

SUMMARY
Although persistent elevations in circulating glucose concentrations promote compensatory increases in pancreatic islet mass, unremitting insulin resistance causes a deterioration in beta cell function that leads to the progression to diabetes. Here we show that mice with a knockout of the CREB coactivator CRTC2 in beta cells have impaired oral glucose tolerance due to decreases in circulating insulin concentrations. CRTC2 was found to promote beta cell function in part by stimulating the expression of the transcription factor MafA. Chronic hyperglycemia disrupted cAMP signaling in pancreatic islets by activating the hypoxia inducible factor (HIF1)-dependent induction of the Protein Kinase A Inhibitor beta (PKIB), a potent inhibitor of PKA catalytic activity. Indeed, disruption of the PKIB gene improved islet function in the setting of obesity. These results demonstrate how cross-talk between nutrient and hormonal pathways contributes to loss of pancreatic islet function.
Graphical abstract
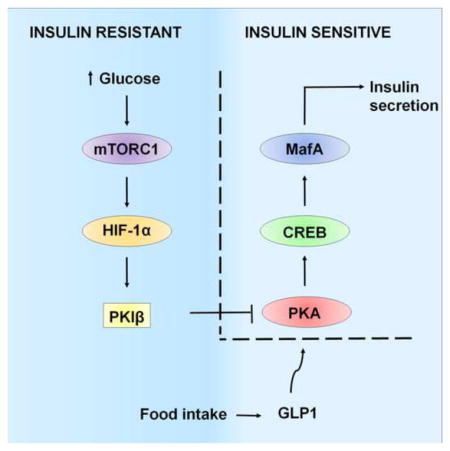
INTRODUCTION
The second messenger cAMP has been shown to mediate effects of incretin hormones, in part through induction of the Ser/Thr protein kinase PKA. Blocking cAMP signaling in beta cells through targeted disruption of the Gsα subunit of the heterotrimeric G protein leads to severe glucose intolerance and beta cell apoptosis (Xie et al., 2007). Conversely, mutations that increase PKA activity, either via disruption of the R1α regulatory subunit (Song et al., 2011) or via a gain of function mutation in the PKA catalytic subunit (Kaihara et al., 2013), enhances insulin secretion.
cAMP promotes cellular gene expression via the PKA-mediated phosphorylation of the CREB family of activators and via the de-phosphorylation of the cAMP Regulated Transcriptional Coactivators (CRTCs). Following its activation, CREB has been found to promote islet function in part by upregulating the insulin receptor substrate 2 (IRS2) in beta cells (Jhala et al., 2003) (Park et al., 2006). Although deletion of the single family member CREB1 alone has no effect on beta cell function under normal chow conditions (Shin et al., 2014), expression of a dominant negative CREB polypeptide A-CREB, which blocks all three family members (CREB1, ATF1, CREM), leads to severe hyperglycemia due in part to reductions in glucose stimulated insulin secretion (Jhala et al., 2003).
IRS2-dependent increases in insulin signaling are thought to promote islet growth through the activation of mTORC1 complexes and subsequent induction of the hypoxia inducible factor HIF1 (Van de Velde et al., 2011) (Gunton et al., 2005). Although the mTORC1-HIF pathway appears to be critical for adaptive expansion of pancreatic islet mass, beta cell function deteriorates in the setting of chronic insulin resistance (Zhao et al., 1998). Based on the ability for GLP1 agonists to improve beta cell function in this setting, we examined the potential role of CREB and CRTC2 in mediating these effects.
RESULTS
Exposure of INS1 insulinoma cells to the cAMP agonist Forskolin (FSK) promoted the phosphorylation of CREB at Ser133 and the dephosphorylation of CRTC2 within 30 minutes (figure 1A). By contrast, exposure to depolarizing concentrations of KCl (40mM) stimulated CREB phosphorylation to a lesser extent. Under basal conditions, CRTC2 was sequestered in the cytoplasm of beta cells from cultured islets; exposure to FSK triggered CRTC2 nuclear translocation (figure 1B).
Figure 1. Impaired glucose tolerance in mice with a beta cell specific knockout of CRTC2.

A. and B. Dephosphorylation and nuclear translocation of CRTC2 in INS1 (A) and mouse pancreaitc islets (B) following exposure to cAMP agonist forskolin (FSK) or KCl. C. Oral glucose tolerance testing (OGTT) of mice with a knockout of CRTC2 in beta cells (MIP-T2KO) relative to control littermates (* p < 0.05; ** p < 0.01; *** p < 0.001; n=10). D. Insulin tolerance testing of control and MIP-T2KO mice. E. Circulating insulin concentrations in MIP-T2KO and control control littermates 10 minutes following administration of glucose (** p < 0.01: n=10). F. Effect of glucose alone (20mM) or plus exendin (10nM) on insulin secretion from primary cultured islets of MIP-T2KO and control littermates (** p < 0.01; n=6). G. Insulin content in pancreatic islets from CRTC2 mutant or control littermates (* p < 0.05; n=6). H. Effect of CRTC2 or dominant negative A-CREB over-expression on insulin secretion from INS1 cells (* p < 0.05, *** p < 0.001; n=5). Data are shown as mean ± s.e.m.
