

Abstract
Introduction: This paper aims to evaluate the toxicity profile of additive gemcitabine to adjuvant taxane-based chemotherapy in breast cancer patients. Methods: Patients enrolled in this open-label randomized controlled Phase III study were treated with 3 cycles of epirubicin-fluorouracil-cyclophosphamide (FEC) chemotherapy followed by 3 cycles of docetaxel with those receiving 3 cycles of FEC followed by 3 cycles of gemcitabine-docetaxel (FEC-DG). 3690 patients were evaluated according to National Cancer Institute (NCI) toxicity criteria (CTCAE). The study medications were assessed by the occurrence of grade 3–4 adverse events, dose reductions, postponements of treatment cycles and granulocyte colony-stimulating factor (G-CSF) support. Results: No differences in neutropenia or febrile neutropenia were demonstrated. However, thrombocytopenia was significantly increased with FEC-DG treatment (2.0 vs. 0.5 %, p < 0.001), as was leukopenia (64.1 vs. 58.5 %, p < 0.001). With FEC-DG significantly more G-CSF support in cycles 4 to 6 (FEC-DG: 57.8 %, FEC-D: 36.3 %, p < 0.001) was provided. Transaminase elevation was significantly more common with FEC-DG (SGPT: 6.3 %, SGOT: 2 %), whereas neuropathy (1.2 %), arthralgia (1.6 %) and bone pain (2.6 %) were more common using FEC-D. Dose reductions > 20 % (4 vs. 2.4 %) and postponement of treatment cycles (0.9 vs. 0.4 %) were significantly more frequent in the FEC-DG arm. Eight deaths occurred during treatment in the FEC-DG arm and four in the FEC-D arm. Conclusion: The addition of gemcitabine increased hematological toxicity and was associated with more dose reductions and postponements of treatment cycles.
Key words: adjuvant chemotherapy, gemcitabine, toxicity, hematological, breast cancer
Abstract
Zusammenfassung
Einleitung: Die vorliegende Studie untersucht das Toxizitätsprofil nach der zusätzlichen Gabe von Gemcitabin in Kombination mit einer adjuvanten Taxan-basierten Chemotherapie bei Patientinnen mit Brustkrebs. Methode: Es handelt sich hier um eine randomisierte kontrollierte Phase-III-Open-Label-Studie. Alle in der Studie aufgenommenen Patientinnen erhielten 3 Zyklen Epirubicin-Fluorouracil-Cyclophosphamid (FEC) gefolgt von 3 Zyklen Docetaxel bzw. 3 Zyklen FEC gefolgt von 3 Zyklen Gemcitabin-Docetaxel (FEC-DG). Die Daten von insgesamt 3690 Patientinnen wurden mittels der Toxizitätskriterien (CTCAE) des nationalen Krebsinstituts der USA (National Cancer Institute [NCI]) ausgewertet. Kriterien für die Auswertung der Studienmedikation waren das Auftreten unerwünschter Ereignisse 3. oder 4. Grades, Dosisreduktionen, Verschiebungen nachfolgender Behandlungszyklen und Granulozytenkolonie-stimulierender Faktor (G-CSF)-Gabe. Ergebnisse: Es zeigten sich keine Unterschiede zwischen den beiden Gruppen hinsichtlich der Entwicklung einer Neutropenie oder einer febrilen Neutropenie. Dagegen war die Thrombozytopenie nach der Behandlung mit FEC-DG signifikant erhöht (2,0 vs. 0,5 %, p < 0,001), und die Leukopenie trat in dieser Gruppe ebenfalls deutlich häufiger auf (64,1 vs. 58,5 %, p < 0,001). Der Einsatz von G-CSF-Präparaten in den Zyklen 4–6 war deutlich höher in der FEC-DG-Gruppe (FEC-DG: 57,8 %, FEC-D: 36,3 %, p < 0,001). Ein Transaminasenanstieg kam wesentlich häufiger in der FEC-DG-Gruppe vor (SGPT: 6,3 %, SGOT: 2 %), während Neuropathien (1,2 %), Arthralgien (1,6 %) und Knochenschmerzen (2,6 %) häufiger in der FEC-D-Gruppe auftraten. Dosisreduktionen > 20 % (4 vs. 2.4 %) sowie eine Verschiebung nachfolgender Behandlungszyklen (0,9 vs. 0,4 %) kamen deutlich häufiger im FEC-DG-Studienarm vor. Während der Behandlung starben 8 Patientinnen im FEC-DG-Studienarm und 4 im FEC-D-Studienarm. Schlussfolgerung: Die zusätzliche Gabe von Gemcitabin hat die hämatologische Toxizität erhöht und war mit mehr Dosisreduktionen und mehr Verschiebungen nachfolgender Behandlungszyklen assoziiert.
