


Abstract
Genome sequence compositions and epigenetic organizations are correlated extensively across multiple length scales. Replication dynamics, in particular, is highly correlated with GC content. We combine genome-wide time of replication (ToR) data, topological domains maps and detailed functional epigenetic annotations to study the correlations between replication timing and GC content at multiple scales. We find that the decrease in genomic GC content at large scale late replicating regions can be explained by mutation bias favoring A/T nucleotide, without selection or biased gene conversion. Quantification of the free dNTP pool during the cell cycle is consistent with a mechanism involving replication-coupled mutation spectrum that favors AT nucleotides at late S-phase. We suggest that mammalian GC content composition is shaped by independent forces, globally modulating mutation bias and locally selecting on functional element. Deconvoluting these forces and analyzing them on their native scales is important for proper characterization of complex genomic correlations.
INTRODUCTION
The GC content of a genome is one of its most fundamental sequence features, and probably the most studied one. GC content variation between and within species is observed throughout the tree of life, and correlates with numerous organismal and molecular features including the time of replication (ToR) (1). The diversity of genomic GC content motivated several theories proposing evolutionary scenarios underlying its origins and potential functional impact. Early theories on the so-called isochore phenomenon (2) were later been refined or replaced with models evaluating the effect of the dynamics of mutation spectra and factors affecting it (3,4), the role of GC-biased gene conversion (gBGC) (5,6), the activity of repetitive elements (7) and the role of putative selective forces (8,9). Recent findings are mostly in the favor of the gBGC model (10–14). Only recently, it became possible to study detailed genomic maps covering not only sequences, but also functional and epigenomic information at the single base pair level (15) and reassess the links between GC content, ToR, evolution and functional context.
To properly characterize GC content variation within genomes, and in particular within large and complex genomes such as human or mouse, it is essential to address the multiple scales of genome structure and function that can affect it. Advances in genomics (16), functional genomics (15) and chromosomal structure (17–19), indicated that genomes are composed of a patchwork of functional and non-functional/uncharacterized elements that are embedded within multi-scale, organized genomic territories. For example, functional elements (transcription factor binding sites, or exons) are tens to hundreds of bases long, transcriptional units can span thousands to ten thousands base pairs, and replication domains (20,21) or topological domains (22,23) appear on scales of hundred thousand to millions of base pairs. At each of these scales, different evolutionary forces affect GC content, and the complex hierarchical organization of the genome entangles these forces together. For example, early DNA replication domains are associated with topological domains (19,24) that are typically located away from the nuclear lamina (17), and contain a relatively higher density of exons or other functional elements (1). Each of these factors may contribute to increase in GC content. Regions located sub-telomerically, on the other hand, are prone to high recombination rates and biased gene conversion, while also accumulating more repetitive sequences than other genomic regions (25). Using previously unavailable comprehensive maps of functional elements, physical chromosomal architectures and replication landscapes, it is now possible to study GC content variation simultaneously at all scales, thereby leading to better understanding of the primary and secondary factors driving it.
In this work, we use a multi-scale approach to explore the relations between genomic GC content and potential evolutionary factors driving it. Most notably, we consider ToR domains at scales of megabases and selection on functional elements at scales of few hundred bases. Using comprehensive functional genomics maps, we can decouple the evolutionary forces on these two scales, and demonstrate them to work independently to decrease GC content in late-replicating domains and increase it in domains that are rich in functional elements (and therefore, indirectly, in early replicating domains). Analysis of divergence and polymorphism, coupled with exploration of the relation between ToR and GC content in Hi-C domains clearly indicate that replication-mediated decrease in GC content of late replicating domains cannot be linked with biased gene conversion or selection (that shares common allele-frequency signatures (5,6)). Furthermore, direct measurement of free dNTP pools along S-phase suggests that changes in nucleotide availability may result in asymmetric mutation spectrum during replication. The changes in the dNTP pools also provide a possible non-selective mechanism explaining this phenomenon. The multi-scale framework is thereby providing a simple explanation for the origin of GC content variation in mammalian genomes and for the strong correlation between time of replication and nucleotide composition. It suggests that the common models for GC content variation in mammals are true mainly for early replicating regions, whereas in late replicating regions GC-disfavoring mutation rates is the dominant evolutionary force.
MATERIALS AND METHODS
Sequence, time of replication and epigenomic data
Sequence and genomic positions of exons and repetitive elements were downloaded from UCSC genome browser (26). Molt4 and FFT ToR data (27) were downloaded from GSE17235. Repli-seq data (28) were downloaded from SRA (SRP012560) and mapped using bowtie to hg18. For each S phase section (S1, S2, S3, S4) and each tissue (BG02, H0287, GM06990, BJ, K562, TL010), data replicates were combined and averaged over 10kbp bins. The coordinates of genomic regions with constitutive ToR over 26 distinct human cell types were taken from Rivera-Mulia et al. (29). DNase-I data of 46 ENCODE tissues, as well as H3K4me1 and H3K27Ac and H3K27me3 (HelaS3) data were downloaded from ENCODE (15). LaminB1 DamID data were downloaded from UCSC genome browser. ORC1 ChIP-seq data and Repli-seq (Hela) (30) were downloaded from GEO (GSE37583). Bubble-seq origins of replication data (31) were downloaded from GEO (GSE38809). Nucleosome occupancy of Gaffney et al. (32) data were downloaded from GEO (GSE36979). Two replicates of CTCF ChIP-Seq data on Hela cells were downloaded from GEO (GSM749729 and GSM749739) and combined. Weakly mappable sites were defined as genomic coordinates with Duke-35 values <0.8 (downloaded from UCSC genome browser) after smoothing over sliding window of 500 bp. Hi-C raw data (IMR90) (23) were downloaded from SRA (SRP010370) and renormalized according to Yaffe and Tanay 2011 (33). Domain borders were extracted as described in Sexton et al. (22).
