

Abstract
Purpose
Develop a Cremophor® and solvent free formulation of paclitaxel using amphiphilic block co-polymer micelles of poly(ethylene glycol)-b-poly(ε-caprolactone) (PEG-b-PCL) and characterize their release, solubility, cytotoxicity, tolerability, and disposition.
Methods
Hydrophobic prodrugs of paclitaxel were synthesized via DCC/DMAP or anhydride chemistry to overcome the poor loading (<1% w/w) of paclitaxel in micelles of PEG-b-PCL. Micelles were prepared by a co-solvent extraction technique. A micellar formulation of paclitaxel prodrug (PAX7′C6) was dosed intravenously to rats (10 mg/kg) and compared to Taxol® (paclitaxel in CrEL:EtOH) and PAX7′C6 in CrEL:EtOH as controls at the same dose. Pharmacokinetic parameters and tissue distribution were assessed.
Results
Paclitaxel prodrugs had solubilities >5 mg/ml in PEG-b-PCL micelles. Resulting PEG-b-PCL micelles contained 17-22% w/w prodrug and were less than 50 nm in diameter. PEG-b-PCL micelles released paclitaxel prodrugs over several days, t1/2>3 d. Only the 7′derivative of paclitaxel with the shortest acylchain 7′hexonoate (PAX7′C6) maintained cytotoxic activity similar to unmodified paclitaxel. PAX7′C6 micelles demonstrated an increase in area under the curve, half-life, and mean residence time while total clearance and volume of distribution decreased.
Conclusions
Paclitaxel prodrugs in PEG-b-PCL micelle nanocarriers augment the disposition and increase tolerability making further studies on tumor efficacy warranted.
Keywords: nanocarrier, paclitaxel, pharmacokinetics, polymer micelle, prodrug
Introduction
Paclitaxel has demonstrated efficacy in the treatment of a variety of cancers [1–4]. However, the poor patient tolerability and life-threatening hypersensitivity reactions to the current Taxol® formulation (paclitaxel in Cremophor® EL: Ethanol—CrEL:EtOH) have driven development of safer, improved, and less antagonistic delivery systems. For instance, the Food and Drug Administration (FDA) has recently approved an albumin-coated nanoparticle paclitaxel formulation with decreased toxicity, but it has been reported that albumin nanoparticles are rapidly cleared by the mono-nuclear phagocyte system [5,6].
Amphiphilic block copolymer (ABC) micelles are an intriguing group of self-assembled nanocarriers for drug delivery, owing to the prospects of biocompatibility, large solubilization capacity for poorly water soluble molecules, and potential for passive drug targeting to solid tumors via the enhanced permeability and retention (EPR) effect [7]. Poly(ethylene glycol)-block-poly(ε-caprolactone) (PEG-b-PCL) copolymers are especially promising due to their kinetic and thermodynamic stability (critical micelle concentration typical <1 μM), biocompatibility, and both components (poly(ethylene glycol)— PEG and poly(ε-caprolactone)—PCL) are FDA approved for use in humans. The utilization of ABC micelles has been effective in encapsulating different lipophilic drug molecules without the inclusion of potentially harmful surfactants and excipients such as Cremophor® EL and ethanol [8–10].
Poly(ethylene glycol)-block-poly(ε-caprolactone) (PEG-b-PCL) copolymers have demonstrated sustained circulation in rodents and biocompatibility [11]. PEG-b-PCL micelles are good candidates for poorly water soluble molecules in preclinical development, requiring drug solubilization, and optimized delivery. We have previously demonstrated that PEG-b-PCL can solubilize hydrophobic drugs such as amphotericin B, rapamycin, and geldanamycin prodrugs, owing to the highly hydrophobic nature of PCL [7–10,12,13]. However, they are unable to sufficiently solubilize paclitaxel, without extensive cross-linking of the core [14], which may incur manufacturing and regulatory issues.
We hypothesized that rendering paclitaxel more hydrophobic and bulky by the synthesis of acyl ester prodrugs of paclitaxel would increase the incorporation of the drug into PEG-b-PCL micelles and result in sustained release. This hypothesis emerges from an estimation of drug-PCL compatibility based on the Flory-Huggin drug-PCL parameter, often used to gauge polymer–solvent interaction [9,15].
The aims of these investigations are to overcome the solubilization problems of paclitaxel (Fig. 1, compound 1)by synthesis and encapsulation of novel acyl ester prodrugs of paclitaxel. Subsequently, we determined the ability to retain in vitro anti-cancer activity and the pharmacokinetics and tissue distribution of the acyl ester prodrug of paclitaxel—PAX7′C6 (Fig. 1, compound 4a), our lead candidate, solubilized in PEG-b-PCL compared to Taxol® (paclitaxel in Cremophor CrEL: EtOH) and PAX7′C6 (in CrEL:EtOH) as control formulations.
Fig. 1.

Paclitaxel prodrug synthesis scheme.
Materials and Methods
Paclitaxel Prodrug Synthesis
Hydrophobic prodrugs of paclitaxel were synthesized according to the procedure of Ali and co-workers [16]. Briefly, paclitaxel was conjugated at the 2′ and 7′ hydroxyl to fatty acids of increasing length (up to 16 carbons) via DCC/DMAP or anhydride chemistry [16] (Fig. 1). Synthesis of 6 derivatives of paclitaxel 1 was undertaken and included 7′hexonatepaclitaxel 4a, 7′dodeconate-paclitaxel 4b, 7′palmitate-paclitaxel 4c, 2′hexonate-paclitaxel 5a, 2′ dodeconate-paclitaxel 5b, and 2′ palmitate-paclitaxel 5c (Fig. 1). Examples of the synthesis procedures for 7′palmitate-paclitaxel 4c and 2′palmitate-paclitaxel 5c are given below.
Synthesis of 7-palmitate-paclitaxel 4c
The method for synthesis of 7-palmitate-paclitaxel 4c is described below. Synthesis of 4a–b were according to the same procedure, with substitution of the appropriate fatty anhydride.
2-TBS-paclitaxel 2
To a solution of paclitaxel 1 (300 mg, 0.35 mmol) in 1.2 ml dry DMF, TBDMSCl (158.84 mg, 1.053 mmol) and imidazole (59.80 mg, 0.8783 mmol) were added. The reaction mixture was stirred at room temperature for 12 h. The resulting solution was reduced to dryness in vaccuo, redissolved in 2 ml CH2Cl2, washed with saturated NH4Cl (5 mlx/1) followed by water (5 ml × 1), and the organic layer dried over Na2SO4. Removal of the solvent followed by preparatory TLC on silica (1:1 EtOAc:hexane) provided 2 as a white solid (310.42 mg, 95% yield). 1H NMR (400 MHz, CDCl3) δ 0.5 (s, 9H, tert-butyl), 1.10 (s, 3H, H17), 1.22 (s, 3H, H16), 1.76 (s, 3H, H19), 1.93 (s, 3H, H18), 1.92–2.14 μm, 2H, H6), 2.3 and 2.56 (m, 2H, H14), 2.58 (s, 3H, 4-Ac), 3.91 (d, J = 6.9 Hz, 1H, H3), 4.23 (d, J = 8.1 Hz, 1H, H20), 4.30 (d, J = 1.8 Hz, 1H, 10-OH), 4.35 (d, J = 8.1 Hz, 1H, H20), 4.42 (dd, J = 6.6 and 10.8 Hz, 1H, H7), 4.68 (d, J = 2.1 Hz, 1H, H2′), 4.98 (dd, J = 1.5 and 9.3 Hz, 1H, H5), 5.13 (d, J = 1.8 Hz, 1H, H10), 5.69 (d, J = 6.9 Hz, 1H, H2), 5.73 (dd, J = 1.8 and 9 Hz, 1H, H3′), 6.34 (t, J = 8.7 Hz, 1H, H13), 7.11 (d, J = 9 Hz, 1H, NH), 7.33–8.16 (m, 15H).
2-TBS-7-palmitate-paclitaxel 3
To a solution of 2 (50 mg, 0.053 mmol) in 1 ml dry toluene, palmitic anhydride (38.3 mg, 0.0774 mmol) was added. The reaction mixture was stirred at 90°C for 18 h. The resulting solution was washed with 1-M HCl (5 ml × 1) followed by water (5 ml × 1), and the organic layer was dried over Na2SO4. Removal of the solvent followed by preparatory TLC on silica (1:1 EtOAc:hexane) provided 3 as a white solid (25 mg, 41% yield).
1H NMR (400 MHz, CDCl3) d 0.5 (s, 9H, tert-butyl), 0.88 (t, 3H, CH3), 1.10 (s, 3H, H17), 1.22 (s, 3H, H16), 1.76 (s, 3H, H19), 1.93 (s, 3H, H18), 1.92–2.14 (m, 2H, H6), 2.3 and 2.56 (m, 2H, H14), 2.58 (s, 3H, 4-Ac), 3.91 (d, J = 6.9 Hz, 1H, H3), 4.23 (d, J = 8.1 Hz, 1H, H20), 4.30 (d, J = 1.8 Hz, 1H, 10-OH), 4.35 (d, J = 8.1 Hz, 1H, H20), 4.42 (dd, J = 6.6 and 10.8 Hz, 1H, H7), 4.68 (d, J = 2.1 Hz, 1H, H2′), 4.98 (dd, J = 1.5 and 9.3 Hz, 1H, H5), 5.13 (d, J = 1.8 Hz, 1H, H10), 5.69 (d, J = 6.9 Hz, 1H, H2), 5.73 (dd, J = 1.8 and 9 Hz, 1H, H3′), 6.34 (t, J = 8.7 Hz, 1H, H13), 7.11 (d, J = 9 Hz, 1H, NH), 7.33–8.16 (m, 15H).
