

Summary
Compelling evidence suggests that interleukin (IL)‐17 and IL‐17‐producing cells play a pivotal role in the pathogenesis of primary Sjögren's syndrome (pSS). We investigated phenotypical and functional effects of the anti‐CD20 antibody rituximab (RTX) on circulating and glandular IL‐17‐producing T cells in pSS. RTX is able to deplete glandular IL‐17+ CD3+CD4–CD8– double‐negative (DN) and CD4+ Th17 cells as well as circulating IL‐17+ DN T cells. A fraction of glandular and circulating IL‐17+ DN cells and CD4+ T helper type 17 (Th17) cells co‐expresses CD20 on the cell surface explaining, at least in part, such depletive capacity of RTX. The exposure to RTX does not rescue the in‐vitro corticosteroid resistance of IL‐17+ DN T cells. Our results support further the therapeutic role in pSS of RTX that, despite its B cell specificity, appears able to also hamper IL‐17‐producing T cells in this disease.
Keywords: rituximab, salivary glands, Sjögren's syndrome, Th17 cells
Introduction
Primary Sjögren's syndrome (pSS) is a heterogeneous systemic autoimmune disease characterized primarily by a focal chronic inflammation of exocrine glands. However, a subgroup of pSS patients experience extraglandular involvement, leading to a worsening of disease prognosis 1, 2, 3. Furthermore, 5% of patients may develop lymphoma, which represents the most severe complication of the disease 4. Although the pathogenesis of pSS is not clarified completely, it is well established that an aberrant autoimmune response with T and B lymphocyte hyperactivity and autoantibody production represents the crucial mechanism for the induction of autoimmune epithelitis and the perpetuation of inflammation. Moreover, several lines of evidence in animal models and pSS patients suggest that the proinflammatory cytokine interleukin (IL)‐17 and, therefore, IL‐17‐producing T helper cells (Th17) play a pivotal role in the induction and perpetuation of chronic inflammation occurring in this disease 5, 6, 7. We have previously demonstrated that besides CD3+CD4+ T helper type 17 (Th17) cells, a small CD3+ T cell subset lacking both CD4 and CD8 on the cell surface (double‐negative, DN), is expanded in the peripheral blood (PB), infiltrates minor salivary glands (MSGs) of patients with pSS and produces IL‐17 spontaneously 8. On the contrary, DN T cells are absent in MSGs of subjects with sicca symptoms but without any clinical, serological or histological features of pSS, highlighting their pathogenic role in this disease. Similarly, DN T cells have been identified in cervical lymph nodes, spleen and submandibular salivary glands of mouse models of SS 9, 10. Finally, the MSG‐DN cell percentage is correlated directly with the severity of glandular involvement and with ectopic lymphoid neogenesis, being higher in MSGs that display more severe glandular damage and germinal centre (GC)‐like structures 11. Unlike DN T cells from healthy donors (HD), we demonstrated previously that circulating pSS‐DN T cells appear to be resistant to corticosteroids (CS) and to the immune‐modulatory effects of mesenchymal stem cells (MSCs) in vitro 8, 12.
To date, therapeutic approaches in pSS are mainly empirical, due to the limited evidence available for most drugs and to the difficulties in elaborating solid therapeutic recommendations 13, 14. Because mounting evidence points to a central role for B cells in pSS pathogenesis, B cell depletion with the anti‐CD20 antibody rituximab (RTX) is also being evaluated as a treatment in this disease 15, 16. Studies that enumerated circulating lymphocyte subpopulations in patients with autoimmune diseases treated with RTX have been focusing mainly on the B cell compartment, due to the mechanism of action of this compound. However, it has been observed that RTX is also able to deplete circulating total CD3+ T cells 17 as well as circulating and synovial Th17 cells in rheumatoid arthritis (RA) 18, 19. With regard to pSS, although a previous study reported that the overall percentage of circulating effector T cells did not change significantly after RTX treatment 20, the observation that RTX reduces glandular IL‐17 and IL‐22 expression 21, 22 raised the hypothesis that at least some pathogenic T cell subpopulations may be targeted.
The aims of the present study were to investigate the effects of RTX on glandular and circulating IL‐17‐producing T cell subsets in pSS patients and to identify the phenotypical and functional features underlying such modulation in an attempt to strengthen the rational of RTX employment in this disease.
Materials and methods
Patients
Twenty‐two female patients with early and active pSS were treated with either RTX (12 patients) or disease‐modifying anti‐rheumatic drugs (DMARDs) (10 patients). These patients were part of a previously reported trial and fulfilled the American European Consensus Group criteria for pSS, including histopathological criteria 23, 24. Briefly, inclusion criteria were: (i) pSS diagnosis, (ii) recent onset of the disease (maximum 2 years from the onset of first symptoms related to the disease) and (iii) active disease, as defined by values >50 mm for two of four visual analogical scales (VAS; 0–100 mm): global disease activity (including extraglandular manifestations), pain, sicca symptoms and fatigue, and a EULAR Sjögren's syndrome disease activity index (ESSDAI) chosen arbitrarily ≥6 25. Patients in the RTX arm received an infusion of 1000 mg rituximab (Mabthera®; Hoffmann‐La Roche AG, Basel, Switzerland) at days 1 and at 15 to complete a course of therapy. This course was repeated every 24 weeks. During the study period, all patients received two courses of therapy with RTX and a stable dose of prednisone. To minimize side effects, all patients were pretreated with methylprednisolone (40 mg intravenously), paracetamol (1000 mg orally) and chlorpheniramine (10 mg intravenously). Patients in the DMARD arm were treated with one or more DMARDs (hydroxychloroquine, methotrexate, cyclosporin) that are largely used in this disease, although none of these has been licensed for pSS, in association with a stable dose of prednisone, according to clinical manifestations.