Functional density track and masking of functional elements
Functional sites were extracted by merging the genomic intervals of the top 1 percentiles of DNase-I sensitivity and H3K4me1 and H3K27Ac ChIP-seq values across all 46 tissues (after merging replicates) (15) and combined with weakly mappable (see above) and exon intervals. These masking parameters were chosen to cover most of the functional regions in the genome, such that the masked list will be dominated by non-functional regions. A list of the residual ‘masked’ non-functional genomic intervals can be downloaded from http://compgenomics.weizmann.ac.il/tanay/?page_id=667. Functional density was defined as the fraction of basepairs covered with functional sites as defined above. For masking the genome and focusing mostly on non-functional regions, we used an extended set of functional elements with relaxed definition (top 4 percentiles) of DNase-I hypersensitive sites, H3K4me1 and H3K27Ac peaks. Masked GC values per genomic bin, were defined as the number of G and C nucleotide divided by the total number of basepairs of non-functional elements within the bin.
Evolutionary analysis
We estimated the number of GC gain (T/A→G/C) and GC loss (G/C→A/T) substitutions at non-CpG contexts and CpG deamination across the primate phylogeny (Marmoset, Rhesus, Orangutan, Chimp and Human) in 100 kbp genomic bins using our previously published context-dependent evolutionary model (34). Specifically, we obtained the following statistics:
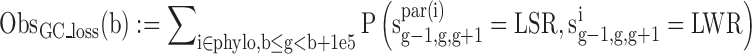 |
 |
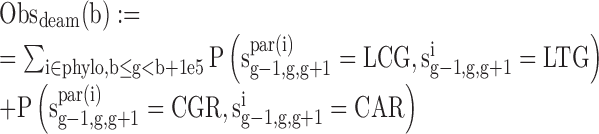 |
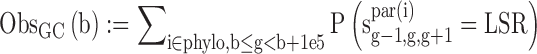 |
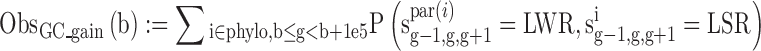 |
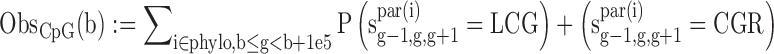 |
where sig-1,g,g+1 is the sequence of node i at positions g-1,g,g+1.S = G/C, W = A/T, L = A/C/G/T, R = A/C/G/T while LS! = CG and SR! = CG, b is the start position of the genomic bin, phylo contains all the node in the phylogeny tree, par(i) is the parent of node i.
Using these statistics we estimated the substitution probabilities per genomic bin:
 |
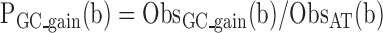 |
 |
Distributions of PGC_loss and PGC_gain at different loci are presented in Figure 3A and B while Pdeam(b) distribution is presented in Supplementary Figure S8A.
Figure 3.

Evolutionary analysis of GC dynamics betweenand within species. A) GC gain substitutions vs. ToR. Shown are primates GC gaining substitution rates (y-axis) vs. ToR (x-axis), for the whole intergenic genome (g) or functionally masked (m) regions. B) GC loss substitutions rate vs. ToR. Shown are primates GC losing substitution rates (y-axis) vs. ToR (x-axis), for the whole intergenic genome (g) or functionally masked (m) regions. C) GC substitution bias vs. ToR. Shown is the rate of GC gaining substitutions divided by the sum of rates of GC gaining and GC losing substitutions for intergenic (left) or functionally masked (right) regions. D-F) Frequency of low frequency (rare) alleles involved in GC gaining and GC losing as a function of ToR. Shown are frequency of low frequency GC gaining alleles (dashed line) and GC losing alleles (solid line) for the whole intergenic genome (left) and for functionally masked regions (right) vs. ToR in limphoblasts (molt4) (D), embryonic stem cells (BG02) (E) and in limphoblasts (molt4) when restricting to genomic regions with constitutive ToR as defined in Rivera-Mulia et al (29). (F) All statistics are shown for 5 equally sized ToR percentile bins (0, 0.2, 0.4, 0.6, 0.8, 1.0). Error bars represent binomial confidence interval with 95% significance.
Human heterozygosity data (35) were downloaded from http://hapmap.ncbi.nlm.nih.gov/downloads/genotypes. GC gain SNPs were defined as loci where the major allele is A/T and the minor allele is G/C. Similarly GC loss SNPs were defined as SNPs with major allele of G/C and minor allele is A/T. Low heterozygosity SNPs were defined as SNPs with heterozygous frequency <0.15 which is equivalent to derived allele frequency of ∼0.08.
Cell culture
L1210 lymphocytic leukemia cells (ATCC CCL219) were cultured in CO2-independent L-15 medium (BI) supplemented with Glucose (Sigma), penicillin, streptomycin, L-Glutamine, sodium pyruvate and 10% v/v heat-inactivated (56°C, 30 min) FBS (BI). Balb/3T3 (American Type Culture Collection number CCL-163) cells were cultured in DMEM (Sigma) supplemented with penicillin, streptomycin and 10% v/v heat-inactivated (56°C, 30 min) horse serum (Life Technologies, Inc.)
Cell cycle synchronization and dNTP measurements
L1210 cells were synchronized using the ‘baby machine’ device, newborn cells were isolated from the ‘baby machine’, as previously described in (36). A fraction of every experiment was stained with PI and scanned using FACScan (BD) to validate synchronization level. 3T3 cells were synchronized by serum starvation as described in (37). Synchronization was checked using CCA-l flow cytometer according to the instructions of the manufacturer (Partec GmbH). dNTPs were extracted as previously described in (38). Separation and quantification of dNTPs and NTPs employing HPLC were carried out as described in (39). The experiments were repeated two and five times for 3T3 and L1210, respectively.
RESULTS
GC content is correlated with replication dynamics at multiple scales
Replication dynamics is known to be correlated with GC content at scales ranging from hundreds of KB to 1 MB (Figure 1A,(1,20,40)) but less is known about its association with the fine-grained variation of GC content visible on scales of 1–10 kb (Supplementary Figure S1A). To characterize GC content variation simultaneously at multiple genomic scales we computed and visualized domainograms (41) summarizing the distribution of GC content in scales varying between 1 kb and 4 Mb using vertically stacked color-coded bands (Figure 1B and Supplementary Figure S1B–D). This analysis showed that while the different scales are correlated, the GC content of many small genomic regions is opposing to the trend of the large-scale domains in which they reside. On the other hand, domainograms from data on ToR (27) (Figure 1C and Supplementary Figure S1B–D, red—early replicating, blue—late replicating regions) demonstrated that replication dynamics variation is observed on 100 kb or higher scales, partly due to experimental reasons, but mostly due to the biological mechanisms involved.
