

Summary
Mycobacterium leprae infection gives rise to the immunologically and histopathologically classified spectrum of leprosy. At present, several tools for the stratification of patients are based on acquired immunity markers. However, the role of innate immunity, particularly the complement system, is largely unexplored. The present retrospective study was undertaken to explore whether the systemic levels of complement activation components and regulators can stratify leprosy patients, particularly in reference to the reactional state of the disease. Serum samples from two cohorts were analysed. The cohort from Bangladesh included multi‐bacillary (MB) patients with (n = 12) or without (n = 46) reaction (R) at intake and endemic controls (n = 20). The cohort from Ethiopia included pauci‐bacillary (PB) (n = 7) and MB (n = 23) patients without reaction and MB (n = 15) patients with reaction. The results showed that the activation products terminal complement complex (TCC) (P ≤ 0·01), C4d (P ≤ 0·05) and iC3b (P ≤ 0·05) were specifically elevated in Bangladeshi patients with reaction at intake compared to endemic controls. In addition, levels of the regulator clusterin (P ≤ 0·001 without R; P < 0·05 with R) were also elevated in MB patients, irrespective of a reaction. Similar analysis of the Ethiopian cohort confirmed that, irrespective of a reaction, serum TCC levels were increased significantly in patients with reactions compared to patients without reactions (P ≤ 0·05). Our findings suggests that serum TCC levels may prove to be a valuable tool in diagnosing patients at risk of developing reactions.
Keywords: complement, leprosy, reactions
Introduction
Leprosy is a chronic debilitating disease caused by Mycobacterium leprae, an intracellular parasite with tropism for macrophages and Schwann cells. Effective treatment with multi‐drug therapy (MDT) has reduced the prevalence around the world, although new case detection has remained stable at approximately 200 000 per annum. Nevertheless, the disease is still endemic in several parts of the world, including parts of Bangladesh, Ethiopia, Brazil and Nepal (http://www.who.int/mediacentre/factsheets/).
Although the infection is asymptomatic for a prolonged period, the disease eventually presents with nerve damage, which is the major cause of patients' disability and deformities. On the basis of clinical, histopathological and immunological criteria, leprosy is recognized as a spectral disease 1. The interindividual variablity of acquired immune response to M. leprae and its antigens dictates the clinical, histopathological and immunological spectrum of leprosy 2. As a result, it is now well established that the leprosy spectrum fluctuates between two poles: tuberculoid leprosy (TT), with a strong M. leprae‐specific T helper type 1 (Th1) cell‐mediated immunity associated with negligible bacillary load, and lepromatous leprosy (LL) with a strong antibody response to M. leprae, phenolic glycolipid‐1 (PGL‐I) accompanied by complete absence of a M. leprae‐specific Th1 response. The polar LL patients show high bacillary load in relation to T cell anergy to M. leprae not only in the lesions, but also in other tissues in a disseminated manner. Between the two polar forms, the majority of patients belong to immunologically unstable borderline categories that are classified as borderline tuberculoid (BT), mid‐borderline (BB) and borderline lepromatous (BL), with variable degrees of bacillary load with an increasing trend from BT towards BL/LL. On the basis of bacillary indices of the lesions LL, together with BB and BL, are grouped collectively as multi‐bacillary (MB), whereas the BT and TT forms are grouped as pauci‐bacillary (PB) 1.
MDT is effective in clearing bacilli load in leprosy patients to a large extent as, in the majority of MB patients, the dead bacilli are cleared steadily. However, a considerable number of patients show a changing clinical and immunohistopathological status in the course of the disease as well as during and post‐treatment either as a result of treatment or as a natural evolution of the disease. Such episodic disease status is recognized widely as a reactional state, resulting in clinical and pathological alterations accompanied by exacerbation of tissue, particularly nerve damage 3, 4.
The change in immunological response results in one of two types of reactions: (i) reversal reaction (RR, also called type 1 reaction) encountered primarily with patients with BT and BL category or (ii) erythema nodosum leprosum (ENL, also called type 2 reaction), especially in the borderline and lepromatous region of the spectrum. Both these episodic reactions appear to be due to the persistence of antigens such as lipoarabinomannan (LAM) or PGL‐I 5. Interestingly, the localization of persisting M. leprae antigens in leprosy patients with nerve damage was also demonstrated by Shetty et al. 6.
In general, RR or type 1 reactions are due to the polarization of M. leprae‐specific T cell activity with the cytokine characteristic of the Th1 profile 7, 8, and usually occur early in the course of treatment and result in an increased cellular immune response to mycobacterial antigens.
Conversely, ENL or type 2 reactions are due to the increased T cell‐dependent antibody production (specifically to M. leprae antigens or to treatment drugs) resulting in immune complex formation and complement activation 9, 10, 11, 12, 13.
Accordingly, efforts to establish sets of biomarkers for laboratory diagnosis and prognosis of leprosy spectrum and leprosy reactions has concentrated on acquired immunity‐based cytokine and antibody profiling of the patients 14, 15, 16, 17. In contrast, biomarkers of innate immunity in leprosy pathomechanism have received little attention. Indeed, studies linking biomarkers of innate immunity with regard to the role of complement in leprosy disease state, particularly to the reactional state, are rare in literature.
