

Abstract
Purpose
Lysophosphatidic acid (LPA), which is present in ascites of ovarian cancer patients, stimulates expression of vascular endothelial growth factor (VEGF). VEGF is essential for the development and abdominal dissemination of ovarian cancer. We examined how LPA drives VEGF expression to gain a better understanding of tumor angiogenesis under normoxic conditions.
Experimental Design
ELISA, Northern blotting, immunoblotting, quantitative PCR, and promoter reporter analysis in combination with small interfering RNA and pharmacologic inhibitors were used to examine LPA-induced VEGF expression and the underlying mechanisms.
Results
LPA stimulated expression of multiple VEGF variants. A 123-bp fragment proximal to the transcriptional initiation site was identified to be functional promoter region responsible for the response to LPA. The fragment harbors consensus sites for several transcription factors including c-Myc and Sp-1 but not hypoxia-inducible factor-1. Blockade of Rho, ROCK, or c-Myc reduced LPA-dependent VEGF production and promoter activation, suggesting that the G12/13-Rho-ROCK-c-Myc cascade partially contributes to VEGF induction by LPA. More significantly, the multiple Sp-1 sites within the responsive region of the VEGF promoter were essential for LPA-mediated transcription. LPA induced Sp-1 phosphorylation and DNA-binding and transcriptional activities. The silencing of Sp-1 expression with small interfering RNA or inhibition of Sp-1 with pharmacologic inhibitors blocked VEGF production induced by LPA.
Conclusions
LPA stimulates hypoxia-inducible factor-1-independent VEGF expression to promote tumor angiogenesis through activation of the c-Myc and Sp-1 transcription factors.
Lysophosphatidic acid (LPA), a naturally occurring phospholipid, affects many biological processes including neurogenesis, angiogenesis, would healing, immunity, and carcinogenesis (1, 2). LPA is a ligand of several G protein-coupled receptors LPA1/Edg-2, LPA2/Edg-4, LPA3/Edg-7, LPA4/GPR23/p2y9, LPA5/GPR92, LPA6/GPR87, and LPA7/p2y5. The LPA1, LPA2, and LPA3 receptors are members of the endothelial cell differentiation gene (Edg) family, sharing 50% to 57% homology in their amino acid sequences (3–5). LPA4, a member of the purinergic receptor family, and the related LPA5, LPA6, and LPA7 were structurally distant from the Edg LPA1 to LPA3 receptors (6–9). These LPA receptors couple to multiple G proteins, G12/13, Gi, Gq, and probably Gs, which, in turn, activate diverse pathways including Gq-mediated stimulation of phospholipase C, Gi-mediated activation of the Ras-mitogen-activated protein kinases and phosphatidylinositol-3-kinase, and G12/13-mediated activation of RhoA. Activation of these signaling events downstream of LPA receptors culminates in cytoskeleton remodeling, cell proliferation, survival, and migration (1, 2, 10).
In addition to these well-established biological functions, LPA is a potent modulator of gene expression, especially in cancer cells. LPA target genes include those involved in the inflammation, angiogenesis, and cancer progression such as interleukin (IL)-6 (11), IL-8 (11), growth-regulated oncogene α (12), cyclooxygenase-2 (13, 14), vascular endothelial growth factor (VEGF; refs. 15–17), and urokinase plasminogen activator (18). Although post-transcriptional protection of mRNA stability may be partially responsible for the sustained induction of some of these genes by LPA (14), the major input of induction is driven by transcriptional activation (11, 12, 14, 15). LPA may contribute to cancer development and progression through regulating expression of its target genes, resulting in a more angiogenic and metastatic microenvironment for tumor cells.
Angiogenesis is necessary for tumor growth and metastasis (19, 20). The development of new blood vessels depends on angiogenic factors released from tumor cells and/or cells in the tumor microenvironment. VEGF, also known as vascular permeability factor, is one of the most potent angiogenic factors involved in tumor angiogenesis (21, 22). VEGF mRNA and protein are extensively expressed in various human malignancies including ovarian cancer (21–23). Through increasing vascular permeability, VEGF also plays a pivotal role in the formation of ascites in ovarian cancer (23, 24). In addition, overexpression of VEGF is linked to tumor progression and poor prognosis of various tumors (23, 25).
A major trigger of VEGF expression in advanced tumors is hypoxia, a condition caused by an imbalance in oxygen supply and consumption. The hypoxia-induced up-regulation of VEGF is considered to be primarily mediated through hypoxiainducible factors (HIF; refs. 26, 27). HIF-1 is a heterodimeric basic helix-loop-helix transcription factor composed of two subunits, HIF-1α and HIF-1β. HIF-1α is the key regulatory component, which is rapidly degraded in normoxic conditions but stabilized and activated in hypoxia (26, 27). The HIF complex recognizes a consensus hypoxia response element in the promoter of VEGF and a broad range of other HIF target genes (26, 27). However, recent evidence suggests that HIF-1α deficient or silenced cells still showed a significant, albeit reduced, induction of VEGF in response to hypoxia (28, 29). The VEGF promoter can be activated by hypoxia when canonical hypoxia response elements are mutated or deleted in human cancer cell lines (30, 31). Furthermore, HIF-1 does not seem to play a major role in regulation of VEGF expression in normoxic conditions. These findings underscore the complexity of transcriptional regulation of VEGF.
LPA induces VEGF expression in ovarian and other cancer cell lines (15, 16). Because ascites of ovarian cancer patients is a LPA-enriched environment (32), the ability of LPA to up-regulate VEGF expression implies an in vivo function of LPA in regulation of VEGF expression in cancer patients. A recent observation suggests that LPA triggers VEGF expression in ovarian cancer cells via induction and activation of HIF-1α (16). Another study suggests the synergism between hypoxia and LPA in modulation of gene expression and other cellular responses (33). To elucidate the underlying mechanism for the effect of LPA on VEGF expression, we analyzed the human VEGF promoter and roles of potential transcription factors involved in LPA induction of VEGF in ovarian cancer cells. Our results indicate that Sp-1 and c-Myc mediate HIF-1α-independent induction of VEGF by LPA in ovarian cancer cells.
Materials and Methods
Materials
1-Oleoly (18:1) LPA was obtained from Avanti Polar Lipids. Before use, LPA was dissolved in PBS containing 0.5% fatty acid-free bovine serum albumin (BSA). BSA and protease inhibitor cocktail tablets were purchased from Roche. 32P and [α-32P]dCTP were purchased from Perkin-Elmer and Amersham Biosciences, respectively. Plasmid DNA was purified using the endo-free purification kit (Qiagen). Luciferase assay reagents were obtained from Promega. Pharmacologic inhibitors of ROCK (Y-27632), c-Myc (10058-F4), Sp-1 (mithramycin), and anti-tubulin α antibody were obtained from Calbiochem. Another Sp-1 inhibitor betulinic acid and anti-Sp-1 monoclonal antibody for the supershift assay were obtained from Sigma. All oligonucleotides and primers were synthesized by Operon Biotechnologies. Anti-HIF-1α monoclonal antibody was obtained from BD Transduction Laboratories. Anti-HIF-2α antibody was purchased from Abcam. The antibodies against Sp-1 and Rho for Western blotting were purchased from Santa Cruz Biotechnology. Trizol and cell culture reagents were obtained from Invitrogen. Bovine fetal serum was from Biomeda.