Figure 1.

Decoupling multi-scale correlations between GC content, time of replication (ToR) and functional elements. (A) Correlation between GC content and ToR at large genomic scale. Shown is a density plot of GC versus ToR binned for 200 kb bins. (B and C) Multi-scale visualization of GC content and ToR. Domainograms of GC content (B) and ToR (C) across a section of chromosome 3. For every genomic coordinate (x-axis), shown are color-coded (blue–low/ late, red–high/early) averaged values over surrounding windows at multiple scales (y-axis), ranging from 1 to 2048 kb. (D) Correlation between GC content and ToR at a small genomic scale. Shown is a density plot of GC versus ToR binned for 2 kb bins. (E) Multi-scale visualization of functional density. Similarly to B and C shown is domainogram of functional density at multiple scales (y-axis) across the same genomic section (x-axis) (F) GC Correlation versus scale. Shown are Spearman correlations (y-axis) between GC content and ToR (black) or between GC content and functional elements density (red) at different genomic scales (x-axis) (G) genomic GC content versus ToR. Boxplot visualization of the distribution of genomic (All) and masked genomic (Masked) GC content (100 kb) versus ToR.
As expected (42), other large-scale epigenomic features, including Lamina proximity (Supplementary Figure S2A and B), repressive histone modification density (Supplementary Figure S2C and D), and smaller scale nucleosome occupancy (Supplementary Figure S2E and F) showed trends similar to replication dynamics. The multiscale correlation between the different epigenomic features, and the multiscale nature of GC content heterogeneity in the genome should be interpreted carefully, since trends in one scale can residually be observed in smaller or larger scales (Figure 1D). Identification of the evolutionary or mechanistic forces underlying those multiscale correlations must involve separation of scales. For example, variation in larger scales (replication domains, topological domains) must be stratified when analyzing the factors affecting smaller scales (functional elements, exons). Similarly, the cumulative effects of local elements must be controlled for when testing larger scale trends. This prompted us to develop statistical approaches for dissecting local from global GC content variation using additional genomic and epigenomic annotations.
Detailed maps of functional elements separate local from global GC content variation
We used genome-wide maps of exons and putative local functional elements as derived by the ENCODE project (15) in an attempt to separate local variation in GC content from the megabase scale effects described above. We identified one million elements with either DNase-I hypersensitivity or significant enrichment in enhancer-linked histone marks, covering 416 Mb in total with elements ranging in size between 50 and 900 bps. We combined these genomic regions with documented exons (235000 elements covering 74 Mb), and used the resulting labels to compute the approximate density of functionally constrained sequences over the entire genome. Multi-scale visualization of this density track (Figure 1E and Supplementary Figure S3A–C) suggested that functional density is heterogeneous at multiple scales (Supplementary Figure S3D), and is strongly correlated, as was previously shown (1,20,21,28) with both GC content and ToR (Figure 1F and Supplementary Figure S3E–G). Systematic analysis of the correlation between ToR or functional density with GC content in scales ranging from 200 bp to 5 Mbp (Figure 1F) indicated that although both features are highly linked with GC content (spearman rho >0.8), the correlation with functional density is observed at much smaller scales than the correlation with time of replication. Given that ToR and functional density are also highly correlated with each other on the MB scale (Supplementary Figure S3E and F), we next wished to understand if the association between ToR and GC will be preserved even when functional elements are controlled for.
We computed the GC content distribution in genomic regions classified according to their ToR, with and without excluding an extended set of the sequences that we permissively associated with putative function (3.2 M elements, covering a total of 1.1 Gb). We also filtered all repeat-masked sequences to allow further evolutionary analysis downstream (35 million elements covering a total of 1782 MB) (Supplementary Figure S3G). As shown in Figure 1G, when working in the 100 kbp scale, we detected significant decrease in GC content for late replicating sequences even when masking functional elements, (medians 0.326 and 0.338 for the 0–20 and 20–40 percentile ranges, P << 10−10, Mann–Whitney). On the other hand, the increase in GC content for early replicating regions was softened significantly following functional masking, (medians of masked GC increases in 3.2 instead of 6.6% without masking between the median of the 80–100 percentiles and the 60–80 percentiles, P << 10−10, KS test). Further analysis showed that the correlation between masked GC and ToR remain robust when stratifying on nucleosome occupancy levels (Supplementary Figure S3H). In summary, following filtering of putative functional elements, the correlation between ToR and GC content remains highly significant (Supplementary Figure S3I), but its structure is altered and can be attributed mainly to links between late replicating regions and low GC content.
GC content at function-masked sequences is associated with chromatin organization
The size of replication domains is variable, hence any selection of a fixed genomic scale (i.e. fixed genomic bins) may introduce biases when analyzing the relation between ToR and other attributes. To study large-scale GC content, ToR and functional densities further without assuming an arbitrary genomic scale, we renormalized Hi-C data and identified topologically associated domains (TADs) from chromosomal contacts map of IMR90 cells (23) (Supplementary Figure S4A). These domains represent coherent and large-scale building blocks of mammalian chromosomes (22,23) and therefore provide a natural framework for studying large-scale genomic effects. Analysis of the correspondence between ToR, GC content and functional density across Hi-C domains reconfirms that late replicating domains are associated with lower GC content, even when functional elements are masked (Figure 2A and B). On the other hand, the observation of higher GC content in early replicating domains is dependent on contributions from local functional elements that are more abundant in such domains.
Figure 2.

GC and time of replication of topological domains (A) Correlation between GC content and ToR binned by Hi-C domains. Shown is a density plot of GC versus ToR over chromosomal domains. (B) GC content versus ToR binned by Hi-C domains. Boxplot visualization of the distribution of genomic (All) and masked genomic (Masked) GC content versus ToR across chromosomal domains. (C) Hi-C map and projected GC profiles. Chromatin interaction intensity matrix around late replicating domain of chromosome 2 is shown (bottom, strong interaction levels—orange, weak interaction levels—blue) with linear profiles of functional density, ToR, GC content and inferred evolutionary rates of GC gain and GC loss that correspond to the same genomic section (top).
