

Abstract
Introduction
Despite increased screening rates and advances in targeted therapy, colorectal cancer (CRC) remains the third leading cause of cancer-related mortality. CRC models that recapitulate key features of human disease are essential to the development of novel and effective therapeutics. Classic methods of modeling CRC such as human cell lines and xenograft mice, while useful for many applications, carry significant limitations. Recently developed in vitro and in vivo models overcome some of these deficiencies and thus can be utilized to better model CRC for mechanistic and translational research.
Areas Covered
The authors review established models of in vitro cell culture and describe advances in organoid culture for studying normal and malignant intestine. They also discuss key features of classic xenograft models and describe other approaches for in vivo CRC research, including patient-derived xenograft, carcinogen-induced, orthotopic transplantation, and transgenic mouse models. We also describe mouse models of metastatic CRC.
Expert opinion
No single model is optimal for drug discovery in CRC. Genetically engineered models overcome many limitations of xenograft models. Three-dimensional organoids can be efficiently derived from both normal and malignant tissue for large-scale in vitro and in vivo (transplantation) studies, and are thus a significant advance in CRC drug discovery.
Keywords: Colorectal cancer mouse models, intestinal organoid, patient-derived xenograft, transgenic colorectal cancer mouse model
1. Introduction
Colorectal cancer (CRC) is the third most common cancer worldwide and confers significant morbidity and mortality [1]. Incidence and mortality are decreasing in the United States due to a combination of enhanced screening and improved treatment; however, despite these gains, five-year survival of metastatic (i.e., Stage IV) CRC remains less than 10% [1]. The discovery and preclinical testing of novel therapeutic strategies requires the use of in vitro and in vivo models of colorectal cancer. However, of candidate therapies selected for clinical trials based on encouraging preclinical data, only approximately 5% percent demonstrate clinical efficacy in Phase III trials [2]. The failure of the drug development process can be at least partially attributed to the use of preclinical models that poorly recapitulate the disease. In this review, we will discuss the limitations and appropriate use of cell and animal models in CRC research.
2. In Vitro Models
Typically, drug discovery begins with mechanistic and efficacy studies in cell-based models. The most common in vitro models include human and mouse cancer cell lines. Recently, three-dimensional “organoid” culture systems have been described that model cancer. Practically, robust in vitro systems are essential for mechanistic studies that identify pathways or targets for cancer therapy.
2.1 Human Cell Lines
Cell lines have played a significant role in elucidating signaling pathways in cancer since the derivation of the HeLa cervical cancer line in 1951. Primary tumors, upon surgical removal, are digested into individual cells and cultured on plastic dishes. Many human CRC cell lines are commercially available and have been extensively used in drug discovery [3]. Cell lines are relatively easy and inexpensive to use, and provide rapid experimental results. Most of the widely used cell lines have been genomically characterized and represent the genetic landscape of human CRC. Thus, a panel of lines with activating KRAS mutations can be easily compared to a panel lines with wild-type KRAS. Alternatively, cell lines can be genetically manipulated through homologous recombination, short hairpin RNA (shRNA) gene knockdown, or CRISPR-Cas9 gene editing. Many assays that evaluate antitumor efficacy can be automated, which aids in drug discovery as multiple agents can be concurrently tested against a range of cell lines.
There are several important limitations to cancer cell lines. Cell lines represent a clonal population of tumor cells that are naturally selected to grow in culture plates and media, and thus likely differ substantially from the original tumor. Cell lines do not recapitulate the functional and genetic heterogeneity of human cancers, which is a significant factor in resistance to targeted therapies [4]. Additionally, there have been reports of cross contamination of one cell line into another [5]; a CRC cell line known as WiDr was thought to be unique until chromosome analysis proved it to be the HT-29 line [6]. Modern authentication techniques and use of cells from repositories (e.g., the American Type Culture Collection or ATCC) reduce the likelihood of cell line cross-contamination. Finally, cell lines are difficult to create from individual patient tumors and cannot be derived from matching normal tissue. Thus, traditional cancer cell lines are not well suited for personalized clinical application.
2.2 Mouse Cell Lines
CRC cell lines are also available from murine sources. Among the commonly used mouse cell lines are MC38, an adenocarcinoma cell line derived from a C57BL/6 mouse [7] and CT26, derived from a BALB/c mice [8]. These cell lines were developed in the 1970s after repeated subcutaneous injection of the carcinogen 1,2-dimethylhydrazine dihydrochloride and repeated rectal administration of the carcinogen N-nitro-N-methylurethane, respectively. They are easy to culture and readily available from repositories. They are most often used for syngeneic subcutaneous transplantation studies (i.e., MC38 cells into C57BL/6 recipient mice and CT26 cells into BALB/c mice) to study tumor growth in the setting of an intact immune system. Our group derived primary cell lines from genetically engineered colorectal tumors. These primary cell lines have the advantage of being low passage, and thus less likely to have acquired additional mutations in vitro or been subjected to contamination. The gene expression profiles of these Kras mutant and wild-type lines closely reflect the profiles of human KRAS mutant and wild-type CRC tumors, which suggests that they are a useful in vitro model of human disease [9]. There are important disadvantages to the use of mouse CRC cell lines for drug development. First, there are far fewer murine lines available compared to human lines. Second, mouse cell lines that model less common cancer mutations are generally unavailable. Finally, murine cells are not as well functionally and genetically characterized as human cell lines.
2.3 Mouse Organoids
A key limitation of traditional monolayer cell culture is that normal intestine cannot be efficiently cultured. The Clevers lab recently described a three-dimensional culture model in which murine intestinal crypts (which contain self-renewing stem cells and adjacent niche Paneth cells) are cultured in three-dimensional collagen gel and specialized media containing Wnt3, the Wnt activator R-spondin1, the BMP inhibitor Noggin, and other growth factors. The crypts grow into “mini-intestines” or organoid structures that contain stem cells and the differentiated cell types of the intestine. Alternatively, FACS-sorted stem cells from Lgr5-EGFP-creERT2 mice (in which Lgr5+ stem cells are labeled with GFP) can be cultured as organoids. While single Lgr5+ stem cells can form organoids, culture efficiency is markedly increased in the presence of niche Paneth cells that provide endogenous Wnt3 ligand to support stem cell function [10]. Primary intestinal organoids have been maintained in culture for greater than 1.5 years [11]. Murine colon organoids have also been reported by the Clevers lab [12]. Intestinal organoids have been extensively used as in vitro models of normal intestinal function. An alternative approach to generating organoids involves culture of minced neonatal intestinal tissue within a collagen gel with an air-liquid interface; stromal cells in the tissue provide Wnt and other supportive signals [13].
The majority of CRCs are initiated by truncating APC mutations in intestinal stem cells, which lead to activation of Wnt signaling. Colorectal cancer organoids have been derived from murine Apc-deficient intestinal adenomas. Unlike normal intestinal organoids, cancer organoids exhibit endogenous Wnt activation, and thus do not require supplemental Wnt or R-spondin1 ligand to grow. Apc can also be deleted in vitro in Apcflox/flox villin-CreER mice with 4-OH-Tamoxifen, or in organoids derived from wild-type mice by shRNA knockdown or CRISPR/Cas9 editing; selection of cells with Apc loss requires only removal of Wnt and R-spondin1 from culture media [14–16]. Additional mutations associated with CRC have been modeled by infection with retrovirus encoding mutant KrasG12D or shRNA-mediated deletion of the tumor suppressor genes Tp53 or Smad4 [17]. A distinct advantage over conventional cell culture for drug discovery is that normal and cancer organoids can be compared side-by-side with candidate pharmacologic agents.
2.4 Human Organoids
Human intestinal and colon tissue can be cultured as three-dimensional organoids from colonoscopy biopsy specimens, surgical resections, or even single human EphB2+ stem cells [11, 18]. Human intestinal crypts are more difficult to grow than murine crypts and require supplementation with inhibitors of the Alk and p38 pathways for long-term culture, in addition to Wnt, R-spondin1, and Noggin. Organoids derived from human colon biopsies can undergo at least 100 population doublings in culture, thus allowing for adequate numbers of organoids for experimental study [11]. The development of a cell line that produces Wnt3, R-spondin1, and Noggin may substantially reduce the cost of large-scale genetic loss of function or small molecule screens of human intestinal organoids [19].