GLP1 and other incretin hormones are released from intestinal cells only following nutrient ingestion (Holst et al., 2011), so we analysed effects of oral glucose tolerance testing (OGTT) in mice with a beta cell specific knockout of CRTC2 (MIP-T2KO; figure S1A). Although they were almost comparable to control littermates by intraperitoneal glucose tolerance test (IPGTT), MIP-T2KO mice showed impaired glucose tolerance by OGTT test (figure 1C; figure S1B). Indeed, we observed similar differences between control and MIP-T2KO mice following intra-peritoneal (IP) injection of exendin-4 in addition to glucose (figure S1C). Arguing against an increase in insulin resistance, circulating glucose concentrations decreased identically in MIP-T2KO mice and control littermates following insulin administration (figure 1D). Rather, MIP-T2KO mice secreted less insulin in response to oral glucose gavage, suggesting that beta cell function is disrupted in these animals (figure 1E).
We evaluated effects of CRTC2 on insulin secretion from cultured islets in response to cAMP. Following 10 minutes exposure to exendin-4 plus high glucose (20mM), insulin release from MIP-T2KO islets was reduced 30% (figure 1F) relative to control islets. Exposure to glucose alone stimulated insulin secretion comparably in cultured islets from wild-type and MIP-T2KO mice (figure 1F). Insulin (Ins1, Ins2) gene expression and protein content were also modestly decreased in MIP-T2KO islets (figure 1G; figure S1D). Conversely, over-expression of CRTC2 in INS1 insulinoma cells enhanced insulin secretion in response to GLU/FSK. These effects were blocked by expression of a dominant negative CREB polypeptide called A-CREB (figure 1H).
We performed transcriptome-wide studies to identify CREB target genes that promote insulin expression and secretion. This analysis revealed a number of FSK-inducible genes that contain CREB binding sites and that are down-regulated following expression of A-CREB inhibitor. Amongst these, we identified MafA, a beta cell restricted factor that regulates the expression of insulin and a number of genes associated with beta cell maturity and insulin exocytosis (Abdulahad et al., 2012; Lee et al., 2014; Martin et al., 2012; Selmi, 2012; Tu et al., 2012) (figure 2A; table S1). In addition, exposure to FSK also stimulated the expression of a subset of synaptotagmins (Syt4, Syt5, Syt7), proteins that function as major calcium sensors for synaptic vesicles (Iezzi et al., 2004; Iezzi et al., 2005) (figure S1E).
Figure 2. CRTC2 Stimulates MafA Expression.

A. RNAseq analysis of INS1 cells exposed to FSK. Top genes upregulated 2-fold or better indicated. Effect of dominant negative A-CREB expression shown. B. Immunohistochemical assay of MafA staining in islets from pancreatic sections of control and MIP-T2KO mice. Right, relative number of MafA positive cells in control and MIP-T2KO sections shown (** p < 0.01; n=5). C. Effect of CRTC2 over-expression on MafA protein amounts in INS1 cells. Exposure to FSK indicated. D. Chromatin immunoprecipitation (ChIP) assay of INS1 cells showing amounts of CREB and CRTC2 recruited to the MafA promoter under basal conditions and following exposure to FSK (*** p < 0.001; n=6). E. Effect of MafA depletion on insulin secretion from INS1 cells (* p < 0.05, ** p < 0.01; n=6). F. Effect of adenoviral GFP control or MafA over-expression on insulin secretion from MIP-T2KO mice islets compared to control (** p < 0.01; n=6). Data are shown as mean ± s.e.m.
Realizing the importance of MafA for insulin expression and secretion, we addressed the potential role of this transcription factor in mediating effects of CREB on beta cell function. Exposure of isolated mouse islets to FSK increased the expression of MafA (figure S1F) which promote beta cell maturation. Indeed, islet MafA expression was reduced by two-thirds in pancreatic islets from MIP-T2KO mice relative to control animals by immunohistochemical analysis (figure 2B). As a result, beta cell maturity gene expression (Glp1r, Pc, Pdx1) was also down-regulated in CRTC2 mutant islets (figure S1F). Conversely, CRTC2 overexpression in INS1 cells increased MafA expression (figure 2C).
We evaluated whether MafA is a direct target gene for CREB and CRTC2. Supporting this notion, the MafA promoter contains a consensus cAMP response element (CRE) that is constitutively occupied by CREB in INS1 cells (figure 2D, figure S1G). In line with its cytoplasmic sequestration in unstimulated cells (Screaton et al., 2004), amounts of CRTC2 over the MafA promoter were low under basal conditions but increased following FSK treatment, when CRTC2 is translocated to the nucleus. Inhibiting CREB occupancy, through A-CREB over-expression, down-regulated MafA gene in INS1 cells (figure S1H).
We tested whether MafA activity contributes to cAMP-dependent increases in insulin secretion. In line with this notion, RNAi-mediated depletion of MafA decreased insulin content and secretion from INS1 cells while MafA over-expression increased them (figure 2E; figure S1I and J). Indeed, adenoviral expression of MafA restored insulin secretion to wild-type levels in CRTC2 mutant pancreatic islets exposed acutely to glucose plus exendin (figure 2F). Taken together, these results demonstrate that CRTC2 promotes beta cell function in part by upregulating MafA in response to cAMP.
Although initially compensated by an increase in pancreatic islet mass, prolonged insulin resistance causes an impairment in beta cell function that is thought to reflect the down-regulation of certain beta cell factors including MafA. Having seen that CREB and CRTC2 promote MafA expression in response to cAMP signals, we tested effects of insulin resistance on this pathway in pancreatic islets. By contrast with the robust upregulation of MafA and other CREB target genes by FSK in pancreatic islets from lean mice, FSK had only modest effects on islets from high fat diet (HFD) mice (figure 3A; figure S2A,B). Similarly, effects of FSK on insulin secretion were attenuated in HFD islets relative to control (figure S2C). In principle, the loss of CREB activity in HFD fed mice could reflect chronic increases in a number of nutrients including circulating glucose or free fatty acids. To test the role of hyperglycemia in this process, we performed high carbohydrate diet (HCD) feeding studies. Although it had no effect on body weight, HCD feeding impaired glucose tolerance and insulin secretion after only 8 weeks (figure S2D–G). Similar to islets from HFD mice, pancreatic islets from HCD animals showed only modest increases in insulin secretion and CREB target gene expression following exposure to FSK (figure S2H and I).