The complement system is an integral part of innate immunity, comprising more than 30 serum and cell‐associated proteins, and plays an important role in host immunity and inflammation 18. Its activation and regulation occurs via multiple pathways. Complement activation can be triggered by antigen–antibody complexes (classical pathway), foreign surfaces (alternative pathway) or bacterial sugars (lectin pathway). Regardless of the trigger, activation results in the cleavage of C3, generating the anaphylatoxin C3a and the opsonin C3b, the latter of which binds pathogens, thereby mediating clearance by phagocytes. C3b is also required for the formation of the C5 convertase to cleave C5 into C5a and C5b. C5b initiates activation of the terminal pathway, which results in the formation of the membrane attack complex (MAC) comprising a heteropolymer of C5b, C6, C7, C8 and multiple C9 molecules that forms transmembrane channels in the target cell, resulting in lysis.
Deposits of MAC or the soluble terminal complement complex (TCC) were demonstrated in association with damaged nerve in leprosy patients 19. In this context, we recently showed the association between persistence of the M. leprae antigen LAM and complement activation in the damaged nerve. This finding suggests strongly that complement activation plays a causal role in nerve damage in leprosy. Because nerve damage is prominent in reactional episodes, it is rational to speculate that complement activation and formation of TCC or MAC can be valuable in diagnosing leprosy patients without reaction from those with reactions. There have been only a few studies reported in the literature on the serology of complement activation in leprosy 20, 21, 22, 23. Previous serological studies showed (1) reduced complement hemolytic activity and reduced levels of C4 in LL patients, suggesting consumption of complement in the circulation via activation of the classical or lectin pathway 23, 24; (2) an increased C1q binding activity (the initiator of classical pathway activation) in LL patients with ENL reactions, suggesting involvement of the classical pathway in these patients, and increased C3d levels in 70% of patients with ENL and 18% of patients with uncomplicated LL. Such findings suggested that C3d could be a marker of complement activation, which may be of some practical interest in the early diagnosis of the reactional state 12. Thus, taking the literature findings with our own, we decided to reinvestigate the use of serological markers of complement activation in leprosy reaction, which is the major cause of leprosy‐associated nerve damage.
In this retrospective study we used a state‐of‐the‐art multiplex assay set (Meso Scale) for the quantification of complement activation products and regulators in serum of patients and controls to test whether the quantification of complement products might be valuable in diagnosing leprosy patients without reaction from those with reactions.
Materials and methods
Serum samples
Serum samples were obtained from the stored serum bank collected for a prospective cohort study in two leprosy endemic regions, Bangladesh and Ethiopia 25. Ethical approval of the study protocol was obtained through appropriate ethics committees and written informed consent was obtained from the patients before the samples were collected.
These samples were transported to the Leiden University Medical Centre, Leiden (the Netherlands) for storage. It should be stated with clarity that the serum samples were collected and stored under conditions found suitable for another study 25; complement assay requires the special addition of ethylenediamine tetraacetic acid (EDTA), which was not included in the collection of these sera. We have refrained from dealing with the types of reactions, as the patients' samples were obtained from archive of stored specimens and not with the aim of longitudinal study. Serum samples were stored at −20ºC or below and shipped frozen on dry ice.
Briefly, in this study we used biobanked serum samples of untreated leprosy patients without clinical reactions (and before MDT) and newly diagnosed patients who visited clinics with reactions, and sera were collected in a similar fashion and before starting treatment. In addition, we had the opportunity to follow‐up four of these patients with reaction and also collected samples after the completion of treatment. Serum samples from the Bangladesh cohort consisted of MB patients with (n = 12) or without (n = 46) reactions at intake and endemic controls lacking clinical signs and symptoms of leprosy or TB (n = 20); a replication cohort comprised serum samples of PB (TT and BT) (n = 7), multi‐bacillary (BL and LL) without (n = 23) and with reaction (n = 15) from Ethiopia (Table 1). No endemic control samples were tested for the Ethiopian cohort.
Table 1.
Demographic and clinical data of cases and controls for serological studies.
| Number of cases | Female/male ratio | Age (years) | Ethnicity | Leprosy type |
|---|---|---|---|---|
| 20 | 7 : 13 | 22–45 | Bangladesh | Endemic controls |
| 46 | 12 : 22 | 20–41 | Bangladesh | MB, no reaction |
| 47 | 5 : 14 | 18–44 | Bangladesh | MB, reaction |
| 7 | 3 : 4 | 24–46 | Ethiopia | PB (TT/BT), no reaction |
| 23 | 10 : 13 | 16–41 | Ethiopia | MB (BL/LL), no reaction |
| 15 | 3 : 13 | 18–44 | Ethiopia | MB, reaction |
MB = multi‐bacillary; PB = paucibacillary; BL = borderline lepromatous; TT = tuberculoid leprosy; LL = lepromatous leprosy; BT = borderline tuberculoid.
Meso Scale discovery (MSD) platform
All the complement assays were performed on a novel multiplex developed using the Meso Scale Discovery Platform (MSD; Gaithersburg, MD, USA; www.mesoscale.com). The multiplex set comprised C1s, the activation markers C4d, Bb, iC3b and TCC and the regulator Factor H (FH) (using FH Y402, FH H402 monoclonal antibodies which quantifies the concentration of total FH, as described previously 26) and clusterin (Table 2).
Table 2.
Antibodies for the Meso Scale discovery (MSD) assays.
| Assay | Coating antibody (source) | Detection antibody (source) |
|---|---|---|
| C1s | M81 (Hycult) | F33 (in‐house, B. P. M.) |
| C4d | Neo C4d (A251, Quidel) | C4d (A213, Quidel) |
| Bb | Neo Bb (A252, Quidel) | JC1 (in‐house, B. P. M.) |
| iC3b | Neo iC3b (Hycult) | C3‐30 (in‐house, B. P. M.) |
| TCC | aE11 (Hycult) | E2 (in‐house, B. P. M.) |
| FH Y402 | MBI‐6 (in‐house, B. P. M.) | Ox‐24 |
| FH H402 | MBI‐7 (in‐house, B. P. M.) | Ox‐24 |
| Clusterin | Polyclonal anti‐apolipoprotein J (AB825, Millipore) | MBI‐40 (in‐house, B. P. M.) |
TCC = terminal complement complex; FH = Factor H.