7-palmitate-paclitaxel 4c
To a solution of 3 (25 mg, 0.211 mmol) in 1 ml of THF, 5 drops of 1-M TBAF (tetrabutylamoniumfloride) in THF was added. The reaction mixture was stirred at room temperature for 1 h. The resulting solution was reduced to dryness in vaccuo, redissolved in 2 ml CH2Cl2, washed with water (5 ml × 1), and the organic layer dried over Na2SO4. Removal of solvent followed by preparatory TLC on silica (1: 1 EtOAc:hexane) provided 4c as a white solid (20 mg, 90% yield). 1H NMR (400 MHz, CDCl3) d 0.88 (t, 3H, CH3), 1.10 (s, 3H, H17), 1.22 (s, 3H, H16), 1.76 (s, 3H, H19), 1.93 (s, 3H, H18), 1.92–2.14 (m, 2H, H6), 2.3 and 2.56 (m, 2H, H14), 2.58 (s, 3H, 4-Ac), 3.91 (d, J = 6.9 Hz, 1H, H3), 4.23 (d, J = 8.1 Hz, 1H, H20), 4.30 (d, J = 1.8 Hz, 1H, 10-OH), 4.35 (d, J = 8.1 Hz, 1H, H20), 4.42 (dd, J = 6.6 and 10.8 Hz, 1H, H7), 4.68 (d, J = 2.1 Hz, 1H, H2′), 4.98 (dd, J = 1.5 and 9.3 Hz, 1H, H5), 5.13 (d, J = 1.8 Hz, 1H, H10), 5.69 (d, J = 6.9 Hz, 1H, H2), 5.73 (dd, J = 1.8 and 9 Hz, 1H, H3′), 6.34 (t, J = 8.7 Hz, 1H, H13), 7.11 (d, J = 9 Hz, 1H, NH), 7.33–8.16 (m, 15H).
Synthesis of 2-palmitate-paclitaxel 5c
The method for synthesis of 2-palmitate-paclitaxel 5c is described below. Synthesis of 5a–b were according to the same procedure, with substitution of the appropriate fatty anhydride.
2-palmitate-paclitaxel 5c
To a solution of paclitaxel 1 (100 mg, 0.117 mmol) in 1.5 ml dry toluene was added palmitic anhydride (115.79 mg, 0.234 mmol) and DMAP (11.435 mg, 0.0936 mmol). The reaction mixture was stirred at room temperature for 12 h. The resulting solution was washed with a 1-M HCl (5 ml × 1) and water (5 ml × 1), and the organic layer was dried over Na2SO4. Removal of solvent followed by preparatory TLC (1:1 EtOAc:hexane) provided 5c as a white solid (60 mg, 47% yield). 1H NMR (400 MHz, CDCl3) d 0.87 (t, 3H, CH3), 1.10 (s, 3H, H17), 1.22 (s, 3H, H16), 1.76 (s, 3H, H19), 1.93 (s, 3H, H18), 1.92–2.14 (m, 2H, H6), 2.3 and 2.56 (m, 2H, H14), 2.58 (s, 3H, 4-Ac), 3.91 (d, J = 6.9 Hz, 1H, H3), 4.23 (d, J = 8.1 Hz, 1H, H20), 4.30 (d, J = 1.8 Hz, 1H, 10-OH), 4.35 (d, J = 8.1 Hz, 1H, H20), 4.42 (dd, J = 6.6 and 10.8 Hz, 1H, H7), 4.68 (d, J = 2.1 Hz, 1H, H2′), 4.98 (dd, J = 1.5 and 9.3 Hz, 1H, H5), 5.13 (d, J = 1.8 Hz, 1H, H10), 5.69 (d, J = 6.9 Hz, 1H, H2), 5.73 (dd, J = 1.8 and 9 Hz, 1H, H3′), 6.34 (t, J = 8.7 Hz, 1H, H13), 7.11 (d, J = 9 Hz, 1H, NH), 7.33–8.16 (m, 15H).
Preparation and Characterization of Prodrug Loaded PEG-b-PCL Micelles
Paclitaxel prodrug loaded PEG-b-PCL micelles were prepared by dissolving PEG-b-PCL (5000:10500, Mw/Mn 1.11, JCS Biopolytech Inc., Toronto, Ontario Canada) and pro-drug in a minimum volume of acetone and adding drop-wise to vigorously stirred ddH2O using a syringe pump. The organic solvent was then removed by stirring under an air purge. Where stated, samples were further concentrated by prolonged evaporation under an air purge. After removing the organic solvent, PEG-b-PCL micelles were passed through a 0.22-μm polyestersulfone filter to remove insoluble material and unincorporated drug [8]. In a typical experiment, 1 μM of PEG-b-PCL was dissolved in 0.75 ml of dry acetone and added dropwise (50 μl/min) to 2 ml of ddH2O yielding 0.5-μM PEG-b-PCL micelles after removing the volatile organic solvent.
The incorporation of prodrugs into PEG-b-PCL micelles was verified by equivalent retention times in ultraviolet (UV) and refractive index (RI) chromatographs from gel permeation chromatography. PEG-b-PCL micelles were injected on an OHpak SB-806M GPC column (20-μl injections, 0.5-μM PEG-b-PCL, 0.75 ml/min of ddH2O, 10°C; Shodex, Kawasaki, Japan) and detected by refractive index (RI) and UV absorbance (228 nm). Prodrug loading into PEG-b-PCL micelles was quantitatively determined by reverse-phase HPLC (Alltech Econosphere 3-μm 4.6 × 50 mm) using a 0.01% (v/v) trifluoroacetic acid (TFA)—acetonitrile (ACN) gradient (40– 100% ACN, 50°C, 228-nm detection and trans-stilbene as internal standard). Hydrodynamic diameters of PEG-b-PCL micelles were determined by dynamic light scattering (DLS; NICOMP 380 ZLS, Particle Sizing Systems, Santa Barbara, CA). Data were analyzed by intensity-weighted Gaussian distribution fitting (NICOMP version 1.76). Measurements were made for a minimum of 10 min or at least 100 × 105 counts in channel 1.
PEG-b-PCL Micelle Prodrug Release Studies
Release experiments were based on the methodology of Eisenberg and coworkers [17] with modifications for temperature and pH control. Micelle prodrug solutions were prepared at 0.5 μM (PEG-b-PCL basis) with 20% w/w prodrug as above, and 0.5 ml of each solution was diluted to 2.5 ml with ddH2O and injected into 10,000 MWCO dialysis cassettes (Pierce, Rockford, IL; n = 4). As a control, an equal quantity of prodrug dissolved in 100 μl of EtOH was injected into separate cassettes (n = 3). Dialysis cassettes were placed in a well-mixed temperature controlled water bath at 37°C, overflowed with ddH2O so that the bath volume was refreshed every 15 to 20 min. Peristaltic pumps under computer control separately injected 50 g/l solutions of tribasic and monobasic phosphate to maintain pH at 7.4±0.05 (apparatus built in-house). At fixed time points, dialysis cassette volumes were made up to 2.5 ml with ddH2O, 100-μl aliquots l aliquots withdrawn, and prodrug concentrations determined by reverse-phase HPLC (see supra).
Octanol-Water Partition Coefficients
Octanol-water partition coefficients (log Po/w) of paclitaxel prodrugs were determined indirectly by microemulsion electrokinetic chromatography (MEEKC) based on the technique of Klotz et al.[18]. Running buffer was prepared by titration of 25-mM sodium phosphate monobasic with 50-mM sodium tetraborate to pH 7.00, and 1.44 g of sodium dodecyl sulfate, 6.49 g of 1-butanol, and 0.82 g of heptane were made up to 100 ml with phosphate-borate buffer. The running buffer was ultrasonicated for 30 min in a closed 250-ml flask in ice water (G112SP1 Special Ultrasonic Cleaner, Laboratory Supplies Company Inc., Hicksville, NY). Longer times may be required to obtain a stable emulsion with lower power ultrasonicators. Compounds and standards (n = 3) were dissolved in the running buffer (0.05 mg/ml) with 0.5 μl/ml of nitromethane and 0.5 μl/ml of 1-phenyldodecane by ultrasonication (10 min) in a closed tube and centrifuged (16,000×g, 3 min) to degas. A BioFocus 3000 capillary electrophoresis system (Bio-Rad, Hercules, CA) equipped with a 50-μm ID × 37-cm uncoated fused-silica column (Polymicron Technologies LLC, Phoenix, AZ) was used for MEEKC experiments. The column was prewashed with 1-M NaOH for 5 min and before runs with 0.1-M NaOH for 1 min, ddH2O for 1 min, and running buffer for 1 min at 100 psi (690 kPa). Running conditions were 10 kV (ca. 30–35 μA, 30 min/run) at 20°C with 1-psi·s injections (6.9 kPa·s) and detection at 210 and 232 nm. Log Po/w and retention factors, k′, were calculated using the equations:
where tr, t0, and tme are retention times of the prodrug, nitromethane, and 1-phenyldodecane, respectively. Fitting parameters a and b were determined by linear regression of known standards: pyridine, phenol, benzoic acid, anisole, benzene, toluene, dodecanoic acid, benzopyrene, and pyrene (R2 = 0.996, Excel® 2003, Microsoft Corp.).
Paclitaxel 7′C6 (PAX7′C6) Formulation in PEG-b-PCL Micelles
Paclitaxel 7′C6 (PAX7′C6) loaded PEG-b-PCL micelles were prepared as described above. The drug solution was made isotonic with 300 mg dextrose (5% w/v) and then filter-sterilized. The control formulations of paclitaxel in Cremophor CrEL:EtOH) and PAX7′C6 (in CrEL:EtOH) were prepared by dissolving paclitaxel in ethanol (3 mg/ml) and adding to a solution of ethanol (final 1.5 mg/ml paclitaxel) [19]. The resulting mixture was vortexed until clear and passed through a 0.45-μm PTFE syringe filter. PAX7′C6 was solubilized to 1.33 mg/ml in PEG-b-PCL. Taxol® (30 mg/ml paclitaxel in Cremophor CrEL:EtOH) and PAX7′C6 (30 mg/ml PAX7′C6 in CrEL: EtOH) were formulated at 1.5 mg/ml by dilution in 0.9% saline. Immediately before injection, the concentrations of paclitaxel and PAX7′C6 were verified by reverse-phase HPLC as above.