Figure 3. Disruption of the Beta cell CREB Pathway in chronic hyperglycemia.

A. Relative induction of CREB target genes by FSK in cultured pancreatic islets from normal chow (NC) or high fat diet (HFD) fed mice. mRNA amounts for NR4A2 and MafA shown (** p < 0.01; n=6). B. Effect of prolonged exposure to high glucose on FSK-induced increases in MafA protein amounts. C. Effect of chronic high glucose and adenoviral MafA expression on insulin secretion from INS1 cells (*** p < 0.001; n=6). D. Immunoblots showing effect of prolonged high glucose exposure on CREB phosphorylation (left) and on cellular PKA activity (right) in primary cultured mouse islets exposed to FSK. Effect of glucose on phosphorylation of LKB1 at a consensus PKA site (Ser431) also shown. E. RNAseq analysis of INS1 cells showing effect of prolonged exposure to high glucose on downregulation of cAMP inducible genes. Effect of FSK shown. F. Effect of high fat diet (HFD) feeding on PKIB mRNA (left) and protein expression (right) in mouse islets (** p < 0.01; n=4). G. Effect of chronic high glucose treatment on nuclear shuttling of PKA catalytic subunit in INS1 cells exposed to FSK. Cytoplasmic (top) and nuclear (bottom) fractions shown. Data are shown as mean ± s.e.m.
We wondered whether exposure of cultured pancreatic islets to high concentrations of glucose mimics effects of hyperglycemia on the CREB pathway. Supporting this idea, prolonged exposure of either INS1 cells or cultured pancreatic islets to high glucose (20mM, 72 hours) inhibited the expression of MafA and other CREB target genes in cells exposed to FSK (figure 3B; figure S3A and B). Exposure to 10mM glucose was not sufficient for PKIB induction in INS1 cells. Indeed, FSK-dependent increases in insulin secretion were substantially down-regulated in INS1 cells exposed to high-glucose and FSK (figure 3C). The impairment in insulin secretion under chronic high glucose conditions was partially rescued by MafA over-expression in both INS1 cells and primary islets (figure 3C; figure S3C).
We addressed the mechanism by which high glucose exposure attenuates CREB activity. Although total amounts of CREB were unchanged, high glucose treatment disrupted the FSK-induced phosphorylation of CREB in both INS1 cells and pancreatic islets (figure 3D; figure S3D). Moreover, phospho-CREB amounts were also down-regulated in cultured islets from HFD and HCD mice (figure S3E and F) Arguing against an effect on adenyl cyclase or phosphodiesterase activities, exposure to FSK increased intracellular cAMP concentrations comparably in control and high glucose treated INS1 cells (figure S3G). Rather, cellular PKA activity was substantially reduced in islets chronically exposed chronic glucose, as visualized in immunoblot assays with phospho-PKA substrate antiserum (figure 3D). Consistent with its effects on CREB phosphorylation, prolonged exposure of INS1 cells to high glucose largely inhibited the induction of cAMP responsive genes (figure 3E, table S2).
Having seen that chronic glucose exposure disrupts PKA activity without affecting cAMP accumulation, we considered the involvement of a Protein Kinase A Inhibitor (PKI) in this process. Consisting of three closely related polypeptides (PKIA, PKIB, PKIG) the PKIs have been shown to bind with high specificity and affinity to PKA; they also contain a potent nuclear export signal that maintains PKA in the cytoplasm (Taylor et al., 2005) (Fantozzi et al., 1994; Wen et al., 1994; Wen et al., 1995). PKIB was selectively upregulated in INS1 cells and cultured pancreatic islets following prolonged exposure to high glucose (figure S4A,B). Indeed, PKIB mRNA and protein amounts were also increased in pancreatic islets but not other metabolic tissues from HFD and HCD fed mice relative to controls (figure 3F; figure S4C,D).
In immunohistochemical studies, we detected PKIB in beta cells of the pancreatic islets, but not in surrounding glucagon producing alpha cells or in acinar cells of the exocrine pancreas (figure S4E). Similar to its effects in mouse islets, chronic glucose exposure also promoted PKIB expression in human islets, thereby attenuating CREB phosphorylation and target gene expression (figure S4F–J).
Based on its proposed role in nuclear export, we reasoned that PKIB may reduce CREB phosphorylation by blocking the nuclear accumulation of PKA. Supporting this idea, exposure to FSK promoted an increase in nuclear amounts of PKA catalytic subunit in INS1 cells maintained under low glucose conditions but not in cells maintained on high glucose (figure 3G).
Although PKI potently inhibits PKA activity, the importance of this pathway for insulin secretion is unclear, as increases in circulating glucose modulate insulin secretion primilary through calcium signaling. In that event, the PKIB would cause only modest changes in circulating glucose and insulin concentrations. To evaluate the effects of this inhibitor on glucose homeostasis, we employed a double stranded AAV8 vector expressing PKIB under control of the mouse insulin promoter, which targets transgene expression specifically to beta cells. Over-expression of AAV8-encoded PKIB in MIN6 insulinoma cells reduced PKA activity and correspondingly disrupted CREB phosphorylation in response to FSK treatment (figure 4A). Following intra-peritoneal administration, a control AAV-MIP-EGFP virus was expressed in pancreatic islets but not other tissues such as liver and testis (figure 4B). Ad libitum circulating glucose concentrations increased progressively over a 6 weeks period in mice expressing AAV encoded PKIB (figure 4C,D). Although their body weights were identical to controls, PKIB-over-expressing animals became relatively glucose intolerant and they had lower circulating concentrations of insulin in response to oral glucose tolerance testing, indicating that PKIB expression is sufficient to disrupt beta cell function (figure. 4E,F).
