerases hasten the forward reaction with a post-chemistry conformational change (i.e., opening) that deters pyrophosphorolysis (i.e., reverse reaction), thereby pulling the reaction forward. At the same time, the closed enzyme conformation protects the product of single-nucleotide DNA gap filling (i.e., nicked DNA) by sheltering the DNA nick. Unrepaired single-strand DNA breaks could initiate a cascade of cellular events that can lead to cell death (75). Thus, the stability of the closed post-catalytic complex, as well as the rate of opening, must be modulated to accommodate downstream events such as DNA ligation of a properly base paired nick or removal of an aberrant DNA terminus (i.e., 8-oxoG). The closed enzyme form binds correctly paired DNA more tightly (32), and time-lapse crystallography indicates that the lifetime of the closed post-catalytic form is significantly longer with an undamaged Watson–Crick terminal base pair compared to an aberrant base pair (i.e., mismatch or damaged base pair) (16,41). Accordingly, the conformational state of the pol-β product complex could signal downstream events. For example, the closed conformation would recruit DNA ligase to facilitate the completion of base excision repair. In contrast, the open conformation would deter DNA ligation and recruit a proofreading exonuclease to correct an aberrant DNA terminus. Abnormalities at DNA nicks promote abortive ligation and stabilize the cytotoxic nick (76–77). Thus, the structural and energetic features revealed here help explain aspects of a complex surveillance system evolved by pol-β and the cellular machinery to handle oxidative lesions.
Supplementary Material
Acknowledgments
Research support from Philip Morris USA Inc. and Philip Morris International to T.S. is gratefully acknowledged. Computations were made possible by NYU's HPC cluster as well as using the resources of the CCNI supported by the New York State Foundation for Science, Technology and Innovation (NYSTAR), and the Dell computer cluster by New York University Information Technology Services (NYU ITS).
Footnotes
Present address: Bret D. Freudenthal, Department of Biochemistry and Molecular Biology, University of Kansas Medical Center, Kansas City, KS 66160, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Philip Morris USA Inc; Intramural Research Program of the NIH (in part); National Institute of Environmental Health Sciences [Z01-ES050158 and Z01-ES050159 to S.W. and was in association with NIH grant 1U19CA105010]. Funding for open access charge: NIH.
Conflict of interest statement. None declared.
REFERENCES
- 1.Wilson S.H. Mammalian base excision repair and DNA polymerase β. Mutat. Res. 1998;407:203–215. doi: 10.1016/s0921-8777(98)00002-0. [DOI] [PubMed] [Google Scholar]
- 2.Kunkel T.A. DNA Replication Fidelity. J. Biol. Chem. 2004;279:16895–16898. doi: 10.1074/jbc.R400006200. [DOI] [PubMed] [Google Scholar]
- 3.Seeberg E., Eide L., Bjoras M. The base excision repair pathway. Trends Biochem. Sci. 1995;20:391–397. doi: 10.1016/s0968-0004(00)89086-6. [DOI] [PubMed] [Google Scholar]
- 4.Friedberg E.C. DNA damage and repair. Nature. 2003;421:436–460. doi: 10.1038/nature01408. [DOI] [PubMed] [Google Scholar]
- 5.Jackson S.P., Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoeijmakers J.H.N. DNA Damage, Aging, and Cancer. Engl. J. Med. 2009;361:1475–1485. doi: 10.1056/NEJMra0804615. [DOI] [PubMed] [Google Scholar]
- 7.An C.L., Chen D.S., Makridakis N.M. Systematic biochemical analysis of somatic missense mutations in DNA polymerase β found in prostate cancer reveal alteration of enzymatic function. Hum. Mutat. 2011;32:415–423. doi: 10.1002/humu.21465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dalal S., Chikova A., Jaeger J., Sweasy J.B. The Leu22Pro tumor-associated variant of DNA polymerase beta is dRP lyase deficient. Nucleic Acids Res. 2008;36:411–422. doi: 10.1093/nar/gkm1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lang T.M., Dalal S., Chikova A., DiMaio D., Sweasy J.B. The E295K DNA Polymerase Beta Gastric Cancer-Associated Variant Interferes with Base Excision Repair and Induces Cellular Transformation. Mol. Cell. Biol. 2007;27:5587–5596. doi: 10.1128/MCB.01883-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Starcevic D., Dalal S., Sweasy J.B. Is there a link between DNA polymerase beta and cancer? Cell Cycle. 2004;3:998–1001. [PubMed] [Google Scholar]