The entire study was approved by the local Ethic Committee (Local Health Authority ASL 1 Avezzano‐Sulmona‐L'Aquila Ethics Committee) and performed according to the Good Clinical Practice guidelines. All patients provided written informed consent in accordance with the declaration of Helsinki.
Cell isolation and flow cytometry
Blood samples were collected at baseline (before starting the first course of RTX (T0) and at months 3 (T3) and 6 (before starting the second course of RTX, T6) for patients in the RTX arm. Similarly, blood samples were collected at T0, before starting DMARDs, T3 and T6 for patients in the DMARD arm. Peripheral blood mononuclear cells (PBMCs) were isolated by gradient separation (Lympholyte; Cedarlane, Burlington, ON, Canada). MSGs from five of the 22 treatment‐naive pSS patients were washed extensively and mashed gently in phosphate‐buffered saline to obtain cell suspensions. Phenotypical characterization of IL‐17‐producing T cell subsets was performed as described elsewhere 8, 9, 10, 11. For surface staining, phycoerythrin (PE), fluorescein isothiocyanate (FITC) or phycoerythrin‐cyanin 7 (PE‐Cy7)‐labelled anti‐human CD3, CD4, CD8, CD20, and the respective isotypes, were used (BD Biosciences, San Jose, CA, USA). For intracellular staining, cells were stimulated for 6 h in vitro with 25 ng/ml phorbol 12‐myristate 13‐acetate (PMA), 1 mg/ml ionomycin and 0·1 mg/ml brefeldin in complete medium, fixed with 4% paraformaldehyde after surface staining and subsequently permeabilized with 0·1% saponin blocking buffer. Alexa Fluor 647‐labelled anti‐human IL‐17, RAR‐related orphan receptor‐γt (ROR‐γt) and their isotypes were used (BD Biosciences). Up to four different fluorochromes were used in the same vial and debris were excluded by back‐gating to CD3+ T cells in forward‐/side‐scatter (FSC/SSC) plots. Samples were analysed using fluorescence activated cell sorter (FACS)Calibur flow cytometer and CellQuest ProTM software (BD Biosciences).
Serum IL‐17 assessment
IL‐17 concentration in serum samples obtained at T0, T3 and T6 was assessed with the human IL‐17 Quantikine enzyme‐linked immunosorbent assay (ELISA) kit (R&D Systems, Minneapolis, MN, USA), according to the manufacturer's instructions.
Histological evaluation
MSGs from all patients treated with either RTX (no. 12) or DMARDs (no. 10) who underwent biopsy at T0 and after two courses of therapy (month 12, T12) were evaluated. As reported previously 4, focus score was assessed in haematoxylin and eosin (H&E)‐stained sections. In addition, cellular infiltrate, T and B cell compartmentalization and lymphoid organization with the presence of GC‐like structures were assessed by immunofluorescence staining of sequential sections. T and B lymphocytes were identified by double staining with anti‐CD3 and anti‐CD20 antibodies. Monoclonal mouse anti‐human CD3 with secondary antibody goat anti‐mouse/Alexa Fluor‐555 and monoclonal mouse anti‐human CD20 with biotin goat anti‐mouse immunoglobulin (Ig)G and streptavidin/Alexa Fluor‐488 (1 : 200) were employed. Lymphoid‐like organization was assessed by the identification of a follicular dendritic cell (FDC) network with positive staining for CD21 using mouse anti‐human CD21 (1 : 20) followed by secondary antibody goat anti‐mouse/Alexa Fluor‐488. A biopsy was considered GC‐positive (GC+) if displaying at least one focus with lymphoid‐like organization within normal MSG tissue. Focal infiltrates without lymphoid organization were classified as GC‐negative (GC–). Moreover, double immunofluorescence staining aimed to localize and quantify DN T cells was performed with monoclonal rabbit anti‐human CD3, mouse anti‐human CD4 and mouse anti‐human CD8 primary antibodies followed by anti‐rabbit/Alexa Fluor‐488 and antibody anti‐mouse/Alexa Fluor‐568 11. Images were acquired using an Olympus BX53 fluorescence microscope with CellSens software (Olympus America Inc., Center Valley, PA, USA). All sections were analysed randomly by two expert observers, blinded to clinical and molecular data. Each sample was evaluated independently, and any discrepancies were resolved by consensus.
Cell culture
Untouched peripheral blood mononuclear cells (PBMCs) from six patients in the RTX arm were isolated at baseline, T3 and T6 and cultured for 5 days with 20 U recombinant human IL‐2 on cross‐linked anti‐CD3‐coated plates in the presence or absence of dexamethasone phosphate (Dex) (Soldesam). As negative control, cells were cultured with complete medium alone. Cell viability before and after culture was determined by Trypan blue staining 8. Phenotypical analysis of IL‐17‐producing T cell subsets after culture was performed as above.
Statistical analysis
All data analysis was performed using IBM spss version 21·0 (IBM Corp., Armonk, NY, USA). One‐way analysis of variance for repeated‐measures and multiple‐comparison post‐hoc tests were employed to calculate differences between baseline and following time‐points. For those analyses where only two time‐points were available, the Wilcoxon's matched‐pairs test was used. The comparison between the two treatment groups at T0, T3 and T6 was performed with the Mann–Whitney U‐test. Unless specified otherwise, all values are indicated as mean ± standard error of the mean (s.e.m.). The significance level was two‐sided and set at P < 0·05.