Figure 4. PKIB Disrupts Beta Cell Function.

A. Immunoblots showing effect of dsAAV-MIP-PKIB expression on PKA activity using phospho-PKA substrate antibody (left) or phospho-CREB and phospho LKB antiserum (right) in MIN6 cells (n=4). B. Immunohistochemical analysis of GFP expression from AAV encoded EGFP virus in testis, liver and pancreas 10 days after injection (n=3). C. and D. Body weight (C) as well as circulating ad libitum glucose concentrations in mice infected with PKIB or control EGFP (D) (** p < 0.01, *** p < 0.01; n=5) E. and F. Circulating glucose (OGTT) and insulin concentrations in mice following oral glucose administration (F) (** p < 0.01, *** p < 0.01; n=5). Data are shown as mean ± s.e.m.
We used mice with a knockout of PKIB to determine whether the upregulation of this inhibitor in response to high fat diet feeding contributes to the deterioriation in pancreatic islet function. Although they were otherwise unremarkable (Belyamani et al., 2001), NC-fed PKIB KO mice were modestly more glucose tolerant relative to wild-type littermates by OGTT (figure S5A). When placed on a HFD, however, PKIB mutants had lower fasting blood glucose concentrations and were substantially more glucose tolerant (figure 5A, figure S5 B–D). Moreover, circulating insulin concentrations rose 3-fold in PKIB knockout mice following glucose administration, but they remained unchanged in controls (figure 5B). Similarly, MafA expression was strongly reduced in pancreatic sections from control mice following HFD-feeding, but it remained high in PKIB KO mice (figure 5C). Taken together, these results demonstrate that the upregulation of PKIB in insulin resistance disrupts beta cell CREB activity and insulin secretion.
Figure 5. Knockout of PKIB Improves Pancreatic Islet Function in insulin resistance.
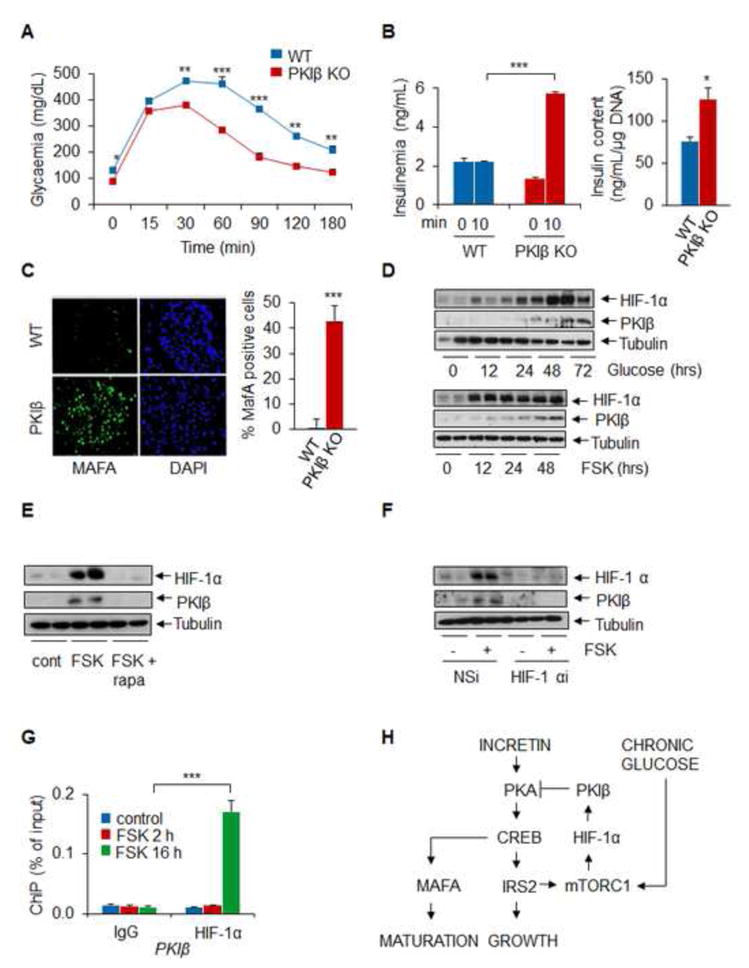
A. OGTT of PKIB knockout and control littermates maintained on a HFD for 12 weeks. (** p < 0.01, *** p < 0.01; n=6) B. Circulating insulin concentrations in control and PKIB mutant littermates under basal conditions and 10 minutes following oral glucose administration. Right, relative insulin content in isolated pancretic islets (* p < 0.05, *** p < 0.01; n=6). C. Immunohistochemical analysis showing MafA staining in pancreatic sections from HFD-fed control mice and PKIB−/− littermates. D. Time course of PKIB mRNA accumulation in INS1 cells exposed to FSK (n=4). E. Effect of mTORC1 inhibitor rapamycin on PKIB mRNA amounts in INS1 cells exposed to FSK for 14 hours (n=4). F. Effect of HIF-1α RNAi-mediated knockdown on PKIB mRNA levels in INS1 cells (n=4). G. ChiP assays showing effect of short (2hr) or long term (16 hrs) exposure to FSK on HIF-1α occupancy over the PKIB promoter (*** p < 0.001; n=6). H. Feedback regulation of beta cell CREB activity in insulin resistance. Glucose and incretin dependent increases in CREB/CRTC2 activity during feeding promote the expression of IRS2, which in turn mediates induction of mTORC1 complexes. When hyperglycemia is prolonged, increases in HIF-1 protein trigger expression of PKIB, which down-regulates the CREB/CRTC2 pathway by binding to and inhibiting PKA. Data are shown as mean ± s.e.m.