All the in‐house antibodies were established initially as enzyme‐linked immunosorbent assays (ELISAs) for evaluating the level of complement activation products on the MSD platform. The assays were validated extensively, and the results were described in detail previously 26, 27.
Antibody‐coated plates were blocked with bovine serum albumin (BSA)/EDTA/phosphate‐buffered saline (PBS), serum samples and standards diluted in BSA/EDTA/PBS added to wells and incubated for 1 h at room temperature (RT) on a shaker. Plates were washed; the detecting antibody cocktail was added to the plate (Table 2) and incubated for 1 h at RT on a shaker. After washing, the reading buffer (R92TC‐2; Meso Scale Diagnostics, Rockville, MD, USA) was added and plates were read on a MSD Sector Imager 6000 instrument. The data were analysed using SoftMax Pro 4·6 Enterprise Edition (Molecular Devices LLC, Sunnyvale, CA, USA).
Statistical analysis
Data analysis was performed using the GraphPad Prism version 5·0 (GraphPad Software Inc., San Diego, CA, USA) statistical package. Student's t‐test was performed for statistical analyses comparing two groups. For comparison of more than two groups, one‐way analysis of variance (anova) with Bonferroni's multiple comparison post‐hoc test was used when the data were distributed normally. For non‐normally distributed data the Kruskal–Wallis test was used. Differences were considered statistically significant when P ≤ 0·05. The data are presented and expressed as standard error (s.e.) of the mean.
Results
Complement activation and regulation in MB leprosy patients with or without reaction from Bangladesh
Selected complement components, activation products and regulators were measured in serum samples of leprosy patients with or without a reaction from Bangladesh and endemic controls collected under similar conditions, and all samples were collected before the start of MDT. Analytes were selected based on different parts of the complement activation cascade in order to assess the contributions of different activation pathways. Complement components, regulators and activation products were measured on a novel multiplex built on the MSD platform. The Bangladesh leprosy population showed no significant difference in serum C1s values compared to controls (P = 0·442) (data are shown in Table 3). Levels of the classical/lectin pathway activation fragment C4d (P < 0·001 without R; P < 0·001 R) and the alternative pathway fragment Bb (P < 0·01 without R; P < 0·001 R) were increased significantly in MB leprosy patients with or without reaction compared to endemic controls (Fig. 1a,b). C4d levels were also increased significantly in patients with reaction compared to patients without a reaction (P < 0·05) (Fig. 1a). The increased serum levels of C4d in the leprosy reaction patients suggest the involvement of the classical and/or lectin pathway in the reaction process.
Table 3.
Complement levels in the Bangladeshi cohort.
| Endemic controls | MB patients without reaction | MB patients with reaction | ||||
|---|---|---|---|---|---|---|
| Bangladeshi cohort | Mean (mg/l) | SE (mg/l) | Mean (mg/l) | SE (mg/l) | Mean (mg/l) | SE (mg/l) |
| C1s | 94·69 | 2·77 | 99·41 | 3·07 | 90·66 | 4·84 |
| C4d | 3·46 | 0·39 | 8·83 | 0·77 | 13·41 | 1·60 |
| Bb | 16·53 | 1·92 | 24·26 | 0·98 | 28·12 | 1·96 |
| iC3b | 5·42 | 0·63 | 8·63 | 0·81 | 12·05 | 2·71 |
| TCC | 1·87 | 0·15 | 2·27 | 0·09 | 2·56 | 0·10 |
| Factor H | 253·7 | 15·82 | 277·3 | 11·28 | 231·6 | 21·97 |
| Clusterin | 297·3 | 19·74 | 398·0 | 10·15 | 378·9 | 15·87 |
Figure 1.
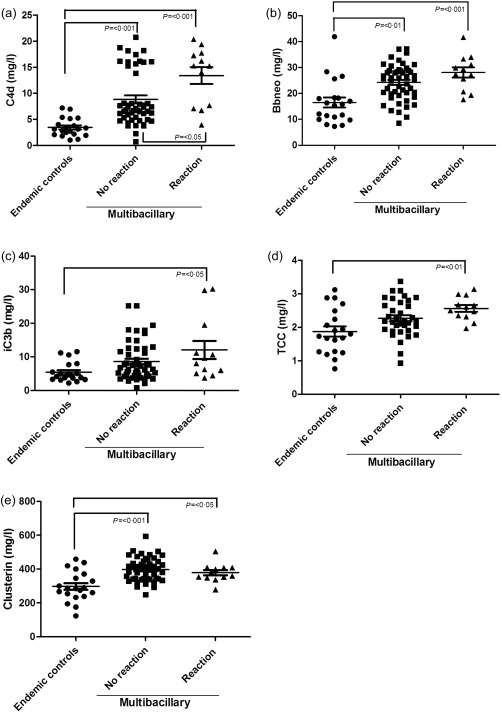
Complement activation and regulation in multi‐bacillary leprosy patients with and without reactions from Bangladesh. Meso Scale discovery (MSD) platform for measuring complement activation products C4d (a), mid‐borderline (Bb) (b), inactive complement component 3b (iC3b) (c), terminal complement complex (TCC) (d) and clusterin (e) in serum from endemic controls (n = 20) and multi‐bacillary leprosy patients with (n = 12) or without (n = 46) reaction at intake, showing a significant increase in multi‐bacillary leprosy patients with and without reaction compared controls for all measured components (P < 0·05). Complement component 4 deposition (C4d) levels are increased specifically and significantly in patients with reaction compared to those without reaction (b) [mean C4d no reaction 8·83 mg/l, standard error (s.e.) = 0·77] versus mean C4d reaction 13·41 mg/l (s.e. = 1·60); P < 0·05]. In addition, clusterin levels were increased significantly in serum of multi‐bacillary leprosy patients compared to endemic controls, but no difference between patients with or without reactions (e) [mean clusterin no reaction 398·0 mg/l (s.e. = 10·15); P < 0·001 versus mean clusterin reaction 378·9 mg/l (s.e. = 15·87); P < 0·05]. The error bars represent the standard error of the mean (s.e.).
