

Abstract
Primitive neuroectodermal tumors (PNET) are highly malignant, yet relatively uncommon neoplasms of the central nervous system. Although a host of different parts of the nervous system can be affected, intramedullary location of PNET is extremely rare. Most reports on intramedullary PNET have reported central PNET (cPNET); peripheral PNET (pPNET) affecting intramedullary spinal location is extremely rare. Till now, seven such cases of intramedullary pPNET have been described in medical literature in English. Here, we report an 11-year-old boy with cervicomedullary junction intramedullary pPNET who presented with intratumoral bleed, wherein the clinical presentation and radiological features gave us no clue preoperatively about the underlying diagnosis. In this report, we additionally review certain salient aspects of this dreaded disease in light of the existing evidence.
Keywords: Intramedullary, intratumoral bleed, peripheral primitive neuroectodermal tumors (pPNET), surgery
INTRODUCTION
Primitive neuroectodermal tumors (PNET) are highly malignant neoplasms sharing common histologic features. First reported by Earle and Hart in 1973, these tumors are derived from the common neuroepithelial cells capable of differentiating into glial cells, neurons, or melanocytes.[1] Two variants are recognized, namely central PNET (cPNET) and peripheral PNET (pPNET) (although, this demarcation is pathological and lacks clinical significance).
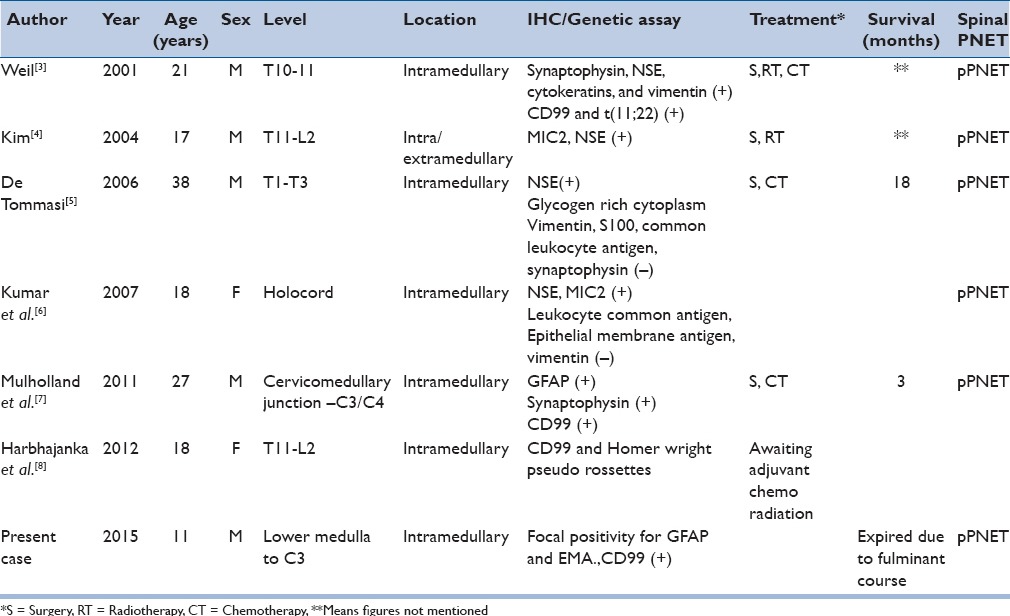
PNET involving the spine is an even rarer occurrence.[2] Spinal PNETs can be either primary or may develop secondary to subarachnoid dissemination of intracranial PNET. These can be extradural, intradural, or rarely, purely intramedullary in location. Intramedullary PNET is more frequent in children or adolescent age groups and extremely rare. Only seven cases of pPNET involving primarily the spinal cord have been described [Table 1].[3,4,5,6,7,8]
Table 1.
Peripheral PNET involving the spinal cord
We present here, a case of cervicomedullary junction intramedullary pPNET. The unique nature of this case is the location and presentation (intratumoral bleed) with histology of a peripheral variety of PNET. To our knowledge, intramedullary peripheral PNET with intratumoral bleed has not been reported before.
CASE REPORT
An 11-year-old boy presented with tingling paresthesias involving all four limbs of a 20-day-duration, followed by progressive quadriparesis involving upper limbs worse than lower limbs. There was a recent onset lower cranial nerve paresis with nasal regurgitation of meals.
Magnetic resonance imaging (MRI) of the cervical spine with lower brainstem revealed an intramedullary mass of heterogenous intensity on T1 and T2 sequences. The lesion was isointense with a hypointense center on T1 sequences, extending from the lower medulla to C3 and a caudal syrinx till the lower border of C6. There was a moderately strong enhancement of the peripheral part; however, the central T1/T2 hypointense area did not change signal on contrast [Figure 1a–d]. A working diagnosis of ependymoma was contemplated. After admission, the patient had acute deterioration with worsening quadriparesis and difficulty in respiration (pentaplegia). He was immediately placed on ventilator support under the cover of the steroid (methylprednisolone: Bolus, 30 mg/kg followed by 5.4 mg/kg/h infusion) and urgently taken up for surgery.