In the setting of insulin resistance, chronic hyperglycemia is thought to promote beta cell hypertrophy in part through the induction of the energy sensing kinase mTOR. Indeed, prolonged exposure of pancreatic islets to glucose or FSK triggers the mTORC1-dependent activation of HIF-1α (Van de Velde et al., 2011) in beta cells, prompting us to evaluate the role of this pathway in mediating the induction of PKIB. Exposure of INS1 cells to glucose or FSK increased HIF-1α protein amounts after 12–24 hours followed by increases in PKIB protein amounts (figure 5D).
Consistent with a requirement for mTOR activity, exposure to the mTORC1 inhibitor rapamycin effectively blocked the upregulation of PKIB in cells exposed to FSK (figure 5E). Moreover, RNAi-mediated depletion of HIF-1α also disrupted PKIB induction, whereas HIF-1α over-expression potentiated it (figure 5F, figure S5E,F). The effect of HIF on PKIB expression appears direct; HIF1 protein amounts over consensus binding sites on the PKIB promoter increased following prolonged but not short term FSK treatment of INS1 cells by ChIP assay (figure 5G). Indeed, slective induction of endogenous HIF1, by administration of the prolyl hydroxylase inhibitor DMOG, also upregulated PKIB expression, demonstrating that HIF is sufficient for induction of PKIB in beta cells (figure S5G,H).
DISCUSSION
Taken together, our results show how CRTC2 mediates effects of incretin hormones through upregulation of MafA and other CREB target genes that promote insulin gene expression and secretion (figure 5H). In response to nutrient stress, GLP1 and glucose promote pancreatic islet function through increases in mTORC1 activity that culminate in the induction of the HIF pathway. When insulin resistance is prolonged, HIF1 inhibits CREB activity by stimulating the expression of PKIB and blocking the activation of PKA (figure 5H). Although prolonged exposure to FSK led to induction of PKIB in cultured islets, chronic treatment of insulin-resistant mice with GLP-1R agonist improved glucose intolerance in vivo (Irwin et al., 2009). Incretin signaling accounts for a large fraction of post-prandial glucose disposal, and the attenuation of this pathway in type II diabetes is thought to contribute significantly to the increased glucose excursions that are characteristic of this disease (Holst et al., 2011). Indeed, a sizable percentage of type II diabetic patients appear to be unresponsive to GLP1 agonist therapy (Buysschaert et al., 2012; Hall et al., 2013; Preumont et al., 2012), potentially reflecting the upregulation of PKIs. Although the mechanism is unclear, PKIA also appears to be upregulated in pancreatic islets from type II diabetic individuals (Gunton et al., 2005). Reducing the expression of PKIs in beta cells may provide therapeutic benefit in this setting.
EXPERIMENTAL PROCEDURES
Animals
All studies except for high fat and high carbohydrate diets studies were performed using 10 to 12 weeks old males mice in a C57Bl6 background. Animals were adapted to their environment for 1 week before studies and were housed in colony cages with a 12 hours light/12 hours dark cycle in a temperature-controlled environment. C57BL6 were purchased from Jackson laboratories. For high fat diet studies, C57BL6 mice were fed a high fat diet for 20 weeks beginning at 8 weeks of age (60 % kcal fat, D12492, Research Diet Inc). Age-matched C57BL6 on normal chow were used as controls. For high carbohydrate diet studies, 8 weeks old males C57BL6 where maintain in diet with 73% carbohydrate during 10 weeks (AIN-93M, Resarch Diet Inc). PKIB mice were purchased from MMRRC (B6.129P2-Pkibtm1Idz/Mmmh). MIP-CreERT mice were a gift from Dr. Phillipson of the University of Chicago. The CRTC2flox/flox mice are homozygous for a “floxed” CRTC2 allele in which CRTC2 exons 1 and 5 are flanked by loxP target sites for Cre recombinase. Animal studies were approved by the Institutional Animal Care and Use Committee (IACUC) at the Salk Institute.
In vivo analysis
At 10 weeks of age, CRTC2 flox/flox MIP-Cre/ERT mice (MIP-T2 KO mice) were fed with tamoxifen (sigma, T5648) dissolved in corn oil at 100 mg/kg body weight (once per day for 5 days). Following tamoxifen administration, the mice were housed for 5–10 days before being used and analyzed for Cre-recombinase activity. MIP-T2 KO mice fed with corn oil were used as controls. Following an overnight fast, mice were administered glucose (1g/Kg) by oral gavage or by i.p., and glucose levels were measured every 30 minutes over a 3 hours period. Insulin tolerance was tested by i.p. injection of 6-hour fasted mice with 1U/Kg of insulin (Humulin R, Eli Lilly) followed by blood glucose measurements every 15 minutes until 1 hour. For insulin dosage, blood was taking from the tail vein after 10 minutes of glucose gavage. dsAAV infections were carried out at a dose of a 4×1011 genomes (vg) per mouse. Viruses were administrated intraperitoneally in a total volume of 800 μL of sterile saline solution containing 5% sorbitol.