Organoid cultures are used to model CRC, either directly from cancer tissue or from genetic manipulation of normal intestinal tissue. Similar to murine Apc-deficient organoids, human cancer organoids exhibit activated Wnt signaling and thus do not require Wnt for growth and long-term passage [11]. CRISPR/Cas9-mediated editing of APC, followed by deletion of SMAD4, and TP53, and introduction of activating mutations in KRAS and PIK3CA results in isogenic human organoids that model the adenoma-carcinoma sequence in human CRC [20, 21]. The Clevers lab has developed a library of organoids grown from human CRCs and matching adjacent normal colon for in vitro drug testing; agents with efficacy in the tumor organoid that are not toxic to the normal organoid can be selected for further study [22]. For example, one patient’s CRC organoid was exquisitely sensitive to Wnt inhibition, which was predicted by a mutation in the Wnt negative feedback regulator RNF43. Organoids were successfully derived from approximately 90% of tumor specimens. Thus, organoid culture of malignant and normal colorectal tissue is a novel platform for translating tumor genetic data into personalized therapy. Organoids could also conceivably be cultured from different sections of tumors to model tumor heterogeneity, or from primary and metastatic sites to identify mechanisms of cancer metastasis.
3. In Vivo Transplant Models
Despite the many advantages of in vitro cancer models, in vivo systems are essential to assess the role of the tumor microenvironment, host immune system, and angiogenesis in tumor response to therapy. The most common in vivo model is the murine xenograft. Recently, patient derived xenografts and orthotopic transplant models have been developed to overcome deficiencies of the xenograft.
3.1 Cell Line Xenografts
Injection of human colorectal cancer cells subcutaneously into an immunodeficient mouse will typically result in growth of tumor at the injection site. Commonly used mouse strains include classically nude (athymic) and severe combined immunodeficient (SCID) mice, which are devoid of T lymphocytes [23] or both B and T lymphocytes, respectively [24]. NOD/SCID mice, unlike SCID mice, also have deficient NK cells. This model is commonly used for cancer drug discovery due to the lack of significant technical skill required to implant tumors, the straightforward monitoring of tumor growth by the naked eye, and the reasonable cost of maintaining colonies, and reasonable tumor latency of a few weeks [25].
Xenograft models have significant disadvantages. Injection of a large, homogenous cell population in order to form tumor does not reflect the cellular heterogeneity seen in most human CRCs (Figure 1a). The use of immunodeficient animals eliminates the important role of the host immune system in tumorigenesis. Given the difference in species, interactions between cancer cells and stromal cells cannot be studied. In addition, cells in xenografts are typically injected into the subcutaneous space, a microenvironment that is very different than the intestine. Finally, tumors derived from transplanted CRC cell lines do not recapitulate the histological features of human cancer, although transplanted cancer organoids partially overcome this problem. Perhaps due to these factors, drug response in xenograft models correlate poorly with drug response in clinical trials [26]. Recently, transplant models have been developed that attempt to overcome some of these limitations.
Figure 1. Histology of in vivo tumors of commonly used in vivo tumor models.

A – Hemotoxylin & Eosin (H&E) stain of xenograft tumor formation after injection of human SW480 colorectal cells into an immunodeficient mouse. Note the homogenous cell population. Reprinted from [99] with permission of Nature Publishing Group B – H&E stain of an adenoma seen in the small bowel of an ApcMin mouse. C – H&E stains of a patient derived xenograft. a is the primary, patient tumor and b is the tumor after 11 generations of passage in mouse. Note the preservation of tumor structure after passaging. From [100] with permission of PLoS One. D – H&E stain of an Apc and Tp53-deficient murine colonic tumor with features of high-grade dysplasia. E – H&E stain of an murine CRC cell line orthotopic transplant tumor. Murine MC38 colon cancer cells were injected via mouse colonoscope. From [36] with permission of PLoS One. F – H&E stain of liver metastasis. A murine cell lime derived from an Apc, Kras and Tp53-mutant transgenic mouse were injected into the spleen of a wild type C57BL/6J mouse. Four weeks later, liver was explanted. * metastatic tumor, ** surrounding healthy liver.
3.2 Organoid Xenografts
Xenograft transplantation of Apc-deficient murine colon organoids with additional driver mutations has been reported. Combinatorial Apc, Tp53, KrasG12D, and Smad4 mutations result in tumors with histology similar to invasive adenocarcinoma, although adenoma organoids deficient in Apc alone do not successfully engraft [17]. Xenograft transplantation can also be performed under the kidney capsule, which permits metastasis to the liver. The Sato laboratory compared kidney capsule engraftment and liver metastasis of CRISPR/Cas9-engineered human CRC organoids (i.e., editing of the APC, TP53, SMAD4, KRAS, and/or PIK3CA genes from normal colonic organoids) and organoids derived from metastatic CRC. Benign engineered adenomas (i.e., APC deficient) did not engraft, whereas organoids with additional driver mutations successfully formed tumors. In addition, only metastatic CRC organoids, not engineered organoids, efficiently metastasized to the liver [20]. In a related study, the Clevers lab found that CRISPR-mediated deletion of APC, TP53, and SMAD4, and activation of KRASG12D, was required for efficient tumor formation in human small intestinal organoid xenografts. All colon organoids with mutations in APC, KRAS, and TP53 (with or without SMAD4 deletion) formed adenomas upon transplantation. Histologically, only xenografts with all four mutations exhibited small areas of invasive carcinoma; xenografts with fewer mutations formed cystic structures that lacked important features of colon adenocarcinoma [21]. These studies demonstrate the potential application of CRC organoid xenografts for research and personalized therapy. However, the clinical utility of CRC xenograft engraftment may be limited by low engraftment rates for less advanced cancers. Further research is required to evaluate the transplantation efficiency and histological features of patient-derived CRC organoid xenografts.
3.3 Patient Derived Xenografts
In the Patient Derived Xenograft (PDX) model, portions of patient tumor tissue are obtained during surgery and implanted into an immunodeficient mouse (P0). Once the tumor grows it is surgically removed and implanted into other mice (P1). This is repeated until enough animals are obtained for the experiment (i.e., P2, P3, etc.). Unlike traditional xenografts, in PDXs tumor stroma will grow with the tumor cells, thus allowing for tumor-stroma cross-talk (Figure 1c) [27]. Both cellular and molecular tumor heterogeneity of tumor is more pronounced compared with monoclonal xenografts. PDXs conserve important aspects of tumor histology, vascularity and architecture of primary CRCs. Important driver mutations appear to be consistent along passages of PDXs, such as KRAS and PIK3CA [28] and establishment of PDX models that reflect the heterogeneity of CRC is feasible [29, 30]. PDX models predict clinical response to therapy better than traditional xenografts [26, 31, 32]. An important application of PDX is personalized cancer treatment. Following surgery, an individual patient’s cancer can, following surgery, be implanted into a mouse, passaged, and studied with various chemotherapy agents to determine a clinical approach to treat the patient [32]. PDXs may have an important role in the care of patients with rare tumor types or combination of driver mutations where there is no established treatment regimen, or for patients with resistance to established therapy [33].
Despite excitement about their potential, PDX models carry important disadvantages. Like traditional xenografts, PDX models require the use of immunodeficient host animals. As with cell line xenografts, a “more” immunodeficient mouse (e.g., NOD/SCID) may be better suited for engraftment than a “lesser” immunodeficient mouse (e.g., nude). While most cell lines implanted into xenografts are well defined, primary tumors used in PDXs require additional molecular characterization. It has been shown that human tumor stroma is initially preserved after transplantation but is slowly replaced by murine stroma with time; the significance of this is somewhat unclear [27]. Despite the promise of PDX for personalized medicine, the reported tumor initiation rate for PDX is approximately 70%, which may limit clinical utility [34]. Additionally, tumor implantation and screening potential therapies may require six or more months. Finally, PDX models are expensive for clinical application and are typically not covered by private or national health plans.
3.4 Orthotopic Transplant Models
A major disadvantage of traditional and patient-derived xenografts is that the microenvironment of the subcutaneous space differs greatly from that of the colon. Thus, orthotopic transplant models have been developed in which CRC cells are directly implanted into the colon. Implantation of tumor cells can occur via a laparoscopy directly into the serosal surface of the cecum. This approach results in both a primary tumor and possible metastasis to local lymphatics, lung, and liver [35]. Orthotopic transplantation of as few as 1,000 cells into the distal colon, via enema or colonoscopy-guided mucosal injection, results in tumors that can be monitored by optical colonoscopy (Figure 1e) [9, 36]. With colonoscopy, tumor growth can be longitudinally determined by the degree of luminal obstruction and response to therapy assessed with serial biopsy. Human colon cancer cell lines are implanted into immunodeficient mice, while mouse cell lines are transplanted into syngeneic recipient mice. Intestinal organoids can also be transplanted into the colon; administration of dextran sulfate sodium (DSS) to induce colitis allows successful rectal delivery and engraftment of normal organoids [37]. This approach could be used to orthotopically transplant malignant murine or human intestinal organoids for validation of drug targets or study of mechanisms of carcinogenesis.