Schlüsselwörter: adjuvante Chemotherapie, Gemcitabin, Toxizität, hämatologisch, Brustkrebs
Introduction
Although adjuvant chemotherapy with anthracyclines and taxanes reduces mortality and the risk of recurrence for patients with high-risk breast cancer, such treatment is often associated with life-threatening side effects 1, 2, 3, 4, 5, 6. Anthracyclines are known for their cardiotoxicity 7, 8, 9. Other common side effects of anthracyclines are myelosuppression, febrile neutropenia, mucositis, nausea, vomiting and alopecia 10. Common side effects of taxanes are neutropenia, anemia, neurological symptoms, stomatitis and dermal afflictions 11, 12.
Gemcitabine is a pyrimidine analogue antimetabolite drug that has been used as an alternative in the treatment of metastatic breast cancer 13, 14, 15. It is known as an option for combination therapy because of its mechanism of action, toxicity profile, additive or synergistic activity in vitro and a lack of cardiotoxicity. Drug combinations with vinorelbine, cisplatin, 5-fluorouracil, taxanes and anthracyclines have yielded overall response rates of 58 to 92 % as a first-line treatment 16, 17. It is profitable for pretreated anthracycline and/or taxane resistant breast cancer patients 18, 19. It is used in cases of local relapse after neoadjuvant chemotherapy 20, 21. The hematological toxicity of gemcitabine includes leukopenia, anemia and thrombocytopenia. Non-hematological side effects are nausea, vomiting, diarrhea, obstipation, mucositis and loss of appetite. Other side effects are transaminitis, alkaline phosphatase and bilirubin elevation. Respiratory complications include dyspnea, bronchospasm and interstitial pneumonitis 22, 23, 24.
This study aims to focus in detail on the evaluation of the toxicity profile of adding gemcitabine to taxane-based therapeutic regimens.
Patients and Methods
Eligibility criteria
Eligible women had operable breast cancer with clear surgical margins (R0), metastases to the axillary nodes or were node negative with a high-risk profile (> pT2, G3, age < 35 years, negative hormonal receptors). Surgical treatment was either mastectomy or lumpectomy with sentinel lymph node biopsy and with or without axillary dissection. Patients with all molecular breast cancer subtypes were eligible (luminal A, luminal B, HER-2 subtype, triple negative). Radiotherapy was applied after breast-conserving surgery or at high risk of local recurrence. Chemotherapy was started at least six weeks after surgery. Patients were required to provide written informed consent before being registered.
Inclusion criteria were an Eastern Cooperative Oncology Group (ECOG) Performance Status < 2, white blood cell count (WBC) ≥ 3.0 × 109/l, platelet count ≥ 100 × 109/l, bilirubin levels within normal range and transaminase and alkaline phosphatase levels within 1.5 of the upper limit of normal as measured by the referring laboratory. Patients were required to be free of metastasis as evaluated by chest x-ray, bone scintigraphy and liver ultrasound. In premenopausal women a pregnancy test was obligatory.
The study was approved on 25.08.2005 by 37 German ethical boards (lead ethical board: Ludwig-Maximilians-University Munich) and conducted in accordance with the Declaration of Helsinki.
Study design
The SUCCESS-A study (clinical trial.gov registration ID NCT02181101; EudraCT 2005–000490-21) is an open-label, prospective, randomized, controlled Phase III study comparing disease-free survival of high-risk early breast cancer patients receiving adjuvant sequential cyclophosphamide/taxane-based chemotherapy with or without gemcitabine (first randomization), and disease-free survival of patients receiving 2 years vs. 5 years of zoledronic acid treatment (second randomization; see Fig. 1 for the complete study design and supplement for CONSORT statement).
Fig. 1.