Visualization of Hi-C contact maps around repressed chromosomal domains together with projected profiles of ToR and masked GC content (Figure 2C and Supplementary Figure S4B) showed that as was suggested before, repressive domains are of typically low GC content and late replication time (42–44). Interestingly, while chromosomal domains are sharply defined by Hi-C, ToR and GC content decrease gradually over hundreds of kilobases into the repressed domain. Averaged profiles of GC, masked GC and ToR show coupling of the decrease in masked GC within the repressive domain (∼33.6% to ∼32.6% over ∼600kb) and a gradual transition of ToR (Supplementary Figure S5A–F). The relative uniform ToR within chromosomal domains and the gradual change in the ToR between the domains is typical to the replication program which is a patchwork of constant ToR regions (CTRs) that are connected by temporal transition regions (TTRs) (19,28,45,46). Analysis of CTRs and TTRs (Supplementary Figure S6), as well as study of CTCF bindings that demarcate domain borders (Supplementary Figure S7A), indicate that the correlation between ToR and masked GC content holds at both constant and non-constant ToR regions.
Genomic elements located away from active replication origins should show a gradual change in their ToR, merely due to the time the replication process takes, thus in order to further study the spatial association between ToR and GC content, we re-analyzed data of origins of replication inferred from Orc1 Chip-seq or bubble-seq experiments (30,31). We stratified sites according to their minimal distance to an implicated replication origin and studied the average GC content in each stratum. Similar to repressive domain borders, time of replication together with GC content and masked GC content decrease as the distance to Orc1/bubble-seq peaks increase (Supplementary Figure S7B and C). Interestingly, similar coupling is observed when computing the decrease of masked GC content around DNase I hypersensitive sites (DHSs) (Supplementary Figure S7D), probably reflecting the tight connection between DNA replication origins and DNA accessibility (47). These results suggest that the link between the ToR and the GC content exists even in regions in which the ToR depends most probably only on its distance from an origin of replication. In conclusion, sites of genomic transition such as borders of topological domains, or loci with gradual, or origin-like replication dynamics support the direct linkage between late ToR and decreased GC content. The increase in GC content in early replicating domains, on the other hand, is tightly correlated with higher density of functional elements and not neccesarily ToR.
Neutral evolutionarily regimes are likely drivers of GC content decrease in late replicating domains
The evolutionary processes underlying the emergence and maintenance of variable GC content may involve different forces. All of these must however end up affecting the rate of substitutions gaining and losing GC nucleotides. Analysis of the context-dependent substitution spectrum over non-CpG contexts (34) in Hi-C domain borders (Figure 2C and Supplementary Figure S5) and globally across genomic regions stratified according to ToR (27) showed that the rate of both GC gain and GC loss are higher in late replicating sequences (Figure 3A and B). This effect could not be explained by higher frequencies of deamination of methylated CpGs in late replicating regions (Supplementary Figure S8A) since this nucleotide context was excluded from the analysis. This result holds even when filtering putative functional elements. This increase, however, is mostly observed in the lower 40 percentiles of the ToR distribution, whether masking of functional elements is applied or not. Interestingly, the rate of GC loss is approximately two times higher than the rate of GC gain substitutions, and combining both substitution processes together result in a predicted potential bias toward decreasing GC content in late replicating regions (Figure 3C and Supplementary Figure S8B).
We next wished to further investigate the source of the above replication linked substitution regimes. Is it a result of selective forces, evolutionary dynamics with selection-like properties such as biased gene conversion, or a consequence of changes in the mutational spectrum itself? To differentiate these scenarios, we analyzed SNP heterozygosity data, quantifying the frequency of low-allele frequency SNPs gaining or losing GC nucleotides in different time of replication regimes. When such analysis is performed without masking functional sequences (Figure 3D–F and Supplementary Figure S8C–I, left), a clear trend separating derived alleles losing or gaining GC is observed. Notably, such difference occurs almost only in early replicating regions, whereas in late replicating regions there is no difference. Selection and biased gene conversion are both predicted to change the allele frequency spectrum in affected loci. Thus, our observation for the early replicating regions confirms previous reports (10–13,48). Indeed, functional masking, eliminates this trend (Figure 3D–F and Supplementary Figure S8C–I, right). On the other hand, late replicating domains show little difference in the rare allele frequency between alleles losing or gaining GC. This is even more pronounced in the masked genome in which no difference was observed. Thus, our data suggest that selection or biased gene conversion are not the major forces driving changes in the balance between GC gaining and GC losing substitutions at late replicating sequences (Figure 3A–C). Neutral effects, and most notably changes in the mutation spectrum that are linked with replication dynamics therefore emerge as key factors shaping ToR-linked variation in GC content in late replicating regions.
Changes in dNTP pool along the cell cycle
Changes in the mutation spectrum between early and late replicating regions suggest that in germline cells there are differences along S phase in either replication errors or their repair. One potential source for differential replication errors is a change in the relative levels of dNTPs along S. We hypothesized that if regions that replicate at different times are exposed to different environments and in particular to variable dNTP concentrations, they may accumulate mutations at different rates (49,50). Indeed, it is well established that imbalanced dNTP pool affects mutation types and rate both in vitro and in vivo (50,51). In order to explore if this can be a potential explanation for the differences in the mutation spectrum along S, we decided to measure the dNTPs pool along the cell cycle in two types of tissue cultured cells that can be easily synchronized without affecting directly dNTP pool levels. To this end, we synchronized L1210 cells using ‘baby machine’ (36) and applied serum starvation for synchronizing BALB\3T3 cells. We then isolated nucleotides and measured dNTP concentrations. As have been shown previously (reviewed in Mathews et al. (52,53)), the four dNTPs are present in vivo at different concentrations, with dGTP concentration much lower than the others. However, we also found that in both cell lines examined, dATP and dTTP concentrations increase along S phase whereas dGTP concentration was uniform and dCTP concentration was either uniform (L1210) or slightly decreased (BALB\3T3) along S phase (Figure 4 and Supplementary Figure S9 and 10). In spite of the difference between the two cell lines in dCTP concentration, both demonstrate an increase in the AT to GC ratio during S phase progression. These data demonstrate that the ratio between AT to GC deoxynucleotides may vary significantly (up to 2-fold) along S phase at least in some tissue culture cell types, and therefore may also occur in the germline. Such changes in the dNTP pool can affect ToR-linked mutation bias and even small differences in the mutation spectrum may lead to large differences in the GC content during evolution.