Results
Clinical efficacy of RTX in early active pSS
Table 1 displays clinical, serological and histological features of patients at baseline. Table 2 shows clinical outcomes of patients treated with either RTX or DMARDs. In line with the results from the original study cohort 23, the reduction of ESSDAI scores were faster (T6 versus T12) and more pronounced (P < 0·05 comparing T6, T9 and T12 values of the two treatment arms) in RTX‐treated compared to DMARD‐treated patients. All other domains resulted significantly lower in both treatment groups at T6 and T12 compared to the corresponding T0. Again, the effect of RTX on such domains was more pronounced with respect to DMARDs (P < 0·05 comparing T6 and T12 values of the two treatment arms).
Table 1.
Demographic, clinical and histological characteristics of the patient cohort.
| Treatment group | ||
|---|---|---|
| DMARDs | RTX | |
| Number of patients | 10 | 12 |
| Age (years) | 42 (30–54) | 41 (29–52) |
| Sex (female) | 12 (100) | 12 (100) |
| Disease duration (months) | 14 (8–20) | 13 (6–19) |
| Parotid gland enlargement | 10 (100) | 11 (92%) |
| Pulmonary involvement | 1 (10) | 1 (10) |
| Neurological involvement | 3 (30) | 2 (17) |
| Renal involvement | 0 | 0 |
| Autoimmune cytopenia | 5 (50) | 6 (50) |
| Myositis | 0 | 0 |
| Arthritis | 8 (80) | 9 (75) |
| Lymphadenopathy | 10 (100) | 10 (83) |
| ESSDAI | 18·7 (6–41) | 19·5 (6–41) |
| Prednisone, mean dose in mg | 12·5 | 12·5 |
| DMARDs (HCQ/MTX/CsA) | 12 (9/8/2) | 0 (0/0/0) |
| Focus score | 1·7 (1·2–3·1) | 1·8 (1·3–3·4) |
Data presented as mean (range) or number (%) unless otherwise stated. CsA = cyclosporin; DMARDs = disease‐modifying anti‐rheumatic drugs; ESSDAI = European League Against Rheumatism (EULAR) Sjögren's syndrome disease activity index; HCQ = hydroxychloroquine; MTX = methotrexate; RTX = rituximab.
Table 2.
Clinical outcomes of RTX‐ and DMARDs‐treated patients.
| RTX‐treated (n = 12) | DMARDs‐treated (n = 10) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| T0 | T3 | T6 | T9 | T12 | T0 | T3 | T6 | T9 | T12 | |
| ESSDAI | 20·36 ± 3·4 | 14·27 ± 3·1 | 9·818 ± 1·8* | 9·273 ± 1·5* | 8·182 ± 1·4* | 19·83 ± 3·1 | 17·67 ± 2·6 | 17·08 ± 2·8 | 14.92 ± 1.9 | 13.50 ± 1.9* |
| GDA VAS | 91·10 ± 2·7 | 72·70 ± 4·6 | 64·00 ± 4·5* | n.a. | 54·00 ± 4·7* | 91·22 ± 3 | 82·11 ± 3·8 | 73·44 ± 4·9* | n.a. | 65.22 ± 6.2* |
| Pain VAS | 71·13 ± 8·7 | 55·00 ± 7·4 | 53·50 ± 7·5* | n.a. | 44·13 ± 8·6* | 70·13 ± 9 | 59·88 ± 7·9 | 56·75 ± 7·9* | n.a. | 51.38 ± 7.6* |
| Fatigue VAS | 87·36 ± 3·6 | 65·91 ± 3·7 | 55·45 ± 5·1* | n.a. | 50·91 ± 5·2* | 88·10 ± 4·1 | 77·40 ± 5·1 | 68·70 ± 5·8* | n.a. | 62.10 ± 6.3* |
| Dryness VAS | 75·11 ± 8·1 | 53·67 ± 6·9 | 50·89 ± 6·4* | n.a. | 42·56 ± 7·7* | 76·80 ± 7·6 | 68·90 ± 8·4 | 61·40 ± 8·6* | n.a. | 58.1 ± 8.2* |
All values are displayed as mean ± standard error of the mean. ESSDAI = European League Against Rheumatism (EULAR) Sjögren's syndrome disease activity index; GDA = global disease activity; VAS = visual analogical scale; n.a. = not available. *P < 0·05 compared to respective T0.
Circulating and glandular DN T cells are depleted by RTX
We first sought to investigate short‐term effects of RTX and DMARDs treatment on circulating IL‐17‐producing cell subsets in pSS. In line with our previous observations 8, 11, the percentages of circulating IL‐17+ cells among CD4+ and DN T cells at T0 in all the 22 patients were 14 ± 3·1% and 65 ± 1·9%, respectively. At T0, IL‐17+ DN T and CD4+ Th17 cell percentages did not differ significantly in the two groups of treatment. Three months after the first infusion of RTX, a significant decrease was observed of the circulating IL‐17+ DN T cell percentage compared to T0, while at month 6 an increase of this cell subset could be detected with a restoration or even a further increase compared to baseline levels (Fig. 1a). Conversely, CD4+ Th17 cells were reduced slightly but not significantly at T3 and increased enormously at T6 (Fig. 1b). Of note, no modifications in the PB IL‐17+ DN T and CD4+ Th17 cell percentages at T3 and T6 were observed in patients treated with DMARDs (Fig. 1c–d). Moreover, at T3, as expected, both IL‐17+ DN T and CD4+ Th17 cells were significantly lower in the RTX group compared to the DMARDs group (P < 0·001 and P < 0·05, respectively). At T6, IL‐17+ DN T cells did not differ significantly in the two treatment groups, while CD4+ Th17 cells were higher in the RTX group compared to the DMARDs group (P < 0·001). Representative flow cytometry plots are displayed in Fig. 1e–i.