4. Carcinogen Induced Mouse Models
Colonic tumors can be induced in mice with 1,2-dimethylhyrazine (DMH), or its metabolite, azoxymethane (AOM). AOM is N-oxidated and hydroxylated in the liver, then excreted in the bile. In the colon, microbial flora then metabolize the agent into a form that promotes carcinogenesis [38]. AOM administration results in tumors that often have mutations in Kras and Ctnnb1 (encoding beta-catenin) [39]. However, mucosal invasion and distant metastasis is rarely seen in this model. A commonly used mouse CRC cell line, MC38, was obtained from a DMH-induced tumor [7]. A limitation of AOM is that a latency period of up to 30 weeks may be required for tumor formation. An alternative method is the administration of AOM together with dextran sulfate sodium (DSS), an agent that causes colitis [40]. DSS alone can cause tumorigenesis; however, the incidence is low if not combined with AOM. This combination lowers latency time to 10 weeks and is used to model inflammatory bowel disease-associated CRC with a tumor incidence of close to 100% [41]. Typically, DSS is administered orally via drinking water and AOM via sequential intraperitoneal injection [42]. Other carcinogens are used either alone or with DSS to model CRC, including heterocyclic amines, aromatic amines, and alkylnitrosamide compounds; however, DMH and AOM are the best characterized cancer-inducing agents [7, 43]. Carcinogen induced models have been used for chemoprevention studies in CRC, including research on COX-2 inhibitors and Peroxisome Proliferator-Activated Receptor (PPAR) ligands such as pioglitazone and rosiglitazone [44, 45].
Advantages to carcinogen-induced models include high reproducibility, straightforward and inexpensive tumor initiation, low multiplicity, the ability to monitor tumors in the distal colon with optical colonoscopy, and the ability to induce tumors in mice of different genetic backgrounds. Thus, whereas xenograft and genetically engineered models are genetically homogeneous, tumorigenesis in multiple murine strains, including wild mice, can be studied using carcinogens to model the genetic diversity of human cancer. However, the AOM/DSS model may be more suited as a model of inflammatory bowel disease-associated CRC, which constitutes a minority of CRCs, rather than sporadic CRC.
5. Genetically Engineered Mouse Models
Genetically engineered mice are powerful models, as they recapitulate adenoma or carcinoma formation in the native, immunocompetent colon microenvironment. Since the initial discovery of the ApcMin mouse in 1990, numerous additional genetically engineered models have been added to the armamentarium of colon cancer modeling.
5.1 Germline Apc Mutant Models
The ApcMin mouse was discovered as a result of N-ethyl-N-nitrosourea (ENU) mutagenesis screening [46]. During the mutagenesis screen, mice were subjected to ENU, an alkylating agent. One phenotype was anemia that was associated with a heterozygous nonsense mutation at codon 850 in the Apc gene, the mouse equivalent of human APC, and acquired homozygous mutations in intestinal polyps [47]. This mouse was named Min (Multiple intestinal neoplasia) as it spontaneously develops 30 or more polyps by 4–6 months of age (Figure 1b). Thus, the mouse is an excellent model of familial adenomatous polyposis (FAP), which is caused by a germline heterozygous truncating APC mutations and early onset polyposis from subsequent loss of the second APC allele [48]. Almost all tumors in the ApcMin mouse are adenomas by histology; carcinoma and metastasis are rare. Additionally, as the polyps develop predominantly in the small intestine, tumors cannot be monitored via colonoscopy. The ApcMin mouse led to the critical discovery of the central role of APC in intestinal tumorigenesis: inactivation of APC results in nuclear localization of beta-catenin and transcription of Wnt target genes, in particular MYC [49, 50]. The ApcMin mouse has been used in hundreds of chemoprevention, therapeutic and mechanistic studies [51]. Since the development of the ApcMin mouse, other mice bearing targeted, truncating germline mutations in Apc have been created which vary in the number and location of intestinal lesions but produce polyps with similar histological features [52–62].
5.2 Other Germline Genetically Engineered Models of CRC
Mutations in DNA mismatch repair genes, predominantly in MSH2 and MLH1, cause hereditary nonpolyposis colorectal cancer (HNPCC), also known as Lynch Syndrome, and are characterized by microsatellite instability. Mice deficient in Mlh1, Msh2, Msh3, Msh6 or Pms2 develop tumors in the small bowel as well as lymphomas and skin tumors [63–66]. These models may be helpful when dealing with cancer subtypes of human disease such as medullary, mucinous, undifferentiated and signet-ring carcinomas, as they display high levels of microsatellite instability [52, 67]. Mismatch repair-deficiency increases adenoma formation in an Apc deficient background [68, 69].
TGF-β activates the SMAD4 pathway and is mutated in 10–35% of colorectal cancers, usually late in cancer progression. Germline SMAD4 deletion is responsible for familial juvenile polyposis, an inherited disease characterized by early onset gastrointestinal polyps and cancers; Smad4+/− ApcMin mice exhibit greater invasion and transplant efficiency compared to ApcMin controls [70, 71]. Tgfb1 deletion in the immunocompromised Rag2 strain results in cecal and colon neoplasm without Apc loss [72, 73]. Knockout of Smad3, a downstream protein from TGF-β, results in mucinous carcinoma in the colon [74].
5.3 Conditional or inducible Apc deletion Models
The Cre recombinase system is used to conditionally target gene deletion or activation to the intestine, thus avoiding lethality and extra intestinal manifestations that may be present with germline mutations. This can be achieved by crossing mice with an intestine-specific cre with Apc floxed mice. Constitutive and tamoxifen-inducible Villin-cre mice are widely used to limit deletion of Apc and other genes to the intestinal epithelium and model intestinal tumorigenesis [75]. The Lgr5-EGFP-creERT2 mouse marks Lgr5+ stem cells, which are located at the base of intestinal crypts and are the cells of origin of intestinal cancer; conditional deletion of Apc in Lgr5+ stem cells leads to small intestinal polyposis [14, 76]. Apc loss in a quiescent intestinal stem cell marker, Lrig1, induces colorectal adenomas with high-grade dyplasia that are seen on colonoscopy [77]. Other cre mouse lines that are used to model CRC (with varying polyp burden in small intestine vs. colon) include Fabp-Cre [59], Ah-Cre (Cyp1A promoter) [78], CDX2P-NLS-Cre [79], and CAC-Cre (carbonic anhydrase I gene promoter, expressed only in the large intestine) [80]. The addition of LSL-KrasG12D/+ to Apcloxp14/+ CAC-Cre results in an average of 4.3 colon adenomas (but not carcinomas) per mouse [81].
Another colon-specific CRC model involves exogenous delivery of adeno-cre to the colons of Apcloxp14/loxp14 mice. This can be achieved by rectal enema [82] or, more efficiently, by restricting adeno-cre infection to the distal colon with colonic clips placed during laparotomy [83]. Surgical delivery of adeno-cre results in reproducible formation of 1–4 adenomas in the distal colon. Apc-null tumors are monitored longitudinally by optical colonoscopy to assess therapeutic response; tumor biopsy before and after treatment is used to determine tumor histology, genotype, mRNA or protein expression. Tp53 loss or Kras or Braf mutation in the setting of Apc deficiency results in invasive adenocarcinoma in a subset of tumors (Figure 1d) [9, 83–85]. The Kras mutant murine model recapitulates the gene expression patterns and prognosis of Kras mutant CRC patient cohorts, which suggests that it is an excellent resource for drug discovery [86].
A novel approach to studying gene function during defined time periods is to use a mouse with cre-dependent expression of rtTA3, providing induction of TRE-controlled transgenes in the presence of doxycycline [87]. For example, inducible expression of Cas9 permits CRISPR-mediated editing of Apc and subsequent tumorigenesis. This model can be used to assess the role of up to six genes in tumorigenesis, perhaps most efficiently tumor suppressors, with limited off-target effects [88].