Cell culture
INS-1 insulinoma cells were cultured in RPMI (corning cellgro) containing 10% fetal bovine serum (Gemini Bio Products), 100 μg/mL penicillin-streptomycin and 1 mM sodium pyruvate (corning cellgro). MIN6 cells were grown in DMEM (corning cellgro) with 10 % FBS, 100 μg/mL penicillin- streptomycin and 70 uM beta-mercaptoethanol. For chronic glucose experiments, cells were maintained under low (2.8mM) or high (20mM) glucose for 3 days. Exendin-4 (10nM), Forskolin (10 μM), KCl (40mM), DMOG (1 mM) and rapamycin (50nM) were added to cells as indicated.
cAMP measurement
Cellular cAMP levels were measured using an ELISA kit (Cayman Chemical Company) according to manufacturer’s instructions.
RNA interference
The sequences of the oligonucleotides used for target rat mafA were as follow : 5′-GAGGAUCUGUAVUGGAUGA-3′ and 5′ UCAUCCAGUACAGAUCCUC-3′. As for a negative control, RNA duplexes targeting GFP were used: 5′-GCAAGCUGACCCUGAAGUUC-3′; and 5′-GAACUUCAGGGUCAGCUUGC-3′. After annealing 100 pmol of synthetic RNA duplex using Lipofectamine 2000 reagent (Invitrogen) per well (6 well plates) and cells were harvest 48 hours later.
Islets isolation and human islets
Islets isolation was performed as described previously (Shapiro et al., 1996). Briefly, pancreata from 10 to 12 weeks old mice were injected with liberase (0.2mg/mL, Roche Applied Science) and digest at 37°C for 15 minutes. Preparations were washed with Hank’s buffered salt solution and the dissociated islets were purified on histopaque gradients (Sigma Aldrich) and cultured in RPMI with 10% FBS for 3 days before testing. Human islets were supplied by the Integrated islets Distribution Program (IIDP) (http://iidp.coh.org/). Donor information is listed in Table 3.
Insulin secretion
INS-1 cells or primary islets were starved 2 hours in Krebs-Ringer Bicarbonate Hepes buffer (KRBH) containing 0.2% BSA and exposed to 2.8mM or 20mM glucose with or without FSK (10μM) or exendin 4 (10nM) for 1 hour. Insulin release and content were measured using the ultrasensitive insulin ELISA kit (Mercodia). Results are presented as insulin secretion (ng/mL) per hour normalized to insulin content. Insulin content is normalized to DNA. Insulin dosage in vivo, was assayed using the ultra sensitive mouse insulin ELISA (Crystal Chem).
Adenoviruses
For adenoviruses construction, cDNA were subcloned in the pAdTRACK vector. Rat PKIB cDNA was obtained by PCR using primers 5′-CATCTCGAGATGAGGACAGATTC (sense) and 5′-CATGGTACCTTATTTGT CTTCGTCTAG (anti-sense), which introduces XhoI and KpnI sites (underlined) respectively. Complete viral vectors were generated by homologous recombination with the AdEASY vector as described (Koo et al., 2005). The MafA adenovirus was a gift from Dr. Matsuoka TA. Adenoviruses were then produced in MGH cells and purified using CsCl gradients. dsAAV-MIP-EGFP was a gift from Dr. Paul D. Robbins of the University of Pittsburg. Mouse PKIB cDNA was obtained by PCR using primers 5′-CATACCGGTATGAGGACAGATTCATCAGA (sens) and 5′-CATGCGGCCGCTCATTTTCCTTCATTTAG (anti-sens), which introduces AgeI and NotI (underlined) respectively. The dsAAV virus expressing mouse PKIβ was generated by excising EGFP with restriction enzymes AgeI and NotI. Recombinant dsAAV vectors were generating according to the triple transfection protocol using AAV8 serotype.
Real-Time Quantitative PCR and RNAseq
Total RNAs from cells or primary islets were extracted using Trizol and cDNA was generated using the Transcriptor First Strand cDNA Synthesis kit (Roche Applied Science). cDNAs were quantified on a lightcycler 480 instrument (Roche Applied Science). Gene expression data were presented relative to the expression of housekeeping gene L32 (rat samples) and 18S (mouse and human islets). Primer sequences are listed in Table 4. RNA-Seq libraries were prepared using the mRNA isolation protocol and the NEBNext-Ultra kits from New England Biolabs following the manufacturer’s protocols. Libraries were quantitated by Qubit (Invitrogen), and run on a MiSeq instrument with paired-end 75bp reads using v3 chemistry (Illumina). Data were analyzed by tophat2 and cuffdiff against the mouse mm10 genome build. The GEO accession number for RNAseq studies is GSE60158.
Chromatin Immunoprecipitation
INS-1 cells were plated on 15 cm dishes and exposed to forskolin as specifed. Chromatin immunoprecipitation with HIF-1α, CREB, P-CREB and CRTC2 antisera was performed as described (Ravnskjaer et al., 2007). Oligonucleotides used for ChIP analysis are listed in Table 5.
Protein analysis
Total protein from cultured cells or primary islets was extracted in a Tris-HCl buffer containing 0.5% NP40, protease and phosphatase inhibitors. Proteins were quantified using Bradford reagent and separated using SDS-PAGE. For cellular fractionation, cells were resuspended in hypotonic lysis buffer (40mM TrisHCl PH 7.4, 10mM NaCl, 1mM EDTA with DTT and protease inhibitors). Cells were lysed using a dounce homogenizer and centrifuged. Cytosolic supernatants were collected. Nuclear pellets were washed 3 times and resuspended in nuclear extraction buffer (40mM TrisHCl PH 7.4, 420mM NaCl, 10% Glycerol, 1 mM EDTA). Samples were sonicated and centrifuged. Nuclear supernatants were collected.