The levels of the activation pathway fragment iC3b and the terminal pathway activation marker TCC were raised significantly in leprosy patients with a reaction compared to endemic controls (P < 0·05 and P < 0·01, respectively), showing further evidence for increased complement activation in reactions (Fig. 1c,d). Four patients from Bangladesh who were followed‐up after treatment showed that the TCC levels stay high even after treatment (Supporting information, Fig. S1). This suggests that treatment with either multi‐drug therapy or steroids does not lower complement serum levels in reaction patients.
Serum levels of FH, the principle plasma regulator of the alternative pathway, and clusterin, a plasma regulator of the TCC, were also analysed in serum samples of MB patients with or without reaction from Bangladesh. Total FH levels did not show a significant difference between leprosy patients with or without a reaction compared to controls (P = 0·149) (data are shown in Table 3).
Clusterin levels in leprosy patients with or without reaction were significantly higher than endemic controls (P < 0·05 without R; P < 0·001 with R) (Fig. 1e), but not different between the patient groups.
TCC levels are increased in serum of MB leprosy patients with reaction compared to patients without reaction from Ethiopia
In the preceding section, results using the leprosy serum samples from Bangladesh data indicate that levels of complement activation products were higher in patients with reaction compared to those without – significantly for C4d, iC3b and TCC. We therefore suggest that these markers could be used as an indicator for identifying patients with reactions. In order to substantiate this finding we measured the levels of these complement activation products in serum samples of an Ethiopian leprosy cohort. These samples were also collected before the start of MDT and classified as PB (TT and BT) or MB (BL and LL) leprosy patients without reaction and MB patients with reaction. In these samples, like those of Bangladesh, no significant difference was found in the level of C1s in serum of MB compared to PB patients without reaction (P = 0·384) (data are shown in Table 4). Serum levels of the complement activation products in MB patients compared to PB patients without a reaction were increased significantly for C4d (P = 0·04), Bb (P = 0·03) iC3b (P = 0·02) and TCC (P = 0·003) (Supporting information, Fig. S2). No significant difference was found in the levels of C4d and Bb in the MB leprosy patients without reaction compared to the patients with a reaction (P = 0·392 and P = 0·143, respectively) (data is shown in Table 4). Levels of the common complement pathway marker iC3b were not significantly different between MB leprosy patients without reaction compared to those with a reaction (Fig. 2a). However, levels of TCC in Ethiopian cohort MB patients with a reaction were raised significantly in comparison to PB (P < 0·01) patients and to MB patients without reaction (P < 0·05) (Fig. 2b). This latter result confirms independently that raised plasma levels of TCC represent a potential tool for identifying leprosy patients with reactions.
Table 4.
Complement levels in the Ethiopian cohort.
| Pauci‐bacillary (PB) patients without reaction | Multi‐bacillary (MB) patients without reaction | MB patients with reaction | ||||
|---|---|---|---|---|---|---|
| Ethiopian cohort | Mean (mg/l) | s.e. (mg/l) | Mean (mg/l) | s.e. (mg/l) | Mean (mg/l) | s.e. (mg/l) |
| C1s | 112·4 | 9·71 | 128·2 | 8·66 | 134·2 | 8·59 |
| C4d | 17·99 | 1·99 | 27·15 | 4·04 | 36·73 | 4·93 |
| Bb | 24·91 | 2·39 | 28·62 | 1·99 | 36·75 | 2·72 |
| iC3b | 13·15 | 1·94 | 22·39 | 1·92 | 14·80 | 2·84 |
| TCC | 2·02 | 0·23 | 4·03 | 0·28 | 7·49 | 1·48 |
| Factor H | 385·0 | 26·85 | 301·9 | 14·15 | 381·1 | 22·61 |
| Clusterin | 457·9 | 34·08 | 493·8 | 21·94 | 656·1 | 43·70 |
Figure 2.
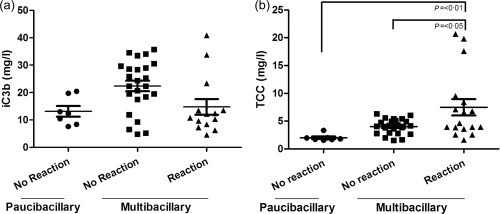
Terminal complement complex (TCC) levels are increased in serum of multibacillary leprosy patients with reaction compared to patients from Ethiopia without reaction. Meso Scale discovery (MSD) assay for measuring the common complement pathway activation fragment iC3b (a) and the terminal pathway activation marker TCC (b), in serum samples of pauci‐bacillary (n = 7) and multi‐bacillary patients without a reaction (n = 23) or with a reaction (n = 15). Although no significant difference was found in the levels of iC3b in serum of pauci‐bacillary and multi‐bacillary patients without and with a reaction, TCC levels were increased significantly in multi‐bacillary leprosy patients with a reaction comparted to pauci‐bacillary and multi‐bacillary patients without a reaction [mean TCC no reaction 4·03 mg/l [standard error (s.e.) = 0·28) versus mean TCC reaction 7·49 mg/l (s.e. = 1·48); P < 0·01 and P < 0·05]. The error bars represent the standard error of the mean (s.e.).