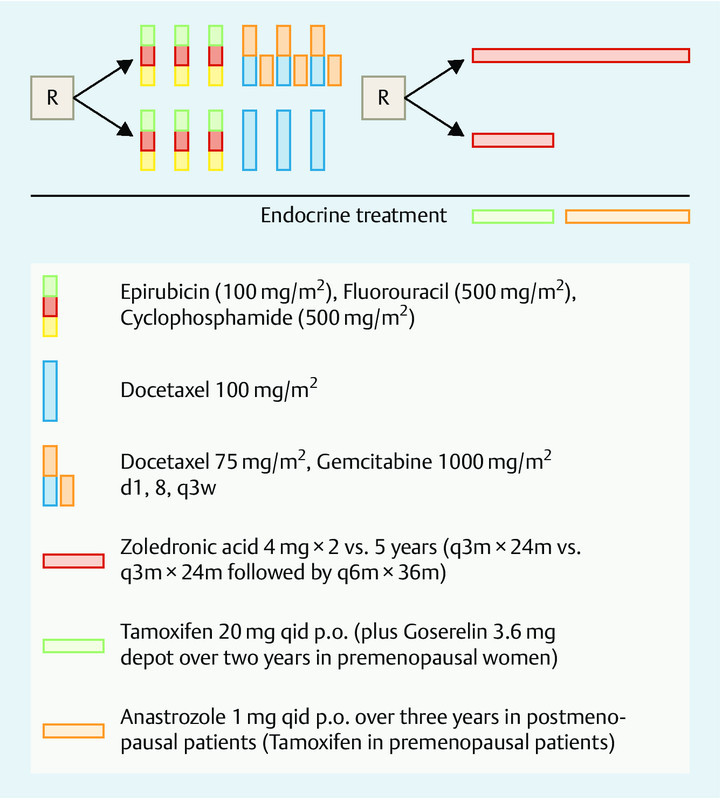
Study design of the randomized SUCCESS-A trial.
Treatment regimens
All patients first received three full cycles of epirubicin (100 mg/m2)-fluorouracil (500 mg/m2)-cyclophosphamide (500 mg/m2) (FEC) chemotherapy, followed by either three cycles of full-dose docetaxel (100 mg/m2; D) for patients in the FEC-D arm or 3 cycles of gemcitabine (1000 mg/m2 d1, d8) and dose-reduced docetaxel (75 mg/m2, d1) (DG) for patients in the FEC-DG arm. After the completion of chemotherapy, the patients were further randomized to receive either 2 years of zoledronic acid treatment (4 mg i. v. every 3 months) or 5 years of zoledronic acid treatment (4 mg i. v. every 3 months for two years, followed by 4 mg i. v. every 6 months for the duration of additional three years). During the zoledronic acid treatment period, patients received daily 500 mg calcium p. o. and 400 IE vitamin D p. o.
Patients with positive hormone receptor status (≥ 10 % positively stained cells for estrogen and/or progesterone) of the primary tumor received tamoxifen treatment 20 mg p. o. per day for 2 years, after the end of chemotherapy. Subsequent to chemotherapy, postmenopausal patients with positive hormone receptor status were treated with anastrozole 1 mg p. o. for additional 3 years; premenopausal patients continued tamoxifen treatment for additional 3 years. In addition to tamoxifen, all patients with positive hormone receptor status of the primary tumor and under the age of 40 or restart of menstrual bleeding within 6 months after the completion of cytostatic treatment or with premenopausal hormone levels (luteinizing hormone [LH] < 20 mIE/ml, follicle stimulating hormone [FSH] < 20 mIE/ml and estradiol [E2] > 20 pg/ml) received goserelin 3.6 mg subcutaneously every 4 weeks over a period of 2 years following chemotherapy. In patients with clearly HER-2 overexpressing tumors (immunohistochemistry score 3+ or fluorescent in situ hybridization [FISH] positive) therapy with trastuzumab was applied for a period of one year every three weeks after the completion of chemo- and radiotherapy (dosage: 8 mg/kg loading dose followed by 6 mg/kg body weight).
To decrease toxicity, dexamethasone, 2-mercaptoethanesulfonate-sodium (mesna), and 5-HT-3-antagonists were provided. To guarantee the maximal blockage of receptors, the 5-HT-3-antagonist was administered intravenously 15 minutes before the initial cytostatic agent was given. To increase the patientsʼ tolerance of cytotoxic agents and the efficacy of 5-HT3-receptor-antagonists, dexamethasone was provided at a dose of 8 mg i. v. before every chemotherapy application. Moreover during cycle 4–6 dexamethasone was provided at a dose of 8 mg p. o. in the evening after chemotherapy and in the morning and evening before and after the day of chemotherapy. To prevent hemorrhagic cystitis during cyclophosphamide therapy, the bladder protectant mesna and adequate hydration were provided.