Figure 1.

On MRI of the cervicomedullary junction, a T1 (a) and T2 (b) heterogenous mass is seen expanding the lower medulla and the upper cervical spinal cord till the level of C3. The mass has hypo intense areas on both T1/T2 images (a and b). On contrast image, there is peripheral rim like enhancement of the mass without any enhancement of the centre (c). The mass is almost centrally located inside the neuraxis (d). The tumor showed pleomorphic cells displaying hyper chromatic nuclei, scant cytoplasm with brisk mitosis on H & E staining using 40x magnification (e). There was a strong membraneous positivity noted for CD99 (f)
C1-4 laminectomy with the removal of the posterior margin of the foramen magnum was done. The cord was expanded from the cervicomedullary junction to C4 without any surface changes. Dorsal midline myelotomy was done and the tumor was seen as a heterogeneous mass with a dusky red and friable appearance. It was moderately vascular with areas of thrombosed veins and hemorrhage within. While toward the lower pole, it was tenacious, minimally vascular, and gritty at places. The plane of demarcation was poor throughout. Near-total removal was achieved with an intraoperative impression of high grade lesion. The patient succumbed to sepsis, secondary to chest infection.
Histopathologically, the tumor was densely cellular with brisk mitosis and large areas of necrosis focally interspersed within discohesive groups of cells displaying high nuclear-cytoplasmic (N/C) ratio, moderately pleomorphic hyperchromatic nuclei, scanty to moderate cytoplasm. Occasional multinucleate tumor giant cells and few bizarre cells were noted [Figure 1e] as well. Cells were diffusely reactive for vimentin and focally reactive for smooth muscle antigen, epithelial membrane antigen, and glial fibrillary acidic protein. There was strong reactivity for CD99 molecule [Figure 1f]. Hence, a diagnosis of pPNET was made, although full battery of immunohistochemical and other tests could not be performed.
DISCUSSION
Hart & Earle first described PNET as a highly malignant and undifferentiated small round cell tumor of the central nervous system in 1973.[1] These tumors arise from the multipotent progenitor cells.
Two distinct varieties of PNET are known, namely cPNET and pPNETs. The cPNET variety is derived from the precursor cells of the central nervous system and pPNET developed from neural crest cells.[9] The cPNET behave similar to medulloblastomas while pPNET is grouped with Ewing's sarcoma family and managed accordingly. The cPNET are associated with cerebrospinal fluid (CSF) dissemination and distant metastases with pPNET.
PNET rarely affect the spine and may involve extradural, intradural extramedullary, as well as intramedullary compartments.[2] PNETs arise de novo in the spine or be secondary to drop metastasis. Hence, screening MRI of the entire craniospinal axis needs to be performed. Evaluation of metastasis elsewhere needs to be done with positron emission tomography (PET) scan. Due to lack of suspicion of such a rare condition, a preoperative craniospinal screening MRI or PET scan of the whole body was not done. In the absence of cranial symptoms or a known primary, we would like to believe that our case was a primary intramedullary PNET.
Pure intramedullary location is extremely uncommon as far as PNETs are concerned. We could encounter only one previous report of cervicomedullary junction involvement in a patient with type 1 neurofibromatosis (NF1).[7] Primary intramedullary PNETs are usually cPNETs. Ours is the eighth report of intramedullary pPNET with the second instance of cervicomedullary involvement. The patient presented with intratumoral bleeding, which has not been previously reported.
Immunohistochemistry is very crucial in subclassification of PNETs.[10] Chromosomal translocations t(11;22)(q24;q12) and variants such as t(21;22), t(7;22), t(17,22) are associated with pPNET while nonrandom genetic gains and losses are seen with cPNET (17q abnormalities being usually absent). The question is whether positivity for cluster of differentiation (CD) 99 and friend leukemia integration 1 transcription factor (FLI 1) is only pathognomonic of pPNETs. The fact is, although pPNETs typically show CD 99 and FLI 1 positivity, there are other tumors that can show positivity for the two markers. Tumors such as rhabdomyosarcomas, lymphoblastic lymphomas/leukemias, synovial sarcomas, solitary fibrous tumors, and neuroendocrine tumors may demonstrate CD99 positivity.[11] On the other hand, vascular tumors and lymphoblastic lymphoma may show FLI 1 positivity.[11] Hence, demonstration of chromosomal translocation and rearrangement at t(11;22) (q24;q12) using fluorescence in situ hybridization (FISH) technique is required to conclusively differentiate the two varieties of PNET.
Radiologically, these tumors are T1 hypointense and T2 hyperintense with moderate enhancement. Imaging features lack differentiation from more common intramedullary masses such as ependymoma and astrocytoma. The findings in our patient seemed to resemble intramedullary ependymoma with central location, caudal syrinx, and heterogeneous enhancement. However, the interior of the tumor appeared hypointense on both T1 and T2 and this part failed to enhance after contrast administration. These radiological findings were possibly suggestive of hemosiderin deposits from prior hemorrhage inside the tumor. During surgery, we found that the tumor was purely intramedullary, unlike the only other report of cervicomedullary pPNET (with NF-1), where the tumor was partly extramedullary.[7] The presence of intratumoral bleeding and thrombosed veins intraoperatively aroused a suspicion of glioblastoma. Bleeding inside an intramedullary PNET, to our knowledge, has not been previously reported.