Discussion
Nerve damage leading to deformity is a major problem in the course of leprosy and progression of the disease among susceptible hosts. In the absence of the peripheral neuropathy, leprosy would be an innocuous inflammatory skin disease rather than one that, even today in the 21st century, is one of the most stigmatized diseases, often associated with severe social repercussions for the patient 28, 29. The aetiology of leprosy reactions and nerve damage has been attributed largely to the consequence of granulomatous reactions due to the host immune response to M. leprae. Variability in the hosts' immune response has a causal relationship to the spectral pathology of the disease. Timely diagnosis followed by optimum and evidence‐based treatment would reduce risks of permanent tissue damage and assist regeneration of the damaged tissue 30. Despite continued efforts in the refinement of diagnostic methods reactions are often misdiagnosed, even by experienced health workers and clinicians 31. Unquestionably, a reliable test or combination of tests for the prompt diagnosis of reactions, particularly RR or type 1, would contribute positively to the clinical outcome of the patients. Biomarkers for identification of patients developing reactions have focused on circulating parameters of adaptive immunity, such as antibodies, cytokines and chemokines, as well as gene expression profiles 32. However, a recent report described the importance of innate immunity in orchestrating leprosy immunopathology and in the initiating phase of host defence against the pathogen 33. Innate immune effectors include cellular systems that are now being studied in relation to mycobacterial pathology, and also humoral effectors, including the complement system, are critical in initiating and orchestrating the innate immune response to infection 34, 35. These findings imply that the analysis of the complement system might be of value for the diagnosis and prognosis of leprosy disease status.
It is now well appreciated that complement activation can be induced by pathogen‐associated molecular patterns (PAMPs) 18, 34, 35, 36. Particularly in the context of leprosy, we showed recently that the membrane attack complex (MAC) of complement is generated upon cognate interaction of the axon of the peripheral nerve with the M. leprae‐specific PAMP LAM 19. In addition, we showed that MAC formation results in nerve damage, and its inhibition is neuroprotective in a mouse model for M. leprae‐induced nerve damage. We also demonstrated that MAC is deposited in nerve lesions of leprosy patients, paralleling the observed nerve degeneration. Our findings indicate an important role for complement in the disease, and therefore we examined whether increased systemic levels of complement activation could be detected in leprosy patients and whether complement products and regulators might be useful markers of the leprosy disease state.
The present study reports, for the first time, the quantification of a panel of complement activation products and regulators in serum of leprosy patients in multiplexed assays on the Meso Scale Discovery (MSD) platform. The study was performed retrospectively using serum samples from PB and MB patients with or without reaction collected in Ethiopia and Bangladesh for a previous study 25.
Here we show significantly increased levels of complement activation products C4d and Bb in serum of Bangladeshi leprosy patients compared to endemic controls. The increased levels of C4d in the patients supports activation of the lectin and/or classical pathway of complement, while elevated Bb supports a significant involvement of the alternative pathway in leprosy patients. Earlier studies had already suggested a role for the classical pathway of the complement system in leprosy across the spectrum by measuring the ability of circulating immune complexes isolated from sera of leprosy patients to activate complement, but none of these studies measured pathway‐specific activation products 23, 24, 37.
Bacterial antigens either, in the free form or by slow multiplication of bacilli in the reaction patients, could initiate complement activation via the lectin pathway, initiated by the binding of the pattern recognition molecule mannose‐binding lectin (MBL) to bacterial surfaces 38. In support of the involvement of the lectin pathway in MB leprosy patients, one study found that MBL serum levels were increased significantly in the LL form of leprosy compared to other leprosy types 23. However, it should be noted that the association of MBL with leprosy pathophysiology is controversial, because some investigators found increased levels of MBL in the patients undergoing reaction. For this reason, no confirmatory conclusion can be drawn from our present limited study on the role of MBL pathway in contributing a higher level of complement activation products in leprosy reactions.iC3b levels in the Bangladeshi population were increased significantly in leprosy patients with reaction compared to controls. The levels were also higher compared to patients without a reaction, although not significant.
The present study is limited in identifying the major contribution of either the classical or lectin pathways for complement activation, TCC levels in serum of leprosy patients from Bangladesh were significantly higher in patients with reaction compared to endemic controls. No significant difference was found between leprosy patients without reactions and endemic controls for TCC. This reflects that complement TCC level may be considered as a promising marker for patients developing a reaction. It also suggests that treatment with either MDT or steroids does not lower complement activation in reaction patients, reinforcing the possibility that complement contributes to the nerve damage in these patients.