Cell culture
The sources of ovarian cancer cell lines used in the study were described previously (11, 12). These cells were cultured in RPMI 1640 supplemented with 10% fetal bovine serum, 100 units/mL penicillin, and 100 μg/mL streptomycin. All cell lines were frozen at early passages and used for <10 weeks in continuous culture.
Quantification of VEGF in culture supernatants
Ovarian cancer cell lines were plated in 6-well plates and grown to 70% confluence in complete medium. The cells were then starved for 24 h before stimulation with LPA or vehicle for the specified periods. The levels of VEGF present in culture supernatants were quantified using a VEGF ELISA kit (R&D Systems).
Northern blotting
Total cellular RNA was extracted from cell lines using the Trizol reagent following instructions of the supplier (Invitrogen). RNA samples were electrophoresed on agarose gel containing formaldehyde, stained with ethidium bromide, and transferred to N+ hybrid nylon. RNA was immobilized with UV cross-linking, prehybridized, and hybridized to 32P-labeled cDNA probes at 65°C overnight in a hybridization buffer (1% BSA, 0.5 mol/L NaH2PO4, 1 mmol/L EDTA, 7% SDS, and 10 μg/mL salmon sperm DNA). The cDNA of the human VEGF gene (gift of Dr. R. Jaffe, University of California-San Francisco) was used as a probe for hybridization. The 32P-deoxy-CTP-labeled DNA probe was prepared using the High Prime labeling system (Roche). Equal loading of RNA samples was confirmed by rehybridization to the cDNA of 18S rRNA (American Type Culture Collection).
Western blotting
Cells were lysed in SDS sample buffer or in ice-cold X-100 lysis buffer [1% Triton X-100, 50 mmol/L HEPES (pH 7.4), 150 mmol/L NaCl, 1.5 mmol/L MgCl2, 1 mmol/L EGTA, 10% glycerol, 100 mmol/L NaF, 10 mmol/L sodium pyrophosphate, and protease inhibitor cocktail]. Total cellular proteins were resolved by SDS-PAGE, transferred to Immun-Blot membrane (polyvinylidene difluoride; Bio-Rad), and immunoblotted with antibodies following the protocols of manufacturers. Immunocomplexes were visualized with an enhanced chemiluminescence detection kit (Amersham) using the horseradish peroxidase-conjugated secondary antibodies (Cell Signaling).
Quantitative PCR
mRNAs of VEGF variants were quantified by quantitative reverse transcription. cDNA was synthesized from total cellular RNA (1 μg, random primers) using the Superscript reverse transcription kit (Invitrogen). The TaqMan quantitative PCR kits for each VEGF isoform without cross-reaction were synthesized by Applied Biosystems. A common forward primer 5′-ATCTTCAAGCCATCCTGTGTGC-3′ and fluorescent hybridization probe 5′-AGTGTGTGCCCACTGAGGAGTCC-3′, both in exon 3, were used. Each splice variant was amplified with specific reverse primers spanning the variant specific exon boundaries (34): exon 5/8 for VEGF121 (5′-TGCGCTTGTCACATTTTTCTTG-3′), exon 5/7 for VEGF165 (5′-CAAGGCCCACAGGGATTTTC-3′), and exon 6/7 for VEGF189 (5′-CACAGGGAACGCTCCAGGAC-3′).
Luciferase vectors, deletion, and site-directed mutagenesis
The luciferase vectors containing 2018 to +50 (pGL2-VEGF2018-Luc) or −25 to +50 (pGL2-VEGF25-Luc; refs. 15, 26, 30) were kindly provided by Dr. R. Jaffe. The Sp-1 responsive luciferase vector (pGL2-4xSp1-TKLuc) was generated by cloning four repeats of the Sp-1 consensus sequence (TCGGGGCGGGGCG) into the NheI and HindIII sites in front of the herpes simplex virus thymidine kinase gene promoter (−35 to +50) in the pGL2-TK-Luc vector. The −708 to +50, −232 to +50, and −123 to +50 fragments were PCR amplified from pGL2-VEGF2018-Luc and inserted into the pGL2-basic vector to generate pGL2-VEGF708-Luc, pGL2-VEGF232-Luc, and pGL2-VEGF123-Luc. The promoter sequences in these plasmids were verified by automatic sequencing. The Sp-1 and c-Myc consensus sites within pGL-VEGF123-Luc were converted into inactive sequences using site-directed mutagenesis. The four potential Sp-1 consensus sequences designated Sp-1-I, Sp-1-II, Sp-1-III, and Sp-1-IV were mutated from CCCCGCCCCC (Sp-1-I), CGGGGCGG GC (Sp-1-II), GGGGGCGG GG (Sp-1-III), and CGGGCGGGGC (Sp-1-IV) to CCCCGAACCC, CGGAACGGGC, GGGAACGGGG, and CGGGAAGGGC, respectively. The c-Myc-binding site located at −56 was changed from CATGCG to CAAAAG.
For luciferase assays, ovarian cancer cell lines were transfected with luciferase vectors using TransIT-TKO (Mirus Bio). About 48 to 60 h after transfection, the cells were starved for 24 to 48 h before stimulation with LPA or vehicle for 6 h. Cell extracts were prepared and assayed for luciferase activity using the luciferase assay kit (Promega).
Rho activation
Activation of Rho was analyzed by GST pull-down assays (35). The cells were grown in 10 cm dishes to subconfluence, starved overnight, and stimulated with LPA or vehicle for the indicated periods. The cells were lysed in magnesium-containing lysis buffer [25 mmol/L HEPES (pH 7.5), 150 mmol/L NaCl, 1% NP40, 10% glycerol, 10 mmol/L MgCl2, 1 mmol/L EDTA, 1 mmol/L sodium orthovanadate, 10 μg/mL leupeptin, 10 μg/mL aprotinin, and 10 mmol/L NaF]. Clarified lysates were incubated for 1 h at 4°C with GST-Rhotekin-RBD (residues 7-89; ref. 35) produced in Escherichia coli and immobilized on glutathione-coupled Sepharose beads. Beads were washed in magnesium-containing lysis buffer three times, eluted with SDS sample buffer, and analyzed by Western blotting using rabbit anti-RhoA antibody.
Small interfering RNA
The human c-Myc small interfering RNA (siRNA; sense 5′-AACAGAAAUGUCCUGAGCAAUTT-3′ and antisense 5′-AUUGCUCAGGACAUUUCUGUUTT-3′) and nontarget control siRNA were obtained from Cell Signaling. The c-Myc siRNA was cotransfected along with the pGL-VEGF123-Luc vector using TransIT-TKO according to the manufacturer (Mirus Bio). About 48 h after transfection, the cells were starved for 24 h before stimulation with LPA or vehicle for 6 h. Cell extracts were prepared and assayed for luciferase activity.