6. Metastatic models
Given that the majority of CRC deaths are from metastatic disease, a great deal of effort has gone into development of mouse models that mimic the metastatic spread of tumor cells. Colonic delivery of adeno-Cre to Apcloxp14/loxp14 LSL-KrasG12D/+ mice (described above) is reported to result in liver metastases that are high-grade carcinomas in 20% of mice 24 weeks following adeno-cre injection [67, 83]. VillinCre LSL-KrasG12D Tgfbr2E2flx/E2flx mice develop lymph node and lung metastases from primary colon tumors via a beta-catenin-independent mechanism [89]. However, CRC metastases in these models are however infrequent and thus may not be practical for drug testing or large-scale experiments.
The major site of CRC metastasis is the liver [1]. One method of assessing the growth of colon cancer in the liver is direct inoculation of tumor cells into the liver parenchyma [90]. This is the most direct method of studying the metastatic progression of colorectal cancer in the liver as it directly assesses colonization competence of tumor cells [91]. Hematogenous seeding of tumors via the portal vein through splenic injection is an alternative route (Figure 1f). In such models, tumor cells are injected into the spleen via laparotomy (the spleen can be removed, partially removed, or left in place after injection), after which growth of liver metastasis follows within a few weeks [92]. The splenic injection approach can be performed with human CRC cells into immunodeficient mice or with murine cell lines into syngeneic mice; metastasis rate is affected by the tumor cell line and recipient mouse strain [93]. This model relies heavily on the latter events of the metastatic cascade (mainly seeding and extravasation); however, it does so at very high efficiency and thus can reliably be used for experiments with multiple therapeutic conditions or where the number of animals used in a study is limited. Alternatively, tumor cells can be directly injected into the portal vein [93].
Orthotopic transplantation models of CRC metastasis have also been developed. Tumors injected into the serosal surface of the cecum develop metastasis at relatively low rate, but passage of tumor cells through a xenograft host increases lymph node and lung metastases formation rate to approximately 10% in Kras wild-type cells [94, 95]. Orthotopic injection of murine tumor cells into the rectal mucosa of syngeneic recipient mice produces a liver metastasis rate of only 3.3% 50 days after transplantation [96]. Metastasis formation in cell line models can be confirmed and monitored with the use of fluorescence and/or luciferase-tagged cells [97]. The Lipkin lab recently reported a novel approach to establishing orthotopic CRC metastatic cancers by hijacking the ability of chemokine receptor 9 (CCR) to target lymphocytes to the intestine and colon [98]. Human CRC cell lines engineered to conditionally express CCR9 form intestinal tumors following intravenous injection in immunodeficient mice. Silencing of CCR9 expression results in liver metastasis formation. In summary, many CRC metastasis models have been reported and are potentially useful for preclinical drug discovery research.
7. Conclusion
In the past few decades, many CRC models have been developed to aid research and drug discovery. Cell lines, xenografts, organoid culture, and genetically engineered mice offer the ability to model the genetic, histologic, and molecular features of human CRC. While no single model is optimal, each has advantages and disadvantages that must be considered by the investigator.
8. Expert Opinion
The historically poor correlation between drug efficacy in animal models and clinical trials suggests that innovative in vitro and in vivo models are needed to develop novel colorectal cancer (CRC) treatments. Advantages and disadvantages of each model are described in Table 1. Key historical developments in CRC modeling include: the development of the HeLa cancer cell line (1951) followed by derivation of many human CRC cell lines; discovery of the ApcMin genetically engineered model (1990); discovery that loss of Apc in intestinal stem cells causes carcinogenesis (2009); first report of murine intestinal organoids (2009), successful organoid culture from murine and human CRCs (2011); development of a cell line based orthotopic transplant CRC model (2011); and report of a tissue “biobank” of human CRC organoids for drug discovery (2015). Here, we offer our recommendations on the use of these models in drug development.
Table 1.
Overview of the advantages and disadvantages of in vitro and in vivo colorectal cancer models.
| Model type | Model | Advantages | Disadvantages |
|---|---|---|---|
| In vitro | Cell lines | • Easy to culture • Many well-characterized lines available • Easy to model multiple genetic mutations |
• Monoclonal cells poorly represent heterogeneity of tumor |
| Organoids | • Possibility for personalized treatment • Ability to model normal intestinal tissue • Easy to model multiple genetic mutations |
• Can be costly to culture |
|
| In vivo | Xenografts Organoid xenograft Patient-derived xenograft |
• Easy to establish, rapid tumor onset • Can be used with most human cell lines • Recapitulates histology of human CRC • Can be derived from patient tumor or from genetically altered normal tissue for personalized therapy • Therapy tailored for individual patients • Tumor histology and genetic heterogeneity is similar to original tumor |
• Requires immunodeficient host • Non-colon microenvironment • Homogeneous cell population does not reflect tumor heterogeneity • Requires immunodeficient host • Can be used with most human cell lines • Requires immunodeficient host • Potentially long tumor latency |
| Orthotopic transplantation | • Uses the same microenvironment (colon) as human cancer • If transplanted into distal colon, optical colonoscopy can be used to monitor growth and response to therapy |
• Technically challenging to perform • If human cells are used, requires immunodeficient animal |
|
| Carcinogen-induced | • Near 100% efficiency in tumor formation • Ability to induce tumors in most mouse strains • Correct tumor microenvironment • Recapitulates human adenoma histology |
• Models inflammatory bowel disease-mediated CRC rather than sporadic CRC • Long tumor latency |
|
|
In vivo metastasis |
Genetically engineered Genetically engineered (adeno- cre model) Splenic transplantation CCR9-mediated liver metastasis |
• Correct tumor microenvironment • Recapitulates human adenoma histology • Histologically accurate liver metastases • Reproducible liver metastasis or seeding • Liver metastasis form from primary intestinal tumors |
• Long tumor latency • Time consuming and expensive to model multiple mutations • Most tumors are adenomas, not carcinomas • Long tumor latency and poor penetrance make this model impractical for large- scale study • Requires technically challenging surgery in Apcloxp14/loxp14LSL- KrasG12D/+ mice [83] • Does not model true metastasis from primary tumor • Requires genetically manipulated cell lines |
Human cell line and xenograft models are the mainstay of therapeutic studies due to low cost and ease of use, but poorly reproduce the heterogeneity of human cancer. Since human cell lines have been extensively studied as in vitro and in vivo models, they will continue to play an important role in cancer biology and drug discovery research. Orthotopic transplantation of human cell lines into the cecum is not widely used because of technical challenges and difficulty in tumor monitoring. Cell line transplantation into the colonic mucosa is a promising new approach that will require further optimization before it can be applied in CRC research. We believe that cell line xenografts will gradually be replaced with patient-derived xenografts or patient-derived organoid xenografts for personalized medicine applications.
The ApcMin genetically engineered model of CRC and related Apc-deficiency models have been extensively used in drug discovery and mechanistic studies. However, the key limitations that have prevented genetically engineered models from replacing xenografts include long tumor latency and need for expensive and time consuming gene targeting and breeding to study the effects of genetic mutations on tumorigenesis. Therefore, while genetically engineered models will continue to be essential for mechanistic studies, they will not replace the low cost and ease of use of xenograft models. Carcinogen-induced models share similar advantages and disadvantages with genetically engineered models, with the additional limitation that the initiating event is a chemical insult instead of loss of Apc function (as in most human CRCs). CRISPR/Cas9-based gene targeting in the germline may reduce the time and expense of studying tumor suppressor gene function in vivo.
A major recent innovation in CRC modeling is the ability to recapitulate the ultrastructure of normal and malignant intestine in three-dimensional culture using defined media conditions (i.e., organoid or mini-intestine culture). Small intestinal or colonic organoids can be derived from essentially any mouse model or patient, and therefore have far-reaching implications for drug discovery applications. In particular, human organoids are ideal for personalized medicine because individual patient cancers and control normal tissues can be propagated indefinitely for genetic, functional, and drug response studies in culture and, potentially, xenograft transplantation assays. Thus, we believe that as the costs of organoid research decrease, organoid culture will gradually replace other mouse CRC models for many applications.