- 11.Iwanaga A., Ouchida M., Miyazaki K., Hori K., Mukai T. Functional mutation of DNA polymerase β found in human gastric cancer – inability of the base excision repair in vitro. Mutat. Res. 1999;435:121–128. doi: 10.1016/s0921-8777(99)00036-1. [DOI] [PubMed] [Google Scholar]
- 12.Nicolay N.H., Helleday T., Sharma R.A. Biological relevance of DNA polymerase beta and translesion synthesis polymerases to cancer and its treatment. Curr. Mol. Pharmacol. 2012;5:54–67. [PubMed] [Google Scholar]
- 13.Balbo P.B., Wang E.C., Tsai M.D. Kinetic mechanism of active site assembly and chemical catalysis of DNA polymerase β. Biochemistry. 2011;50:9865–9875. doi: 10.1021/bi200954r. [DOI] [PubMed] [Google Scholar]
- 14.Tanabe K., Bohn E.W., Wilson S.H. Steady-state kinetics of mouse DNA polymerase β. Biochemistry. 1979;18:3401–3406. doi: 10.1021/bi00582a029. [DOI] [PubMed] [Google Scholar]
- 15.Sawaya M.R., Pelletier H., Kumar A., Wilson S.H., Kraut J. Crystal structure of rat DNA polymerase beta: evidence for a common polymerase mechanism. Science. 1994;264:1930–1935. doi: 10.1126/science.7516581. [DOI] [PubMed] [Google Scholar]
- 16.Freudenthal B., Beard W.A., Shock D., Wilson S.H. Observing a DNA polymerase choose right from wrong. Cell. 2013;154:157–168. doi: 10.1016/j.cell.2013.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arora K., Schlick T. In silico evidence for DNA polymerase-beta's substrate-induced conformational change. Biophys. J. 2004;87:3088–3099. doi: 10.1529/biophysj.104.040915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arora K., Schlick T. Conformational transition pathway of polymerase β DNA upon binding correct incoming substrate. J. Phys. Chem. B. 2005;109:5358–5367. doi: 10.1021/jp0446377. [DOI] [PubMed] [Google Scholar]
- 19.Beard W.A., Wilson S.H. Structure and mechanism of DNA polymerase β. Biochemistry. 2014;53:2768–2780. doi: 10.1021/bi500139h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Radhakrishnan R., Schlick T. Correct and incorrect nucleotide incorporation pathways in DNA polymerase β. Biochem. Biophys. Res. Commun. 2006;350:521–529. doi: 10.1016/j.bbrc.2006.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alberts I.L., Wang Y., Schlick T. DNA polymerase β catalysis: are different mechanisms possible? J. Amer. Chem. Soc. 2007;129:11100–11110. doi: 10.1021/ja071533b. [DOI] [PubMed] [Google Scholar]
- 22.Foley M., Arora K., Schlick T. Intrinsic motions of DNA polymerases underlie their remarkable specificity and selectivity and suggest a hybrid substrate binding mechanism in innovations in biomolecular modeling and simulations. R. Soc. Chem. Lond. 2012;2:81–110. [Google Scholar]
- 23.Sawaya M.R., Prasad R., Wilson S.H., Kraut J., Pelletier H. Crystal structures of human DNA polymerase β complexed with gapped and nicked DNA: evidence for an induced fit mechanism. Biochemistry. 1997;36:11205–11215. doi: 10.1021/bi9703812. [DOI] [PubMed] [Google Scholar]
- 24.Li Y., Korolev S., Waksman G. Crystal structures of open and closed forms of binary and ternary complexes of the large fragment of Thermus aquaticus DNA polymerase I: structural basis for nucleotide incorporation. EMBO J. 1998;17:7514–7525. doi: 10.1093/emboj/17.24.7514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Doublie′ S., Ellenberger T. The mechanism of action of T7 DNA polymerase. Curr. Opin. Struct. Biol. 1998;8:704–712. doi: 10.1016/s0959-440x(98)80089-4. [DOI] [PubMed] [Google Scholar]
- 26.Kiefer J.R., Mao C., Braman J.C., Beese L.S. Visualizing DNA replication in a catalytically active Bacillus DNA polymerase crystal. Nature. 1998;391:304–307. doi: 10.1038/34693. [DOI] [PubMed] [Google Scholar]