The human Sp-1 and HIF-1α SMARTpool siRNA and the control siRNAs were obtained from Dharmacon. The siRNAs (2.25 μg) were transfected into ovarian cancer cell lines (1.25-1.5 × 106 cells) with Amaxa Nucleofector II (Kit T, program T32). The transfected cells were cultured in 12-well plates in complete medium. After 48 h, the cells were starved overnight in serum-free RPMI 1640 and stimulated with LPA for 14 to 16 h before culture supernatants were collected for quantification of VEGF. The efficiency of siRNA knockdown was confirmed by Western blotting analysis of cell lysates.
Electrophoretic mobility shift assay
The Sp-1 consensus oligonucleotides (sense 5′-GGATTCGATCGGGGCGGGGCGGAG-3′ and antisense 5′-GGCTCCGCCCCGCCCCGATCGAAT-3′) and the Sp-1 mutant form (sense 5′-GGATTCGATCGGTTCGGGGCGGAG-3′ and antisense 5′-GGCTCCGCCAA GCCCCGATCGAAT-3′) were annealed in 20 mmol/L Tris (pH 7.4), 1 mmol/L DTT, 50 mmol/L NaCl, and 10 mmol/L MgCl2. Annealed oligonucleotides were labeled at the 3′-ends with [α-32P]dCTP with Klenow. For the gel shift assay, binding reaction was done by incubating 4 μg nuclear proteins in 20 mmol/L HEPES (pH 8.0), 10% glycerol, 1 μg poly(dI-dC), and 300 μg/mL BSA in a final volume of 25 μL for 10 min at 25°C. The labeled oligonucleotides or cold competitor in the presence or absence of anti-Sp-1 monoclonal antibody (1 μg) was added to the reaction and incubated for an additional 15 min at 25°C before electrophoresis on 5% native polyacrylamide gels. The gels were dried and subjected to autoradiography using a Phosphorimager.
Statistics
All numerical data were presented as mean ± SD. The statistical significance of differences was analyzed using Student’s t test where P < 0.05 was considered statistically significant.
Results
Stimulation of VEGF expression by LPA in ovarian cancer cells
LPA increased VEGF expression in ovarian cancer cell lines (15, 16). In light of the copresence of LPA and ovarian tumor cells in ascites of ovarian cancer patients (32), LPA may act as a physiologic stimulus of VEGF expression in vivo. Therefore, a better understanding of this action of LPA may yield novel insights into the mechanism of tumor angiogenesis in ovarian cancer. We examined VEGF production in ovarian cancer cell lines cultured in 6-well plates treated with 10 μmol/L LPA for 16 h. VEGF levels in culture supernatants were measured with quantitative ELISA kits. As shown in Fig. 1A, LPA significantly stimulated VEGF production in Caov-3, OVCAR-3, and Dov-13 cells. The most responsive cell line Caov-3 expresses high levels of the LPA1 and LPA2 receptors (11, 14). Different from induction of other LPA target genes such as cyclooxygenase-2 and IL-8 (11, 14), the effect of LPA on VEGF expression seemed to be a delayed response. An increase in VEGF mRNA levels was obvious after LPA treatment for 2 h, peaking at 4 to 6 h (Fig. 1B), whereas LPA-mediated induction of cyclooxygenase-2 and IL-8 was detectable 0.5 to 1 h after exposure to LPA (11, 14). The different kinetics suggests involvement of distinct mechanisms in LPA induction of VEGF.
Fig. 1.

Induction of multiple isoforms of VEGF by LPA in ovarian cancer cell lines. A, LPA-stimulated secretion of VEGF. Caov-3, OVCAR-3, and Dov-13 cells were plated in 6-well plates, cultured to ~60% confluence, starved for 24 h and then stimulated with LPA (10 μmol/L) or vehicle (BSA control) for 16 h. VEGF levels (pg/mL) in culture supernatants were quantified using VEGF ELISA kits. B, LPA-induced VEGF mRNA expression. Caov-3 cells were starved and treated with LPA for 6 h at indicated concentrations (left) or treated with 10 μmol/L LPA for different periods (h; right). Total cellular RNA was isolated and analyzed by Northern blotting using 32P-labeled VEGF cDNA as a probe. The locations of 28S and 18S RNA were indicated. The membrane was reprobed with GAPDH to confirm comparable loading among samples. C, Caov-3 and OVCAR-3 cells were treated with 10 μmol/L LPA for 4 h before RNA preparation. The mRNA levels of three major VEGF variants (VEGF121, VEGF165, and VEGF189) were determined by quantitative PCR using quantitative PCR primers specific for each of the VEGF isoform as detailed in Materials and Methods. For this and the following illustrations, all numerical data are mean ± SD, representative of three independent assays. *, P < 0.05, statistical significant differences.
VEGF variants induced by LPA
There are many VEGF isoforms of different lengths of amino acids derived from alternative splicing from a single gene (34). The three major isoforms of VEGF are those composed of 121 amino acids (VEGF121), 165 amino acids (VEGF165), and 189 amino acids (VEGF189; ref. 34). Due to differential representation of basic residues encoded by exons 6 and 7, these VEGF isoforms differ in their heparin-binding properties and membrane association (34). VEGF121, which lacks the basic residues of both exons, does not bind heparin-containing cell surface proteoglycan (36) and is present in a secretive form. VEGF165 may partially bind to the cell surface or the extracellular matrix because cationic residues in exon 7 enable it to bind heparin. VEGF189 containing both exons 6 and 7 has the highest affinity for heparin and therefore is fully cell associated (34, 36). In addition to the difference in membrane association, there is also evidence that certain VEGF isoforms correlate with cancer patient outcomes or disease stages (37). It is therefore important to profile VEGF isoforms induced by LPA, which could not be revealed by ELISA quantification of secreted VEGF present in culture medium.
We designed TaqMan quantitative PCR specific for VEGF121, VEGF165, or VEGF189 to examine expression of their mRNAs. Caov-3 cells were treated with 10 μmol/L LPA or vehicle for 4 h and total cellular RNA was analyzed by quantitative PCR. As shown in Fig. 1C, LPA treatment led to induction of each of the three isoforms of VEGF in Caov-3 and OVCAR-3 cells. About 4- to 6-fold increases in VEGF121 and VEGF165 were observed in LPA-treated cells. VEGF189 was increased 1- to 3-fold by LPA.
HIF-1α–independent induction of VEGF by LPA
Hypoxia is a major environmental factor to trigger transcriptional activation of VEGF primarily through inducing HIF (26, 27). We examined whether LPA-induced VEGF expression involves HIF-1α or other HIF isoforms such as HIF-2α. Cobalt, a transition metal, mimics hypoxia by stabilizing HIF (38). Therefore, we treated Caov-3 and OVCAR-3 cells with LPA, CoCl2, or LPA + CoCl2. As shown in Fig. 2A, LPA (10 μmol/L, 16 h) stimulated a weak induction of HIF-1α when compared with the extremely strong effect of cobalt on HIF-1α. HIF-2α was induced by cobalt but not by LPA in these cell lines (Fig. 2A).
Fig. 2.