Cell Cytotoxicity
The 6 derivatives of paclitaxel were screened for preservation of activity by incubation with each prodrug in MDA-MB-231 (Her-2/Neu negative, Estrogen receptor negative Human Breast Adenocarcinoma) and MCF-7 (Her-2/Neu negative, Estrogen receptor positive Human Breast Adenocarcinoma) cell lines. Paclitaxel and PAX7′C6 (0.5–5,000 ng/ml) were further tested in the cancer cell lines, SK-BR-3 (Her-2/Neu positive, Estrogen receptor negative Human Breast Adenocarcinoma) and +SA (WAZ-2T; Spontaneous Mouse Breast Cancer Cell Line). PAX7′C6 in PEG-b-PCL micelles was incubated with MDA-MB-231 cells (0.5–5,000 ng/ml). All cell lines were purchased from the American Type Culture Association (ATCC, Rockville, MD). MDA-MB-231 and SKBR-3 cell lines were maintained in McCoy's 5A medium and supplemented with 10% heat-inactivated fetal bovine serum (FBS), penicillin-streptomycin solution (10 ml/l), and HEPES (6.0 g/l). MCF-7 were maintained in RMPI 1640 supplemented with 10% fetal bovine serum, 100 IU penicillin, and 100 μg/ml streptomycin, and 2 mM l-glutamine. +SA cell line was maintained in Dulbecco's Modified Eagle Medium (DMEM) and supplemented with 10% heat-inactivated fetal bovine serum (FBS), penicillin-streptomycin solution (10 ml/l), HEPES (2.4 g/l), and sodium pyruvate (110.4 mg/l). All cell lines were incubated at 37°C in a 5% CO2 atmosphere. MDA-MB-231, and +SA cell lines was seeded at 5,000 cells/well while SK-BR-3 cell line was seeded at 15,000 cells/well and MCF-7 were seeded at 3,000 cells/well in 96-well plates.
Alamar Blue Assay
Alamar Blue (resazurin) fluorescent dye is a facile and accurate assay to determine the cytotoxicity of many cell lines including the four cancer cell lines used in the present study [20]. The resazurin non-fluorescent compound is metabolized into the fluorescent compound resorufin by intact and viable cells. This emission of fluorescence can be quantified using a cell plate reader and the number of viable cells following treatment can be determined. Cells were counted and seeded on 96 well plates. The seeded cells were incubated at 37°C in a 5% CO2 atmosphere for 24 h. On the day of the experiment, paclitaxel and PAX7′C6 was dissolved in DMSO while the PAX′7C6 formulation was dissolved in water. The formulation and each compound were diluted in medium to yield concentrations of 0.5, 1, 5, 10, 50, 100, 500, 1000, and 5000 ng/ml. Following aspiration of the medium, cells were treated with either paclitaxel, PAX7′C6, or PAX7′C6 PEG-b-PCL micelles. Additional cells were treated with either DMSO diluted in medium, water diluted in medium (for micelle comparison), or medium only as controls. Treated and control cells were incubated at 37°C in a 5% CO2 atmosphere for 72 h. After cell plates were removed from the incubator, alamar blue (resazurin) fluorescent dye was added directly to the media (20 μl) to give a final concentration of 10% alamar blue in each well. Cell plates were incubated at 37°C in a 5% CO2 atmosphere for an additional 3 h. Following incubation, cell plates were placed in a darkened environment for 15 min at room temperature. Next, the cell plates were placed into the Cytoflour®4000 fluorescence multi-well plate reader (Applied Biosystems, USA). Fluorescence was read at an excitation of 485 nm and an emission of 530 nm. The viable cell number (as a percent of control) in each cell line exposed to varying concentrations of paclitaxel prodrug and paclitaxel were measured. The IC50 values for each cell line were determined by using pharmacodynamic modeling using WinNonlin® software (Ver. 5.1).
Surgical Procedures
Male Sprague-Dawley rats (200–240 g) were obtained from Simonsen Labs (Gilroy, CA, USA) and given food (Purina Rat Chow 5001) and water ad libitum in our animal facility for at least 3 days before use. Rats were housed in temperature-controlled rooms with a 12 h light/dark cycle. The day before the pharmacokinetic experiment, the right jugular veins of the rats were catheterized with sterile silastic cannula (Dow Corning, Midland, MI, USA) under halothane (Sigma Chemical Co. St. Louis Mo. USA) anesthesia. This involved exposure of the vessel prior to cannula insertion. After cannulation, the Intramedic PE-50 polyethylene tubing (Becton, Dickinson and Company, Franklin Lakes, NJ, USA) connected to the cannula was exteriorized through the dorsal skin. The cannula was flushed with 0.9% saline. The animals were transferred to metabolic cages and fasted overnight. Animal use protocols were approved by The Institutional Animal Care and Use Committee at Washington State University, in accordance with “Principles of laboratory animal care” (NIH publication No. 85-23, revised 1985).
Pharmacokinetic Study
Sprague-Dawley male rats with body weights ranging from 200 to 240 g were used to examine the effect of solubilizing vehicle on the pharmacokinetics of PAX7′C6 compared to Taxol® (paclitaxel in Cremophor CrEL:EtOH) and PAX7′C6 in CrEL:EtOH as controls. The rats were housed in a temperature-controlled room with a 12 h light/dark cycle for at least 1 week. The day before the pharmacokinetic experiment, the rats were cannulated as described above. Each of the animals was placed in separate metabolic cages, allowed to recover overnight, and fasted for 12 h before dosing. On the days of the experiment, the animals were dosed intravenously (10 mg/kg) with PAX7′C6 formulated in PEG-b-PCL nanocarrier, Taxol® (paclitaxel in Cremophor CrEL:EtOH), or PAX7′C6 (in CrEL:EtOH; n = 10 for each treatment group). The rats were given between 0.5 and 1 ml of each formulation as IV bolus. The utilized dose was based on previous pharmacokinetics studies that administered Taxol® and other similar delivery systems of paclitaxel [21–23]. After dosing, serial blood samples (∼0.30 ml) were collected from the cannula at 0, 1 min, and 30 min, then 1, 2, 4, 6, 12, 24, and 48 h after IV administration, and the cannula flushed with 0.9% saline. After dosing and after each serial blood sampling, blinded observers were present to record any visible behavior, bleeding, or change in overall appearance of the animal as signs of acute toxicity. Each blood sample was collected into regular polypropylene microcentrifuge tubes and following centrifugation, the serum was collected and stored at −70°C until analyzed. Urine samples were also collected at 0, 2, 6, 12, 24, 48, and 72 h following IV administration and were stored at −70°C until analyzed.
Biodistribution Studies
To assess the effect of formulation on the tissue distribution of PAX7′C6, rats (n = 10 for each group, 200– 240 g) were cannulated and intravenously administered with Taxol® (paclitaxel in Cremophor CrEL:EtOH), PAX7′C6 in CrEL:EtOH, or PAX7′C6 in PEG-b-PCL micelles all equivalent to the previous pharmacokinetic studies. At either 6 or 24 h after formulation injection, each animal (n = 5 for each time point) was anaesthetized and exsanguinated by cardiac puncture. Brain, lungs, heart, liver, spleen, kidneys, urinary bladder, muscle as well as samples of whole blood and serum were collected. Tissue samples were blotted with paper towel, washed in ice-cold saline, bottled to remove excess fluid, weighed and rapidly frozen in liquid nitrogen, pulverized to a fine powder with a mortar and pestle under liquid nitrogen and stored at −70°C until assessed for drug concentrations by HPLC analysis.
Pharmacokinetic Analysis
Pharmacokinetic analysis was performed using data from individual rats for which the mean and standard error of the mean (SEM) were calculated for each group. The elimination rate constant (KE) was estimated by linear regression of the blood or plasma concentrations in the log-linear terminal phase. In order to estimate the serum concentrations (C0) immediately after nanoformulated PAX7′C6, PAX7′C6 in CrEL:EtOH or Taxol® (paclitaxel in CrEL:EtOH) IV dosing, a two-compartmental model was fitted to the serum concentration versus time data using WinNonlin® software (Ver. 5.1). The estimated C0 was then used with the actual measured serum concentrations to determine the area under the concentration-time curve (AUC). The AUC0-∞ was calculated using the combined log-linear trapezoidal rule for data from time of dosing to the last measured concentration, plus the quotient of the last measured concentration divided by KE. Non-compartmental pharmacokinetic methods were used to calculate mean residence time (MRT by dividing AUMC0-∞ by AUC0-∞), clearance (CL by dividing dose by AUC0-∞) and volume of distribution (Vdβ by dividing CL by KE). Based on the cumulative urinary excretion, the fraction excreted in urine [Fe by dividing the total cumulative amount of paclitaxel excreted in urine (ΣXu) by the dose], renal clearance (CLr by multiplying Fe by CL), hepatic clearance (CLhepatic by subtracting CLr from CL), extraction ratio [ER by dividing CLhepatic by hepatic flow (Q)]. The mean hepatic blood flow (Q) is approximately 3.22 l/h/kg [24]. Using the hematocrit in rat (24) of 0.48, this yields a mean hepatic plasma flow of 1.74 l/h/kg. Therefore, depending on serum or whole blood the correct hepatic flow (Q) needs to be employed.
Data Analysis
Compiled data were presented as mean and standard error of the mean (mean ± SEM). Where possible, the data were analyzed for statistical significance using NCSS Statistical and Power Analysis software (NCSS, Kaysville, UT). General Linear Model (GLM) ANOVA was employed with a value of p < 0.05 being considered statistically significant.
Results
Solubility and Sizing
All of the paclitaxel prodrugs have greatly enhanced solubility in PEG-b-PCL as compared to paclitaxel. Drug-loaded micelles were readily concentrated by evaporation to achieve solubilities >5 mg/ml (Table I). Drug-loaded micelles were 27 to 44 nm in diameter as determined by DLS. Acylation increases solubility in caprolactone micelles. The decrease in the χ parameter as compared to paclitaxel demonstrates greater core-drug compatibility. The Flory-Huggins interaction parameter for these prodrug and PCL was calculated (Table I). Drugs with relatively small interaction parameters, χdrug-PCL <1, are well solubilized (>1:1 mmol drug: mol PCL) by PEG-b-PCL or PEG-b-PCl-b-PEG nanocarrier systems whereas drugs with larger interaction parameters, such as paclitaxel, are very poorly solubilized by PEG-b-PCL micelles. The oil-water and n-octanol partitioning behavior of drugs can be useful in predicting a priori the in vivo tissue distribution [25]. All of the fatty acid prodrugs had similar lipophilic, partitioning into the oil phase as compared to paclitaxel (Table I). Actually loadings are reported in Table I.