Management strategies employed by different authors have largely been extrapolated from studies on medulloblastomas.[2,10] Because these tumors are poorly demarcated from the normal cord substance, decompression under constant monitoring using somatosensory evoked potentials/motor evoked potentials (SSEP/MEP) is recommended. Adjuvant therapy with a combination of radiochemotherapy has met with moderate success. Because radiotherapy (RT) can be detrimental to the cord and more so in the pediatric age group, intensity modulated radiation (IMRT), and proton beam RT have received attention. Couman et al. advocated proton beam RT, considering its safety profile compared to conventional cobalt-based radiotherapy.[10] The role of chemotherapy is to prevent systemic relapse, reduce craniospinal cerebrospinal fluid (CSF) dissemination, and at times to delay RT. Prognosis of intramedullary PNET is mostly dismal.[2]
Intramedullary pPNET involving the cervicomedullary junction is an extremely rare entity in the differential diagnosis of intramedullary tumors in children. As of now, maximal safe resection followed by aggressive chemoradiotherapy appears to be the standard treatment. Prognosis is generally poor but each and every case needs to be reported to increase our understanding of this rare, albeit, a highly malignant disease.
Financial support and sponsorship
Nil.
Conflicts of interest
The authors have no conflict of interest to declare.
REFERENCES
- 1.Hart MN, Earle KM. Primitive neuroectodermal tumors of the brain in children. Cancer. 1973;32:890–7. doi: 10.1002/1097-0142(197310)32:4<890::aid-cncr2820320421>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 2.Fujisawa H, Kaneko T, Tohma Y, Kida S, Kaizaki Y. Central nervous system primitive neuroectodermal tumor of spinal cord developing 20 years after curative treatment of pineal tumor. Neurol Med Chir (Tokyo) 2011;51:596–9. doi: 10.2176/nmc.51.596. [DOI] [PubMed] [Google Scholar]
- 3.Weil RJ, Zhuang Z, Pack S, Kumar S, Helman L, Fuller BG, et al. Intramedullary Ewing sarcoma of the spinal cord: Consequences of molecular diagnostics. Case Report. J Neurosurg. 2001;95(Suppl):270–5. doi: 10.3171/spi.2001.95.2.0270. [DOI] [PubMed] [Google Scholar]
- 4.Kim YW, Jin BH, Kim TS, Cho YE. Primary intraspinal primitive neuroectodermal tumor at conus medullaris. Yonsei Med J. 2004;45:533–8. doi: 10.3349/ymj.2004.45.3.533. [DOI] [PubMed] [Google Scholar]
- 5.De Tommasi A, De Tommasi C, Occhiogrosso G, Cimmino A, Parisi M, Sanguedolce F, et al. Primary intramedullary primitive neuroectodermal tumor (PNET) - Case report and review of the literature. Eur J Neurol. 2006;13:240–3. doi: 10.1111/j.1468-1331.2006.01183.x. [DOI] [PubMed] [Google Scholar]
- 6.Kumar R, Reddy SJ, Wani AA, Pal L. Primary spinal primitive neuroectodermal tumor: Case series and review of the literature. Pediatr Neurosurg. 2007;43:1–6. doi: 10.1159/000097517. [DOI] [PubMed] [Google Scholar]
- 7.Mulholland CB, Barkhoudarian G, Cornford ME, McBride DQ. Intraspinal primitive neuroectodermal tumor in a man with neurofibromatosis type 1: Case report and review of the literature. Surg Neurol Int. 2011;2:155. doi: 10.4103/2152-7806.86835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harbhajanka A, Jain M, Kapoor SK. Primary spinal intramedullary primitive neuroectodermal tumor. J Pediatr Neurosci. 2012;7:67–9. doi: 10.4103/1817-1745.97631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gollard RP, Rosen L, Anson J, Mason J, Khoury J. Intramedullary PNET of the spine: Long-term survival after combined modality therapy and subsequent relapse. J Pediatr Hematol Oncol. 2011;33:107–12. doi: 10.1097/MPH.0b013e3181f84b7f. [DOI] [PubMed] [Google Scholar]
- 10.Coumans JV, Walcott BP, Nahed BV, Oh KS, Chi AS. Multimodal therapy of an intramedullary cervical primitive neuroectodermal tumor in an adult. J Clin Oncol. 2012;30:e15–8. doi: 10.1200/JCO.2011.38.6474. [DOI] [PubMed] [Google Scholar]
- 11.Mardekian SK, Gandhe A, Miettinen M, Pack S, Curtis MT, Abdullaev Z. Two cases of spinal, extraosseous, intradural Ewing's sarcoma/peripheral neuroectodermal tumor: Radiologic, pathologic, and molecular analysis. J Clin Imaging Sci. 2014;4:6. doi: 10.4103/2156-7514.126050. [DOI] [PMC free article] [PubMed] [Google Scholar]