Supportive measures were taken according to the study protocol. No upfront granulocyte colony stimulating factor (G-CSF) support was provided. G-CSF at a dose of 34 million IU/d was provided as a secondary prophylaxis on days 5 to 10 or until leukocyte levels were normal to patients with febrile neutropenia (i.e., temperature > 38.5 °C, absolute neutrophil count [ANC] < 0.5 × 109/l, requiring hospitalization and intravenous antibiotics), isolated neutropenia (ANC < 0.5 × 109/l, > five days), severe neutropenia (ANC < 0.1 × 109/l) and any cases in which leukopenia led to the postponement of treatment cycles. Once one of these criteria were met, in all subsequent cycles a secondary prophylactic G-CSF administration at a dose of 34 million IU/d was provided from day 5 to day 10, or until the number of leukocytes had exceeded the nadir of 5000/ul.
Facultative prophylactic antibiotic therapy with oral fluoroquinolones (e.g. ciprofloxacin 500 mg, p. o., 2×/d, levofloxacin 500 mg, p. o., 1×/d) was offered to patients with grade 3 and 4 neutropenia. Antibiotic prophylaxis was recommended to patients with a neutrophil granulocyte cell count (ANC) of less than 0.5 × 109/l. Once started, facultative antibiotic therapy and antibiotic prophylaxis were provided until the requirements for resuming chemotherapy were met (ANC ≥ 1.5 × 109/l or WBC ≥ 3.0 × 109/l). Patients with febrile neutropenia were assessed according to their NCI score (Common Terminology Criteria for Adverse Events of the National Cancer Institute, CTCAE V2.0). Patients with febrile neutropenia (> 38.0 °C) were hospitalized for isolation, empiric i. v. antibiotic therapy and diagnostic workup. During febrile neutropenia the duration of antibiotic therapy was dependent on the individual course of disease and the secondary complications.
Chemotherapy cycles could be delayed for a maximum of two weeks. This also applied to gemcitabine (d1 and d8). If the eighth day in a cycle was delayed for less than a week, the subsequent cycle was administered according to the therapy protocol. If the treatment was delayed one week or longer, the next cycle was not administered according to the study protocol date, but two weeks after the date of the delayed administration. The requirements for resuming chemotherapy were ANC ≥ 1.5 × 109/l or WBC ≥ 3.0 × 109/l and thrombocytes ≥ 100 × 109/l.
In patients receiving secondary G-CSF application during cycle 4–6, gemcitabine was only applied on day 8 if the requirements for resuming chemotherapy were met (ANC ≥ 1.5 × 109/l or WBC ≥ 3.0 × 109/l). No G-CSF application was provided on the day of chemotherapy. During all cycles dose reduction was mandatory when neutropenia persisted despite G-CSF application.
Gastrointestinal toxicity and mucositis at NCI grade 3 led to dose reduction by one level. Chemotherapy was terminated with mucositis or vomiting of NCI grade 4. Neurological toxicity of NCI grade 2 led to a dose reduction of one level. Chemotherapy was terminated in patients who suffered major arrhythmias that required treatment or who experienced relevant left ventricular ejection fraction reduction; however, treatment was continued under cardiac monitoring in cases of minor cardiac symptoms (benign arrhythmias, isolated asymptomatic ventricular extrasystoles).
Randomization, data collection and statistical considerations
Overall, 3754 patients at 271 study centers were randomized from September 2005 until March 2007 in the SUCCESS-A trial. The trial was completed in September 2013. The safety and toxicity analyses reported here were performed based on the safety population, defined as patients who were treated with at least one cycle of FEC chemotherapy (n = 3690).
Toxicity was evaluated once before applying each chemotherapy cycle and once 28 days after chemotherapy. Cases of NCI toxicity grade 4 or death were communicated to the investigator. Patients were followed at the study sites at three monthly intervals for the first three years and every six months thereafter, including clinical examination (each visit), mammography (every six months) and symptom-driven examinations, if necessary. Data was obtained from the electronic case report form of the SUCCESS study. High data quality was ensured by electronic data management including automated randomization, tests for plausibility as well as regular monitoring visits to the study site by an independent contract research organization.