Table I. Solubility Parameters and Sizing of PEG-b-PCL Micelles Loaded with Paclitaxel Prodrugs.
| Prodrug | δ drug (J/cm3)1/2 | Vdrugcm3/mol | Diameter (intensity), nmc | χ drug-PCL | log Po/w | Prodrug: caprolactone mmol:mola | Prodrug w/w %a | Solubilized mg/mla,b |
|---|---|---|---|---|---|---|---|---|
| Paclitaxel 1 | 26.7 | 498 | – | 8.59 | 4.40±0.06 | <1 | – | <0.2 |
| 4a | 24.5 | 604 | 34±4 | 4.55 | 4.43±0.06 | 36.5 | 17.1 | 1.55±0.04 (5.1±0.5) |
| 4b | 23.5 | 700 | 27±5 | 3.14 | 4.59±0.18 | 31.8 | 16.4 | 1.47±0.03 (2.2±0.5) |
| 4c | 23.0 | 765 | 44±2 | 2.43 | 4.48±0.06 | 33.3 | 21.6 | 1.62±0.03 (3.0±0.9) |
| 5a | 24.5 | 604 | 32±0 | 4.55 | 4.45±0.03 | 33.4 | 17.8 | 1.42±0.11 |
| 5b | 23.5 | 700 | 28±0 | 3.14 | 4.49±0.03 | 34.0 | 17.3 | 1.57±0.02 |
| 5c | 23.0 | 765 | 37±6 | 2.43 | 4.51±0.04 | 40.0 | 19.8 | 1.85±0.05 |
Solubility and encapsulation based on 20% w/w prodrug loading in 0.5-mM PEG-b-PCL micelles. Results are given ± standard deviation (n = 3).
Results in parentheses are after evaporation to 25% of original volume and refiltration (0.22-μm).
Hydrodynamic diameters from DLS with Gaussian intensity weighing of drug loaded micelles prepared at 20% w/w drug; standard deviations are of the size distribution.
Release of PAX7′C6 from PEG-b-PCL Micelles
We investigated the in vitro release kinetics of paclitaxel prodrug PAX7′C6 from PEG-b-PCL micelles at a physiological pH of 7.4 and the nominal body temperature of 37°C. Micelles released the prodrug slowly over the course of several days (Fig. 2). Release half-lives of the PAX7′C6 from PEG-b-PCL micelles was slow and without a significant burst effect. Less than 50% of PAX7′C6 was released over a period of 2 weeks.
Fig. 2.

In vitro release of PAX7′C6 prodrug from 0.5-mM PEG-b-PCL micelles in ddH2O at 37°C and pH 7.4. (mean ± SD, n = 4) Micelles were prepared with 20% w/w; actual incorporations are reported in Table I. An equal quantity of prodrug dissolved in 100 μl of ethanol and injected into a separate cassette was a control.
Cytotoxicity
Only the 7′derivative of paclitaxel with the shortest acyl-chain 7′hexonoate (PAX7′C6) 4a, maintained activity similar to unmodified paclitaxel 1 in MDA-MB-231 and MCF-7 cells. The 2′derivatives (5a, 5b, 5c), 7′dodeconoate (4b)and 7′palmitate (4c) were not active, i.e. IC50 >1 μM (Table II). For this reason, further studies were pursued using only the PAX7′C6 prodrug.
Table II.
Prodrug cytotoxicity against SK-BR-3, MDA-MB-231, MCF-7, and +SA Breast Cancer Cell Lines. Cells Were Incubated 72 Hours with each Prodrug and Cell Viability was Assessed Through Alamar Blue Assay (n = 4)
| IC50 (nM ± SD) | ||||
|---|---|---|---|---|
|
|
||||
| SK-BR-3 | MDA-MB-231 | MCF-7 | + SA | |
| Paclitaxel 1 | 213 ± 92 | 326 ± 128a | 47.0 ± 18.1 | 93.9 ± 42.9 |
| PAX7′C6 4a | 91.7 ± 37.5 | 288 ± 94 | 60.1 ± 26.2 | 129 ± 28 |
| 4b | >1 μM | >1 μM | >1 μM | >1 μM |
| 4c | >1 μM | >1 μM | >1 μM | >1 μM |
| 5a | >1 μM | >1 μM | >1 μM | >1 μM |
| 5b | >1 μM | >1 μM | >1 μM | >1 μM |
| 5c | >1 μM | >1 μM | >1 μM | >1 μM |
| PAX7′C6 in micelles | – | 114 ± 30a | – | – |
Denotes statistical significant difference (P < 0.05) between paclitaxel and PAX7′C6, or paclitaxel and PAX7′C6 in micelles.
The paclitaxel prodrug, PAX7′C6, showed activity in the nanomolar range with IC50's similar to paclitaxel in all breast cancer cells tested (Table II). In Fig. 3, we report the activity in MDA-MB-231 cells as a representative graph. In both SKBR-3 and MDA-MB-231, PAX7′C6, showed similar activity with no statistically significant differences (IC50 = 91.7 nM and 288 nM, respectively) to paclitaxel (IC50 = 213 and 326 nM). PAX7′C6 was also found active in +SA (WAZ-2) murine breast cancer cells with an IC50 of 129 nM which was comparable to paclitaxel at an IC50 of 93.9 nM. Solubilization of PAX7′C6 in PEG-PCL micelles did not alter activity in vitro in MDA-MB-231 cells (IC50 = 114 nM; Fig. 3).
Fig. 3.

Anti-Cancer Activity of Control and PAX7′C6 formulations in A) SK-BR-3 cells, B) MDA-MB-231 cells, and C) +SA cells (mean ± SEM, n = 4).
Pharmacokinetics of Polymeric Micellar PAX-7C6 After Intravenous Administration
The serum paclitaxel and PAX7′C6 concentration vs time profiles observed for the three formulations were not parallel (Fig. 4). It was observed that Taxol® (paclitaxel in CrEL:EtOH) was rapidly cleared from the body allowing its detection up to 6 h. The paclitaxel concentrations after the prodrug PAX7′C6 was dissolved in CrEL:EtOH demonstrated rapid clearance and no detection of paclitaxel 6 h post-dosing. However, when PAX7′C6 was loaded in PEG-b-PCL micelles a sustained release and conversion of paclitaxel was observed up to 24 h having higher concentrations than the paclitaxel concentrations in the Taxol® and PAX7′C6 in CrEL:EtOH controls (Fig. 4). This indicates that the micellar system provides an improved delivery and a more sustained release. Also, the concentrations of PAX7′C6 prodrug were detected in serum, and the concentrations in CrEL:EtOH were significantly lower compared to the PEG-b-PCL formulation. Furthermore, the concentrations of prodrug (PAX7′C6) after dissolution in CrEL:EtOH were detected up to 12 h, while PAX7′C6 after loading in PEG-b-PCL micelles was detected in serum up to 48 h. There were significant differences in serum elimination phase t1/2 of paclitaxel: the serum half-life of paclitaxel from PAX7′C6 in PEG-b-PCL (5.602 ± 1.656 h) being significantly higher than both Taxol® and paclitaxel from PAX7′C6 in CrEL:EtOH (1.548 ± 0.122 h and 1.333 ± 0.055 h, respectively). Higher elimination phase t1/2 of the prodrug (PAX7′C6) was also observed after loading in PEG-b-PCL (6.946 ± 0.479 h) compared to the prodrug dissolved in CrEL:EtOH (5.131 ± 0.989 h; Table III).
Fig. 4.

Concentration-time profile in serum of paclitaxel after iv administration of control Taxol® (paclitaxel in Cremophor EL:Ethanol: filled squares); paclitaxel (filled circles) and PAX7′C6 (open circles) after iv administration of control PAX7′C6 in Cremophor EL:Ethanol: and paclitaxel (filled triangles) and PAX7′C6 (open triangles) after iv administration of PAX7′C6 in PEG-b-PCL. The intravenous dose for all the formulations was 10 mg/kg to rats (mean ± SEM, n = 10 per group).
Table III.
Pharmacokinetics of Paclitaxel After iv Administration of Control Taxol® (Paclitaxel in Cremophor EL:Ethanol); Paclitaxel and PAX7′C6 After iv Administration of Control PAX7′C6 in Cremophor EL:Ethanol; and Paclitaxel and PAX7′C6 after iv Administration of PAX7′C6 in PEG-b-PCL. The Intravenous Dose for all the Formulations was 10 mg/kg to Rats (mean±SEM, n =10 per group)
| Pharmacokinetic Parameter | Paclitaxel from Taxol® Control | Paclitaxel from PAX7′C6 in CrEL:EtOH | Paclitaxel from PAX7′C6 in PEG-b-PCL | PAX7′C6 from PAX7′C6 in CrEL:EtOH | PAX7′C6 from PAX7′C6 in PEG-b-PCL |
|---|---|---|---|---|---|
| AUCinf (μgh/ml) | 7.194 ± 1.495 | 1.519 ± 0.235a | 51.113 ± 1.362b,c | 16.247 ± 3.062 | 392.758 ± 31.501d |
| Vdβ (L/kg) | 3.377 ± 0.687 | 13.125 ± 1.498a | 1.602 ± 0.504b,c | 4.678 ± 0.765 | 0.259 ± 0.030d |
| CLrenal (L/h/kg) | 0.900 ± 0.203 | 0.635 ± 0.072 | 0.014 ± 0.002b,c | 0.004 ± 0.001 | 3.378 × 10−06 ± 4.291 × 10−07d |
| CLhepatic (L/h/kg) | 0.655 ± 0.220 | 6.274 ± 1.094a | 0.182 ± 0.004b,c | 0.656 ± 0.121 | 0.026 ± 0.002d |
| CLtot (L/h/kg) | 1.556 ± 0.400 | 6.909 ± 1.070a | 0.196 ± 0.005b,c | 0.661 ± 0.122 | 0.026 ± 0.002d |
| Fe (%) | 58.913 ± 5.953 | 9.695 ± 1.805a | 6.959 ± 0.851b | 0.656 ± 0.081 | 0.014 ± 0.003d |
| KE (h−1) | 0.453 ± 0.033 | 0.522 ± 0.022 | 0.145 ± 0.036b,c | 0.145 ± 0.027 | 0.101 ± 0.007 |
| t1/2 (h) serum | 1.548 ± 0.122 | 1.333 ± 0.055 | 5.602 ± 1.656b,c | 5.131 ± 0.989 | 6.946 ± 0.479 |
| t1/2 (h) urine | 10.932 ± 2.091 | 10.898 ± 1.526 | 15.827 ± 0.808b,c | 7.042 ± 0.290 | 17.102 ± 1.947d |
| MRT (h) | 1.100 ± 0.214 | 1.912 ± 0.103a | 4.291 ± 0.767b,c | 7.635 ± 1.107 | 4.138 ± 0.380d |
| Extraction ratio (ER) | 0.377 ± 0.126 | 3.606 ± 0.628a | 0.105 ± 0.002b,c | 0.377 ± 0.070 | 0.015 ± 0.001d |
Denotes statistical significant difference (P< 0.05) between paclitaxel after iv administration of control Taxol® (paclitaxel in Cremophor EL:Ethanol) and paclitaxel after iv administration of control PAX7′C6 in Cremophor EL:Ethanol.