- 27.Koshland D.E. The Key-Lock theory and the induced fit theory. Angew. Chem. Int. Ed. Engl. 1994;33:2375–2378. [Google Scholar]
- 28.Beard W.A., Wilson S.H. Structural insights into DNA polymerase β fidelity: hold tight if you want it right. Chem. Biol. 1998;5:R7–R13. doi: 10.1016/s1074-5521(98)90081-3. [DOI] [PubMed] [Google Scholar]
- 29.Doublié S., Sawaya M.R., Ellenberger T. An open and closed case for all polymerases. Structure. 1999;7:R7–R13. doi: 10.1016/S0969-2126(99)80017-3. [DOI] [PubMed] [Google Scholar]
- 30.Radhakrishnan R., Arora K., Wang Y., Beard W.A., Wilson S.H., Schlick T. Regulation of DNA repair fidelity by molecular checkpoints: "Gates" in DNA polymerase beta's substrate selection. Biochem. 2006;45:15142–15156. doi: 10.1021/bi061353z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schlick T., Arora K., Beard W.A., Wilson S.H. Perspective: pre-chemistry conformational changes in DNA polymerase mechanisms. Theor. Chem. Acc. 2012;131:1287–1300. doi: 10.1007/s00214-012-1287-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beard W.A., Shock D.D., Wilson S.H. Influence of DNA structure on DNA polymerase β active site function: extension of mutagenic DNA intermediates. J. Biol. Chem. 2004;279:31921–31929. doi: 10.1074/jbc.M404016200. [DOI] [PubMed] [Google Scholar]
- 33.Ziech D., Franco R., Georgakilas A.G., Georgakila S., Malamou-Mitsi V., Schoneveld O., Pappa A., Panayiotidis M.I. The role of reactive oxygen species and oxidative stress in environmental carcinogenesis and biomarker development. Chem. Biol. Interact. 2010;188:334–339. doi: 10.1016/j.cbi.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 34.Azzam E.I., Jay-Gerin J.P., Pain D. Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury. Cancer Lett. 2012;327:48–60. doi: 10.1016/j.canlet.2011.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Migliore L., Coppede F. Environmental-induced oxidative stress in neurodegenerative disorders and aging. Mutat. Res. 2009;674:73–84. doi: 10.1016/j.mrgentox.2008.09.013. [DOI] [PubMed] [Google Scholar]
- 36.Fraga C.G., Shigenaga M.K., Park J.W., Dega P., Ames B.N. Oxidative damage to DNA during aging:8-hydroxy-2'-deoxyguanosine in rat organ DNA and urine. PNAS. 1990;87:4533–4537. doi: 10.1073/pnas.87.12.4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ames B.N., Gold L.S. Endogenous mutagens and the causes of aging and cancer. Mutat. Res. 1991;250:3–16. doi: 10.1016/0027-5107(91)90157-j. [DOI] [PubMed] [Google Scholar]
- 38.Ames B.N., Shigenaga M.K., Hagen T.M. Oxidants, antioxidants, and the degenerative diseases of aging. PNAS. 1990;90:7915–7922. doi: 10.1073/pnas.90.17.7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oda Y., Uesugi S., Ikehara M., Nishimura S., Kawase Y., Ishikawa H., Inoue H., Ohtsuka E. NMR studies of a DNA containing 8-hydroxydeoxyguanosine. Nucleic Acids Res. 1991;19:1407–1412. doi: 10.1093/nar/19.7.1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Freudenthal B.D., Beard W.A., Wilson S.H. DNA polymerase minor groove interactions modulate mutagenic bypass of a templating 8-oxoguanine lesion. Nucleic Acids Res. 2013;41:1848–1858. doi: 10.1093/nar/gks1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Freudenthal B., Beard W.A., Perera L., Shock D., Kim T., Schlick T., Wilson S.H. Uncovering the polymerase-induced cytotoxicty of an oxidized nucleotide. Nature. 2015;517:635–639. doi: 10.1038/nature13886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Batra V.K., Shock D.D., Beard W.A., McKenn C.E., Wilson S.H. Binary complex crystal structure of DNA polymerase β reveals multiple conformations of the templating 8-oxoguanine lesion. PNAS. 2012;109:113–118. doi: 10.1073/pnas.1112235108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yamtich J., Sweasy J.B. DNA polymerase Family X: Function, structure, and cellular roles. Biochim. Biophys. Acta. 2010;1804:1136–1150. doi: 10.1016/j.bbapap.2009.