Histology
After antigen retrieval, 10 μm frozen pancreatic sections were incubated with the indicated antibodies overnight and with fluorophore conjugated secondary antibody and DAPI for 1 h. Sections were mounted with PBS 70% and analysed in a Zeiss VivaTome.
Antisera
Antibodies used for immunoblotting, ChIP and Immunofluorescence are indicated in alphabetical order: glucagon (G2654, Sigma Aldrich), HIF-1 α (10006421, Cayman chemical), insulin (180067, ZYMED laboratories), MafA (NB400-137, Novus), PKAα cat (sc-903, Santa Cruz Biotechnology Inc.), PKIB (NBP1-74255, Novus Biologicals), PKIB (IF) (NBP1-55720, Novus Biologicals), pLKB1 (pSer428) (C67A3, Cell Signalling Technology), Phospho PKA substrates (100G7E, Cell Signalling Technology), Tubulin (05-829, Millipore). For CREB, pCREB (pSer133), and CRTC2 detection, rabbit polyclonal antibodies were raised against their respective antigens.
Statistical analysis
All mice used in experiments were around 10–12 weeks old except mice in HFD or HCD. Whenever possible, littermates of appropriate genotype were used as age-matched controls. The number of mice per experiment was limited by the availability of the required genotype and age.
Criteria of exclusion were: 1) gender and age 2) evident signs of disease. 3) spontaneous natural death during the experiment. Sample size (number of mice, islets and cells) is within the range of published literature. All results are presented as means ± SEM (standard error of mean). Statistical analysis were performed with unpaired Student’s t-test. Differences were considered statistically significant at p<0.05 (* p < 0.05; ** p < 0.01 and *** p < 0.001).
Supplementary Material
Acknowledgments
We thank Mark Huising for helpful discussioins. This work was supported by NIH grants (R01-DK049777, R01-DK083834, and R01-DK091618 (MM)) and P01-DK049210 (KK), the Clayton Foundation for Medical Research, the Kieckhefer Foundation, and the Leona M. and Harry B. Helmsley Charitable Trust grant #2012-PG-MED002.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdulahad WH, Kroese FG, Vissink A, Bootsma H. Immune regulation and B-cell depletion therapy in patients with primary Sjogren’s syndrome. Journal of autoimmunity. 2012;39:103–111. doi: 10.1016/j.jaut.2012.01.009. [DOI] [PubMed] [Google Scholar]
- Belyamani M, Gangolli EA, Idzerda RL. Reproductive function in protein kinase inhibitor-deficient mice. Mol Cell Biol. 2001;21:3959–3963. doi: 10.1128/MCB.21.12.3959-3963.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buysschaert M, Paris I, Selvais P, Oriot P, Preumont V. Glycemic control and weight changes in patients with type 2 diabetes intensified to three insulin regimens after therapeutic failure to exenatide. Acta clinica Belgica. 2012;67:250–254. doi: 10.2143/ACB.67.4.2062668. [DOI] [PubMed] [Google Scholar]
- Fantozzi DA, Harootunian AT, Wen W, Taylor SS, Feramisco JR, Tsien RY, Meinkoth JL. Thermostable inhibitor of cAMP-dependent protein kinase enhances the rate of export of the kinase catalytic subunit from the nucleus. J Biol Chem. 1994;269:2676–2686. [PubMed] [Google Scholar]
- Gunton JE, Kulkarni RN, Yim S, Okada T, Hawthorne WJ, Tseng YH, Roberson RS, Ricordi C, O’Connell PJ, Gonzalez FJ, et al. Loss of ARNT/HIF1beta mediates altered gene expression and pancreatic-islet dysfunction in human type 2 diabetes. Cell. 2005;122:337–349. doi: 10.1016/j.cell.2005.05.027. [DOI] [PubMed] [Google Scholar]
- Hall GC, McMahon AD, Dain MP, Wang E, Home PD. Primary-care observational database study of the efficacy of GLP-1 receptor agonists and insulin in the UK. Diabetic medicine : a journal of the British Diabetic Association. 2013;30:681–686. doi: 10.1111/dme.12137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holst JJ, Knop FK, Vilsboll T, Krarup T, Madsbad S. Loss of incretin effect is a specific, important, and early characteristic of type 2 diabetes. Diabetes Care. 2011;34(Suppl 2):S251–257. doi: 10.2337/dc11-s227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iezzi M, Kouri G, Fukuda M, Wollheim CB. Synaptotagmin V and IX isoforms control Ca2+-dependent insulin exocytosis. J Cell Sci. 2004;117:3119–3127. doi: 10.1242/jcs.01179. [DOI] [PubMed] [Google Scholar]
- Iezzi M, Theander S, Janz R, Loze C, Wollheim CB. SV2A and SV2C are not vesicular Ca2+ transporters but control glucose-evoked granule recruitment. J Cell Sci. 2005;118:5647–5660. doi: 10.1242/jcs.02658. [DOI] [PubMed] [Google Scholar]
- Irwin N, Hunter K, Frizzell N, Flatt PR. Antidiabetic effects of sub-chronic activation of the GIP receptor alone and in combination with background exendin-4 therapy in high fat fed mice. Regul Pept. 2009;153:70–76. doi: 10.1016/j.regpep.2008.11.007. [DOI] [PubMed] [Google Scholar]