Toxicities were calculated as proportion of patients randomized to a treatment arm that experienced a grade 3–4 adverse event during the course of the treatment (i.e. cumulative over all six treatment cycles). Furthermore, to facilitate assessment of adverse events more specifically related to the addition of gemcitabine, toxicities were also calculated as proportion of patients randomized to a treatment arm that experienced a grade 3–4 adverse event during the docetaxel vs. docetaxel plus gemcitabine treatment cycles only (i.e. cumulative over cycles 4–6).
Summary tabulations of numbers and percentages within each category are presented for all categorical variables, while age, a non-normally distributed continuous variable, is reported with medians and ranges. Comparisons of the frequency of toxicities or supportive treatments between the two arms were conducted with the χ2 test or Fisherʼs exact test (in case expected frequencies in single cells of cross tabulations were 5 or less). All statistical tests were two-sided, and p values of less than 0.05 were considered significant (i.e., no adjustments of significance levels for multiple comparisons were made). Statistical analyses were performed with IBM SPSS Statistics, Version 21.0 (IBM Corp., New York, USA).
Results
Of the 3690 patients, 1861 patients were randomized to the FEC-D arm and 1829 patients were randomized to the FEC-DG arm. Complete, monitored toxicity data for the duration of chemotherapy treatment were available for all patients of the safety population.
The median age in both treatment arms was fifty-three years (Table 1). More than half of all patients (58 %) were postmenopausal. Ductal invasive carcinomas were observed in 82 % and lobular carcinomas were observed in 11 %. 52 % had carcinomas measuring between two and five centimeters (T2), and 42 % had tumors smaller than two centimeters. T3 and T4 tumors were observed in less than 7 %. Histological grade 3 carcinomas were found in approximately 48 % of the patients. The majority had a positive estrogen receptor status (66 %) as well as progesterone receptor status (59 %), and a negative HER2 status (75 %). Most patients received breast conserving surgery (71 %) and radiotherapy (85 %). The two randomization arms were well-balanced with regard to tumor and patient characteristics (Table 1).
Table 1 Distribution of patient and tumor characteristics by randomization arm.
| Total (n = 3 690) | Randomization arm | |||
|---|---|---|---|---|
| FEC-D (n = 1 861) | FEC-DG (n = 1 829) | |||
| Age (years) | median | 53.0 | 54.0 | 53.0 |
| range | 21–86 | 21–86 | 22–85 | |
| Menopausal status | premenopausal | 1 542 (41.8 %) | 765 (41.1 %) | 777 (42.5 %) |
| postmenopausal | 2 148 (58.2 %) | 1 096 (58.9 %) | 1 052 (57.5 %) | |
| Tumor size | pT1 | 1 538 (41.7 %) | 765 (41.1 %) | 773 (42.3 %) |
| pT2 | 1 901 (51.5 %) | 964 (51.8 %) | 937 (51.2 %) | |
| pT3 | 194 (5.3 %) | 105 (5.6 %) | 89 (4.9 %) | |