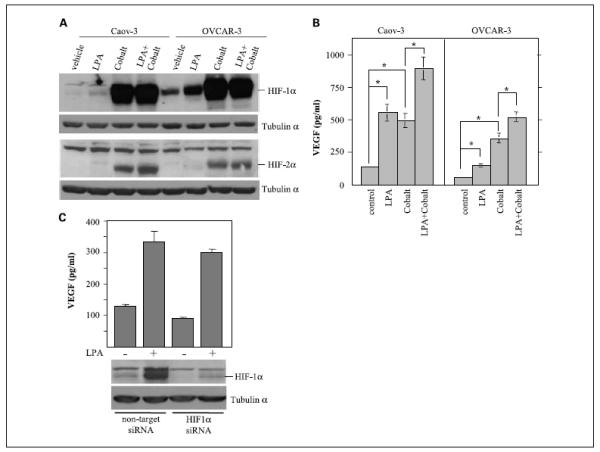
HIF-1α-independent induction of VEGF by LPA. Caov-3 and OVCAR-3 cells were starved and exposed for 16 h to LPA (10 μmol/L), CoCl2 (150 μmol/L), or LPA (10 μmol/L) + CoCl2 (150 μmol/L). Cell lysates were analyzed for HIF-1α and HIF-2α expression by immunoblotting (A) and culture supernatants were collected for ELISA quantification of VEGF (pg/mL; B). C, Caov-3 cells treated with the HIF-1α siRNA or with nontarget control siRNA were starved and stimulated with or without LPA (10 μmol/L) for 14 h before ELISA analysis of VEGF in culture supernatants. HIF-1α siRNA efficiency was confirmed by immunoblotting analysis of cell lysates.
We further analyzed VEGF levels present in culture supernatants of these cells. Both LPA and cobalt induced VEGF production. In Caov-3 cells, LPA elicited a stimulatory effect on VEGF comparable with that of cobalt, whereas, in OVCAR-3 cells, cobalt was more potent than LPA in increasing VEGF levels. Interestingly, in both cell lines, there was an additive effect on VEGF production between LPA and cobalt (Fig. 2B), consistent with utilization of disparate signaling processes by LPA and cobalt in promoting VEGF production.
To determine whether the weakly induced HIF-1α plays a role in stimulation of VEGF expression by LPA, we down-regulated HIF-1α expression with siRNA. Immunoblotting analysis of cell lysates confirmed strong suppression of HIF-1α in both LPA-treated and untreated cells (Fig. 2C). Although the knockdown of HIF-1α was associated with an appreciable decrease in basal VEGF levels in unstimulated control cells, LPA-induced VEGF expression over the basal levels was not significantly affected by siRNA suppression of HIF-1α (Fig. 2C), suggesting that other transcriptional pathways are involved in LPA induction of VEGF.
Promoter sequences required for LPA-mediated induction of VEGF
To characterize the effect of LPA on VEGF expression, we analyzed the VEGF promoter activity in LPA- and vehicle-treated ovarian cancer cell lines transfected with pGL2-VEGF2018-Luc containing the −2018 to +50 VEGF promoter sequences. The transfected cells were starved and then incubated with LPA for 6 h before the cell lysates were prepared and assayed for luciferase activity. LPA induced dose-dependent, multi-fold increases in luciferase activity in pGL2-VEGF2018-Luc-transfected Caov-3, OVCAR-3, and Dov-13 cells (Fig. 3A), suggesting transcriptional activation of the VEGF promoter by LPA.
Fig. 3.

Activation of VEGF transcription by LPA. A, LPA-stimulated activation of the VEGF promoter. Caov-3, OVCAR-3, or Dov-13 cells were transfected with pGL2-VEGF2018-Luc, starved for 1 day, and stimulated with LPA for 6 h at the indicated concentrations. The luciferase activities were analyzed with luciferase assay kits (Promega). Results were presented as relative light units (RLU). B, VEGF promoter sequences responsible for LPA stimulation were mapped to a 123-bp fragment proximal to the initiation site. Caov-3 cells were transfected with a series of deletion mutants containing 2018, 708, 232, 123, or 25 bp fragments of the VEGF promoter. LPA-stimulated luciferase activity was determined as in A. Data are presented as relative percentages with the activity of cells transfected with pGL2-VEGF2018-Luc defined as 100%. In this and the following figures, luciferase activities were normalized to β-galactosidase activity in the cells cotransfected with pCMVβ-gal.
To define the functional promoter region responsible for LPA stimulation, we made a series of deletions to remove the 5′-flanking sequences. LPA-induced luciferase activities in Caov-3 cells transfected with the deletion mutants (pGL2-VEGF708-Luc, pGL2-VEGF232-Luc, pGL2-VEGF123-Luc, and pGL2-VEGF25-Luc) were analyzed and compared as shown in Fig. 3B. A full response remained when the promoter sequence was eliminated to 123 bp upstream of the initiation site. A drastic decrease in LPA-stimulated luciferase activity was seen in pGL2-VEGF25-Luc-transfected cells, indicating that the response elements lie within the −123 to 25 bp fragment. The results are consistent with our previous observation that the regulatory sequences of other LPA target genes are usually close to the transcription initiation sites (11, 13). Interestingly, the 123-bp fragment of the VEGF promoter does not harbor any potential HIF-1-binding sites. The well-defined HIF-1 consensus sequence is located at around −975 of the VEGF promoter (26, 28).
Involvement of the Rho-ROCK-c-Myc cascade in LPA-induced VEGF expression
The 123-bp fragment of the VEGF promoter contains consensus sites of several well-defined transcription factors that have been implicated in HIF-1α-independent VEGF induction by other stimuli or in other cell models. These include the binding sites for c-Myc (location −56) and Ap-2 (location −87) and four GC-rich boxes for Sp-1 (Sp-1-I, Sp-1-II, Sp-1-III, and Sp-1-IV).
To evaluate the involvement of the c-Myc in up-regulation of VEGF by LPA, we mutated the c-Myc site from CATGCG to CAAAAG of pGL2-VEGF123-Luc and compared the LPA-induced luciferase activity with that of wild-type construct. The results shown in Fig. 4A indicated that abrogation of the c-Myc site led to a partial yet significant decrease in the promoter activity. Similarly, knockdown of c-Myc expression with siRNA (Fig. 4B) or treatment with the c-Myc-specific inhibitor 10058-F4 (Fig. 4C) also reduced the luciferase activity induced by LPA. These results establish that c-Myc contributes to induction of VEGF by LPA.
Fig. 4.

Input of c-Myc in transcriptional activation of VEGF. A, mutation of the c-Myc-binding site decreased VEGF promoter activity stimulated by LPA. Caov-3 cells were transfected with the wild-type reporter vector (pGL2-VEGF123-Luc) or c-Myc mutant vector (pGL2-VEGF123Mycmu-Luc). The transfected cells were starved and treated with LPA (10 μmol/L) or vehicle (BSA control) for 6 h and analyzed for luciferase activity. B, down-regulation of c-Myc expression attenuated VEGF promoter activity induced by LPA. Caov-3 cells were transfected with pGL2-VEGF123-Luc along with c-Myc-specific siRNA or nontarget control siRNA. LPA-stimulated luciferase activity was measured as in A. C, blockade of c-Myc with a specific inhibitor reduced the VEGF promoter activity. Caov-3 cells transfected with pGL2-VEGF123-Luc were stimulated in the absence or presence of 10058-F4 (2.5 μmol/L) and analyzed for luciferase activity.