- Jhala US, Canettieri G, Screaton RA, Kulkarni RN, Krajewski S, Reed J, Walker J, Lin X, White M, Montminy M. cAMP promotes pancreatic beta-cell survival via CREB-mediated induction of IRS2. Genes Dev. 2003;17:1575–1580. doi: 10.1101/gad.1097103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaihara KA, Dickson LM, Jacobson DA, Tamarina N, Roe MW, Philipson LH, Wicksteed B. beta-Cell-specific protein kinase A activation enhances the efficiency of glucose control by increasing acute-phase insulin secretion. Diabetes. 2013;62:1527–1536. doi: 10.2337/db12-1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo SH, Flechner L, Qi L, Zhang X, Screaton RA, Jeffries S, Hedrick S, Xu W, Boussouar F, Brindle P, et al. The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature. 2005;437:1109–1111. doi: 10.1038/nature03967. [DOI] [PubMed] [Google Scholar]
- Lee Y, Collins M, Kuchroo VK. Unexpected targets and triggers of autoimmunity. Journal of clinical immunology. 2014;34(Suppl 1):S56–60. doi: 10.1007/s10875-014-0040-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin F, Apetoh L, Ghiringhelli F. Controversies on the role of Th17 in cancer: a TGF-beta-dependent immunosuppressive activity? Trends Mol Med. 2012;18:742–749. doi: 10.1016/j.molmed.2012.09.007. [DOI] [PubMed] [Google Scholar]
- Park S, Dong X, Fisher TL, Dunn S, Omer AK, Weir G, White MF. Exendin-4 uses Irs2 signaling to mediate pancreatic beta cell growth and function. J Biol Chem. 2006;281:1159–1168. doi: 10.1074/jbc.M508307200. [DOI] [PubMed] [Google Scholar]
- Preumont V, Hermans MP, Bergman M, Buysschaert M. Predictive factors associated with primary failure to exenatide and non goal attainment in patients with type 2 diabetes. Acta clinica Belgica. 2012;67:411–415. doi: 10.2143/ACB.67.6.2062705. [DOI] [PubMed] [Google Scholar]
- Ravnskjaer K, Kester H, Liu Y, Zhang X, Lee D, Yates JR, 3rd, Montminy M. Cooperative interactions between CBP and TORC2 confer selectivity to CREB target gene expression. Embo J. 2007;26:2880–2889. doi: 10.1038/sj.emboj.7601715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Screaton RA, Conkright MD, Katoh Y, Best JL, Canettieri G, Jeffries S, Guzman E, Niessen S, Yates JR, 3rd, Takemori H, et al. The CREB coactivator TORC2 functions as a calcium- and cAMP-sensitive coincidence detector. Cell. 2004;119:61–74. doi: 10.1016/j.cell.2004.09.015. [DOI] [PubMed] [Google Scholar]
- Selmi C. Autoimmunity in 2011. Clinical reviews in allergy & immunology. 2012;43:194–206. doi: 10.1007/s12016-012-8330-2. [DOI] [PubMed] [Google Scholar]
- Shapiro AM, Hao E, Rajotte RV, Kneteman NM. High yield of rodent islets with intraductal collagenase and stationary digestion--a comparison with standard technique. Cell transplantation. 1996;5:631–638. doi: 10.1177/096368979600500606. [DOI] [PubMed] [Google Scholar]
- Shin S, Le Lay J, Everett LJ, Gupta R, Rafiq K, Kaestner KH. CREB mediates the insulinotropic and anti-apoptotic effects of GLP-1 signaling in adult mouse beta-cells. Molecular metabolism. 2014;3:803–812. doi: 10.1016/j.molmet.2014.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song WJ, Seshadri M, Ashraf U, Mdluli T, Mondal P, Keil M, Azevedo M, Kirschner LS, Stratakis CA, Hussain MA. Snapin mediates incretin action and augments glucose-dependent insulin secretion. Cell Metab. 2011;13:308–319. doi: 10.1016/j.cmet.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SS, Kim C, Vigil D, Haste NM, Yang J, Wu J, Anand GS. Dynamics of signaling by PKA. Biochim Biophys Acta. 2005;1754:25–37. doi: 10.1016/j.bbapap.2005.08.024. [DOI] [PubMed] [Google Scholar]
- Tu E, Ang DK, Bellingham SA, Hogan TV, Teng MW, Smyth MJ, Hill AF, van Driel IR. Both IFN-gamma and IL-17 are required for the development of severe autoimmune gastritis. European journal of immunology. 2012;42:2574–2583. doi: 10.1002/eji.201142341. [DOI] [PubMed] [Google Scholar]
- Van de Velde S, Hogan MF, Montminy M. mTOR links incretin signaling to HIF induction in pancreatic beta cells. Proc Natl Acad Sci U S A. 2011;108:16876–16882. doi: 10.1073/pnas.1114228108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen W, Harootunian AT, Adams SR, Feramisco J, Tsien RY, Meinkoth JL, Taylor SS. Heat-stable inhibitors of cAMP-dependent protein kinase carry a nuclear export signal. J Biol Chem. 1994;269:32214–32220. [PubMed] [Google Scholar]
- Wen W, Meinkoth JL, Tsien RY, Taylor SS. Identification of a signal for rapid export of proteins from the nucleus. Cell. 1995;82:463–473. doi: 10.1016/0092-8674(95)90435-2. [DOI] [PubMed] [Google Scholar]
- Xie T, Chen M, Zhang QH, Ma Z, Weinstein LS. Beta cell-specific deficiency of the stimulatory G protein alpha-subunit Gsalpha leads to reduced beta cell mass and insulin-deficient diabetes. Proc Natl Acad Sci U S A. 2007;104:19601–19606. doi: 10.1073/pnas.0704796104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Cannon B, Nedergaard J. Thermogenesis is beta3- but not beta1-adrenergically mediated in rat brown fat cells, even after cold acclimation. Am J Physiol. 1998;275:R2002–2011. doi: 10.1152/ajpregu.1998.275.6.R2002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.