The ideal CRC model recapitulates essential aspects of human cancer: 1) tumorigenesis in the colon; 2) sequential mutagenesis (i.e., loss of APC, followed by activating KRAS mutations and loss of TP53); 3) histological features of adenoma, invasive adenocarcinoma, and metastasis; 4) immune system interactions between intestinal stroma and tumor; and 5) genetic and functional heterogeneity. The model should also have high tumor penetrance, short tumor latency, and be relatively easy to use. We believe that much of the failure of the drug discovery pipeline can be attributed to deficiencies with commonly used cell line and traditional xenograft models. While no single currently available model achieves all of these goals, we believe that genetically engineered mice should be preferred as in vivo models of tumor initiation and stroma-tumor interaction. A reproducible, efficient genetically engineered model of CRC metastasis is unfortunately not available. For most research and clinical applications, we recommend use of murine and patient-derived organoid cultures, which have been shown to meet many of the above requirements of an ideal CRC model. The CRISPR/Cas9 gene editing system is an important advance for rapid study of gene function in organoids. Human cancer organoids hold great promise for studying tumor heterogeneity in drug response and for selecting targeted therapies for cancer patients. In addition, we anticipate that CRC metastases will soon be modeled with organoid cultures. Further research is needed to define the complementary roles of in vitro patient-derived organoids, organoid xenografts, and patient-derived xenografts in CRC modeling, drug development, and clinical application.
Figure 2. Schematic of the common routes of forming metastasis in modeling metastatic colorectal cancer.
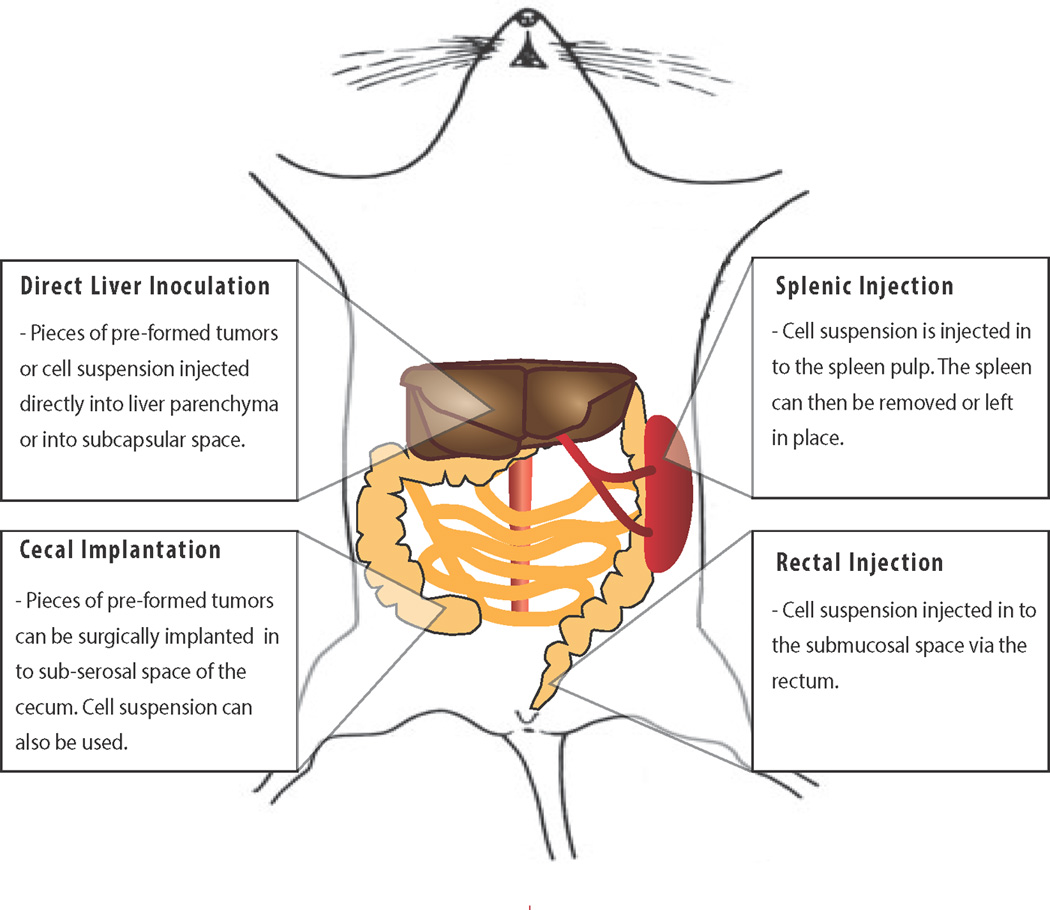
Tumor cells can be directly injected into the liver [90] or into the spleen [92] resulting in efficient metastasis in the liver. Alternatively, cells can be implanted into the cecum [94] or rectum [96], although this is associated with less frequent liver metastasis formation.
Highlights.
Classic colorectal cancer models for drug discovery include cell lines and xenograft animal models. While widely utilized, these models have significant limitations that limit their ability to predict drug response in clinical trials.
Three-dimensional organoid culture systems can be applied for mechanistic studies, candidate drug screening, and cancer therapy selection.
Patient derived xenografts can be used for personalized cancer treatment.
Genetically engineered mouse models are employed in mechanistic studies that may result in novel targets for cancer therapy.
Carcinogen-induced models are useful for studying inflammatory bowel disease-associated colorectal cancer.
CRC in vivo metastasis models are available but not yet widely used in drug discovery.
Acknowledgments
J Roper was supported by the Department of Defense through the Peer Reviewed Cancer Research Program Career Development Award CA120198 and by the Tufts University School of Medicine through the Earl P. Charlton Fund Research Award and Rozan Research Fund Pilot Award. J Roper and OH Yilmaz are supported with a V Scholar Award through the V Foundation for Cancer Research. OH Yilmaz is supported by a National Institutes of Health (NIH) grant DK043351 through the Center for the Study of Inflammatory Bowl Diseases from the Massachusetts General Hospital as well as National Institute of Aging grant NIA R00 AG045144 and NIH Cancer Center Support (core) grant P30-CA14051. D Kedrin is also supported by a National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) T32 grant DK007191.
Footnotes
Financial and Competing Interests Disclosure
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
References
- 1.Torre LA, Bray F, Siegel RL, et al. Global cancer statistics, 2012. CA Cancer J Clin. 2015 Mar;65(2):87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 2.Kola I, Landis J. Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discov. 2004 Aug;3(8):711–715. doi: 10.1038/nrd1470. [DOI] [PubMed] [Google Scholar]
- 3.Shoemaker RH. The NCI60 human tumour cell line anticancer drug screen. Nat Rev Cancer. 2006 Oct;6(10):813–823. doi: 10.1038/nrc1951. [DOI] [PubMed] [Google Scholar]
- 4.Gerlinger M, Rowan AJ, Horswell S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012 Mar 8;366(10):883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.MacLeod RA, Dirks WG, Matsuo Y, et al. Widespread intraspecies cross-contamination of human tumor cell lines arising at source. Int J Cancer. 1999 Nov 12;83(4):555–563. doi: 10.1002/(sici)1097-0215(19991112)83:4<555::aid-ijc19>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 6.Chen TR, Drabkowski D, Hay RJ, et al. WiDr is a derivative of another colon adenocarcinoma cell line, HT-29. Cancer Genet Cytogenet. 1987 Jul;27(1):125–134. doi: 10.1016/0165-4608(87)90267-6. [DOI] [PubMed] [Google Scholar]
- 7.Corbett TH, Griswold DP, Jr, Roberts BJ, et al. Tumor induction relationships in development of transplantable cancers of the colon in mice for chemotherapy assays, with a note on carcinogen structure. Cancer Res. 1975 Sep;35(9):2434–2439. [PubMed] [Google Scholar]
- 8.Brattain MG, Strobel-Stevens J, Fine D, et al. Establishment of mouse colonic carcinoma cell lines with different metastatic properties. Cancer Res. 1980 Jul;40(7):2142–2146. [PubMed] [Google Scholar]
- 9. Martin ES, Belmont PJ, Sinnamon MJ, et al. Development of a colon cancer GEMM-derived orthotopic transplant model for drug discovery and validation. Clin Cancer Res. 2013 Jun 1;19(11):2929–2940. doi: 10.1158/1078-0432.CCR-12-2307. * Development of low passage Kras mutant and Kras wild-type murine colorectal cell lines from genetically engineered mouse models.
- 10.Sato T, Vries RG, Snippert HJ, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009 May 14;459(7244):262–265. doi: 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- 11. Sato T, Stange DE, Ferrante M, et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett's epithelium. Gastroenterology. 2011 Nov;141(5):1762–1772. doi: 10.1053/j.gastro.2011.07.050. * Culture techniques of organoids derived from mouse and human colon and adenoma tissue.