Figure 4.

dNTPs ratio change along S phase. (A and B) The individual change of each dNTP along S phase. L1210 cells (A) were synchronized by Baby Machine, 3T3 cells (B) were synchronized by serum starvation. dNTPs and NTPs were extracted from synchronized cells in multiple points along S phase (the precise time points for early, middle and late S phase were determined according to FACS analysis–Supplementary Figures S9 and S10) and were measured using HPLC. dNTP Quantities were normalized to the total NTP measurements. The averages and standard errors of 2 to 5 independent measurements are shown. (C and D) The dA and dT fraction in the dNTP pool elevates with S phase progression. (dA+dT) to (dG+dC) ratio and standard error at multiple time points in S phase are shown for L1210 cells (C) and 3T3 cells (D). Ratios were calculated from normalized dNTP amounts shown in A and B. Note that the main difference between the two cell lines is in the levels of dCTPs. We do not know the source of this difference.
DISCUSSION
Different models exist for the formation of genomic GC variation, In the last few years most of the findings support the gBGC as being the main process affecting GC content evolution in mammals (10–14,48). Mutation bias and selection are also playing a role (54) and new approaches are needed to understand the full complexity and the interplay between the different evolutionary forces that shape the genomic GC composition. Stratifying to ToR regions enabled us to reveal the major role of mutation bias on late replicating regions.
We present data that support a model for the emergence of GC content variation in mammalian genomes through interplay between local and global evolutionary forces. First, we describe in detail the correspondence between ToR and different scales of GC content in the genome. We show that small scale GC content increase around functional elements, can explain the increase in GC content at early replicating domains, but not the decrease in GC content at late replicating domains. Furthermore, we compute rates of GC gaining and GC losing substitutions and compare them to allele frequency data at GC gaining and GC losing SNPs. This enables us to distinguish selection or biased gene conversion effects on GC content, from neutral and mutation-spectrum linked effects. This analysis shows that functional elements are characterized by substitution dynamics and allele frequencies that indicate selection or gBGC, while the non-functional part of the genome is associated with substitution rate changes but no GC-related changes in allele frequencies. According to the data, changes in the mutation spectrum, are the most likely drivers of the shift in the balance between GC gaining and losing substitution rates that we observed at late replicating regions and repressed topological domains.
What can cause changes in the mutation spectrum? One possibility is that changes in the dNTP pool along S phase may affect the mutation spectrum. For example, relatively higher levels of A and T at late S phase will support C/G to A/T mutations which may cause a gradual shift toward lower GC content. Accumulation of mutations along the evolutionary time scale can lead to large changes in GC content between early and late replicating regions. Indeed, it has been shown that induced imbalanced dNTP pool affects mutation types and rate both in vitro (51) and in vivo (50). Here, by measuring the differential dNTP pool along the S phase in somatic tissues, we observed cell cycle dependent changes in dNTP concentrations. Although the mechanism that governs the change in dATP and dTTP concentrations along S phase is not known yet, its relevance to ToR-coupled mutagenesis can be hypothesized given that there is no excess of dNTPs during the replication process (55,56), and occasional replication errors may be biased according to the available free dNTP pool. Late replicating regions are rich in AT and thus at late S phase, the replication process consumes higher amounts of dATP and dTTP. The dNTP pool is probably regulated to account for the differential need of deoxynucleotides in early and late regions, which may ultimately cause the observed mutation bias. Thus, if the observed changes in dNTP pool along S phase exist also in the germ line, it may lead to changes in the mutation spectrum between early and late replicating regions. Even a small imbalance of the dNTP pool may lead to accumulation of a nucleotide bias in the genome over evolutionary time scale.
Maintenance of the GC skew between early and late replication domains is dependent on mutation rate in the germline. However, due to experimental limitations, we used ToR measurements of multiple somatic tissues (Supplementary Figure S8B–F) but not of germline cells. Finding similar results at all examined tissues strongly suggests that our findings are general and will hold true also for germline ToR data once it will become available. Moreover, restricting our analyses to genomic regions that have a constitutive ToR over 26 distinct human cell types (29) and therefore most probably have the same ToR in germ line tissues, revealed the same (Supplementary Figure S8B and Figure 3F). Similarly, the dNTP pool measurements were carried out in mouse somatic tissue cultured cells (due to the inaccessibility of large amounts of germ line tissues and due to the need to synchronize the cells without perturbing the dNTP pool) that may be different from the human germline. Recent data on ToR in different tissues (29) show that genomic regions that switch their replication time between tissues have more variable GC content than loci with constitutive ToR. One can hypothesize that this may be due to inconsistency with the replication time in the germ line. Taken together, although our dNTP measurements are still far from demonstrating the full mechanism that leads to the ToR-dependent changes in the mutation spectrum, they do provide a possible explanation that was ignored so far. The regulation of the dNTP pool and replication program must therefore be further studied both in germline cells and in cancer and aging tissues of different species in order to understand the scope of this effect.
We have observed a difference in the dNTP concentrations between the two cell lines examined. The differences are especially pronounced for dCTP but exist also for other nucleotides (Figure 4). Such variations are not surprising and were observed before. Indeed a compilation of all published data about the physiological concentrations of purines and pyrimidines (53), revealed big changes in dNTP concentration in different cell lines. Interestingly, similarly to our results, dCTP concentration was found to be the most variant (approximately 500-folds). The variation in the baseline levels of each nucleotide is probably a consequence of differences between cell types in the efficiency of the salvage pathway. Regardless of the individual variation when looking at the AT to GC ratio one can observe an increase along S in both cell types.