Similar to the findings encountered with theBangladeshi cohort, TCC levels in serum samples of leprosy patients from Ethiopia also showed a significant increase in reactions compared to patients without reactions. As complement activation is under control of several regulatory molecules, we also measured the levels of FH, regulator of the alternative pathway of complement, in leprosy patients with or without reactions; previous studies showed that FH is associated with a variety of diseases, including neurodegenerative disease 39, 40, 41, 42. Changes in FH levels might suggest altered regulation of activation of the alternative pathway of complement during disease. We did not detect any significant change in the total level of FH in serum of leprosy patients with and without a reaction. Clusterin, a fluid‐phase regulator of the MAC, is increased in tissue and plasma in different diseases, including neurodegenerative conditions 43, 44, 45, 46, 47, 48. Hence, we determined the circulatory levels of clusterin in leprosy patients in association with disease activities. Clusterin levels were higher in leprosy patients compared to controls. No difference was found in clusterin levels in patients with or without reaction, suggesting no increase in regulatory status of the terminal pathway of complement by clusterin in reaction patients.
Unfortunately, due to the nature of the study (1) being carried out with the samples collected in the field situations and (2) the lack of EDTA in the serum samples, any elaborate interpretation regarding the mechanism of high level of TCC in leprosy patients with reaction should be avoided at this stage of the study. The lack of EDTA in particular might result in in‐vitro generation of TCC, which continues in serum. This limitation is considered equally applicable to all the samples studied; therefore, any comparative values relating the disease state is considered as a true reflection.
Despite such limitations, the present data sets have demonstrated convincingly that systemic activation of complement occurs in leprosy and probably involves multiple pathways. The increased serum levels of TCC and other activation markers in leprosy patients with reaction warrants a prospective and sequential study on complement activation in leprosy, to confirm conclusively whether circulating complement activation products such as TCC can be applied as biomarkers for diagnosis of patients at risk of developing a reaction.
Disclosure
F. B. and V. R. are inventors on a patent that describes the use of inhibitors of the terminal complement pathway for therapeutic purposes. F. B. and V. R. are shareholders of Regenesance BV. The remaining authors declare no disclosures.
Author contributions
P. K. D., V. R. and F. B. formulated the project; A. G. provided access to the stored serum samples collected for a different prospective study; B. P. M. provided access to the in‐house complement assays and advised on the project; S. H. performed assays; N. B. E. I. analysed the data and produced the first draft of the manuscript; P. K. D., F. B. and V. R. assisted N. B. E. I. in generating the final version of the manuscript.
Supporting information
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Fig. S1. Terminal complement complex (TCC) serum levels of leprosy patients followed at reaction and after treatment. TCC serum levels of leprosy patients at reaction do not change after treatment, indicating that treatment does not affect complement activity in leprosy.
Fig. S2. Complement activation in pauci‐bacillary (PB) and multi‐bacillary (MB) leprosy patients from Ethiopia. Meso Scale discovery (MSD) platform for measuring complement activation products C4d (a), mid‐borderline (Bb) (b), inactive complement activation fragment (iC3b) (c) and terminal complement complex (TCC) (d) in serum from PB (n = 7) and MB (n = 23) leprosy patients, showing a significant increase in MB compared to PB leprosy patients for C4d [mean PB = 7·99 mg/l, standard error (s.e.) = 1·99 versus mean MB = 27·15 mg/l, s.e. = 4·04; P = 0·04], Bb (mean PB = 24·91 mg/l, s.e. = 2·39 versus mean MB = 28·62 mg/l, s.e. = 1·99; P = 0.03), iC3b (mean PB = 13·15 mg/l, s.e. = 1·94 versus mean MB = 22·39 mg/l, s.e. = 1·92; P = 0·02) and TCC (mean PB = 2·02 mg/l, s.e. = 0·23 versus MB = 4·03 mg/l, s.e. = 0·28; P = 0.003). The error bars represent the standard error of the mean.
Acknowledgements
The authors would like to thank Dr Sheikh Abdul Hadi (Dhaka), Genet Amare, Haregewoin Yetesha and Alemayehu Kifle (AHRI/ALERT, Addis Ababa), Dr Sayera Banu from the International Center for Diarrhoeal Disease Research Bangladesh, Dhaka (Bangladesh) and Dr Kidist Bobosha from the Armauer Hansen Research Institute, Addis Ababa in Ethiopia for sample collection, without which this study could not have been possible. This work was supported by the Leprosy Foundation of the Netherlands (grant number 701.03.08). A. G. acknowledges the support of the QM Gastmann Wichers Foundation for her participation.