Figure 1.
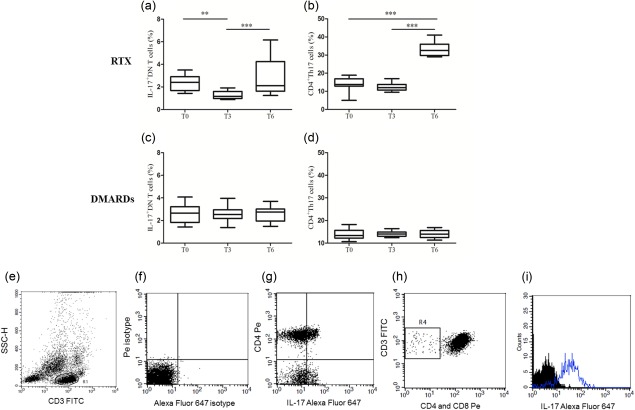
Interleukin (IL)‐17‐producing T cell subsets in the peripheral blood before and after rituximab or disease modifying anti‐rheumatic drugs (DMARDs) treatment in primary Sjögren's syndrome (pSS). Evaluation of circulating IL‐17+ double‐negative (DN) (a–c) and CD4+ T helper 17 (b–d) cells at baseline (T0) and after 3 (T3) and 6 (T6) months of either rituximab (a,b) or DMARDs (c,d) treatment in patients with pSS. The plots display mean ± standard error of the mean. P‐values were calculated with one‐way analysis of variance for repeated‐measures and multiple‐comparison post‐hoc tests. Representative flow cytometry plots are displayed in (e–i). (e) Gate of CD3+ lymphocytes; (f) isotype controls; (g) CD4+ T helper type 17 (Th17) cells; (h) DN T cells; (i) IL‐17 in DN T cells.
In line with our previous observation 11, no association between ESSDAI scores and the percentage of IL‐17‐producing T cells was observed at any time‐points
Serum levels of IL‐17 were undetectable in the majority of patients and we did not observe any differences during the follow‐up period. In particular, neither a change within each study group nor any differences when comparing the two treatment arms at T0, T3 and T6 could be observed.
In all RTX‐treated patients, we noted a disappearance of focal lymphocytic sialadenitis (FLS) and found either a non‐specific chronic sialadenitis (NSCS) or even a full restoration of normal glandular architecture (Fig. 2a,b). Hence, we attempted to clarify whether RTX was able to interfere with glandular IL‐17‐producing T cells. First, as no such data are available so far, we quantified the proportion of IL‐17‐producing T cells among glandular DN and CD4+ cells by flow cytometry in cell suspensions from pSS‐MSGs at baseline. The percentages of IL17+ cells among CD4+ and DN T cells were 70·4 ± 0·8 and 97·9 ± 0·2 (P < 0·0001), respectively. Representative flow cytometry plots are shown in the Supporting information, Fig. S1a–d.
Figure 2.
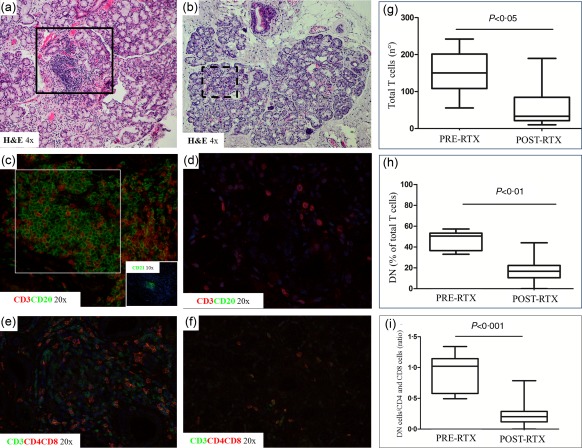
Histological analysis of primary Sjögren's syndrome (pSS) minor salivary gland biopsies before and after rituximab treatment. Histological analysis of minor salivary gland (MSG) biopsy at baseline (T0; a,c,e) and T12 (b,d,f) of pSS patients treated with rituximab; ×4, ×10 and ×20 = level of magnification of the corresponding picture. H&E = haematoxylin and eosin. (g–i) The count of CD3+ T cells (g), the percentage of double‐negative (DN) among total T cells (h) and the ratio of DN and CD4+ and CD8+ T cells (i). The plots display mean ± standard error of the mean. P‐values were calculated with Wilcoxon's matched‐pairs test.
As well as a substantial depletion of CD20‐expressing B cells (Fig. 2c,d), an overall reduction of infiltrating T lymphocytes (Fig. 2e,g) and, in particular, of infiltrating DN T cells, was observed (Fig. 2e,f,h). Among the remaining T cells, the vast majority included non‐DN (namely CD4+ or CD8+) cells, leading to a subsequent reduction of the DN/non‐DN T cell ratio following RTX treatment (Fig. 2i).
Regarding the DMARD arm, neither a reduction of total T cells nor of DN T cells was observed at T12, according with our observation that DMARDs treatment was not able to reduce the inflammatory infiltrate in MSGs (data not shown) 23. Isotype controls for immunofluorescence stainings are depicted in the Supporting information, Fig. S1e,f.