Denotes statistical significant difference (P < 0.05) between paclitaxel after iv administration of control Taxol® (paclitaxel in Cremophor EL:Ethanol) and paclitaxel after iv administration of PAX7′C6 in PEG-b-PCL.
Denotes statistical significant difference (P < 0.05) between paclitaxel after iv administration of control PAX7′C6 in Cremophor EL:Ethanol and paclitaxel after iv administration of PAX7′C6 in PEG-b-PCL.
Denotes statistical significant difference (P < 0.05) between PAX7′C6 after iv administration of control PAX7′C6 in Cremophor EL:Ethanol and PAX7′C6 after iv administration of PAX7′C6 in PEG-b-PCL.
Pharmacokinetic parameters of paclitaxel and PAX7′C6 in PEG-PCL compared to Taxol® and PAX7′C6 in CrEL: EtOH control formulations are presented (Table III). The micelle carrier increased the AUC values in serum of paclitaxel and PAX7′C6 by 7-fold and 24-fold when compared to Taxol® and PAX7′C6 in CrEL:EtOH, respectively. A decrease in total clearance (CLtot) of 8-fold and 25-fold was also observed for paclitaxel and PAX7′C6 from PAX7′C6 in PEG-PCL compared to Taxol® and PAX7′C6 from PAX7′C6 in CrEL:EtOH, respectively. The total clearance (CLtot) value of paclitaxel from PAX7′C6 in CrEL:EtOH (6.909 ± 1.070 l/h/kg) suggests a very rapidly clearance from the serum. The volume of distribution at the elimination phase (Vdβ) in serum was found to decrease by 2-fold and 18-fold for the micelle nanocarrier compared to Taxol® and PAX7′C6 in CrEL:EtOH, respectively. These observed differences in clearance, AUC, volume of distribution, and half-life can be summarized by the differences in the reported mean residence time (MRT). The micelle formulation increased the MRT of paclitaxel by fourfold, while the micellar system decreased the MRT of the PAX7′C6 prodrug in serum by 1.8-fold (Table III).
The mean hepatic blood flow (Q) is approximately 3.22 l/h/kg [24]. Using a hematocrit value in rat [24] of 0.48, yields a mean hepatic plasma flow of 1.74 l/h/kg. While the CLtot value for paclitaxel from Taxol® is 1.556 ± 0.400 l/h/kg, the micellar system reduces the total clearance of paclitaxel by eightfold to 0.196 ± 0.005 l/h/kg. A reduction in the total clearance of 26-fold was also observed for the prodrug PAX7′C6 in PEG-b-PCL compared to the prodrug dissolved in CrEL:EtOH (Table III). Therefore, the nanocarrier induces a longer systemic residence due to a more pronounced sustained release in vivo that allows for the drugs to reside longer in the body (higher MRT values).
The terminal urine half-life of paclitaxel significantly increased after micellar formulation: the PEG-b-PCL formulation increased the half-life (15.827 ± 0.808 h) by 1.4-fold compared to both paclitaxel from Taxol® (10.932 ± 2.091 h) and paclitaxel from PAX7′C6 in CrEL:EtOH (10.898 ± 1.526 h). An increase of 2.4-fold in the urine half-life for the prodrug PAX7′C6 in PEG-b-PCL (17.102 ± 1.907 h) was observed compared to the urine half-life of PAX7′C6 from PAX7′C6 in CrEL:EtOH (7.042 ± 0.290 h; Table III). Three plots are commonly employed to represent urinary excretion. The total amount excreted in urine plot is often used to determine the amount of drug excreted in urine through the length of sampling. The amount remaining to be excreted (ARE) plot is often used to drug excretion in urine over time. In addition, the rate of urinary excretion plot describes elimination rate into urine.
The total amount excreted in urine plot (Fig. 5a) exhibits that paclitaxel from Taxol® is excreted in higher amounts compared to the other formulations and by 24 h post-dose the majority of paclitaxel is excreted. The fraction excreted unchanged (Fe) of paclitaxel from Taxol® (58.913 ± 5.923%) was reduced by sixfold and eightfold compared to paclitaxel from PAX7′C6 in CrEL:EtOH (9.695 ± 1.805%) and paclitaxel from PAX7′C6 in PEG-b-PCL (6.959 ± 0.851%; Table III). This indicates a change in the clearance pattern of paclitaxel from the prodrug and after loading in the micellar system. Furthermore, it can be observed that higher amounts of paclitaxel are released from PAX7′C6 in PEG-b-PCL compared to the amount of paclitaxel released from the prodrug in CrEL:EtOH. The amount of paclitaxel from PAX7′C6 in PEG-b-PCL excreted in urine is relatively constant even 72 h post-dosing compared to the amount of paclitaxel released from PAX7′C6 in CrEL:EtOH that is mostly excreted 24 h post-dosing (Fig. 5a) suggesting the micellar formulation provides a sustained conversion of the prodrug to paclitaxel. The amounts of prodrug (PAX7′C6) excreted as such in urine are significantly lower than the amounts of paclitaxel excreted in urine. The amount of PAX7′C6 from PAX7′C6 in CrEL:EtOH represent only 15.80% of the total amount of paclitaxel excreted in urine. While the amount of PAX7′C6 from PAX7′C6 in PEG-b-PCL represent only 0.05% of the total amount of paclitaxel excreted in urine from the same micellar formulation. Also, the micellar system reduces the Fe value (0.014 ± 0.003%) by 48-fold compared to the Fe value of the prodrug from PAX7′C6 in PEG-b-PCL (0.656 ± 0.081%; Table III).
Fig. 5.

a Total amount excreted in urine plot, b amount remaining to be excreted in urine (ARE) plot, and c urinary rate plot of paclitaxel after iv administration of control Taxol® (paclitaxel in Cremophor EL:Ethanol; filled squares); paclitaxel (filled circles) and PAX7′C6 (open circles) after iv administration of control PAX7′C6 in Cremophor EL:Ethanol; and paclitaxel (filled triangles) and PAX7′C6 (open triangles) after iv administration of PAX7′C6 in PEG-b-PCL. The intravenous dose for all the formulations was 10 mg/kg to rats (mean ± SEM, n = 10 per group).
The amount remaining to be excreted (ARE) plot (Fig. 5b) indicates that paclitaxel from Taxol® follows a rapid initial excretion in urine and 24 h post-dosing most of the paclitaxel is excreted. A similar excretion pattern was observed for paclitaxel from PAX7′C6 in CrEL:EtOH, while paclitaxel from PAX7′C6 in PEG-b-PCL has a steady initial excretion in urine followed by a rapid decline in excretion indicating that at 72 h post-dosing there remains paclitaxel still to be excreted from the micellar system with a consequent longer half-life. These observations correlate with the reported urine terminal half-lives: paclitaxel from Taxol® which has a similar urine terminal half-life (10.932 ± 2.091 h) compared to paclitaxel from CrEL:EtOH (10.898 ± 1.526 h), while the micellar system increases the half-life of paclitaxel by 1.4-fold to 15.827 ± 0.808 h (Table III). The ARE plot also indicates that the prodrug (PAX7′C6) from PAX7′C6 in CrEL:EtOH is mainly excreted in urine 12 h post-dosing, while the ARE plot of the prodrug from PAX7′C6 in PEG-b-PCL exhibits that the prodrug has a more constant steady excretion of the prodrug (Fig. 5b). These differences can be observed in more detail by the significant differences in half-lives in urine, in which micellar system increases the urine t1/2 by 2.5-fold (17.102 ± 1.947 h) compared to the prodrug formulation in CrEL:EtOH (7.042 ± 0.290 h; Table III). The rate plots (Fig. 5c) demonstrate that independent of the formulation system paclitaxel and the prodrug (PAX7′C6) are excreted at the same rate.
Furthermore, as mentioned above it was determined that paclitaxel from Taxol® is mainly cleared by renal clearance (Fe value equal to 58.913 ± 5.923%) and that the prodrug and micellar system change the clearance route to a predominantly non-renal mechanism (Fe values of 9.695 ± 1.805% for paclitaxel from PAX7′C6 in CrEL:EtOH and 6.959 ± 0.851% for paclitaxel from PAX7′C6 in PEG-b-PCL). Therefore, the micellar system invokes a significant change in the pattern of clearance from a predominantly renal clearance to a clearance predominantly via non-renal routes.