References
- 1. Ridley DS, Jopling WH. Classification of leprosy according to immunity. A five‐group system. Int J Lepr Other Mycobact Dis 1966; 34:255–73. [PubMed] [Google Scholar]
- 2. Scollard DM, Adams LB, Gillis TP, Krahenbuhl JL, Truman RW, Williams DL. The continuing challenges of leprosy. Clin Microbiol Rev 2006; 19:338–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ridley DS. Reactions in leprosy. Lepr Rev 1969; 40:77–81. [DOI] [PubMed] [Google Scholar]
- 4. Godal T, Myrvang B, Samuel DR, Ross WF, Lofgren M. Mechanism of ‘reactions’ in borderline tuberculoid (BT) leprosy. A preliminary report. Acta Pathol Microbiol Scand Suppl 1973; 236:45–53. [PubMed] [Google Scholar]
- 5. Verhagen C, Faber W, Klatser P, Buffing A, Naafs B, Das P. Immunohistological analysis of in situ expression of mycobacterial antigens in skin lesions of leprosy patients across the histopathological spectrum. Association of mycobacterial lipoarabinomannan (LAM) and Mycobacterium leprae phenolic glycolipid‐I (PGL‐I) with leprosy reactions. Am J Pathol 1999; 154:1793–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shetty VP, Uplekar MW, Antia NH. Immunohistological localization of mycobacterial antigens within the peripheral nerves of treated leprosy patients and their significance to nerve damage in leprosy. Acta Neuropathol 1994; 88:300–6. [DOI] [PubMed] [Google Scholar]
- 7. Verhagen CE, Wierenga EA, Buffing AA, Chand MA, Faber WR, Das PK. Reversal reaction in borderline leprosy is associated with a polarized shift to type 1‐like Mycobacterium leprae T cell reactivity in lesional skin: a follow‐up study. J Immunol 1997; 159:4474–83. [PubMed] [Google Scholar]
- 8. Sreenivasan P, Misra RS, Wilfred D, Nath I. Lepromatous leprosy patients show T helper 1‐like cytokine profile with differential expression of interleukin‐10 during type 1 and 2 reactions. Immunology 1998; 95:529–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wemambu SN, Turk JL, Waters MF, Rees RJ. Erythema nodosum leprosum: a clinical manifestation of the arthus phenomenon. Lancet 1969; 2:933–5. [DOI] [PubMed] [Google Scholar]
- 10. Das PK, Klatser PR, Pondman KW et al Dapsone and anti‐dapsone antibody in circulating immune complexes in leprosy patients. Lancet 1980; 1:1309–11. [DOI] [PubMed] [Google Scholar]
- 11. Modlin RL. Th1–Th2 paradigm: insights from leprosy. J Invest Dermatol 1994; 102:828–32. [DOI] [PubMed] [Google Scholar]
- 12. Bjorvatn B, Barnetson RS, Kronvall G, Zubler RH, Lambert PH. Immune complexes and complement hypercatabolism in patients with leprosy. Clin Exp Immunol 1976; 26:388–96. [PMC free article] [PubMed] [Google Scholar]
- 13. Kahawita IP, Lockwood DN. Towards understanding the pathology of erythema nodosum leprosum. Trans R Soc Trop Med Hyg 2008; 102:329–37. [DOI] [PubMed] [Google Scholar]
- 14. Stefani MM, Guerra JG, Sousa AL et al Potential plasma markers of Type 1 and Type 2 leprosy reactions: a preliminary report. BMC Infect Dis 2009; 9:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Little D, Khanolkar‐Young S, Coulthart A, Suneetha S, Lockwood DN. Immunohistochemical analysis of cellular infiltrate and gamma interferon, interleukin‐12, and inducible nitric oxide synthase expression in leprosy type 1 (reversal) reactions before and during prednisolone treatment. Infect Immun 2001; 69:3413–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Partida‐Sanchez S, Favila‐Castillo L, Pedraza‐Sanchez S et al IgG antibody subclasses, tumor necrosis factor and IFN‐gamma levels in patients with type II lepra reaction on thalidomide treatment. Int Arch Allergy Immunol 1998; 116:60–6. [DOI] [PubMed] [Google Scholar]
- 17. Iyer A, Hatta M, Usman R et al Serum levels of interferon‐gamma, tumour necrosis factor‐alpha, soluble interleukin‐6R and soluble cell activation markers for monitoring response to treatment of leprosy reactions. Clin Exp Immunol 2007; 150:210–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol 2010; 11:785–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bahia El Idrissi N, Das PK, Fluiter K et al M. leprae components induce nerve damage by complement activation: identification of lipoarabinomannan as the dominant complement activator. Acta Neuropathol 2015; 129:653–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Saitz EW, Dierks RE, Shepard CC. Complement and the second component of complement in leprosy. Int J Lepr Other Mycobact Dis 1968; 36:400–4. [PubMed] [Google Scholar]
- 21. Petchclai B, Chutanondh R, Prasongsom S, Hiranras S, Ramasoota T. Complement profile in leprosy. Am J Trop Med Hyg 1973; 22:761–4. [DOI] [PubMed] [Google Scholar]
- 22. Gelber RH, Drutz DJ, Epstein WV, Fasal P. Clinical correlates of C1Q‐precipitating substances in the sera of patients with leprosy. Am J Trop Med Hyg 1974; 23:471–5. [DOI] [PubMed] [Google Scholar]
- 23. Gomes GI, Nahn EP Jr, Santos RK, Da Silva WD, Kipnis TL. The functional state of the complement system in leprosy. Am J Trop Med Hyg 2008; 78:605–10. [PubMed] [Google Scholar]
- 24. Tyagi P, Ramanathan VD, Girdhar BK, Katoch K, Bhatia AS, Sengupta U. Activation of complement by circulating immune complexes isolated from leprosy patients. Int J Lepr Other Mycobact Dis 1990; 58:31–8. [PubMed] [Google Scholar]
- 25. Khadge S, Banu S, Bobosha K et al Longitudinal immune profiles in type 1 leprosy reactions in Bangladesh, Brazil, Ethiopia and Nepal. BMC Infect Dis 2015; 15:477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hakobyan S, Harris CL, Tortajada A et al Measurement of factor H variants in plasma using variant‐specific monoclonal antibodies: application to assessing risk of age‐related macular degeneration. Invest Ophthalmol Vis Sci 2008; 49:1983–90. [DOI] [PubMed] [Google Scholar]