A subset of glandular and circulating DN and CD4+ T cells co‐expresses CD20 on the cell surface
The different modulation of circulating and glandular DN T and CD4+ Th17 cells induced by RTX prompted us to identify a possible direct mechanism underlying such scenario. On the basis of previously published data in RA 18, 19, we assessed the surface expression of CD20 in T cell subsets isolated from the PB of pSS patients at baseline. The specificity of the staining was verified performing both an isotype control staining that excluded non‐specific fluorescence and the co‐expression of CD19 that was undetectable in CD20+CD4+ and DN T cells. The percentages of CD20+ cells among total circulating CD4+ and DN T cells were 3·8 ± 0·4% and 29 ± 5·9%, respectively (P < 0·0001). However, when the co‐expression of IL‐17 and CD20 was assessed it emerged that, while only a small subgroup of CD4+ Th17 lymphocytes co‐expresses CD20, a significantly higher percentage of IL‐17+ DN T cells displays this molecule on the cell surface (Fig. 3 and Supporting information, Fig. S2a–f). With regard to T cells, only approximately half of CD4+ Th17 cells co‐expressed CD20, while more than 80% of IL‐17+ DN T cells co‐expressed this surface molecule (Fig. 3). Following RTX treatment, a significant reduction of the CD20+ fraction of both IL‐17+ DN and CD4+ Th17 cells was observed (Fig. 3). A reduction of CD20 mean fluorescence intensity (MFI) in IL‐17+ DN cells was also observed (Supporting information, Fig. S2g). According to the aforementioned evidence that DMARDs do not reduce PB IL‐17+ DN T and CD4+ Th17 cell percentages at T3 and T6, no modulation of CD20 expressing cells could be detected.
Figure 3.
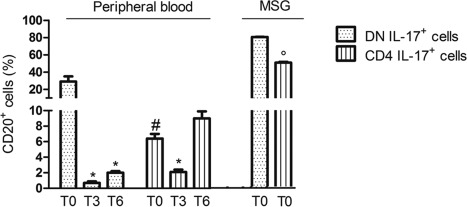
CD20 expression on primary Sjögren's syndrome (pSS) circulating and glandular interleukin (IL)‐17‐producing T cell subsets. The expression of CD20 surface levels was assessed on circulating IL‐17+ double‐negative T and T helper type 17 (Th17) cells in the peripheral blood at baseline (T0) and after 3 (T3) and 6 (T6) months of rituximab (left). P‐values were calculated with one‐way analysis of variance for repeated‐measures and multiple‐comparison post‐hoc tests. CD20 surface level was also assessed on glandular IL‐17+ double‐negative and Th17 cells of treatment‐naive patients (right). P‐value was calculated with the Mann–Whitney U‐test. The plots display mean ± standard error of the mean.
Rituximab is not able to rescue corticosteroid resistance of pSS IL‐17+ DN T cells
Subsequently, we aimed to investigate whether, besides a reduction of the number of circulating DN T cells, RTX may be also able to rescue their resistance to Dex that we reported previously in treatment‐naive pSS patients. Therefore, we arranged the same set of cultures of our previous study 8, plating PBMCs of patients at T0 and after 3 and 6 months of RTX treatment. As shown in Fig. 4, although PBMCs were exposed to RTX the DN T cell resistance to Dex, namely the production of IL‐17, remained unchanged compared to baseline. CD4+ Th17 cells responded to Dex independently on being exposed or not to RTX as observed in treatment‐naive pSS 8.
Figure 4.
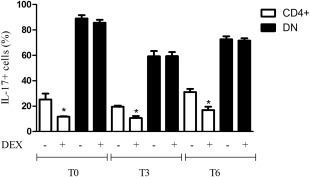
Effects of dexamethasone on CD3‐triggered circulating CD4+ and double‐negative T cells before and after rituximab treatment in primary Sjögren's syndrome (pSS). Circulating double‐negative T cells maintain their resistance to dexamethasone in vitro independently of being exposed to rituximab after 3 (T3) and 6 (T6) months. Conversely, the response of CD4+ cells to dexamethasone is independent of the exposure to RTX in vivo. P‐values were calculated with the Mann–Whitney U‐test. The plots display mean ± standard error of the mean.
Discussion
Although the well‐established role of RTX in the treatment of autoimmune diseases is due to targeting of the B cell compartment 26, several data suggest that this compound may also affect the T cell compartment. In fact, given the widely accepted B/T cell cross‐talk, it has been initially speculated that B cell depletion may lead to indirect T cell impairment due to a subsequent reduction of B cell‐derived proinflammatory cytokines as well as B cell antigen‐presenting activity 27. In the present study we describe for the first time that RTX induces a selective reduction of CD4+ Th17 cells and IL‐17‐producing DN T cells that express CD20 on the cell surface, both in the PB and the MSGs of early active pSS patients.
To date, few studies have investigated the effects of RTX on T lymphocytes and, among these, the majority enrolled patients with RA. In particular, RA patients treated with RTX display a reduction of circulating T lymphocytes, mainly CD4+ T cells 17. Subsequently, Wilk et al. identified a small fraction of circulating CD20+ T lymphocytes in RA patients and demonstrated that they are depleted completely by RTX 28, providing a deeper insight into this issue. Subsequently, a subset of circulating CD20+ T lymphocytes able to secrete IL‐17 in the PB of RA patients has been reported 18. Finally, van de Veerdonk et al. described a reduction of synovial IL‐17 and CD3+ IL‐17+ cells following RTX treatment in RA 19. Very few data are available regarding the role of RTX on the T cell arm of the immune system in pSS. A recent study failed to detect a reduction of total circulating T cells in RTX‐treated pSS patients 20. However, it has been reported that RTX is able to affect IL‐17 in this disease by reducing its mRNA in MSGs 21. Therefore, with regard to the well‐established pathogenic role of IL‐17 and Th17 cells in pSS, our results point to a similar scenario in RA and in pSS and shed some light on the additional therapeutic effects of RTX in this systemic autoimmune disease.