The linkage between ToR and the rates and spectrum of mutations in mammals was recently discussed in several contexts, including cancer (57–60). Here we characterize the multi-scale links between mutation GC bias and ToR and suggest that the implications of such effects on the germ line can explain a significant component of the genomic compositional heterogeneity at the large scale. Somatic cells are also likely to accumulate mutations in a replication-time coupled way, and since large-scale chromosomal organization and functional densities are tightly correlated with the replication process, the mutation spectrum is also expected to be correlated with such functional characteristics of the genome. Intriguingly, the long-term evolutionary effect of this mutational process make different genomic domains highly distinct in their sequence composition, and while our data suggest that the origin of these differences is most likely neutral (or almost neutral), it is far from clear how their accumulation affects genome structure and function. For example, GC rich sequences that evolve at near neutrality in early replicating domains have higher spontaneous rates of generating functional sequences (e.g. binding sites of transcription factors within enhancers) on evolutionary time scale than GC poor sequences in late replicating domains. This can lead to demarcation of the genome in the long run, and support processes of genomic compartmentalization as suggested by us and others (18). As more and more accurate maps of both local and global chromosomal organization are becoming available in multiple species, it can be hoped that more detailed understanding of these processes will become feasible.
Supplementary Material
Acknowledgments
We thank Dr Shlomit Farkash-Amar and Yaara David for assistance with ARTO. Eitan Yaffe and Netta Mendelson-Cohen for help with Hi-C and evolutionary analysis. Dr Dan Lehmann for help in the FACS, Rivera-Mulia and David Gilbert for sharing data and Kenas Wiscott for technical assistance.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Israel Science Foundation [1050/12]; European Research Council [EVOEPIC]; Helen and Martin Kimmel Award; Knut and Alice Wallenberg Foundation; Swedish Cancer Society; Israel Science Foundation [567/10]; European Research Council Starting Grant [281306]. Funding for open access charge: European Research Council Starting Grant [281306].
Conflict of interest statement. None declared.
REFERENCES
- 1.Woodfine K., Fiegler H., Beare D.M., Collins J.E., McCann O.T., Young B.D., Debernardi S., Mott R., Dunham I., Carter N.P. Replication timing of the human genome. Hum. Mol. Genet. 2004;13:191–202. doi: 10.1093/hmg/ddh016. [DOI] [PubMed] [Google Scholar]
- 2.Bernardi G., Olofsson B., Filipski J., Zerial M., Salinas J., Cuny G., Meunier-Rotival M., Rodier F. The mosaic genome of warm-blooded vertebrates. Science. 1985;228:953–958. doi: 10.1126/science.4001930. [DOI] [PubMed] [Google Scholar]
- 3.Wolfe K.H., Sharp P.M., Li W.H. Mutation rates differ among regions of the mammalian genome. Nature. 1989;337:283–285. doi: 10.1038/337283a0. [DOI] [PubMed] [Google Scholar]
- 4.Arndt P.F., Hwa T., Petrov D.A. Substantial regional variation in substitution rates in the human genome: importance of GC content, gene density, and telomere-specific effects. J. Mol. Evol. 2005;60:748–763. doi: 10.1007/s00239-004-0222-5. [DOI] [PubMed] [Google Scholar]
- 5.Galtier N., Piganeau G., Mouchiroud D., Duret L. GC-content evolution in mammalian genomes: the biased gene conversion hypothesis. Genetics. 2001;159:907–911. doi: 10.1093/genetics/159.2.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Duret L., Galtier N. Biased gene conversion and the evolution of mammalian genomic landscapes. Ann. Rev. Genom. Hum. Genet. 2009;10:285–311. doi: 10.1146/annurev-genom-082908-150001. [DOI] [PubMed] [Google Scholar]
- 7.Smith N.G., Webster M.T., Ellegren H. Deterministic mutation rate variation in the human genome. Genome Res. 2002;12:1350–1356. doi: 10.1101/gr.220502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eyre-Walker A., Hurst L.D. The evolution of isochores. Nat. Rev. Genet. 2001;2:549–555. doi: 10.1038/35080577. [DOI] [PubMed] [Google Scholar]
- 9.Chamary J.V., Parmley J.L., Hurst L.D. Hearing silence: non-neutral evolution at synonymous sites in mammals. Nat. Rev. Genet. 2006;7:98–108. doi: 10.1038/nrg1770. [DOI] [PubMed] [Google Scholar]
- 10.Clement Y., Arndt P.F. Meiotic recombination strongly influences GC-content evolution in short regions in the mouse genome. Mol. Biol. Evol. 2013;30:2612–2618. doi: 10.1093/molbev/mst154. [DOI] [PubMed] [Google Scholar]
- 11.Glemin S., Arndt P.F., Messer P.W., Petrov D., Galtier N., Duret L. Quantification of GC-biased gene conversion in the human genome. Genome Res. 2015;25:1215–1228. doi: 10.1101/gr.185488.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kostka D., Hubisz M.J., Siepel A., Pollard K.S. The role of GC-biased gene conversion in shaping the fastest evolving regions of the human genome. Mol. Biol. Evol. 2012;29:1047–1057. doi: 10.1093/molbev/msr279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Katzman S., Capra J.A., Haussler D., Pollard K.S. Ongoing GC-biased evolution is widespread in the human genome and enriched near recombination hot spots. Genome Biol. Evol. 2011;3:614–626. doi: 10.1093/gbe/evr058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Capra J.A., Hubisz M.J., Kostka D., Pollard K.S., Siepel A. A model-based analysis of GC-biased gene conversion in the human and chimpanzee genomes. PLoS Genet. 2013;9:e1003684. doi: 10.1371/journal.pgen.1003684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bernstein B.E., Birney E., Dunham I., Green E.D., Gunter C., Snyder M. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lindblad-Toh K., Garber M., Zuk O., Lin M.F., Parker B.J., Washietl S., Kheradpour P., Ernst J., Jordan G., Mauceli E., et al. A high-resolution map of human evolutionary constraint using 29 mammals. Nature. 2011;478:476–482. doi: 10.1038/nature10530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bickmore W.A., van Steensel B. Genome architecture: domain organization of interphase chromosomes. Cell. 2013;152:1270–1284. doi: 10.1016/j.cell.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 18.Tanay A., Cavalli G. Chromosomal domains: epigenetic contexts and functional implications of genomic compartmentalization. Curr. Opin. Genet. Dev. 2013;23:197–203. doi: 10.1016/j.gde.2012.12.009. [DOI] [PubMed] [Google Scholar]