To understand the signaling network of c-Myc activation, we assessed the role of the Rho-ROCK pathway lying upstream of c-Myc that has been linked to c-Myc-mediated VEGF induction in other cell systems (29, 39). In Caov-3 cells, LPA stimulated potent and sustained increases in Rho-GTPase levels as measured by GST pull-down assays (Fig. 5A). Expression of dominant-negative Rho (N19Rho) or botulinum C3 toxin (40) attenuated LPA-induced VEGF promoter activation by LPA. Inhibition of ROCK with its specific inhibitor Y-27632 also partially decreased luciferase activity from pGL2-VEGF123-Luc. These results are consistent with involvement of the Rho-ROCK-c-Myc pathway in LPA-mediated VEGF expression.
Fig. 5.

Involvement of the Rho-ROCK signaling in transcriptional activation of the VEGF promoter by LPA. A, LPA-stimulated immediate and sustained activation of Rho. Caov-3 cells cultured in 100 mm dishes were starved and stimulated with LPA (10 μmol/L) for the indicated periods (min). The levels of Rho-GTP were determined by GST-Rhotekin pull-down assay as described in Materials and Methods. B, inhibition of Rho activity with expression of C3 toxin or the dominant-negative N19Rho attenuated LPA-induced activation of VEGF promoter. Caov-3 cells were cotransfected with pGL2-VEGF123-Luc along with vectors expressing C3 toxin (left) or N19Rho (right) at a molar ratio 1:3. The transfected cells were starved and stimulated with 10 μmol/L LPA for 6 h and analyzed for luciferase activity as described in Fig. 3. C, inhibition of ROCK with Y-27632 decreased LPA-induced activation of the VEGF promoter. Caov-3 cells transfected with pGL2-VEGF123-Luc were starved and stimulated with LPA (10 μmol/L, 6 h) in the absence or presence of Y-27632 (10 μmol/L) and analyzed for luciferase activity.
Essential role for Sp-1 in LPA induction of VEGF
In addition to the c-Myc site, there are four Sp-1 and one Ap-2 sites clustered between −123 and −50 of the VEGF promoter. Mutation of the Ap-2 site did not compromise activation of the promoter by LPA (data not shown). The Sp-1 transcription factor has been implicated in both basal and stimulated VEGF expression in neoplastic cells (41, 42). To assess the possibility of Sp-1 involvement in LPA-dependent VEGF induction, we made single, double, triple, or all mutations of these sites as described in Materials and Methods. Sp-1-II and Sp-1-III are two most likely Sp-1-binding sites critical for VEGF expression induced by various agents (31, 41–43). Disruption of Sp-1-II or Sp-1-III led to significant decreases in both basal and LPA-stimulated transcription from the 123-bp promoter (Fig. 6A). Simultaneous mutation of the Sp-1-I and Sp-1-II sites led to more reduction in the promoter activity. The triple mutation of the Sp-1-II, Sp-1-III, and the upstream Sp-1-I further reduced the basal and LPA-stimulated activities. When the four Sp-1 sites were all mutated (all mutant), luciferase activity was essentially undetectable in control or LPA-stimulated cells, indicating the loss of the promoter activity by elimination of all potential Sp-1 sites (Fig. 6A).
Fig. 6.

Requirement of the Sp-1 transcription factor for activation of the VEGF promoter and VEGF production. A, Sp-1 sites were essential for LPA-dependent activation of the VEGF promoter. The DNA sequences listed are the VEGF promoter region spanning the four potential Sp-1 sites (underlined). Caov-3 cells were transfected with wild-type pGL2-VEGF123-Luc or Sp-1 mutant vectors. Luciferase activities from wild-type or mutant vectors induced by LPA (10 μmol/L, 6 h). B, LPA-stimulated Sp-1 phosphorylation and Sp-1 DNA-binding and transcriptional activities. Caov-3 cells were starved for 16 h, incubated with phosphate-free DMEM for 20 min, and then labeled with 32P in phosphate-free DMEM for 3 h before incubation with LPA (10 μmol/L) or vehicle (BSA) for 1, 3, or 6 h. Sp-1 was immunoprecipitated from cell lysates, resolved by SDS-PAGE, and transferred to Immun-Blot membrane for autoradiograph followed by immunoblotting for Sp-1 protein (left, top). Sp-1 DNA-binding activity was assessed by electrophoretic mobility shift assay (bottom). Nuclear extracts of Caov-3 cells stimulated for the indicated periods with LPA (10 μmol/L) were incubated with 32P-labeled Sp-1 consensus oligonucleotides (Wt Sp-1) or the inactive mutant form (mutant Sp-1). Reaction mixes were run on 5% native polyacrylamide gels and autoradiographed. The Sp-1 protein levels in nuclear extracts were examined by immunoblotting (left, bottom).The specificity of binding to the 32P-labeled Sp-1 probe was confirmed by inhibition of the binding with the unlabeled oligonucleotide (cold competition; left, bottom) and by a supershift assay with a Sp-1-specific antibody (Sp-1 Ab; right, bottom). LPA-induced Sp-1 transcriptional activity was analyzed by transfection of Caov-3 cells with the Sp-1 reporter pGL2-4×Sp1-TK-Luc. The cells were stimulated with LPA (10 μmol/L, 6 h) before measuring luciferase activity (right, top). C, Sp-1 was necessary for LPA-induced VEGF production. Caov-3 cells treated with the Sp-1 siRNA or nontarget control siRNA were starved and stimulated with LPA (10 μmol/L) for 16 h before ELISA analysis of VEGF present in culture supernatants. Net increases in VEGF production induced by LPA were calculated by subtracting background VEGF levels in unstimulated cells from LPA-induced VEGF production (left). Right, Caov-3 cells were starved and treated with LPA or vehicle for 16 h in the presence or absence of the Sp-1 inhibitor mithramycin (150 nmol/L).
To determine whether LPA activates Sp-1 in ovarian cancer cells, we tested the effect of LPA on phosphorylation of Sp-1, a post-translational modification associated with enhancement of Sp-1 DNA-binding and transcriptional activities (31, 41–43). Indeed, LPA induced time-dependent Sp-1 phosphorylation detectable at 1, 3, and 6 h after addition of LPA (Fig. 6B). We further analyzed Sp-1 DNA-binding activity in LPA-treated cells by electrophoretic mobility shift assay. As shown in Fig. 6B, LPA stimulated Sp-1 DNA binding in a time frame similar to that of Sp-1 phosphorylation induced by LPA. The increases in Sp-1 DNA binding were not accompanied by any augmentation in Sp-1 protein present in nuclear extracts (Fig. 6B), suggesting that Sp-1 was activated by phosphorylation or other post-translational modifications rather than alterations in protein synthesis or translocation. In agreement with increased Sp-1 phosphorylation and DNA binding, LPA also enhanced Sp-1 transcriptional activity as analyzed with the luciferase reporter pGL2-4xSp1-TK-Luc carrying four repeats of the Sp-1 consensus sequence. LPA treatment of Caov-3 cells transfected with the Sp-1 responsive reporter showed >10-fold increases in luciferase activity (Fig. 6B).