| pT4 | 52 (1.4 %) | 26 (1.4 %) | 26 (1.4 %) | |
| unknown | 5 (0.1 %) | 1 (0.1 %) | 4 (0.2 %) | |
| Nodal stage | pN0 | 1 257 (34.1 %) | 623 (33.5 %) | 634 (34.7 %) |
| pN1 | 1 684 (45.6 %) | 868 (46.6 %) | 816 (44.6 %) | |
| pN2 | 507 (13.7 %) | 249 (13.4 %) | 258 (14.1 %) | |
| pN3 | 232 (6.3 %) | 118 (6.3 %) | 114 (6.2 %) | |
| unknown | 10 (0.3 %) | 3 (0.2 %) | 7 (0.4 %) | |
| Histological grading | G1 | 176 (4.8 %) | 79 (4.2 %) | 97 (5.3 %) |
| G2 | 1 754 (47.5 %) | 887 (47.7 %) | 867 (47.4 %) | |
| G3 | 1 756 (47.6 %) | 893 (48.0 %) | 863 (47.2 %) | |
| unknown | 4 (0.1 %) | 2 (0.1 %) | 2 (0.1 %) | |
| Histological type | invasive ductal | 3 027 (82.0 %) | 1 527 (82.1 %) | 1 500 (82.0 %) |
| invasive lobular | 408 (11.1 %) | 207 (11.1 %) | 201 (11.0 %) | |
| other | 251 (6.8 %) | 125 (6.7 %) | 126 (6.9 %) | |
| unknown | 4 (0.1 %) | 2 (0.1 %) | 2 (0.1 %) | |
| Estrogen receptor status | negative | 1 243 (33.7 %) | 605 (32.5 %) | 638 (34.9 %) |
| positive | 2 444 (66.2 %) | 1 254 (67.4 %) | 1 190 (65.1 %) | |
| unknown | 3 (0.1 %) | 2 (0.1 %) | 1 (0.1 %) | |
| Progesterone receptor status | negative | 1 510 (40.9 %) | 760 (40.8 %) | 750 (41.0 %) |
| positive | 2 174 (58.9 %) | 1 096 (58.9 %) | 1 078 (58.9 %) | |
| unknown | 6 (0.2 %) | 5 (0.3 %) | 1 (0.1 %) | |
| Her2 status | negative | 2 748 (74.5 %) | 1 392 (74.8 %) | 1 356 (74.1 %) |
| positive | 877 (23.8 %) | 434 (23.3 %) | 443 (24.2 %) | |
| unknown | 65 (1.8 %) | 35 (1.9 %) | 30 (1.6 %) | |
| Type of surgery | breast conserving | 2 606 (70.6 %) | 1 308 (70.3 %) | 1 298 (71.0 %) |
| mastectomy | 1 083 (29.3 %) | 553 (29.7 %) | 530 (29.0 %) | |
| unknown | 1 (0.0 %) | 0 (0.0 %) | 1 (0.1 %) | |
| Radiotherapy | no | 530 (14.4 %) | 257 (13.8 %) | 273 (14.9 %) |
| yes | 3 148 (85.3 %) | 1 597 (85.8 %) | 1 551 (84.8 %) | |
| unknown | 12 (0.3 %) | 7 (0.4 %) | 5 (0.3 %) | |
Hematological and non-hematological toxicity
NCI grade 3 or 4 toxicities that occurred are depicted in Table 2 for hematological toxicity and in Table 3 for non-hematological toxicity.
Table 2 Most common grade 3–4 hematological toxicity (CTCAE V2.0) according to randomization arm (all cycles, cycles 4–6 only).
| Hematological adverse event | FEC-D% (no. patients) | FEC-DG% (no. patients) | p-valuea |
|---|---|---|---|
| a χ2 test | |||
| All cycles | |||
| Leukopenia | 58.5 (1 089) | 64.1 (1 173) | < 0.001 |
| Anemia | 1.2 (23) | 2.1 (38) | 0.045 |
| Thrombopenia | 0.5 (10) | 2.0 (37) | < 0.001 |
| Neutropenia | 35.4 (659) | 36.9 (675) | 0.345 |
| Febrile neutropenia | 6.8 (127) | 6.0 (109) | 0.283 |
| Cycles 4–6 | |||
| Leukopenia | 45.9 (855) | 52.7 (963) | < 0.001 |
| Anemia | 0.4 (8) | 1.5 (27) | 0.001 |
| Thrombopenia | 0.1 (1) | 1.5 (28) | < 0.001 |
| Neutropenia | 26.1 (485) | 25.1 (459) | 0.502 |
| Febrile neutropenia | 5.0 (93) | 4.2 (76) | 0.221 |
Table 3 Most common grade 3–4 non-hematological toxicity (CTCAE V2.0) according to randomization arm.