We demonstrate that circulating CD4+ Th17 cells and IL‐17‐producing DN T cells of pSS patients express CD20 on the cell surface. We have suggested previously a pathogenic role of DN T cells in pSS, supported by their association with glandular damage and ectopic lymphoneogenesis 11. In addition, as DN T cells are resistant to CS and to the immune modulatory effects of mesenchymal stem cells (MSCs) 8, 12, their targeting for therapeutic purposes is still unclear. In this study, we observed that IL‐17+ DN T cells, and in particular the CD20+ fraction, are reduced after 3 months of RTX therapy and repopulate at T6 similarly to B cells 29. Of interest, although depleted, DN T cells exposed to RTX in vivo behave similarly to that of treatment‐naive patients in vitro, as Dex is not able to affect their production of IL‐17. The slight reduction of circulating CD4+ Th17 cells at T3 appears to be marginal probably because, in proportion to their large number, few co‐express CD20. Conversely, the overall proportion of circulating IL‐17+ DN T cells is lower than CD4+ Th17 cells, but a higher percentage of the former co‐expresses CD20. Although RTX does not reduce circulating CD4+ Th17 cells, their repopulation at T6 is more pronounced compared to that of DN T cells. This may suggest that, on one hand, RTX‐induced T cell depletion is reversible and, on the other hand, IL‐17‐producing CD4+ and DN T cells may share a common ontogenesis 8, 11.
In line with our previous observation in 50 pSS patients with different disease duration 30, the present cohort of patients with early disease displays undetectable serum levels of IL‐17 and, as expected, no modification after RTX treatment was observed.
The differences that we noted in IL‐17‐producing DN T cells and CD4+ Th17 cell behaviour at glandular level deserve some consideration. Although an overall reduction of MSG T cells was noted after two courses of RTX (T12), DN T cells were depleted to a greater extent compared to CD4+ or CD8+ T cells. The evidence that in MSGs more than 80% of IL‐17+ DN and only 50% of CD4+ IL‐17+ co‐express CD20 on the cell surface may account for the different MSG T cell depleting capacity of RTX.
That RTX is able to reduce pSS‐MSG IL‐17 mRNA 21, together with our results, suggests clearly that the reduction of glandular IL‐17 induced by RTX may be also attributable to a depletion of DN T cells. In addition, the disappearance of FLS and GC‐like structures in MSGs of patients treated with RTX, compared to DMARDs‐treated patients, may be the result not only of direct B cell depletion, but also of the direct targeting of IL‐17‐producing T cells that are crucially involved in B/T cell co‐operation 31.
CS represent a widely employed treatment in pSS, mainly for the management of salivary gland swelling and other acute manifestations. As detailed elsewhere, in our study population CS were associated with DMARDs and with RTX infusions 23. Neither CS nor DMARDs seem able to affect circulating IL‐17+ DN T cells and CD4+ Th17 cell subsets in vivo in pSS. We have demonstrated previously that CD4+ Th17, but not DN T cells, isolated from pSS PB are inhibited by Dex, reducing IL‐17 production in vitro 8. In this study, we provided evidence that although able to deplete DN cells, RTX does not rescue their resistance to CS. This finding allows hypothesizing that genetic or epigenetic mechanisms underlying CS resistance of DN T cells need to be elucidated.
Despite their established pathogenic role in pSS, in the present patient cohort we failed to find any associations between IL‐17‐producing T cell subsets and the severity of the clinical picture or specific extraglandular manifestations 32. This observation seems to reinforce the role of IL‐17‐producing T cells in pSS pathogenesis 33. The IL‐17 axis appears to be crucial in the induction as well as the maintenance of pSS independently of the clinical or serological features of the disease. Of note, IL‐17 is expressed highly in the PB of pSS patients with only long‐standing disease 30, while it is already present at glandular level in early pSS 21. Hence, it is conceivable that in patients with early pSS, such as those included in our cohort, the IL‐17 axis is already acting in target organs to induce tissue damage from the very beginning, further underscoring the utility of RTX in early active disease 15, 23.
In conclusion, we are aware that our study may display some limitations. First, this is a pilot study evaluating the biological effects of RTX on T lymphocytes in a small number of pSS patients. Secondly, we evaluated the same cell populations at different time‐points in the PB and MSGs due to ethical reasons for repeated biopsy procedures. However, we provide the novel concept that RTX is also able to reduce IL‐17‐producing T cells in pSS. This may be explained, at least partially, by the fact that IL‐17‐producing T cell subsets, in particular DN T cells, express CD20. In this context, our findings appear to further support a therapeutic role in pSS of RTX that, despite its B cell specificity, may also hamper the pathogenic T cell side of the autoimmune response. Further studies aimed at shedding additional light on the mechanisms involved in RTX‐induced T cell depletion, as well as on the clinical implications of such biological effects, are required.
Disclosure
The authors disclose any financial support or other benefits from commercial sources for the work reported in this paper, or any other financial and non‐financial interests that any of them may have, which could create a potential conflict of interest or the appearance of a conflict of interest with regard to the work.
Author contributions
A. A. and F. C. conceived and designed the study, performed the experiments, analysed data, arranged the images and drafted the manuscript; O. B. and S. C. participated in the experiments and data analysis and drafted the manuscript; E. B. and P. C. recruited pSS patients and healthy donors (HD) collected clinical data and critically revised the manuscript for important intellectual content; P. D. B. performed sampling, participated in the experiments and to manuscript drafting; R.Gi. and R. Ge. participated in the study design and co‐ordination and revised the manuscript critically for important intellectual content. All authors approved the final version of the manuscript.