- 12.Sato T, Clevers H. Growing self-organizing mini-guts from a single intestinal stem cell: mechanism and applications. Science. 2013 Jun 7;340(6137):1190–1194. doi: 10.1126/science.1234852. [DOI] [PubMed] [Google Scholar]
- 13.Ootani A, Li X, Sangiorgi E, et al. Sustained in vitro intestinal epithelial culture within a Wnt-dependent stem cell niche. Nat Med. 2009 Jun;15(6):701–706. doi: 10.1038/nm.1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barker N, Ridgway RA, van Es JH, et al. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature. 2009 Jan 29;457(7229):608–611. doi: 10.1038/nature07602. [DOI] [PubMed] [Google Scholar]
- 15.Onuma K, Ochiai M, Orihashi K, et al. Genetic reconstitution of tumorigenesis in primary intestinal cells. Proc Natl Acad Sci U S A. 2013 Jul 2;110(27):11127–11132. doi: 10.1073/pnas.1221926110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schwank G, Koo BK, Sasselli V, et al. Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell. 2013 Dec 5;13(6):653–658. doi: 10.1016/j.stem.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 17.Li X, Nadauld L, Ootani A, et al. Oncogenic transformation of diverse gastrointestinal tissues in primary organoid culture. Nat Med. 2014 Jul;20(7):769–777. doi: 10.1038/nm.3585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jung P, Sato T, Merlos-Suarez A, et al. Isolation and in vitro expansion of human colonic stem cells. Nat Med. 2011 Oct;17(10):1225–1227. doi: 10.1038/nm.2470. [DOI] [PubMed] [Google Scholar]
- 19.Miyoshi H, Stappenbeck TS. In vitro expansion and genetic modification of gastrointestinal stem cells in spheroid culture. Nat Protoc. 2013 Dec;8(12):2471–2482. doi: 10.1038/nprot.2013.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Matano M, Date S, Shimokawa M, et al. Modeling colorectal cancer using CRISPR-Cas9-mediated engineering of human intestinal organoids. Nat Med. 2015 Mar;21(3):256–262. doi: 10.1038/nm.3802. ** Application of the CRISPR-Cas9 genetic editing system to modify tumor suppressor genes and oncogenes in normal human organoids to produce in vitro cancer organoids and in vivo transplantation models.
- 21. Drost J, van Jaarsveld RH, Ponsioen B, et al. Sequential cancer mutations in cultured human intestinal stem cells. Nature. 2015 Apr 29; doi: 10.1038/nature14415. ** Use of CRISPR-Cas9 genomic editing to modify normal, non-cancer derived intestinal stem cells with Apc, Tp53, Kras and SMAD4. Upon subsequent transplantation into mouse, the modified organoids formed carcinoma.
- 22. van de Wetering M, Francies HE, Francis JM, et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell. 2015 May 7;161(4):933–945. doi: 10.1016/j.cell.2015.03.053. ** Establishment of organoid cultures from patient CRCs and matching normal colon. The established “biobank” was then used for drug screening and comprehensive genetic profiling.
- 23.Croy BA, Linder KE, Yager JA. Primer for non-immunologists on immune-deficient mice and their applications in research. Comp Med. 2001 Aug;51(4):300–313. [PubMed] [Google Scholar]
- 24.Blunt T, Gell D, Fox M, et al. Identification of a nonsense mutation in the carboxyl-terminal region of DNA-dependent protein kinase catalytic subunit in the scid mouse. Proc Natl Acad Sci U S A. 1996 Sep 17;93(19):10285–10290. doi: 10.1073/pnas.93.19.10285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Talmadge JE, Singh RK, Fidler IJ, et al. Murine models to evaluate novel and conventional therapeutic strategies for cancer. Am J Pathol. 2007 Mar;170(3):793–804. doi: 10.2353/ajpath.2007.060929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Voskoglou-Nomikos T, Pater JL, Seymour L. Clinical predictive value of the in vitro cell line, human xenograft, and mouse allograft preclinical cancer models. Clin Cancer Res. 2003 Sep 15;9(11):4227–4239. [PubMed] [Google Scholar]
- 27.Rosfjord E, Lucas J, Li G, et al. Advances in patient-derived tumor xenografts: from target identification to predicting clinical response rates in oncology. Biochem Pharmacol. 2014 Sep 15;91(2):135–143. doi: 10.1016/j.bcp.2014.06.008. [DOI] [PubMed] [Google Scholar]
- 28.Tignanelli CJ, Herrera Loeza SG, Yeh JJ. KRAS and PIK3CA mutation frequencies in patient-derived xenograft models of pancreatic and colorectal cancer are reflective of patient tumors and stable across passages. Am Surg. 2014 Sep;80(9):873–877. [PMC free article] [PubMed] [Google Scholar]
- 29. Burgenske DM, Monsma DJ, Dylewski D, et al. Establishment of genetically diverse patient-derived xenografts of colorectal cancer. Am J Cancer Res. 2014;4(6):824–837. * The authors form a tissue library of PDXs from 14 different patients and use of these models with various therapeutic agents.
- 30.Julien S, Merino-Trigo A, Lacroix L, et al. Characterization of a large panel of patient-derived tumor xenografts representing the clinical heterogeneity of human colorectal cancer. Clin Cancer Res. 2012 Oct 1;18(19):5314–5328. doi: 10.1158/1078-0432.CCR-12-0372. [DOI] [PubMed] [Google Scholar]
- 31.Siolas D, Hannon GJ. Patient-derived tumor xenografts: transforming clinical samples into mouse models. Cancer Res. 2013 Sep 1;73(17):5315–5319. doi: 10.1158/0008-5472.CAN-13-1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hidalgo M, Bruckheimer E, Rajeshkumar NV, et al. A pilot clinical study of treatment guided by personalized tumorgrafts in patients with advanced cancer. Mol Cancer Ther. 2011 Aug;10(8):1311–1316. doi: 10.1158/1535-7163.MCT-11-0233. ** A pilot in personalized cancer treatment. PDX mice were created from patients’ cancers and therapy was based on the response in the PDX mouse.
- 33.Morelli MP, Calvo E, Ordonez E, et al. Prioritizing phase I treatment options through preclinical testing on personalized tumorgraft. J Clin Oncol. 2012 Feb 1;30(4):e45–e48. doi: 10.1200/JCO.2011.36.9678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aparicio S, Hidalgo M, Kung AL. Examining the utility of patient-derived xenograft mouse models. Nat Rev Cancer. 2015 Apr 24;15(5):311–316. doi: 10.1038/nrc3944. [DOI] [PubMed] [Google Scholar]
- 35.Cespedes MV, Espina C, Garcia-Cabezas MA, et al. Orthotopic microinjection of human colon cancer cells in nude mice induces tumor foci in all clinically relevant metastatic sites. Am J Pathol. 2007 Mar;170(3):1077–1085. doi: 10.2353/ajpath.2007.060773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zigmond E, Halpern Z, Elinav E, et al. Utilization of murine colonoscopy for orthotopic implantation of colorectal cancer. PLoS One. 2011;6(12):e28858. doi: 10.1371/journal.pone.0028858. * Use of murine colonoscopy for direct implantation of tumor cells into the mouse colon.