| Non-hematological adverse event | FEC-D% (no. patients) | FEC-DG% (no. patients) | p-valuea |
|---|---|---|---|
| a χ2 test b Fisher exact test | |||
| All cycles | |||
| Alopecia | 0.2 (3) | 0.2 (3) | 1.000b |
| Nausea | 3.3 (61) | 3.7 (67) | 0.522 |
| Fatigue | 2.8 (52) | 2.6 (47) | 0.673 |
| Vomiting | 3.9 (72) | 3.4 (63) | 0.492 |
| Stomatitis | 1.9 (36) | 2.1 (39) | 0.670 |
| SGPT elevation | 2.8 (52) | 6.3 (116) | < 0.001 |
| Constipation | 0.5 (9) | 0.5 (10) | 0.789 |
| SGOT elevation | 1.0 (18) | 2.0 (37) | 0.008 |
| Diarrhea | 2.8 (53) | 3.0 (55) | 0.774 |
| Bone marrow failure | 0.8 (14) | 1.4 (26) | 0.050 |
| Headache | 0.5 (9) | 0.8 (15) | 0.204 |
| Neuropathy | 1.2 (23) | 0.3 (6) | 0.002 |
| General pain | 1.5 (28) | 1.1 (20) | 0.271 |
| Infection | 1.3 (24) | 1.9 (34) | 0.164 |
| Gastrointestinal disorder | 0.8 (15) | 1.3 (24) | 0.133 |
| Arthralgia | 1.6 (29) | 0.7 (12) | 0.009 |
| Bone pain | 2.6 (49) | 1.0 (19) | < 0.001 |
| Cycles 4–6 | |||
| Alopecia | 0.0 (0) | 0.0 (0) | – |
| Nausea | 0.3 (6) | 1.5 (27) | < 0.001 |
| Fatigue | 1.8 (33) | 2.1 (39) | 0.430 |
| Vomiting | 0.4 (7) | 0.6 (11) | 0.326 |
| Stomatitis | 1.5 (27) | 1.6 (30) | 0.641 |
| SGPT elevation | 1.5 (28) | 5.2 (95) | < 0.001 |
| Constipation | 0.3 (5) | 0.3 (5) | 1.000b |
| SGOT elevation | 0.3 (6) | 2.0 (36) | < 0.001 |
| Diarrhea | 2.1 (40) | 2.5 (46) | 0.462 |
| Bone marrow failure | 0.2 (4) | 1.0 (18) | 0.002 |
| Headache | 0.2 (3) | 0.4 (8) | 0.124 |
| Neuropathy | 1.1 (21) | 0.3 (6) | 0.004 |
| General pain | 1.0 (19) | 0.8 (15) | 0.523 |
| Infection | 0.6 (11) | 1.4 (26) | 0.011 |
| Gastrointestinal disorder | 0.7 (13) | 0.9 (16) | 0.544 |
| Arthralgia | 1.3 (25) | 0.6 (11) | 0.022 |
| Bone pain | 2.4 (45) | 0.9 (17) | < 0.001 |
Overall, grade 3 or 4 hematological toxicities were observed more frequently in the FEC-DG arm than in the FEC-D arm (Table 2). While there were no significant differences between the two treatment arms regarding the proportion of patients affected by grade 3 or 4 neutropenia or febrile neutropenia, patients in the FEC-DG arm suffered more often from grade 3 or 4 anemia (2.1 vs. 1.2 %, p = 0.045), leukopenia (64.1 vs. 58.5 %, p < 0.001) and thrombopenia (2.0 vs. 0.5 %, p < 0.001) compared with the FEC-D arm. A separate analysis of grade 3 or 4 hematological toxicities that occurred during cycles 4 to 6 (i.e. during treatment with either docetaxel alone or docetaxel plus gemcitabine) showed that these differences were most likely due to the addition of gemcitabine, as the significant differences regarding anemia, leukopenia and thrombopenia in cycles 4 to 6 closely match the differences observed when toxicities over all 6 cycles were analyzed (Table 2).
Non-hematological toxicity was detected at a lower rate than hematological toxicity in both arms (Table 3). A statistically significant difference in non-hematological toxicity between the two treatment arms was noted with regard to grade 3 or 4 serum glutamic pyruvic transaminase (SGPT) and serum glutamic oxaloacetic transaminase (SGOT) elevation, both of which were significantly higher in the FEC-DG arm (SGPT: 6.3 vs. 2.8 %, p < 0.001; SGOT: 2.0 vs. 1.0 %, p < 0.01). In contrast, grade 3 or 4 neuropathy (1.2 vs. 0.3 %, p = 0.002), arthralgia (1.6 vs. 0.7 %, p = 0.009) and bone pain (2.6 vs. 1.0 %, p < 0.001) occurred significantly more often in the FEC-D arm. No significant differences between FEC-DG and FEC-D were observed for other non-hematological grade 3 to 4 toxicities (Table 3). In general, the frequencies of grade 3 or 4 adverse events observed during cycles 4 to 6 match the cumulative frequencies of grade 3 or 4 adverse events observed during all 6 chemotherapy cycles, indicating that the higher rates of SGPT and SGOT elevation in the FEC-DG arm were indeed due to the addition of gemcitabine, while the higher rates of neuropathy, arthralgia and bone pain in the FEC-D arm were most likely caused by the higher docetaxel dose compared to the FEC-DG arm (Table 3). However, the separate analysis of grade 3 or 4 non-hematological toxicities during cycles 4 to 6 revealed significantly higher rates of grade 3 or 4 nausea (1.5 vs. 0.3 %, p < 0.001) and infection (1.4 vs. 0.6 %, p = 0.01) in the FEC-DG arm, which were not evident when toxicities were analyzed cumulatively over all 6 cycles.