Observations performed by blinded observers reported that almost immediately between 12 h post-IV dosing of Taxol® control formulation and PAX7′C6 in CrEL:EtOH the rats presented nose bleeding, disorientation, heavy breathing, slight decrease in response to sound, and 30% of the Taxol® control subjects died within 48–72 h post-dose. The animals that received the PAX7′C6 in PEG-b-PCL formulation did not display adverse effects for the first 24 h, but demonstrated mild diarrhea and nose bleeding after 48 h post-dose, nonetheless, 100% of the animals survived until the end of the experiment (72 h post-dose).
Biodistribution
Quantifiable amounts of paclitaxel (except in brain in some instances) and PAX7′C6 were observed in all assayed tissues. The tissue collection was performed at 6 h and 24 h post IV administration at 10 mg/kg. The order in concentrations from highest to lowest for the paclitaxel from control Taxol® (paclitaxel in CrEL:EtOH) 6 h post dose was spleen>liver>lung>muscle>heart>urinary bladder>kidney> blood>serum and paclitaxel was not effectively measured in brain (below detectable levels 0.02 μg/ml). The micellar system provided changes in the tissue distribution of paclitaxel and the prodrug PAX7′C6 6 h post dose. The order in concentrations from highest to lowest for the paclitaxel from PAX7′C6 in PEG-b-PCL was liver>brain>kidney>lung> serum>blood>muscle>urinary bladder>heart>spleen. It was also observed that all the tissues presented lower paclitaxel concentrations compared to paclitaxel concentrations from Taxol® control, but the paclitaxel serum concentrations were significantly higher than the control formulation. The order of tissue concentrations from highest to lowest for the PAX7′C6 from PAX7′C6 in PEG-b-PCL was lung>liver>kidney> blood>spleen>urinary bladder>heart>serum>muscle>brain. In this instance it was observed that the concentration of pro-drug in serum and in all the tissues was significantly higher than the concentration of paclitaxel from the control formulation and from the micellar formulation (except in liver which was not significantly different than the concentrations from the control formulation; Table IV).
Table IV. Mean Concentrations (μg/g of Tissue) of Paclitaxel and PAX7′C6 in Rat Tissues (and Tissue to Serum Ratios—Kp Values—in Parenthesis) Measured at 6 h post iv Administrations of Control Taxol® (Paclitaxel in Cremophor EL/Ethanol) and PAX7′C6 in PEG-b-PCL in Plasma after iv Administration (10 mg/kg; mean ± SEM, n =4).
| Tissue | Paclitaxel from Taxol® Control | Paclitaxel from PAX7′C6 in PEG-b-PCL | PAX7′C6 from PAX7′C6 in PEG-b-PCL |
|---|---|---|---|
| Serum | 0.066 ± 0.003 | 0.464 ± 0.095a | 10.062 ± 0.913b,c |
| Whole Blood | 1.463 ± 0.097 (22.110 ± 1.473) | 0.332 ± 0.139a (0.716 ± 0.300)a | 14.730 ± 5.954b,c (1.464 ± 0.592)b |
| Liver | 17.042 ± 5.559 (257.498 ± 83.988) | 1.171 ± 0.184a (2.526 ± 0.396)a | 23.053 ± 7.598 (2.291 ± 0.755)b |
| Kidney | 3.989 ± 1.221 (60.272 ± 18.449) | 0.669 ± 0.147a (1.445 ± 0.317)a | 19.129 ± 4.109b,c (1.901 ± 0.408)b |
| Spleen | 33.419 ± 0.543 (504.942 ± 8.198) | 0.274 ± 0.078a (0.592 ± 0.168)a | 14.397 ± 2.556b,c (1.431 ± 0.254)b,c |
| Brain | <0.02 (<0.30) | 0.736 ± 0.181a (1.587 ± 0.390)a | 0.707 ± 0.191b (0.070 ± 0.019)b,c |
| Muscle | 12.056 ± 3.054 (182.155 ± 46.139) | 0.323 ± 0.025a (0.696 ± 0.054)a | 9.884 ± 1.589c (0.982 ± 0.158)b,c |
| Lung | 16.425 ± 5.098 (248.170 ± 77.028) | 0.606 ± 0.126a (1.307 ± 0.271)a | 33.399 ± 9.000b,c (3.319 ± 0.894)b,c |
| Heart | 11.066 ± 2.561 (167.200 ± 38.698) | 0.294 ± 0.065a (0.635 ± 0.140)a | 12.086 ± 2.503c (1.201 ± 0.249)b,c |
| Urinary bladder | 7.651 ± 0.577 (115.603 ± 8.712) | 0.313 ± 0.072a (0.674 ± 0.155)a | 12.643 ± 2.682b,c (1.256 ± 0.267)b,c |
Denotes statistical significant difference (P < 0.05) between paclitaxel after iv administration of control Taxol® (paclitaxel in Cremophor EL:Ethanol) and paclitaxel after iv administration of PAX7′C6 in PEG-b-PCL.
Denotes statistical significant difference (P < 0.05) between paclitaxel after iv administration of control Taxol® (paclitaxel in Cremophor EL:Ethanol) and PAX7′C6 after iv administration of PAX7′C6 in PEG-b-PCL.
Denotes statistical significant difference (P < 0.05) between paclitaxel after iv administration of PAX7′C6 in PEG-b-PCL and PAX7′C6 after iv administration of PAX7′C6 in PEG-b-PCL.
The tissue distribution changed 24 h post dose demonstrating that the order in concentrations from highest to lowest for the paclitaxel from Taxol® control was lung>spleen>heart>kidney>urinary bladder>liver>muscle>brain>-blood>serum. The micellar system provided changes in the tissue distribution of paclitaxel and the prodrug PAX7′C6 24 h post dose. The order in concentrations from highest to lowest for the paclitaxel from PAX7′C6 in PEG-b-PCL was urinary bladder>lung>heart>liver>spleen>kidney>muscle>-blood> serum, and paclitaxel was also not effectively measured in brain (below detectable levels 0.02 μg/ml). It was also observed that all the tissues and serum presented lower paclitaxel concentrations compared to paclitaxel concentrations from Taxol® control. The order of concentrations from highest to lowest for the PAX7′C6 from PAX7′C6 in PEG-b-PCL was very similar to the one exhibited by paclitaxel from Taxol® control and it was lung>spleen>urinary bladder>kidney> heart>liver>muscle>brain>blood>serum. In this instance, it was observed that the concentrations of prodrug in serum were significantly higher than the concentrations of paclitaxel from both the control formulation and from the micellar formulation. Furthermore, it was observed that the concentrations of prodrug in all the tissues were significantly lower than the concentrations of paclitaxel from both the control formulation and from the micellar formulation (except in brain, muscle, and urinary bladder; Table V).
Table V. Mean Concentrations (μg/g of Tissue) of Paclitaxel and PAX7′C6 in Rat Tissues (and Tissue to Serum Ratios—Kp Values—in Parenthesis) Measured at 24 h Post iv Administrations of Control Taxol® (Paclitaxel in Cremophor EL:Ethanol) and PAX7′C6 in PEG-b-PCL in Plasma after iv Administration (10 mg/kg; mean±SEM, n =4).
| Tissue | Paclitaxel from Taxol® Control | Paclitaxel from PAX7′C6 in PEG-b-PCL | PAX7′C6 from PAX7′C6 in PEG-b-PCL |
|---|---|---|---|
| Serum | 0.036 ± 0.001 | 0.021 ± 0.002a | 0.163 ± 0.012b,c |
| Whole Blood | 0.039 ± 0.001 (1.104 ± 0.065) | 0.074 ± 0.002a (2.984 ± 0.088)a | 0.173 ± 0.032b,c (1.042 ± 0.113)c |
| Liver | 4.801 ± 0.828 (132.581 ± 13.956) | 1.049 ± 0.222a (49.018 ± 2.024)a | 3.140 ± 0.333b,c (17.986 ± 0.803)b,c |
| Kidney | 9.243 ± 1.593 (264.888 ± 36.473) | 1.009 ± 0.002a (48.941 ± 8.316)a | 4.474 ± 0.361b,c (28.164 ± 5.921)b,c |
| Spleen | 20.784 ± 6.064 (593.083 ± 153.607) | 1.034 ± 0.208a (44.401 ± 5.513)a | 7.075 ± 0.541b,c (44.515 ± 9.180)b |
| Brain | 0.239 ± 0.029 (6.950 ± 1.091) | <0.02a (<0.96)a | 1.319 ± 0.021b,c (7.595 ± 0.347)c |
| Muscle | 1.482 ± 0.396 (42.159 ± 13.910) | 0.653 ± 0.045a (31.283 ± 3.234) | 2.136 ± 0.022b,c (13.288 ± 1.613)b,c |
| Lung | 59.914 ± 10.542 (1716.809 ± 242.728) | 1.690 ± 0.114a (82.918 ± 19.619)a | 12.713 ± 1.240b,c (80.212 ± 18.158)b |
| Heart | 13.094 ± 2.659 (354.878 ± 83.265) | 1.056 ± 0.161a (49.869 ± 0.966)a | 3.875 ± 1.288b,c (23.084 ± 4.842)b,c |
| Urinary bladder | 5.841 ± 0.430 (164.041 ± 23.201) | 3.602 ± 0.395a (189.009 ± 38.093) | 6.231 ± 0.321c (38.553 ± 3.115)b,c |
Denotes statistical significant difference (P < 0.05) between paclitaxel after iv administration of control Taxol® (paclitaxel in Cremophor EL:Ethanol) and paclitaxel after iv administration of PAX7′C6 in PEG-b-PCL.
Denotes statistical significant difference (P < 0.05) between paclitaxel after iv administration of control Taxol® (paclitaxel in Cremophor EL:Ethanol) and PAX7′C6 after iv administration of PAX7′C6 in PEG-b-PCL.
Denotes statistical significant difference (P <0.05) between paclitaxel after iv administration of PAX7′C6 in PEG-b-PCL and PAX7′C6 after iv administration of PAX7′C6 in PEG-b-PCL.