- 37.Yui S, Nakamura T, Sato T, et al. Functional engraftment of colon epithelium expanded in vitro from a single adult Lgr5(+) stem cell. Nat Med. 2012 Apr;18(4):618–623. doi: 10.1038/nm.2695. [DOI] [PubMed] [Google Scholar]
- 38.Reddy BS, Weisburger JH, Narisawa T, et al. Colon carcinogenesis in germ-free rats with 1,2-dimethylhydrazine and N-methyl-n'-nitro-N-nitrosoguanidine. Cancer Res. 1974 Sep;34(9):2368–2372. [PubMed] [Google Scholar]
- 39.Kobaek-Larsen M, Thorup I, Diederichsen A, et al. Review of colorectal cancer and its metastases in rodent models: comparative aspects with those in humans. Comp Med. 2000 Feb;50(1):16–26. [PubMed] [Google Scholar]
- 40.Neufert C, Becker C, Neurath MF. An inducible mouse model of colon carcinogenesis for the analysis of sporadic and inflammation-driven tumor progression. Nat Protoc. 2007;2(8):1998–2004. doi: 10.1038/nprot.2007.279. [DOI] [PubMed] [Google Scholar]
- 41.Clapper ML, Cooper HS, Chang WC. Dextran sulfate sodium-induced colitis-associated neoplasia: a promising model for the development of chemopreventive interventions. Acta Pharmacol Sin. 2007 Sep;28(9):1450–1459. doi: 10.1111/j.1745-7254.2007.00695.x. [DOI] [PubMed] [Google Scholar]
- 42.De Robertis M, Massi E, Poeta ML, et al. The AOM/DSS murine model for the study of colon carcinogenesis: From pathways to diagnosis and therapy studies. J Carcinog. 2011;10:9. doi: 10.4103/1477-3163.78279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nishikawa A, Imazawa T, Kuroiwa Y, et al. Induction of colon tumors in C57BL/6J mice fed MeIQx IQ, or PhIP followed by dextran sulfate sodium treatment. Toxicol Sci. 2005 Apr;84(2):243–248. doi: 10.1093/toxsci/kfi079. [DOI] [PubMed] [Google Scholar]
- 44.Clapper ML, Gary MA, Coudry RA, et al. 5-aminosalicylic acid inhibits colitis-associated colorectal dysplasias in the mouse model of azoxymethane/dextran sulfate sodium-induced colitis. Inflamm Bowel Dis. 2008 Oct;14(10):1341–1347. doi: 10.1002/ibd.20489. [DOI] [PubMed] [Google Scholar]
- 45.Osawa E, Nakajima A, Wada K, et al. Peroxisome proliferator-activated receptor gamma ligands suppress colon carcinogenesis induced by azoxymethane in mice. Gastroenterology. 2003 Feb;124(2):361–367. doi: 10.1053/gast.2003.50067. [DOI] [PubMed] [Google Scholar]
- 46.Moser AR, Pitot HC, Dove WF. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science. 1990 Jan 19;247(4940):322–324. doi: 10.1126/science.2296722. [DOI] [PubMed] [Google Scholar]
- 47.Su LK, Kinzler KW, Vogelstein B, et al. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science. 1992 May 1;256(5057):668–670. doi: 10.1126/science.1350108. [DOI] [PubMed] [Google Scholar]
- 48.Groden J, Thliveris A, Samowitz W, et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell. 1991 Aug 9;66(3):589–600. doi: 10.1016/0092-8674(81)90021-0. [DOI] [PubMed] [Google Scholar]
- 49.Sansom OJ, Meniel VS, Muncan V, et al. Myc deletion rescues Apc deficiency in the small intestine. Nature. 2007 Apr 5;446(7136):676–679. doi: 10.1038/nature05674. [DOI] [PubMed] [Google Scholar]
- 50.Morin PJ, Sparks AB, Korinek V, et al. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science. 1997 Mar 21;275(5307):1787–1790. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- 51.Swamy MV, Patlolla JM, Steele VE, et al. Chemoprevention of familial adenomatous polyposis by low doses of atorvastatin and celecoxib given individually and in combination to APCMin mice. Cancer Res. 2006 Jul 15;66(14):7370–7377. doi: 10.1158/0008-5472.CAN-05-4619. [DOI] [PubMed] [Google Scholar]
- 52.Boivin GP, Washington K, Yang K, et al. Pathology of mouse models of intestinal cancer: consensus report and recommendations. Gastroenterology. 2003 Mar;124(3):762–777. doi: 10.1053/gast.2003.50094. [DOI] [PubMed] [Google Scholar]
- 53.Cheung AF, Carter AM, Kostova KK, et al. Complete deletion of Apc results in severe polyposis in mice. Oncogene. 2010 Mar 25;29(12):1857–1864. doi: 10.1038/onc.2009.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Colnot S, Niwa-Kawakita M, Hamard G, et al. Colorectal cancers in a new mouse model of familial adenomatous polyposis: influence of genetic and environmental modifiers. Lab Invest. 2004 Dec;84(12):1619–1630. doi: 10.1038/labinvest.3700180. [DOI] [PubMed] [Google Scholar]
- 55.Fodde R, Edelmann W, Yang K, et al. A targeted chain-termination mutation in the mouse Apc gene results in multiple intestinal tumors. Proc Natl Acad Sci U S A. 1994 Sep 13;91(19):8969–8973. doi: 10.1073/pnas.91.19.8969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kuraguchi M, Wang XP, Bronson RT, et al. Adenomatous polyposis coli (APC) is required for normal development of skin and thymus. PLoS Genet. 2006 Sep 15;2(9):e146. doi: 10.1371/journal.pgen.0020146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Oshima M, Oshima H, Kitagawa K, et al. Loss of Apc heterozygosity and abnormal tissue building in nascent intestinal polyps in mice carrying a truncated Apc gene. Proc Natl Acad Sci U S A. 1995 May 9;92(10):4482–4486. doi: 10.1073/pnas.92.10.4482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Quesada CF, Kimata H, Mori M, et al. Piroxicam and acarbose as chemopreventive agents for spontaneous intestinal adenomas in APC gene 1309 knockout mice. Jpn J Cancer Res. 1998 Apr;89(4):392–396. doi: 10.1111/j.1349-7006.1998.tb00576.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Robanus-Maandag EC, Koelink PJ, Breukel C, et al. A new conditional Apc-mutant mouse model for colorectal cancer. Carcinogenesis. 2010 May;31(5):946–952. doi: 10.1093/carcin/bgq046. [DOI] [PubMed] [Google Scholar]
- 60.Smits R, Kartheuser A, Jagmohan-Changur S, et al. Loss of Apc and the entire chromosome 18 but absence of mutations at the Ras and Tp53 genes in intestinal tumors from Apc1638N, a mouse model for Apc-driven carcinogenesis. Carcinogenesis. 1997 Feb;18(2):321–327. doi: 10.1093/carcin/18.2.321. [DOI] [PubMed] [Google Scholar]
- 61.Roper J, Martin ES, Hung KE. Overview of genetically engineered mouse models of colorectal carcinoma to enable translational biology and drug development. Curr Protoc Pharmacol. 2014;65:14 29 1–14 29 10. doi: 10.1002/0471141755.ph1429s65. [DOI] [PubMed] [Google Scholar]
- 62.Roper J, Hung KE. Priceless GEMMs: genetically engineered mouse models for colorectal cancer drug development. Trends Pharmacol Sci. 2012 Aug;33(8):449–455. doi: 10.1016/j.tips.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 63.Edelmann W, Umar A, Yang K, et al. The DNA mismatch repair genes Msh3 and Msh6 cooperate in intestinal tumor suppression. Cancer Res. 2000 Feb 15;60(4):803–807. [PubMed] [Google Scholar]
- 64.Edelmann W, Yang K, Kuraguchi M, et al. Tumorigenesis in Mlh1 and Mlh1/Apc1638N mutant mice. Cancer Res. 1999 Mar 15;59(6):1301–1307. [PubMed] [Google Scholar]
- 65.Edelmann W, Yang K, Umar A, et al. Mutation in the mismatch repair gene Msh6 causes cancer susceptibility. Cell. 1997 Nov 14;91(4):467–477. doi: 10.1016/s0092-8674(00)80433-x. [DOI] [PubMed] [Google Scholar]
- 66.Reitmair AH, Redston M, Cai JC, et al. Spontaneous intestinal carcinomas and skin neoplasms in Msh2-deficient mice. Cancer Res. 1996 Aug 15;56(16):3842–3849. [PubMed] [Google Scholar]
- 67.Washington MK, Powell AE, Sullivan R, et al. Pathology of rodent models of intestinal cancer: progress report and recommendations. Gastroenterology. 2013 Apr;144(4):705–717. doi: 10.1053/j.gastro.2013.01.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kuraguchi M, Yang K, Wong E, et al. The distinct spectra of tumor-associated Apc mutations in mismatch repair-deficient Apc1638N mice define the roles of MSH3 and MSH6 in DNA repair and intestinal tumorigenesis. Cancer Res. 2001 Nov 1;61(21):7934–7942. [PubMed] [Google Scholar]