Supporting information
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Fig. S1. Representative flow cytometry dot plots of T cells isolated from minor salivary glands of patients with primary Sjögren's syndrome (pSS) (a–d) and isotype controls of tissue immunofluorescence (e–f). (a) CD3+ lymphocytes; (b) CD4+ T helper type 17 (Th17) cells; (c) DN T cells; (d) interleukin (IL)‐17 in double‐negative (DN) T cells; (e) Alexa Fluor 488 isotype control; (f) Alexa Fluor 555 isotype control.
Fig. S2. Representative flow cytometry dot‐plots of co‐expression of CD20 and interleukin (IL)‐17 in CD4+ T helper type 17 (Th17) and double‐negative (DN) T cells. (a) CD3+ lymphocytes; (b) DN T cells, (c) IL‐17+ DN T cells, (d) CD20+ IL‐17+ DN T cells; (e) CD4+ Th17 cells; (f) CD20+ IL‐17+ CD4+ Th17 cells. (g) Mean fluorescence intensity of CD20 before and after treatment with rituximab. Asterisks indicate P‐values less than 0·05 with respect to corresponding T0 P‐values calculated with one‐way analysis of variance for repeated‐measures and multiple‐comparison post‐hoc tests. The plots display mean ± standard error of the mean.
Acknowledgements
This study received no specific grant from any funding agency in the public, commercial or not‐for‐profit sectors.
References
- 1. Ambrosi A, Wahren‐Herlenius M. Update on the immunobiology of Sjogren's syndrome. Curr Opin Rheumatol 2015; 27:468–75. [DOI] [PubMed] [Google Scholar]
- 2. Brito‐Zerón P, Ramos‐Casals M. Advances in the understanding and treatment of systemic complications in Sjögren syndrome. Curr Opin Rheumatol 2014; 26:520–7. [DOI] [PubMed] [Google Scholar]
- 3. Cornec D, Jamin C, Pers JO. Sjögren's syndrome: where do we stand, and where shall we go? J Autoimmun 2014; 51:109–14. [DOI] [PubMed] [Google Scholar]
- 4. Routsias JG, Goules JD, Charalampakis G, Tzima S, Papageorgiou A, Voulgarelis M. Malignant lymphoma in primary Sjögren's syndrome: an update on the pathogenesis and treatment. Semin Arthritis Rheum 2013; 43:178–86. [DOI] [PubMed] [Google Scholar]
- 5. Lin X, Rui K, Deng J et al Th17 cells play a critical role in the development of experimental Sjogren's syndrome. Ann Rheum Dis 2015; 74:1302–10. [DOI] [PubMed] [Google Scholar]
- 6. Nguyen CQ, Hu MH, Li Y, Stewart C, Peck AB. Salivary gland tissue expression of interleukin‐23 and interleukin‐17 in Sjogren's syndrome. Arthritis Rheum 2008; 58:734–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nguyen CG, Yin H, Lee BH, Carcamo WC, Chiorini JA, Peck AB. Pathogenic effect of interleukin‐17A in induction of Sjögren's syndrome‐like disease using adenovirus‐mediated gene transfer. Arthritis Res Ther 2010; 12:R220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Alunno A, Bistoni O, Bartoloni E et al IL‐17‐producing CD4‐CD8‐ T cells are expanded in the peripheral blood, infiltrate salivary glands and are resistant to corticosteroids in patients with primary Sjogren's syndrome. Ann Rheum Dis 2013; 72:286–92. [DOI] [PubMed] [Google Scholar]
- 9. Lin X, Tian J, Rui K et al The role of T helper 17 cell subsets in Sjögren's syndrome: similarities and differences between mouse model and humans. Ann Rheum Dis 2014; 73:e43. [DOI] [PubMed] [Google Scholar]
- 10. Alunno A, Carubbi F, Caterbi S et al The role of T helper 17 cell subsets in Sjögren's syndrome: similarities and differences between mouse model and humans. Ann Rheum Dis 2014; 73:e42. [DOI] [PubMed] [Google Scholar]
- 11. Alunno A, Carubbi F, Bistoni O et al CD4‐CD8‐ T‐cells in primary Sjögren's syndrome: association with the extent of glandular involvement. J Autoimmun 2014; 51:38–43. [DOI] [PubMed] [Google Scholar]
- 12. Alunno A, Montanucci P, Bistoni O et al In vitro immunomodulatory effects of microencapsulated umbilical cord Wharton jelly‐derived mesenchymal stem cells in primary Sjögren's syndrome. Rheumatology (Oxf) 2015; 54:163–8. [DOI] [PubMed] [Google Scholar]
- 13. Ramos‐Casals M, Brito‐Zerón P, Sisó‐Almirall A, Bosch X, Tzioufas AG. Topical and systemic medications for the treatment of primary Sjögren's syndrome. Nat Rev Rheumatol 2012; 8:399–411. [DOI] [PubMed] [Google Scholar]
- 14. Brito‐Zerón P, Sisó‐Almirall A, Bové A, Kostov BA, Ramos‐Casals M. Primary Sjögren syndrome: an update on current pharmacotherapy options and future directions. Expert Opin Pharmacother 2013; 14:279–89. [DOI] [PubMed] [Google Scholar]
- 15. Carubbi F, Alunno A, Cipriani P et al Rituximab in pSS: a ten‐year journey. Lupus 2014; 23:1337–49. [DOI] [PubMed] [Google Scholar]
- 16. Glennie MJ, French RR, Cragg MS, Taylor RP. Mechanisms of killing by anti‐CD20 monoclonal antibodies. Mol Immunol 2007; 44:3823–37. [DOI] [PubMed] [Google Scholar]
- 17. Mélet J, Mulleman D, Goupille P, Ribourtout B, Watier H, Thibault G. Rituximab‐induced T cell depletion in patients with rheumatoid arthritis: association with clinical response. Arthritis Rheum 2013; 65:2783–90. [DOI] [PubMed] [Google Scholar]