- 19.Pope B.D., Ryba T., Dileep V., Yue F., Wu W., Denas O., Vera D.L., Wang Y., Hansen R.S., Canfield T.K., et al. Topologically associating domains are stable units of replication-timing regulation. Nature. 2014;515:402–405. doi: 10.1038/nature13986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Farkash-Amar S., Lipson D., Polten A., Goren A., Helmstetter C., Yakhini Z., Simon I. Global organization of replication time zones of the mouse genome. Genome Res. 2008;18:1562–1570. doi: 10.1101/gr.079566.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hiratani I., Ryba T., Itoh M., Yokochi T., Schwaiger M., Chang C.W., Lyou Y., Townes T.M., Schubeler D., Gilbert D.M. Global reorganization of replication domains during embryonic stem cell differentiation. PLoS Biol. 2008;6:e245. doi: 10.1371/journal.pbio.0060245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sexton T., Yaffe E., Kenigsberg E., Bantignies F., Leblanc B., Hoichman M., Parrinello H., Tanay A., Cavalli G. Three-dimensional folding and functional organization principles of the Drosophila genome. Cell. 2012;148:458–472. doi: 10.1016/j.cell.2012.01.010. [DOI] [PubMed] [Google Scholar]
- 23.Dixon J.R., Selvaraj S., Yue F., Kim A., Li Y., Shen Y., Hu M., Liu J.S., Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–380. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dileep V., Ay F., Sima J., Vera D.L., Noble W.S., Gilbert D.M. Topologically associating domains and their long-range contacts are established during early G1 coincident with the establishment of the replication-timing program. Genome Res. 2015;8:1104–1113. doi: 10.1101/gr.183699.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dreszer T.R., Wall G.D., Haussler D., Pollard K.S. Biased clustered substitutions in the human genome: the footprints of male-driven biased gene conversion. Genome Res. 2007;17:1420–1430. doi: 10.1101/gr.6395807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Karolchik D., Barber G.P., Casper J., Clawson H., Cline M.S., Diekhans M., Dreszer T.R., Fujita P.A., Guruvadoo L., Haeussler M., et al. The UCSC Genome Browser database: 2014 update. Nucleic Acids Res. 2014;42:D764–D770. doi: 10.1093/nar/gkt1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yaffe E., Farkash-Amar S., Polten A., Yakhini Z., Tanay A., Simon I. Comparative analysis of DNA replication timing reveals conserved large-scale chromosomal architecture. PLoS Genet. 2010;6:e1001011. doi: 10.1371/journal.pgen.1001011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hansen R.S., Thomas S., Sandstrom R., Canfield T.K., Thurman R.E., Weaver M., Dorschner M.O., Gartler S.M., Stamatoyannopoulos J.A. Sequencing newly replicated DNA reveals widespread plasticity in human replication timing. Proc. Natl. Acad. Sci. U S A. 2010;107:139–144. doi: 10.1073/pnas.0912402107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rivera-Mulia J.C., Buckley Q., Sasaki T., Zimmerman J., Didier R.A., Nazor K., Loring J.F., Lian Z., Weissman S.M., Robins A.J., et al. Dynamic changes in replication timing and gene expression during lineage specification of human pluripotent stem cells. Genome Res. 2015;8:1103–1091. doi: 10.1101/gr.187989.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dellino G.I., Cittaro D., Piccioni R., Luzi L., Banfi S., Segalla S., Cesaroni M., Mendoza-Maldonado R., Giacca M., Pelicci P.G. Genome-wide mapping of human DNA-replication origins: levels of transcription at ORC1 sites regulate origin selection and replication timing. Genome Res. 2013;23:1–11. doi: 10.1101/gr.142331.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mesner L.D., Valsakumar V., Cieslik M., Pickin R., Hamlin J.L., Bekiranov S. Bubble-seq analysis of the human genome reveals distinct chromatin-mediated mechanisms for regulating early- and late-firing origins. Genome Res. 2013;23:1774–1788. doi: 10.1101/gr.155218.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gaffney D.J., McVicker G., Pai A.A., Fondufe-Mittendorf Y.N., Lewellen N., Michelini K., Widom J., Gilad Y., Pritchard J.K. Controls of nucleosome positioning in the human genome. Plos Genetics. 2012;8:1003036. doi: 10.1371/journal.pgen.1003036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yaffe E., Tanay A. Probabilistic modeling of Hi-C contact maps eliminates systematic biases to characterize global chromosomal architecture. Nat. Genet. 2011;43:1059–1065. doi: 10.1038/ng.947. [DOI] [PubMed] [Google Scholar]
- 34.Cohen N.M., Kenigsberg E., Tanay A. Primate CpG islands are maintained by heterogeneous evolutionary regimes involving minimal selection. Cell. 2011;145:773–786. doi: 10.1016/j.cell.2011.04.024. [DOI] [PubMed] [Google Scholar]
- 35.International HapMap Consortium. The International HapMap Project. Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 36.Thornton M., Eward K.L., Helmstetter C.E. Production of minimally disturbed synchronous cultures of hematopoietic cells. Biotechniques. 2002;32:1098–1105. doi: 10.2144/02325rr05. [DOI] [PubMed] [Google Scholar]
- 37.Chabes A., Thelander L. Controlled protein degradation regulates ribonucleotide reductase activity in proliferating mammalian cells during the normal cell cycle and in response to DNA damage and replication blocks. J. Biol. Chem. 2000;275:17747–17753. doi: 10.1074/jbc.M000799200. [DOI] [PubMed] [Google Scholar]