If LPA induces Sp-1 activation leading to up-regulation of VEGF, manipulation of Sp-1 protein expression or activity could impinge on the action of LPA. To test the possibility, we down-regulated Sp-1 expression in Caov-3 cells with siRNA. As shown in Fig. 6C, silencing of Sp-1 protein expression was associated with ~70% inhibition of LPA-induced VEGF production. Furthermore, mithramycin, an Sp-1 inhibitor that occupies the GC-rich boxes to prevent Sp-1 binding (44), blocked LPA-induced VEGF production by >80% (Fig. 6C). Another Sp-1 inhibitor, betulinic acid, which causes selective proteasome-dependent degradation of Sp-1 (45), also effectively inhibited VEGF production induced by LPA (data not shown). Taken together, these results establish a major role of Sp-1 in LPA-mediated VEGF expression.
Discussion
LPA is present at high levels in ascites of ovarian cancer patients (32). We showed previously that the LPA2 and LPA3 receptor subtypes are overexpressed in significant percentages of primary ovarian cancers and in a majority of ovarian cancer cell lines (10). Recent studies showed that the LPA2 receptor is overexpressed in many other human malignancies such as differentiated thyroid cancer, colon cancer, and mammary ductal carcinomas (46). Besides increased expression of specific LPA receptors, there is also evidence for overexpression of Lyso-PLD/autotaxin (ATX), a key enzyme in LPA production from lysophosphatidylcholine in many human cancers (47). These results indicate that amplification of LPA signaling via specific receptor overexpression or ligand generation is a common event associated with human cancers. However, it remains elusive how LPA participates in oncogenic processes. One possibility is that LPA modifies expression of oncoproteins or factors to create a tumor microenvironment that favors expansion of malignant cells. A major group of LPA target genes are indeed those encoding secreted factors such as IL-6, IL-8, growth-regulated oncogene a, and VEGF that promote tumor angiogenesis or tumor progression (12–17). When LPA2 is transgenically expressed in mouse ovaries, higher levels of VEGF and other LPA-induced factors are produced compared with nontransgenic ovaries (48), further supporting a role for LPA in tumor angiogenesis via up-regulating expression of VEGF and other proangiogenic factors.
In the present study, we used ovarian cancer cells as a model to dissect the signaling mechanism for VEGF induction by LPA. Our results establish that LPA induced transcriptional activation of VEGF independently of HIF-1α, a transcription factor most intimately involved in inducible VEGF expression. This is inconsistent with a recent study in which the effect of high concentrations of LPA (25 μmol/L) on VEGF expression involves HIF-1α induction and activation (16). In our experiments, LPA induced only weak HIF-1α expression compared with cobalt in ovarian cancer cell lines. Down-regulation of HIF-1α decreased the basal but not LPA-stimulated VEGF expression. The VEGF promoter fragment lacking HIF-1α consensus sites was fully capable of driving the response to LPA. These observations suggest that LPA and hypoxia could stimulate VEGF transcription cooperatively through distinct cascades. The ascites of ovarian cancer patients is a hypoxic environment where a large number of ovarian tumor cells reside (32, 33). LPA and hypoxia may thus act in concert to maximize VEGF production that is essential for the abdominal dissemination of ovarian tumor cells (23–25).
Through detailed analysis of the VEGF promoter and functional assays for transcription factors involved, we identified c-Myc and Sp-1 as major mediators of LPA-induced VEGF transcription. The Rho-ROCK-c-Myc pathway has been implicated in HIF-1α-independent induction of VEGF in hypoxia. Our results show that inhibition of each component of the Rho-ROCK-c-Myc pathway partially reduced the response to LPA. In ovarian cancer cell lines, LPA stimulated c-Myc expression and phosphorylation (data not shown), which may enhance c-Myc-mediated transcription. In many types of human cancers, the Myc oncogenes are amplified or overex-pressed. Accumulating evidence supports a correlation of excessive c-Myc with increased VEGF expression and tumor vasculature (49).
Although Sp-1 mediates VEGF expression in other cellular contexts (31, 41–45), linking Sp-1 to LPA-induced VEGF expression is a novel finding. A recent study showed that LPA induced telomerase activity in ovarian cancer cells through Sp-1-binding sites within the proximal 976- to 378-bp regions of the telomerase promoter (50). However, there was no direct evidence that LPA indeed stimulated Sp-1 activity to trigger telomerase up-regulation (50). In our study, we have used multiple approaches to validate the role of Sp-1 in LPA up-regulation of VEGF. The four specific Sp-1-binding sites close to the transcriptional initiation seem to be critical to LPA-mediated activation of the VEGF promoter. Further, LPA stimulated Sp-1 phosphorylation, Sp-1 DNA-binding activity, and transactivation potential. Because Sp-1 phosphorylation at serine and threonine residues is crucial for its binding to target DNA, it is likely that LPA activates Sp-1 through induction of its phosphorylation, although alterations in acetylation or in interaction with other proteins may also modulate Sp-1 transcriptional activity. Consistent with a critical role of Sp-1 in VEGF expression in vivo, previous studies showed that blockade of Sp-1 with pharmacologic inhibitors led to suppression of VEGF expression, tumor angiogenesis, and tumor growth (41–45). Our results presented herein further highlight the importance of targeting Sp-1 as a therapeutic approach to treat ovarian cancer and other malignancies.
Translational Relevance.
The molecular mechanisms governing VEGF expression in normoxic conditions by extracellular mediators such as LPA are poorly understood. We showed that LPA induced VEGF expression through c-Myc and Sp-1 transcription factors independently of HIF-1α in ovarian cancer cells. We further identified the signaling processes upstream of c-Myc and Sp-1 that link to LPA-induced activation of these transcription factors and the subsequent VEGF expression. The results establish a novel mode of VEGF induction through an agonist of G protein-coupled receptors and highlight new strategies to control VEGF expression and tumor angiogenesis.
Acknowledgments
Grant support: NIH/National Cancer Institute grant CA102196, U.S. Department of Defense Ovarian Cancer Award W81XWH0410103, and Massey Cancer Center pilot project grant (X. Fang) and NIH grant P30 CA16059 (Massey Cancer Center of Virginia Commonwealth University).