The tissue to serum ratio (Kp) values for all the tissues after iv administration of Taxol® control ranged from 20–500 6 h post dose, while significant lower Kp values were observed for paclitaxel (0.5–2.5) and PAX7′C6 (0.07–3.3) after administration of PAX7′C6 in PEG–b–PCL (Fig. 6a). These differences in Kp values might be attributed to the differences in disposition and clearance of paclitaxel and the prodrug between the control and the micellar formulations. This can be observed based on the significantly higher concentrations in serum of paclitaxel (0.464 ± 0.095 μg/ml) and PAX7′C6 (10.062 ± 0.913 μg/ml) after administration of the micellar prodrug formulation compared to the paclitaxel serum concentration (0.066 ± 0.003 μg/ml) after administration of control Taxol® formulation (Table IV). Furthermore, 24 h post dose the Kp values for all the tissues after Taxol® control ranged from 1–1,700, while significantly lower Kp values were observed for paclitaxel (3–190) and PAX7′C6 (1-80) after PAX7′C6 in PEG-b-PCL, except for the Kp in blood (Fig. 6b). However, it can be noted at this time point that the serum concentration of paclitaxel after iv administration of control Taxol® (0.036 ± 0.001 μg/ml) was significantly higher than the serum concentration of paclitaxel (0.021 ± 0.002 μg/ml) after administration of the micellar formulation and significantly lower than the PAX7′C6 in PEG-b-PCL (0.163 ± 0.012 μg/ml; Table V).
Fig. 6.
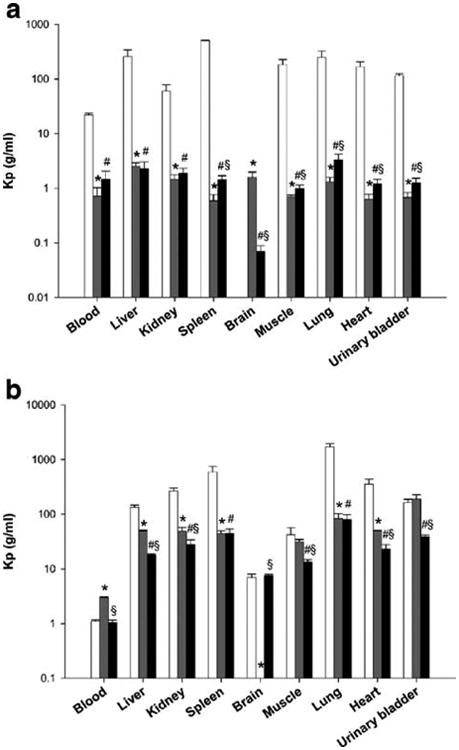
Tissue to serum ratios (Kp) measured at a 6h and b 24 h post-iv administration of paclitaxel (open squares) from control Taxol® (paclitaxel in Cremophor EL:Ethanol), and paclitaxel (grey squares) and PAX7′C6 (filled squares) after iv administration of PAX7′C6 in PEG-b-PCL (10 mg/kg) to rats (mean ± SEM, n = 10 per group). Asterisk denotes statistical significant difference (P < 0.05) between paclitaxel from control Taxol® (paclitaxel in Cremophor EL:Ethanol) and paclitaxel from PAX7′C6 in PEG-b-PCL. Number sign denotes statistical significant difference (P < 0.05) between paclitaxel from control Taxol® (paclitaxel in Cremophor EL:Ethanol) and PAX7′C6 from PAX7′C6 in PEG-b-PCL. Section sign denotes statistical significant difference (P <0.05) between paclitaxel from PAX7′C6 in PEG-b-PCL and PAX7′C6 from PAX7′C6 in PEG-b-PCL.
It was also observed that the concentrations of PAX7′C6 after administration of PAX7′C6 in PEG-b-PCL in the different tissues 6 h and 24 h post dose are similar to the concentrations of paclitaxel after control Taxol®. However, at both time points also significant concentrations of paclitaxel were quantified indicating the conversion of PAX7′C6 to paclitaxel occurred. Furthermore, the significant amounts of PAX7′C6 found in all the tissues indicate that the prodrug is still available for posterior conversion to paclitaxel that would lead to higher residence time and availability to exert its pharmacological activity.
Discussion
Cremophor® EL (polyoxyethylated castor oil) has been related to various side effects such as hypersensitivity reactions, nephrototoxicity, and neuroreactions after intravenous administrations of Taxol® [26]. In order to prevent these reactions, patients need to be premedicated with corticosteroids and antihistamines, and Taxol® has to be administered via a 1–24 h infusion [27,28]. This has led to the development of different modified formulations of paclitaxel that do not contain Cremophor® EL [29–31]. Thus, the objective of this study was to develop novel delivery systems that can act as an effective alternative to DMA, PEG, and Tween 80, Cremophor, or ethanol for paclitaxel solubilization. Further objectives included modifying the rate of release and disposition of paclitaxel, to increase paclitaxel's plasma residence, to ultimately make paclitaxel safer for human administration, and to reduce toxicity-related effects by providing a more effective carrier that can allow for the use of lower doses.
There is precedence of the use of lipophilic prodrugs in oil-based delivery systems that have shown improved solubility [16]. For instance, solubilization of a lipophilic paclitaxel oleate prodrug in a cholesterol nanoemulsion resulted in improved efficacy but high liver uptake of the prodrug [32]. The PEG-b-PCL micelle formulations demonstrated significant differences in pharmacokinetic parameters from the Taxol® control and PAX7′C6 in CrEL:EtOH control formulation (Table III). The significant changes in AUC indicate a higher degree of in vivo stability of the PEG-PCL micelle formulations constituting a long circulating delivery system that facilitates accumulation of drug in tumors by enhanced permeation and retention (EPR) effect [7]. In addition to the EPR effect, it has been observed that nanoparticles <50 nm in diameter rapidly accumulate in vasculated tumors [33] allowing highly localized and improved delivery of paclitaxel.
The solubilization of PAX7′C6 in PEG-b-PCL micelles led to a change in biological fate characterized by an increase in AUC and a reduction in CLtot and (Vdβ) for the solubilized drug. The micellar system increased the volume of distribution (Vdβ) indicating that the prodrug and paclitaxel are extensively distributed. These results suggest a large role played by the carrier system in influencing the distribution of drugs in vivo and how the body handles micellar systems based on their physicochemical properties. Similar increases in volumes of distribution have been previously reported for other carrier-entrapped paclitaxel formulations [21,22,34]. These changes in disposition correlate with the results of the pharmacokinetics studies by Dhanikula et al. [21] who recently reported similar trends in pharmacokinetics and tissue distribution in mice after administration of covalently linked paclitaxel to poloxamer 407, and by Aliabadi et al. [11], who recently reported altered disposition in cyclosporine loaded PEG-b-PCL micelles in rats. Therefore, the encapsulation of the PAX7′C6 paclitaxel prodrug in PEGPCL micelles caused a significant increase in residence time in the serum, longer half-life in urine, and provided a more sustained release in vivo, allowing longer residence and circulation in the body.
Different studies have assessed tissue distribution of Taxol® [35,36] indicating that after intravenous administration it mainly distributes to kidney, lung, heart, and spleen similar to our results indicating uptake by the monomolecular phagocyte system. Furthermore, paclitaxel physically entrapped with poly(d,l-lactide)-b-methoxy PEG micelles exhibited higher concentration in the lung followed by kidney, liver, heart and spleen [34], while paclitaxel conjugated to poly(glutamic acid) reported higher affinity for the liver followed by spleen and kidney [35]compared to Taxol®. Both of these studies using carrier mediated delivery systems compare and parallel our findings. In our study it was observed that conversion of PAX7′C6 prodrug into paclitaxel occurred in the serum and that there was a significant reduction in the distribution to the heart, which may have clinical significance since paclitaxel therapy has been related with cardiotoxicity in combination therapies [37]. In in vitro experiments, both PAX7′C6 alone and in formulation showed activity similar to paclitaxel. Examination of PAX7′C6 pharmacodynamics in rodent xenografts of human breast tumor cells is currently ongoing. Overall, this study indicates that this nanocarrier system coupled with PAX-7′C6 has potential for further pre-clinical and clinical cancer studies.
Conclusions
The PEG-b-PCL polymeric micelle system allowed for the uptake, protection, and retention of the water insoluble prodrug PAX-7′C6 by increasing the half-life, the mean residence time, and the area under the curve, at the same time reducing the volume of distribution, while stabilizing the micellar structure in a biological environment. Based on our results it seems that the PEG-b-PCL micelles might be of the appropriate size and core/shell properties to escape the uptake by the reticuloendothelial system and stay stable during plasma circulation for longer periods more effectively.
Acknowledgments
This research was supported by NIH grant AI-43346-08 and generous grants from Hoffman-La Roche Inc.,Wisconsin Alumni Research Foundation (WARF), and Shimadzu Scientific. MLF was partially supported by a PhRMA postdoctoral fellowship. CMR was partially supported by an AFPE gateway research fellowship.