- 38.Chabes A., Georgieva B., Domkin V., Zhao X., Rothstein R., Thelander L. Survival of DNA damage in yeast directly depends on increased dNTP levels allowed by relaxed feedback inhibition of ribonucleotide reductase. Cell. 2003;112:391–401. doi: 10.1016/s0092-8674(03)00075-8. [DOI] [PubMed] [Google Scholar]
- 39.Hofer A., Ekanem J.T., Thelander L. Allosteric regulation of Trypanosoma brucei ribonucleotide reductase studied in vitro and in vivo. J. Biol. Chem. 1998;273:34098–34104. doi: 10.1074/jbc.273.51.34098. [DOI] [PubMed] [Google Scholar]
- 40.Schubeler D., Scalzo D., Kooperberg C., van Steensel B., Delrow J., Groudine M. Genome-wide DNA replication profile for Drosophila melanogaster: a link between transcription and replication timing. Nat. Genet. 2002;32:438–442. doi: 10.1038/ng1005. [DOI] [PubMed] [Google Scholar]
- 41.de Wit E., Braunschweig U., Greil F., Bussemaker H.J., van Steensel B. Global chromatin domain organization of the Drosophila genome. PLoS Genet. 2008;4:e1000045. doi: 10.1371/journal.pgen.1000045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rhind N., Gilbert D.M. DNA Replication Timing. Cold Spring Harb. Perspect. Med. 2013;3:1–26. doi: 10.1101/cshperspect.a010132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moindrot B., Audit B., Klous P., Baker A., Thermes C., de Laat W., Bouvet P., Mongelard F., Arneodo A. 3D chromatin conformation correlates with replication timing and is conserved in resting cells. Nucleic Acids Res. 2012;40:9470–9481. doi: 10.1093/nar/gks736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baker A., Audit B., Chen C.L., Moindrot B., Leleu A., Guilbaud G., Rappailles A., Vaillant C., Goldar A., Mongelard F., et al. Replication fork polarity gradients revealed by megabase-sized U-shaped replication timing domains in human cell lines. PLoS Comput. Biol. 2012;8:e1002443. doi: 10.1371/journal.pcbi.1002443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Norio P., Kosiyatrakul S., Yang Q., Guan Z., Brown N.M., Thomas S., Riblet R., Schildkraut C.L. Progressive activation of DNA replication initiation in large domains of the immunoglobulin heavy chain locus during B cell development. Mol. Cell. 2005;20:575–587. doi: 10.1016/j.molcel.2005.10.029. [DOI] [PubMed] [Google Scholar]
- 46.Farkash-Amar S., David Y., Polten A., Hezroni H., Eldar Y.C., Meshorer E., Yakhini Z., Simon I. Systematic determination of replication activity type highlights interconnections between replication, chromatin structure and nuclear localization. PLoS One. 2012;7:e48986. doi: 10.1371/journal.pone.0048986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gindin Y., Valenzuela M.S., Aladjem M.I., Meltzer P.S., Bilke S. A chromatin structure-based model accurately predicts DNA replication timing in human cells. Mol. Syst. Biol. 2014;10:722. doi: 10.1002/msb.134859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Capra J.A., Pollard K.S. Substitution patterns are GC-biased in divergent sequences across the metazoans. Genome Biol. Evol. 2011;3:516–527. doi: 10.1093/gbe/evr051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mathews C.K. Deoxyribonucleotides as genetic and metabolic regulators. FASEB J. 2014;9:3832–3840. doi: 10.1096/fj.14-251249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kumar D., Abdulovic A.L., Viberg J., Nilsson A.K., Kunkel T.A., Chabes A. Mechanisms of mutagenesis in vivo due to imbalanced dNTP pools. Nucleic Acids Res. 2011;39:1360–1371. doi: 10.1093/nar/gkq829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kunz B.A., Kohalmi S.E., Kunkel T.A., Mathews C.K., McIntosh E.M., Reidy J.A. International Commission for Protection Against Environmental Mutagens and Carcinogens. Deoxyribonucleoside triphosphate levels: a critical factor in the maintenance of genetic stability. Mutat. Res. 1994;318:1–64. doi: 10.1016/0165-1110(94)90006-x. [DOI] [PubMed] [Google Scholar]
- 52.Mathews C.K., Ji J. DNA precursor asymmetries, replication fidelity, and variable genome evolution. Bioessays. 1992;14:295–301. doi: 10.1002/bies.950140502. [DOI] [PubMed] [Google Scholar]
- 53.Traut T.W. Physiological concentrations of purines and pyrimidines. Mol. Cell. Biochem. 1994;140:1–22. doi: 10.1007/BF00928361. [DOI] [PubMed] [Google Scholar]
- 54.De Maio N., Schlotterer C., Kosiol C. Linking great apes genome evolution across time scales using polymorphism-aware phylogenetic models. Mol. Biol. Evol. 2013;30:2249–2262. doi: 10.1093/molbev/mst131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Poli J., Tsaponina O., Crabbe L., Keszthelyi A., Pantesco V., Chabes A., Lengronne A., Pasero P. dNTP pools determine fork progression and origin usage under replication stress. EMBO J. 2012;31:883–894. doi: 10.1038/emboj.2011.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nicander B., Reichard P. Dynamics of pyrimidine deoxynucleoside triphosphate pools in relationship to DNA synthesis in 3T6 mouse fibroblasts. Proc. Natl. Acad. Sci. U S A. 1983;80:1347–1351. doi: 10.1073/pnas.80.5.1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stamatoyannopoulos J.A., Adzhubei I., Thurman R.E., Kryukov G.V., Mirkin S.M., Sunyaev S.R. Human mutation rate associated with DNA replication timing. Nat. Genet. 2009;41:393–395. doi: 10.1038/ng.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Woo Y.H., Li W.H. DNA replication timing and selection shape the landscape of nucleotide variation in cancer genomes. Nat. Commun. 2012;3:1004. doi: 10.1038/ncomms1982. [DOI] [PubMed] [Google Scholar]
- 59.Liu L., De S., Michor F. DNA replication timing and higher-order nuclear organization determine single-nucleotide substitution patterns in cancer genomes. Nat. Commun. 2013;4:1502. doi: 10.1038/ncomms2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen C.L., Rappailles A., Duquenne L., Huvet M., Guilbaud G., Farinelli L., Audit B., d'Aubenton-Carafa Y., Arneodo A., Hyrien O., et al. Impact of replication timing on non-CpG and CpG substitution rates in mammalian genomes. Genome Res. 2010;20:447–457. doi: 10.1101/gr.098947.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.