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Rivera R, Chun J. Biological effects of lysophospholipids. Rev Physiol Biochem Pharmacol. 2008;160:25–46. doi: 10.1007/112_0507. [DOI] [PubMed] [Google Scholar]
- 2.Mills GB, Moolenaar WH. The emerging role of lysophosphatidic acid in cancer. Nat Rev Cancer. 2003;3:582–91. doi: 10.1038/nrc1143. [DOI] [PubMed] [Google Scholar]
- 3.Hecht JH, Weiner JA, Post SR, Chun J. Ventricular zone gene-1 (vzg-1) encodes a lysophosphatidic acid receptor expressed in neurogenic regions of the developing cerebral cortex. J Cell Biol. 1996;135:1071–83. doi: 10.1083/jcb.135.4.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.An S, Bleu T, Hallmark OG, Goetzl EJ. Characterization of a novel subtype of human G protein-coupled receptor for lysophosphatidic acid. J Biol Chem. 1998;273:7906–10. doi: 10.1074/jbc.273.14.7906. [DOI] [PubMed] [Google Scholar]
- 5.Bandoh K, Aoki J, Hosono H, et al. Molecular cloning and characterization of a novel human G-protein-coupled receptor, EDG7, for lysophosphatidic acid. J Biol Chem. 1999;274:27776–85. doi: 10.1074/jbc.274.39.27776. [DOI] [PubMed] [Google Scholar]
- 6.Noguchi K, Ishii S, Shimizu T. Identification of p2y9/GPR23 as a novel G protein-coupled receptor for lysophosphatidic acid, structurally distant from the Edg family. J Biol Chem. 2003;278:25600–6. doi: 10.1074/jbc.M302648200. [DOI] [PubMed] [Google Scholar]
- 7.Lee CW, Rivera R, Gardell S, Dubin AE, Chun J. GPR92 as a new G12/13- and Gq-coupled lysophosphatidic acid receptor that increases cAMP, LPA5. J Biol Chem. 2006;281:23589–97. doi: 10.1074/jbc.M603670200. [DOI] [PubMed] [Google Scholar]
- 8.Tabata K, Baba K, Shiraishi A, Ito M, Fujita N. The orphan GPCR GPR87 was deorphanized and shown to be a lysophosphatidic acid receptor. Biochem Biophys Res Commun. 2007;363:861–6. doi: 10.1016/j.bbrc.2007.09.063. [DOI] [PubMed] [Google Scholar]
- 9.Pasternack SM, von Kügelgen I, Aboud KA, et al. G protein-coupled receptor P2Y5 and its ligand LPA are involved in maintenance of human hair growth. Nat Genet. 2008;40:329–34. doi: 10.1038/ng.84. [DOI] [PubMed] [Google Scholar]
- 10.Fang X, Schummer M, Mao M, et al. Lysophosphatidic acid is a bioactive mediator in ovarian cancer. Biochim Biophys Acta. 2002;1582:257–64. doi: 10.1016/s1388-1981(02)00179-8. [DOI] [PubMed] [Google Scholar]
- 11.Fang X, Yu S, Bast RC, et al. Mechanisms for lysophosphatidic acid-induced cytokine production in ovarian cancer cells. J Biol Chem. 2004;279:9653–61. doi: 10.1074/jbc.M306662200. [DOI] [PubMed] [Google Scholar]
- 12.Lee Z, Swaby RF, Liang Y, et al. Lysophosphatidic acid is a major regulator of growth-regulated oncogene α in ovarian cancer. Cancer Res. 2006;66:2740–8. doi: 10.1158/0008-5472.CAN-05-2947. [DOI] [PubMed] [Google Scholar]
- 13.Symowicz J, Adley BP, Woo MM, Auersperg N, Hudson LG, Stack MS. Cyclooxygenase-2 functions as a downstream mediator of lysophosphatidic acid to promote aggressive behavior in ovarian carcinoma cells. Cancer Res. 2005;65:2234–42. doi: 10.1158/0008.5472.CAN-04-2781. [DOI] [PubMed] [Google Scholar]
- 14.Oyesanya RA, Lee ZP, Wu J, et al. Transcriptional and post-transcriptional mechanisms for lysophosphatidic acid-induced cyclooxygenase-2 expression in ovarian cancer cells. FASEB J. 2008;22:2639–51. doi: 10.1096/fj.07-101428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu YL, Tee MK, Goetzl EJ, et al. Lysophosphatidic acid induction of vascular endothelial growth factor expression in human ovarian cancer cells. J Natl Cancer Inst. 2001;93:762–8. doi: 10.1093/jnci/93.10.762. [DOI] [PubMed] [Google Scholar]
- 16.Lee J, Park SY, Lee EK, et al. Activation of hypoxia-inducible factor-1α is necessary for lysophosphatidic acid-induced vascular endothelial growth factor expression. Clin Cancer Res. 2006;12:6351–8. doi: 10.1158/1078-0432.CCR-06-1252. [DOI] [PubMed] [Google Scholar]
- 17.Hu X, Mendoza FJ, Sun J, Banerji V, Johnston JB, Gibson SB. Lysophosphatidic acid (LPA) induces the expression of VEGF leading to protection against apoptosis in B-cell derived malignancies. Cell Signal. 2008;20:1198–208. doi: 10.1016/j.cellsig.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 18.Pustilnik TB, Estrella V, Wiener JR, et al. Lysophosphatidic acid induces urokinase secretion by ovarian cancer cells. Clin Cancer Res. 1999;5:3704–10. [PubMed] [Google Scholar]
- 19.Folkman J, Watson K, Ingber D, Hanahan D. Induction of angiogenesis during the transition from hyperplasia to neoplasia. Nature. 1998;339:58–61. doi: 10.1038/339058a0. [DOI] [PubMed] [Google Scholar]
- 20.Weidner N, Semple JP, Welch WR, Folkman J. Tumor angiogenesis and metastasis: correlation in invasive breast carcinoma. N Engl J Med. 1991;324:1–8. doi: 10.1056/NEJM199101033240101. [DOI] [PubMed] [Google Scholar]
- 21.Underiner TL, Ruggeri B, Gingrich DE. Development of vascular endothelial growth factor receptor (VEGFR) kinase inhibitors as anti-angiogenic agents in cancer therapy. Curr Med Chem. 2004;11:731–45. doi: 10.2174/0929867043455756. [DOI] [PubMed] [Google Scholar]
- 22.Ahmed SI, Thomas AL, Steward WP. Vascular endothelial growth factor (VEGF) inhibition by small molecules. J Chemother. 2004;4:59–63. doi: 10.1179/joc.2004.16.Supplement-1.59. [DOI] [PubMed] [Google Scholar]
- 23.Bamberger S, Perrett CW. Angiogenesis in epithelia ovarian cancer. Mol Pathol. 2002;55:348–59. doi: 10.1136/mp.55.6.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mesiano S, Ferrara N, Jaffe RB. Role of vascular endothelial growth factor in ovarian cancer: inhibition of ascites formation by immunoneutralization. Am J Pathol. 1998;153:1249–56. doi: 10.1016/S0002-9440(10)65669-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yamamoto S, Konishi I, Mandai M, et al. Expression of vascular endothelial growth factor (VEGF) in epithelial ovarian neoplasms: correlation with clinicopathology and patient survival, and analysis of serum VEGF levels. Br J Cancer. 1997;76:1221–7. doi: 10.1038/bjc.1997.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Forsythe JA, Jiang BH, Iyer NV, et al. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996;16:4604–13. doi: 10.1128/mcb.16.9.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bruick RK, McKnight SL. A conserved family of prolyl-4-hydroxylases that modify HIF. Science. 2001;294:1337–40. doi: 10.1126/science.1066373. [DOI] [PubMed] [Google Scholar]