Abbreviations
- CDCl3
Deuterated Chloroform
- CH2Cl2
Methylene chloride
- CrEL
Cremophor EL
- DMAP
4-Dimethylaminopyridine
- DMF
N,N-dimethylformamide
- EtOAc
Ethyl acetate
- EtOH
Ethanol
- HCl
Hydrochloric acid
- Na2SO4
Sodium sulfate
- NH4Cl
Ammonium chloride
- PEG-b-PCL
Poly(ethylene glycol)-b-poly(ε-caprolactone)
- TBAF
Tetrabutylamoniumfloride
- TBDMSCl
Tert-butyldimethylsilyl chloride
- TBS
Tetrabromo-2-benzotriazole
- THF
Tetrahydrofuran
- TLC
Thin layer chromatography
References
- 1.Constantinou M, Tsai JY, Safran H. Paclitaxel and concurrent radiation in upper gastrointestinal cancers. Cancer Investig. 2003;21:887–896. doi: 10.1081/cnv-120025092. [DOI] [PubMed] [Google Scholar]
- 2.Retter AS, Figg WD, Dahut WL. The combination of antiangiogenic and cytotoxic agents in the treatment of prostate cancer. Clin Prostate Cancer. 2003;2:153–159. doi: 10.3816/cgc.2003.n.023. [DOI] [PubMed] [Google Scholar]
- 3.Rowinsky EK. Paclitaxel pharmacology and other tumor types. Semin Oncol. 1997;24:S19-11–S19-12. [PubMed] [Google Scholar]
- 4.Treat J, Damjanov N, Huang C, Zrada S, Rahman A. Liposomal-encapsulated chemotherapy: preliminary results of a phase I study of a novel liposomal paclitaxel. Oncology (Williston Park, NY) 2001;15:44–48. [PubMed] [Google Scholar]
- 5.Socinski M. Update on nanoparticle albumin-bound paclitaxel. Clin Adv Hematol Oncol. 2006;4:745–746. [PubMed] [Google Scholar]
- 6.Sparreboom A, Baker SD, Verweij J. Paclitaxel repackaged in an albumin-stabilized nanoparticle: handy or just a dandy? J Clin Oncol. 2005;23:7765–7767. doi: 10.1200/JCO.2005.03.7135. [DOI] [PubMed] [Google Scholar]
- 7.Croyand SR, Kwon GS. Polymeric micelles for drug delivery. Curr Pharm Des. 2006;12:4669–4684. doi: 10.2174/138161206779026245. [DOI] [PubMed] [Google Scholar]
- 8.Forrest ML, Won CY, Malick AW, Kwon GS. In vitro release of the mTOR inhibitor rapamycin from poly(ethylene glycol)-b-poly(epsilon-caprolactone) micelles. J Control Release. 2006;110:370–377. doi: 10.1016/j.jconrel.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 9.Forrest ML, Zhao A, Won CY, Malick AW, Kwon GS. Lipophilic prodrugs of Hsp90 inhibitor geldanamycin for nano-encapsulation in poly(ethylene glycol)-b-poly(epsilon-caprolactone) micelles. J Control Release. 2006;116:139–149. doi: 10.1016/j.jconrel.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 10.Yanez JA, Forrest ML, Ohgami Y, Kwon GS, Davies NM. Pharmacometrics and delivery of novel nanoformulated PEG-b-poly(epsilon-caprolactone) micelles of rapamycin. Cancer chemotherapy and pharmacology. 2007 doi: 10.1007/s00280-007-0458-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aliabadi HM, Brocks DR, Lavasanifar A. Polymeric micelles for the solubilization and delivery of cyclosporine A: pharmacokinetics and biodistribution. Biomaterials. 2005;26:7251–7259. doi: 10.1016/j.biomaterials.2005.05.042. [DOI] [PubMed] [Google Scholar]
- 12.Vakiland R, Kwon GS. PEG-phospholipid micelles for the delivery of amphotericin B. J Control Release. 2005;101:386–389. [PubMed] [Google Scholar]
- 13.Vakiland R, Kwon GS. Poly(ethylene glycol)-b-poly (epsilon-caprolactone) and PEG-phospholipid form stable mixed micelles in aqueous media. Langmuir. 2006;22:9723–9729. doi: 10.1021/la061408y. [DOI] [PubMed] [Google Scholar]
- 14.Shuai X, Merdan T, Schaper AK, Xi F, Kissel T. Core-cross-linked polymeric micelles as paclitaxel carriers. Bioconjug Chem. 2004;15:441–448. doi: 10.1021/bc034113u. [DOI] [PubMed] [Google Scholar]
- 15.Liu J, Xiao Y, Allen C. Polymer-drug compatibility: a guide to the development of delivery systems for the anticancer agent, ellipticine. J Pharm Sci. 2004;93:132–143. doi: 10.1002/jps.10533. [DOI] [PubMed] [Google Scholar]
- 16.Ali S, Ahmad I, Peters A, Masters G, Minchey S, Janoff A, Mayhew E. Hydrolyzable hydrophobic taxanes: synthesis and anti-cancer activities. Anti-Cancer Drugs. 2001;12:117–128. doi: 10.1097/00001813-200102000-00004. [DOI] [PubMed] [Google Scholar]
- 17.Soo PL, Luo L, Maysinger D, Eisenberg A. Incorporation and release of hydrophobic probes in biocompatible polycaprolactone-block-poly(ethylene oxide) micelles: Implications for drug delivery. Langmuir. 2002;18:9996–10004. [Google Scholar]
- 18.Klotz WL, Schure MR, Foley JP. Determination of octanol-water partition coefficients of pesticides by microemulsion electrokinetic chromatography. J Chromatogr. 2001;930:145–154. doi: 10.1016/s0021-9673(01)01171-2. [DOI] [PubMed] [Google Scholar]
- 19.Bai S, Stepkowski SM, Kahan BD, Brunner LJ. Metabolic interaction between cyclosporine and sirolimus. Transplantation. 2004;77:1507–1512. doi: 10.1097/01.tp.0000128372.09220.b5. [DOI] [PubMed] [Google Scholar]
- 20.O'Brien J, Wilson I, Orton T, Pognan F. Investigation of the Alamar Blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur J Biochem/FEBS. 2000;267:5421–5426. doi: 10.1046/j.1432-1327.2000.01606.x. [DOI] [PubMed] [Google Scholar]
- 21.Dhanikula AB, Singh DR, Panchagnula R. In vivo pharmacokinetic and tissue distribution studies in mice of alternative formulations for local and systemic delivery of Paclitaxel: gel, film, prodrug, liposomes and micelles. Curr Drug Discov. 2005;2:35–44. doi: 10.2174/1567201052772852. [DOI] [PubMed] [Google Scholar]
- 22.Crosasso P, Ceruti M, Brusa P, Arpicco S, Dosio F, Cattel L. Preparation, characterization and properties of sterically stabilized paclitaxel-containing liposomes. J Control Release. 2000;63:19–30. doi: 10.1016/s0168-3659(99)00166-2. [DOI] [PubMed] [Google Scholar]
- 23.Straub JA, Chickering DE, Lovely JC, Zhang H, Shah B, Waud WR, Bernstein H. Intravenous hydrophobic drug delivery: a porous particle formulation of paclitaxel (AI-850) Pharm Res. 2005;22:347–355. doi: 10.1007/s11095-004-1871-1. [DOI] [PubMed] [Google Scholar]
- 24.Davies B, Morris T. Physiological parameters in laboratory animals and humans. Pharm Res. 1993;10:1093–1095. doi: 10.1023/a:1018943613122. [DOI] [PubMed] [Google Scholar]
- 25.Poulin P, Schoenlein K, Theil FP. Prediction of adipose tissue: plasma partition coefficients for structurally unrelated drugs. J Pharm Sci. 2001;90:436–447. doi: 10.1002/1520-6017(200104)90:4<436::aid-jps1002>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 26.van Zuylen L, Verweij J, Sparreboom A. Role of formulation vehicles in taxane pharmacology. Invest New Drugs. 2001;19:125–141. doi: 10.1023/a:1010618632738. [DOI] [PubMed] [Google Scholar]
- 27.Greco FA, Thomas M, Hainsworth JD. One-hour paclitaxel infusions: review of safety and efficacy. Cancer J Sci Am. 1999;5:179–191. [PubMed] [Google Scholar]
- 28.Rowinsky EK. The taxanes: dosing and scheduling considerations. Oncology (Williston Park, NY) 1997;11:7–19. [PubMed] [Google Scholar]
- 29.Nuijen B, Bouma M, Schellens JH, Beijnen JH. Progress in the development of alternative pharmaceutical formulations of taxanes. Invest New Drugs. 2001;19:143–153. doi: 10.1023/a:1010682916808. [DOI] [PubMed] [Google Scholar]
- 30.Pawar R, Shikanov A, Vaisman B, Domb AJ. Intravenous and regional paclitaxel formulations. Curr Med Chem. 2004;11:397–402. doi: 10.2174/0929867043455981. [DOI] [PubMed] [Google Scholar]
- 31.Singla AK, Garg A, Aggarwal D. Paclitaxel and its formulations. Int J Pharm. 2002;235:179–192. doi: 10.1016/s0378-5173(01)00986-3. [DOI] [PubMed] [Google Scholar]
- 32.Rodrigues DG, Maria DA, Fernandes DC, Valduga CJ, Couto RD, Ibanez OC, Maranhao RC. Improvement of paclitaxel therapeutic index by derivatization and association to a cholesterol-rich microemulsion: in vitro and in vivo studies. Cancer Chemother Pharmacol. 2005;55:565–576. doi: 10.1007/s00280-004-0930-y. [DOI] [PubMed] [Google Scholar]
- 33.Dreher MR, Liu W, Michelich CR, Dewhirst MW, Yuan F, Chilkoti A. Tumor vascular permeability, accumulation, and penetration of macromolecular drug carriers. J Natl Cancer Inst. 2006;98:335–344. doi: 10.1093/jnci/djj070. [DOI] [PubMed] [Google Scholar]
- 34.Kim SC, Kim DW, Shim YH, Bang JS, Oh HS, Wan Kim S, Seo MH. In vivo evaluation of polymeric micellar paclitaxel formulation: toxicity and efficacy. J Control Release. 2001;72:191–202. doi: 10.1016/s0168-3659(01)00275-9. [DOI] [PubMed] [Google Scholar]
- 35.Li C, Newman RA, Wu QP, Ke S, Chen W, Hutto T, Kan Z, Brannan MD, Charnsangavej C, Wallace S. Biodistribution of paclitaxel and poly(l-glutamic acid)-paclitaxel conjugate in mice with ovarian OCa-1 tumor. Cancer Chemother Pharmacol. 2000;46:416–422. doi: 10.1007/s002800000168. [DOI] [PubMed] [Google Scholar]
- 36.Zhang X, Burt HM, Mangold G, Dexter D, Von Hoff D, Mayer L, Hunter WL. Anti-tumor efficacy and biodistribution of intravenous polymeric micellar paclitaxel. Anti-Cancer Drugs. 1997;8:696–701. doi: 10.1097/00001813-199708000-00008. [DOI] [PubMed] [Google Scholar]
- 37.Salvatorelli E, Menna P, Cascegna S, Liberi G, Calafiore AM, Gianni L, Minotti G. Paclitaxel and docetaxel stimulation of doxorubicinol formation in the human heart: implications for cardiotoxicity of doxorubicin-taxane chemotherapies. J Pharmacol Exp Ther. 2006;318:424–433. doi: 10.1124/jpet.106.103846. [DOI] [PubMed] [Google Scholar]