- 27. Ingram G, Hakobyan S, Hirst CL et al Systemic complement profiling in multiple sclerosis as a biomarker of disease state. Mult Scler 2012; 18:1401–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rafferty J. Curing the stigma of leprosy. Lepr Rev 2005; 76:119–26. [PubMed] [Google Scholar]
- 29. Tsutsumi A, Izutsu T, Islam AM, Maksuda AN, Kato H, Wakai S. The quality of life, mental health, and perceived stigma of leprosy patients in Bangladesh. Soc Sci Med 2007; 64:2443–53. [DOI] [PubMed] [Google Scholar]
- 30. Lockwood DN, Saunderson PR. Nerve damage in leprosy: a continuing challenge to scientists, clinicians and service providers. Int Health 2012; 4:77–85. [DOI] [PubMed] [Google Scholar]
- 31. Raffe SF, Thapa M, Khadge S, Tamang K, Hagge D, Lockwood DN. Diagnosis and treatment of leprosy reactions in integrated services – the patients' perspective in Nepal. PLoS Negl Trop Dis 2013; 7:e2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Geluk A, van Meijgaarden KE, Wilson L et al Longitudinal immune responses and gene expression profiles in type 1 leprosy reactions. J Clin Immunol 2014; 34:245–55. [DOI] [PubMed] [Google Scholar]
- 33. Montoya D, Modlin RL. Learning from leprosy: insight into the human innate immune response. Adv Immunol 2010; 105:1–24. [DOI] [PubMed] [Google Scholar]
- 34. Walport MJ. Complement. First of two parts. N Engl J Med 2001; 344:1058–66. [DOI] [PubMed] [Google Scholar]
- 35. Walport MJ. Complement. Second of two parts. N Engl J Med 2001; 344:1140–4. [DOI] [PubMed] [Google Scholar]
- 36. Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev 2009; 22:240–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ramanathan VD, Thyagi P, Ramanathan U, Katoch K, Ramu G. A sequential study of circulating immune complexes, complement and immunoglobulins in borderline tuberculoid leprosy patients with and without reactions. Indian J Lepr 1998; 70:153–60. [PubMed] [Google Scholar]
- 38. Ip WK, Takahashi K, Ezekowitz RA, Stuart LM. Mannose‐binding lectin and innate immunity. Immunol Rev 2009; 230:9–21. [DOI] [PubMed] [Google Scholar]
- 39. Oksjoki R, Jarva H, Kovanen PT, Laine P, Meri S, Pentikainen MO. Association between complement factor H and proteoglycans in early human coronary atherosclerotic lesions: implications for local regulation of complement activation. Arterioscler Thromb Vasc Biol 2003; 23:630–6. [DOI] [PubMed] [Google Scholar]
- 40. Edwards AO, Ritter R III, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement factor H polymorphism and age‐related macular degeneration. Science 2005; 308:421–4. [DOI] [PubMed] [Google Scholar]
- 41. Pickering MC, Cook HT. Translational mini‐review series on complement factor H: renal diseases associated with complement factor H: novel insights from humans and animals. Clin Exp Immunol 2008; 151:210–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Thambisetty M, Hye A, Foy C et al Proteome‐based identification of plasma proteins associated with hippocampal metabolism in early Alzheimer's disease. J Neurol 2008; 255:1712–20. [DOI] [PubMed] [Google Scholar]
- 43. Rosenberg ME, Silkensen J. Clusterin and the kidney. Exp Nephrol 1995; 3:9–14. [PubMed] [Google Scholar]
- 44. Jenne DE, Tschopp J. Molecular structure and functional characterization of a human complement cytolysis inhibitor found in blood and seminal plasma: identity to sulfated glycoprotein 2, a constituent of rat testis fluid. Proc Natl Acad Sci USA 1989; 86:7123–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Silkensen JR, Schwochau GB, Rosenberg ME. The role of clusterin in tissue injury. Biochem Cell Biol 1994; 72:483–8. [DOI] [PubMed] [Google Scholar]
- 46. Rosenberg ME, Silkensen J. Clusterin: physiologic and pathophysiologic considerations. Int J Biochem Cell Biol 1995; 27:633–45. [DOI] [PubMed] [Google Scholar]
- 47. Nilselid AM, Davidsson P, Nagga K, Andreasen N, Fredman P, Blennow K. Clusterin in cerebrospinal fluid: analysis of carbohydrates and quantification of native and glycosylated forms. Neurochem Int 2006; 48:718–28. [DOI] [PubMed] [Google Scholar]
- 48. Schrijvers EM, Koudstaal PJ, Hofman A, Breteler MM. Plasma clusterin and the risk of Alzheimer disease. JAMA 2011; 305:1322–26. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Fig. S1. Terminal complement complex (TCC) serum levels of leprosy patients followed at reaction and after treatment. TCC serum levels of leprosy patients at reaction do not change after treatment, indicating that treatment does not affect complement activity in leprosy.
Fig. S2. Complement activation in pauci‐bacillary (PB) and multi‐bacillary (MB) leprosy patients from Ethiopia. Meso Scale discovery (MSD) platform for measuring complement activation products C4d (a), mid‐borderline (Bb) (b), inactive complement activation fragment (iC3b) (c) and terminal complement complex (TCC) (d) in serum from PB (n = 7) and MB (n = 23) leprosy patients, showing a significant increase in MB compared to PB leprosy patients for C4d [mean PB = 7·99 mg/l, standard error (s.e.) = 1·99 versus mean MB = 27·15 mg/l, s.e. = 4·04; P = 0·04], Bb (mean PB = 24·91 mg/l, s.e. = 2·39 versus mean MB = 28·62 mg/l, s.e. = 1·99; P = 0.03), iC3b (mean PB = 13·15 mg/l, s.e. = 1·94 versus mean MB = 22·39 mg/l, s.e. = 1·92; P = 0·02) and TCC (mean PB = 2·02 mg/l, s.e. = 0·23 versus MB = 4·03 mg/l, s.e. = 0·28; P = 0.003). The error bars represent the standard error of the mean.