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Y., Reddy S., Beard W.A., Wilson S.H., Schlick T. Differing conformational pathways before and after chemistry for insertion of dATP versus dCTP opposite 8-OxoG in DNA polymerase β. Biophys. J. 2007;92:3063–3070. doi: 10.1529/biophysj.106.092106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang Y.L., Schlick T. Distinct energetics and closing pathways for DNA polymerase β with 8-oxoG template and different incoming nucleotides. BMC Struct. Biol. 2007;7:7–21. doi: 10.1186/1472-6807-7-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shibutani S., Takeshita M., Grollman A.P. Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8−oxodG. Nature. 1991;349:431–434. doi: 10.1038/349431a0. [DOI] [PubMed] [Google Scholar]
- 47.Grollman A.P., Moriya M. Mutagenesis by 8-oxoguanine: An enemy within. Trends Genet. 1993;9:246–249. doi: 10.1016/0168-9525(93)90089-z. [DOI] [PubMed] [Google Scholar]
- 48.Beard W.A., Batra V.K., Wilson S.H. DNA polymerase structure-based insight on the mutagenic properties of 8-oxoguanine. Mutat. Res. 2010;703:18–23. doi: 10.1016/j.mrgentox.2010.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Feller S.E., Zhang Y., Pastor R.W., Brooks B.R. Constant pressure molecular dynamics simulation: the Langevin piston method. J. Chem. Phys. 1995;103:4613–4621. [Google Scholar]
- 50.Darden T., York D., Pedersen L. Particle mesh Ewald: an Nlog(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- 51.Phillips J.C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R.D., Kale L., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Best R.B., Zhu X., Shim J., Lopes P.E.M., Mittal J., Feig M., MacKerell A.D., Jr Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone phi, psi and sidechain chi1 and chi2 dihedral angles. J. Chem. Theory Comput. 2012;8:3257–3273. doi: 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.MacKerell A.D., Jr, Feig M., Brooks C.L. III. Improved treatment of the protein backbone in empirical force fields. J. Amer. Chem. Soc. 2004;126:698–699. doi: 10.1021/ja036959e. [DOI] [PubMed] [Google Scholar]
- 54.MacKerell A.D., Jr, Bashford D., Bellott M., Dunbrack R.L., Evanseck J.D., Field M.J., Fischer S., Gao J., Guo H., Ha S., et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 55.Hart K., Foloppe N., Baker C.M., Denning E.J., Nilsson L., MacKerell A.D., Jr Optimization of the CHARMM additive force field for DNA: Improved treatment of the BI/BII conformational equilibrium. J. Chem. Theory Comput. 2012;8:348–362. doi: 10.1021/ct200723y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Denning E.J., Priyakumar U.D., Nilsson L., MacKerell A.D., Jr Impact of 2-hydroxyl sampling on the conformational properties of RNA: Update of the CHARMM all-atom additive force field for RNA. J. Comput. Chem. 2011;32:1929–1943. doi: 10.1002/jcc.21777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Foloppe N., MacKerell A.D., Jr All-atom empirical force field for nucleic acids: 1) parameter optimization based on small molecule and condensed phase macromolecular target data. J. Comput. Chem. 2000;21:86–104. [Google Scholar]
- 58.MacKerell A.D., Jr, Banavali N. All-atom empirical force field for nucleic acids: 2) application to molecular dynamics simulations of DNA and RNA in solution. J. Comput. Chem. 2000;21:105–120. [Google Scholar]
- 59.Schlick T., Collepardo-Guevara R., Halvorsen L., Jung S., Xiao X. Biomolecularmodeling and simulation: a field coming of age. Quart. Rev. Biophys. 2011;44:191–228. doi: 10.1017/S0033583510000284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brooks B.R., Bruccoleri R.E., Olafson B.D., States D.J., Swaminathan S., Karplus M. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1983;4:187–217. [Google Scholar]
- 61.Stote R.H., Karplus M. Zinc binding in proteins and solution: a simple but accurate nonbonded representation. Proteins. 1995;23:12–31. doi: 10.1002/prot.340230104. [DOI] [PubMed] [Google Scholar]