- 28.Ryan HE, Lo J, Johnson RS. HIF-1 α is required for solid tumor formation and embryonic vascularization. EMBO J. 1998;17:3005–15. doi: 10.1093/emboj/17.11.3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mizukami Y, Fujiki K, Duerr EM, et al. Hypoxic regulation of vascular endothelial growth factor through the induction of phosphatidylinositol 3-kinase/Rho/ROCK and c-Myc. J Biol Chem. 2006;281:13957–63. doi: 10.1074/jbc.M511763200. [DOI] [PubMed] [Google Scholar]
- 30.von Marschall Z, Cramer T, Hocker M, Finkenzeller G, Wiedenmann B, Rosewicz S. Dual mechanism of vascular endothelial growth factor upregulation by hypoxia in human hepatocellular carcinoma. Gut. 2001;48:87–96. doi: 10.1136/gut.48.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pore N, Liu S, Shu HK, et al. Sp1 is involved in Akt-mediated induction of VEGF expression through an HIF-1-independent mechanism. Mol Biol Cell. 2004;15:4841–53. doi: 10.1091/mbc.E04-05-0374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu Y, Gaudette DC, Boynton JD, et al. Characterization of an ovarian cancer activating factor in ascites from ovarian cancer patients. Clin Cancer Res. 1995;1:1223–32. [PubMed] [Google Scholar]
- 33.Kim KS, Sengupta S, Berk M, et al. Hypoxia enhances lysophosphatidic acid responsiveness in ovarian cancer cells and lysophosphatidic acid induces ovarian tumor metastasis in vivo. Cancer Res. 2006;66:7983–90. doi: 10.1158/0008-5472.CAN-05-4381. [DOI] [PubMed] [Google Scholar]
- 34.Park JE, Keller GA, Ferrara N. The vascular endothelial growth factor (VEGF) isoforms: differential deposition into the subepithelial extracellular matrix and bioactivity of extracellular matrix-bound VEGF. Mol Biol Cell. 1993;4:1317–26. doi: 10.1091/mbc.4.12.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ren XD, Schwartz MA. Determination of GTP loading on Rho. Methods Enzymol. 2000;325:264–72. doi: 10.1016/s0076-6879(00)25448-7. [DOI] [PubMed] [Google Scholar]
- 36.Cohen T, Gitay-Goren H, Sharon R, et al. VEGF121, a vascular endothelial growth factor (VEGF) isoform lacking heparin binding ability, requires cell-surface heparan sulfates for efficient binding to the VEGF receptors of human melanoma cells. J Biol Chem. 1995;270:11322–6. doi: 10.1074/jbc.270.19.11322. [DOI] [PubMed] [Google Scholar]
- 37.Cressey R, Wattananupong O, Lertprasertsuke N, Vinitketkumnuen U. Alteration of protein expression pattern of vascular endothelial growth factor (VEGF) from soluble to cell-associated isoform during tumourigenesis. BMC Cancer. 2005;5:128–34. doi: 10.1186/1471-2407-5-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang GL, Semenza GL. Purification and characterization of hypoxia-inducible factor 1. J Biol Chem. 1995;270:1230–7. doi: 10.1074/jbc.270.3.1230. [DOI] [PubMed] [Google Scholar]
- 39.Man K, Su M, Ng KT, et al. Rapamycin attenuates liver graft injury in cirrhotic recipient-the significance of down-regulation of Rho-ROCK-VEGF pathway. Am J Transplant. 2006;6:697–704. doi: 10.1111/j.1600-6143.2005.01231.x. [DOI] [PubMed] [Google Scholar]
- 40.Aktories K, Hall A. Botulinum ADP-ribosyltransferase C3: a new tool to study low molecular weight GTP-binding proteins. Trends Pharmacol Sci. 1989;10:415–8. doi: 10.1016/0165-6147(89)90191-0. [DOI] [PubMed] [Google Scholar]
- 41.Safe S, Abdelrahim M. Sp transcription factor family and its role in cancer. Eur J Cancer. 2005;41:2438–48. doi: 10.1016/j.ejca.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 42.Abdelrahim M, Baker CH, Abbruzzese JL, et al. Regulation of vascular endothelial growth factor receptor-1 expression by specificity proteins 1, 3, and 4 in pancreatic cancer cells. Cancer Res. 2007;67:3286–94. doi: 10.1158/0008-5472.CAN-06-3831. [DOI] [PubMed] [Google Scholar]
- 43.Bradbury D, Clarke D, Seedhouse C, Corbett L, Stocks J, Knox A. Vascular endothelial growth factor induction by prostaglandin E2 in human airway smooth muscle cells is mediated by E prostanoid EP2/EP4 receptors and SP-1 transcription factor binding sites. J Biol Chem. 2005;280:29993–30000. doi: 10.1074/jbc.M414530200. [DOI] [PubMed] [Google Scholar]
- 44.Yuan P, Wang L, Wei D, et al. Therapeutic inhibition of Sp1 expression in growing tumors by mithramycin a correlates directly with potent antiangiogenic effects on human pancreatic cancer. Cancer. 2007;110:2682–90. doi: 10.1002/cncr.23092. [DOI] [PubMed] [Google Scholar]
- 45.Chintharlapalli S, Papineni S, Rapineni S, Ramaiah SK, Safe S. Betulinic acid inhibits prostate cancer growth through inhibition of specificity protein transcription factors. Cancer Res. 2007;67:2816–23. doi: 10.1158/0008-5472.CAN-06-3735. [DOI] [PubMed] [Google Scholar]
- 46.Murph M, Mills GB. Targeting the lipids LPA and S1P and their signalling pathways to inhibit tumour progression. Expert Rev Mol Med. 2007;9:1–18. doi: 10.1017/S1462399407000476. [DOI] [PubMed] [Google Scholar]
- 47.van Meeteren LA, Moolenaar WH. Regulation and biological activities of the autotaxin-LPA axis. Prog Lipid Res. 2007;46:145–60. doi: 10.1016/j.plipres.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 48.Huang MC, Lee HY, Yeh CC, Kong Y, Zaloudek CJ, Goetzl EJ. Induction of protein growth factor systems in the ovaries of transgenic mice overexpressing human type 2 lysophosphatidic acid G protein-coupled receptor (LPA2) Oncogene. 2004;23:122–9. doi: 10.1038/sj.onc.1206986. [DOI] [PubMed] [Google Scholar]
- 49.Knies-Bamforth UE, Fox SB, Poulsom R, Evan GI, Harris AL. c-Myc interacts with hypoxia to induce angiogenesis in vivo by a vascular endothelial growth factor-dependent mechanism. Cancer Res. 2004;64:6563–70. doi: 10.1158/0008-5472.CAN-03-3176. [DOI] [PubMed] [Google Scholar]
- 50.Bermudez Y, Yang H, Saunders BO, Cheng JQ, Nicosia V, Kruk PA. VEGF- and LPA-induced telomerase in human ovarian cancer cells is Sp1-dependent. Gynecol Oncol. 2007;106:526–37. doi: 10.1016/j.ygyno.2007.05.005. [DOI] [PubMed] [Google Scholar]