- 18. Eggleton P, Bremer E, Tarr JM et al Frequency of Th17 CD20+ cells in the peripheral blood of rheumatoid arthritis patients is higher compared to healthy subjects. Arthritis Res Ther 2011; 13:R208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. van de Veerdonk FL, Lauwerys B, Marijnissen RJ et al The anti‐CD20 antibody rituximab reduces the Th17 cell response. Arthritis Rheum 2011; 63:1507–16. [DOI] [PubMed] [Google Scholar]
- 20. Abdulahad WH, Meijer JM, Kroese FG. B‐cell reconstitution and T‐helper‐cell balance after rituximab treatment of active primary Sjogren's syndrome. Arthritis Rheum 2011; 63:1116–23. [DOI] [PubMed] [Google Scholar]
- 21. Ciccia F, Guggino G, Rizzo A et al Rituximab modulates IL‐17 expression in the salivary glands of patients with primary Sjogren's syndrome. Rheumatology (Oxf) 2014; 53:1313–20. [DOI] [PubMed] [Google Scholar]
- 22. Ciccia F, Giardina A, Rizzo A et al Rituximab modulates the expression of IL‐22 in the salivary glands of patients with primary Sjogren's syndrome. Ann Rheum Dis 2013; 72:782–3. [DOI] [PubMed] [Google Scholar]
- 23. Carubbi F, Cipriani P, Marrelli A et al Efficacy and safety of rituximab treatment in early primary Sjögren's syndrome: a prospective, multi‐center, follow‐up study. Arthritis Res Ther 2013; 15:R172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vitali C, Bombardieri S, Jonsson R et al Classification criteria for Sjogren's syndrome: a revised version of the European criteria proposed by the American–European Consensus Group. Ann Rheum Dis 2002; 61:554–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Seror R, Ravaud P, Bowman SJ et al EULAR Sjögren's Syndrome Disease Activity Index (ESSDAI): development of a consensus systemic disease activity index for primary Sjögren's syndrome. Ann Rheum Dis 2010; 69:1103–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Conti F, Perricone C, Ceccarelli F, Valesini G. Rituximab treatment of systemic lupus erythematosus in controlled trials and in clinical practice: two sides of the same coin. Autoimmun Rev 2010; 9:716–20. [DOI] [PubMed] [Google Scholar]
- 27. Datta SK. Anti‐CD20 antibody is an efficient therapeutic tool for the selective removal of autoreactive T cells. Nat Clin Pract Rheumatol 2009; 5:80–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wilk E, Witte T, Marquardt N et al Depletion of functionally active CD20+ T cells by rituximab treatment. Arthritis Rheum 2009; 60:3563–71. [DOI] [PubMed] [Google Scholar]
- 29. Pers JO, Devauchelle V, Daridon C et al BAFF‐modulated repopulation of B lymphocytes in the blood and salivary glands of rituximab‐treated patients with Sjögren's syndrome. Arthritis Rheum 2007; 56:1464–77. [DOI] [PubMed] [Google Scholar]
- 30. Alunno A, Bistoni O, Caterbi S, Bartoloni E, Cafaro G, Gerli R. Serum interleukin‐17 in primary Sjögren's syndrome: association with disease duration and parotid gland swelling. Clin Exp Rheumatol 2015; 33:129. [PubMed] [Google Scholar]
- 31. Hsu HC, Yang P, Wang J et al Interleukin 17‐producing T helper cells and interleukin 17 orchestrate autoreactive germinal center development in autoimmune BXD2 mice. Nat Immunol 2008; 9:166–75. [DOI] [PubMed] [Google Scholar]
- 32. Alunno A, Carubbi F, Bistoni O et al T regulatory and T helper 17 cells in primary Sjögren's syndrome: facts and perspectives. Mediators Inflamm 2015; 2015:243723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Alunno A, Carubbi F, Bartoloni E et al Unmasking the pathogenic role of IL‐17 axis in primary Sjögren's syndrome: a new era for therapeutic targeting? Autoimmun Rev 2014; 13:1167–73. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Fig. S1. Representative flow cytometry dot plots of T cells isolated from minor salivary glands of patients with primary Sjögren's syndrome (pSS) (a–d) and isotype controls of tissue immunofluorescence (e–f). (a) CD3+ lymphocytes; (b) CD4+ T helper type 17 (Th17) cells; (c) DN T cells; (d) interleukin (IL)‐17 in double‐negative (DN) T cells; (e) Alexa Fluor 488 isotype control; (f) Alexa Fluor 555 isotype control.
Fig. S2. Representative flow cytometry dot‐plots of co‐expression of CD20 and interleukin (IL)‐17 in CD4+ T helper type 17 (Th17) and double‐negative (DN) T cells. (a) CD3+ lymphocytes; (b) DN T cells, (c) IL‐17+ DN T cells, (d) CD20+ IL‐17+ DN T cells; (e) CD4+ Th17 cells; (f) CD20+ IL‐17+ CD4+ Th17 cells. (g) Mean fluorescence intensity of CD20 before and after treatment with rituximab. Asterisks indicate P‐values less than 0·05 with respect to corresponding T0 P‐values calculated with one‐way analysis of variance for repeated‐measures and multiple‐comparison post‐hoc tests. The plots display mean ± standard error of the mean.