- 62.MacKerell A.D., Jr Influence of magnesium ions on duplex DNA structural, dynamic and solvation properties. J. Phys. Chem. B. 1997;101:646–650. [Google Scholar]
- 63.Yang L., Arora K., Beard W.A., Wilson S.H., Schlick T. The critical role of magnesium ions in DNA polymerase β's closing and active site assembly. J. Am. Chem. Soc. 2004;126:8441–8453. doi: 10.1021/ja049412o. [DOI] [PubMed] [Google Scholar]
- 64.Bolhuis P.G., Chandler D., Dellago C., Geissler P.L. Transition path sampling: throwing ropes over rough mountain passes, in the dark. Annu. Rev. Phys. Chem. 2002;53:291–318. doi: 10.1146/annurev.physchem.53.082301.113146. [DOI] [PubMed] [Google Scholar]
- 65.Radhakrishnan R., Schlick T. Orchestration of cooperative events in DNA synthesis and repair mechanism unraveled by transition path sampling of DNA polymerase β's closing. PNAS. 2004;101:5970–5975. doi: 10.1073/pnas.0308585101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Radhakrishnan R., Schlick T. Fidelity discrimination in DNA polymerase β: differing closing profiles for a mismatched (G:A) versus matched (G:C) base pair. J. Amer. Chem. Soc. 2005;127:13245–13252. doi: 10.1021/ja052623o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li Y., Gridley C.L., Jaeger J., Sweasy J.B., Schlick T. Unfavorable electrostatic and steric interactions in DNA polymerase β E295K mutant interfere with the enzyme's pathway. J. Amer. Chem. Soc. 2012;134:9999–10010. doi: 10.1021/ja300361r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li Y., Freudenthal B.D., Beard W.A., Wilson S.H., Schlick T. Optimal and variant metal-ion routes in DNA polymerase β's conformational pathways. J. Amer. Chem. Soc. 2014;136:3630–3639. doi: 10.1021/ja412701f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pratt L.R. A statistical method for identifying transition states in high dimensional problems. J. Chem. Phys. 1986;85:5045–5048. [Google Scholar]
- 70.Radhakrishnan R., Schlick T. Biomolecular free energy profiles by a shooting/umbrella sampling protocol, "BOLAS". J. Chem. Phys. 2004;121:2436–2444. doi: 10.1063/1.1766014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kirby T.W., DeRose E.F., Cavanaugh N.A., Beard W.A., Shock D.D., Mueller G.A., Wilson S.H., London R.E. Metal-induced DNA translocation leads to DNA polymerase conformational activation. Nucl Acids Res. 2012;40:2974–2983. doi: 10.1093/nar/gkr1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Batra V.K., Beard W.A., Hou E.W., Pedersen L.C., Prasad R., Wilson S.H. Mutagenic conformation of 8-oxo-7,8-dihydro-2'-dGTP in the confines of a DNA polymerase active site. Nat. Struct. Mol. Biol. 2010;17:889–890. doi: 10.1038/nsmb.1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bakhtina M., Roettger M.P., Kumar S., Tsai M.D. A unified kinetic mechanism applicable to multiple DNA polymerases. Biochemistry. 2007;46:5463–5472. doi: 10.1021/bi700084w. [DOI] [PubMed] [Google Scholar]
- 74.Beard W.A., Shock D.D., Batra V.K., Prasad R., Wilson S.H. Substrate-induced DNA polymerase β activation. J. Biol. Chem. 2014;289:31411–31422. doi: 10.1074/jbc.M114.607432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Horton J.K., Watson M., Stefanick D.F., Shaughnessy D.T., Taylor J.A., Wilson S.H. XRCC1 and DNA polymerase β in cellular protection against cytotoxic DNA single-strand breaks. Cell Res. 2008;18:48–63. doi: 10.1038/cr.2008.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Harris J.L., Jakob B., Taucher-Scholz G., Dianov G.L., Becherel O.J., Lavin M.F. Aprataxin, poly-ADP ribose polymerase 1 (PARP-1) and apurinic endonuclease 1 (APE1) function together to protect the genome against oxidative damage. Hum. Mol. Gen. 2009;18:4102–4117. doi: 10.1093/hmg/ddp359. [DOI] [PubMed] [Google Scholar]
- 77.Çağlayan M., Wilson S.H. Oxidant and environmental toxicant-induced effects compromise DNA ligation during base excision DNA repair. DNA Repair. 2015;35:85–89. doi: 10.1016/j.dnarep.2015.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.