- 69.Shoemaker AR, Haigis KM, Baker SM, et al. Mlh1 deficiency enhances several phenotypes of Apc(Min)/+ mice. Oncogene. 2000 May 25;19(23):2774–2779. doi: 10.1038/sj.onc.1203574. [DOI] [PubMed] [Google Scholar]
- 70.Miyaki M, Kuroki T. Role of Smad4 (DPC4) inactivation in human cancer. Biochem Biophys Res Commun. 2003 Jul 11;306(4):799–804. doi: 10.1016/s0006-291x(03)01066-0. [DOI] [PubMed] [Google Scholar]
- 71.Takaku K, Oshima M, Miyoshi H, et al. Intestinal tumorigenesis in compound mutant mice of both Dpc4 (Smad4) and Apc genes. Cell. 1998 Mar 6;92(5):645–656. doi: 10.1016/s0092-8674(00)81132-0. [DOI] [PubMed] [Google Scholar]
- 72.Engle SJ, Hoying JB, Boivin GP, et al. Transforming growth factor beta1 suppresses nonmetastatic colon cancer at an early stage of tumorigenesis. Cancer Res. 1999 Jul 15;59(14):3379–3386. [PubMed] [Google Scholar]
- 73.Shull MM, Ormsby I, Kier AB, et al. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature. 1992 Oct 22;359(6397):693–699. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhu Y, Richardson JA, Parada LF, et al. Smad3 mutant mice develop metastatic colorectal cancer. Cell. 1998 Sep 18;94(6):703–714. doi: 10.1016/s0092-8674(00)81730-4. [DOI] [PubMed] [Google Scholar]
- 75.el Marjou F, Janssen KP, Chang BH, et al. Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis. 2004 Jul;39(3):186–193. doi: 10.1002/gene.20042. [DOI] [PubMed] [Google Scholar]
- 76.Barker N, van Es JH, Kuipers J, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007 Oct 25;449(7165):1003–1007. doi: 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- 77.Powell AE, Wang Y, Li Y, et al. The pan-ErbB negative regulator Lrig1 is an intestinal stem cell marker that functions as a tumor suppressor. Cell. 2012 Mar 30;149(1):146–158. doi: 10.1016/j.cell.2012.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sansom OJ, Reed KR, Hayes AJ, et al. Loss of Apc in vivo immediately perturbs Wnt signaling, differentiation, and migration. Genes Dev. 2004 Jun 15;18(12):1385–1390. doi: 10.1101/gad.287404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hinoi T, Akyol A, Theisen BK, et al. Mouse model of colonic adenoma-carcinoma progression based on somatic Apc inactivation. Cancer Res. 2007 Oct 15;67(20):9721–9730. doi: 10.1158/0008-5472.CAN-07-2735. [DOI] [PubMed] [Google Scholar]
- 80.Xue Y, Johnson R, Desmet M, et al. Generation of a transgenic mouse for colorectal cancer research with intestinal cre expression limited to the large intestine. Mol Cancer Res. 2010 Aug;8(8):1095–1104. doi: 10.1158/1541-7786.MCR-10-0195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Byun AJ, Hung KE, Fleet JC, et al. Colon-specific tumorigenesis in mice driven by Cre-mediated inactivation of Apc and activation of mutant Kras. Cancer Lett. 2014 Jun 1;347(2):191–195. doi: 10.1016/j.canlet.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shibata H, Toyama K, Shioya H, et al. Rapid colorectal adenoma formation initiated by conditional targeting of the Apc gene. Science. 1997 Oct 3;278(5335):120–123. doi: 10.1126/science.278.5335.120. [DOI] [PubMed] [Google Scholar]
- 83.Hung KE, Maricevich MA, Richard LG, et al. Development of a mouse model for sporadic and metastatic colon tumors and its use in assessing drug treatment. Proc Natl Acad Sci U S A. 2010 Jan 26;107(4):1565–1570. doi: 10.1073/pnas.0908682107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Coffee EM, Faber AC, Roper J, et al. Concomitant BRAF and PI3K/mTOR blockade is required for effective treatment of BRAF(V600E) colorectal cancer. Clin Cancer Res. 2013 May 15;19(10):2688–2698. doi: 10.1158/1078-0432.CCR-12-2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Roper J, Sinnamon MJ, Coffee EM, et al. Combination PI3K/MEK inhibition promotes tumor apoptosis and regression in PIK3CA wild-type, KRAS mutant colorectal cancer. Cancer Lett. 2014 Jun 1;347(2):204–211. doi: 10.1016/j.canlet.2014.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Belmont PJ, Budinska E, Jiang P, et al. Cross-species analysis of genetically engineered mouse models of MAPK-driven colorectal cancer identifies hallmarks of the human disease. Dis Model Mech. 2014 Jun;7(6):613–623. doi: 10.1242/dmm.013904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dow LE, Nasr Z, Saborowski M, et al. Conditional reverse tet-transactivator mouse strains for the efficient induction of TRE-regulated transgenes in mice. PLoS One. 2014;9(4):e95236. doi: 10.1371/journal.pone.0095236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dow LE, Fisher J, O'Rourke KP, et al. Inducible in vivo genome editing with CRISPR-Cas9. Nat Biotechnol. 2015 Apr;33(4):390–394. doi: 10.1038/nbt.3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Trobridge P, Knoblaugh S, Washington MK, et al. TGF-beta receptor inactivation and mutant Kras induce intestinal neoplasms in mice via a beta-catenin-independent pathway. Gastroenterology. 2009 May;136(5):1680–1688. e7. doi: 10.1053/j.gastro.2009.01.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chen HJ, Yang BL, Chen YG, et al. A GFP-labeled human colon cancer metastasis model featuring surgical orthotopic implantation. Asian Pac J Cancer Prev. 2012;13(9):4263–4266. doi: 10.7314/apjcp.2012.13.9.4263. [DOI] [PubMed] [Google Scholar]
- 91.Kuo TH, Kubota T, Watanabe M, et al. Liver colonization competence governs colon cancer metastasis. Proc Natl Acad Sci U S A. 1995 Dec 19;92(26):12085–12089. doi: 10.1073/pnas.92.26.12085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Morimoto-Tomita M, Ohashi Y, Matsubara A, et al. Mouse colon carcinoma cells established for high incidence of experimental hepatic metastasis exhibit accelerated and anchorage-independent growth. Clin Exp Metastasis. 2005;22(6):513–521. doi: 10.1007/s10585-005-3585-0. [DOI] [PubMed] [Google Scholar]
- 93.Bouvet M, Tsuji K, Yang M, et al. In vivo color-coded imaging of the interaction of colon cancer cells and splenocytes in the formation of liver metastases. Cancer Res. 2006 Dec 1;66(23):11293–11297. doi: 10.1158/0008-5472.CAN-06-2662. [DOI] [PubMed] [Google Scholar]
- 94.Alamo P, Gallardo A, Pavon MA, et al. Subcutaneous preconditioning increases invasion and metastatic dissemination in mouse colorectal cancer models. Dis Model Mech. 2014 Mar;7(3):387–396. doi: 10.1242/dmm.013995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Alamo P, Gallardo A, Di Nicolantonio F, et al. Higher metastatic efficiency of KRas G12V than KRas G13D in a colorectal cancer model. FASEB J. 2015 Feb;29(2):464–476. doi: 10.1096/fj.14-262303. [DOI] [PubMed] [Google Scholar]
- 96.Donigan M, Loh BD, Norcross LS, et al. A metastatic colon cancer model using nonoperative transanal rectal injection. Surg Endosc. 2010 Mar;24(3):642–647. doi: 10.1007/s00464-009-0650-9. [DOI] [PubMed] [Google Scholar]
- 97.de Jong GM, Aarts F, Hendriks T, et al. Animal models for liver metastases of colorectal cancer: research review of preclinical studies in rodents. J Surg Res. 2009 Jun 1;154(1):167–176. doi: 10.1016/j.jss.2008.03.038. [DOI] [PubMed] [Google Scholar]
- 98. Chen HJ, Sun J, Huang Z, et al. Comprehensive models of human primary and metastatic colorectal tumors in immunodeficient and immunocompetent mice by chemokine targeting. Nat Biotechnol. 2015 May 25; doi: 10.1038/nbt.3239. ** Use of CRC lines engineered to overexpress CCR9 injected intravenously to form primary intestinal tumors and liver metastasis.
- 99.Williams SA, Anderson WC, Santaguida MT, et al. Patient-derived xenografts, the cancer stem cell paradigm, and cancer pathobiology in the 21st century. Lab Invest. 2013 Sep;93(9):970–982. doi: 10.1038/labinvest.2013.92. [DOI] [PubMed] [Google Scholar]
- 100.Uronis JM, Osada T, McCall S, et al. Histological and molecular evaluation of patient-derived colorectal cancer explants. PLoS One. 7(6):e38422. doi: 10.1371/journal.pone.0038422. [DOI] [PMC free article] [PubMed] [Google Scholar]