During the follow-up period, three cases of leukemia were reported. In the FEC-DG arm, two cases of acute myeloid leukemia occurred. One case of chronic lymphocytic leukemia occurred in the FEC-D arm.
Number of chemotherapy cycles administered and treatment termination
The 3690 patients received a total of 21 428 chemotherapy cycles. In the FEC-DG arm, 10 613 cycles were administered in 1829 patients; in the FEC-D arm, 10 815 cycles were administered in 1861 patients. All six cycles were completed by 3395 (92.0 %) of the patients. Chemotherapy was discontinued prematurely in 162 (8.9 %) of the 1829 patients in the FEC-DG arm and in 133 (7.1 %) of 1861 patients in the FEC-D arm.
Dose reduction, treatment delay, G-CSF support and antibiotic treatment
Dose reductions (Fig. 2, Table 4) occurred significantly more often in the FEC-DG arm compared to the FEC-D arm (FEC-DG: 4.0 % of 10 613 cycles; FEC-D: 2.4 % of 10 815 cycles; p < 0.001; Table 4). In the FEC-DG arm, most of the 427 dose reductions were caused by hematological toxicity (45.9 %), while 33.3 % were due to non-hematological toxicities. In contrast, most of the 261 dose reductions observed in the FEC-D arm were necessitated by non-hematological toxicities (53.6 %), and only 28.7 % of the dose reductions were caused by hematological toxicities.
Fig. 2.
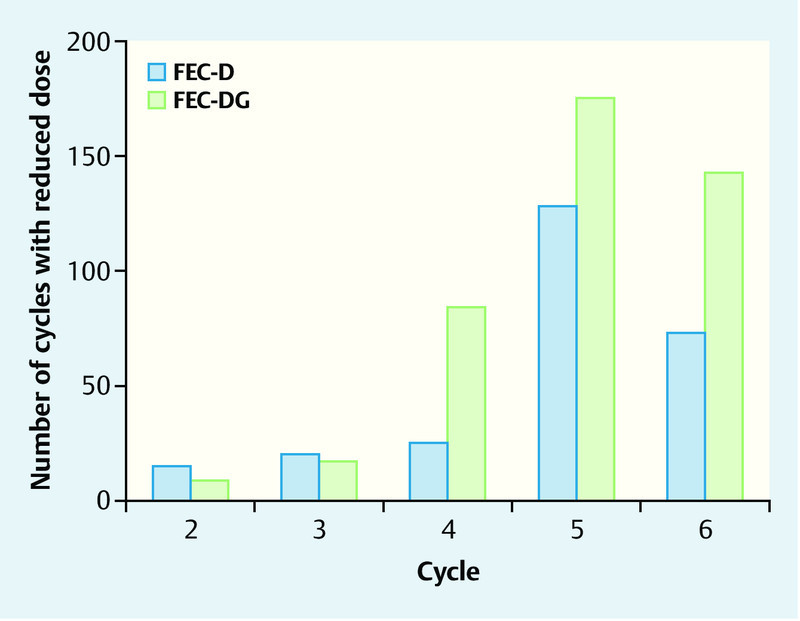
Frequency of chemotherapy dose reductions for single cycles according to treatment arm.
Table 4 Reasons for chemotherapy dose reductions according to treatment arm.
| Reason | Both randomization arms% (n) | FEC-Doc% (n) | FEC-DocG% (n) |
|---|---|---|---|
| Hematological toxicity | 39.4 (271) | 28.7 (75) | 45.9 (196) |
| Non-hematological toxicity | 41.0 (282) | 53.6 (140) | 33.3 (142) |
| Patient request | 3.5 (24) | 2.7 (7) | 4.0 (17) |
| Other | 16.1 (111) | 14.9 (39) | 16.9 (72) |
| Total | 100.0 (688) | 100.0 (261) | 100.0 (427) |
In total, treatment delays of more than two weeks (Fig. 3, Table 5) were documented in 135 (0.6 %) of the 21 428 cycles, with 40 treatment delays in the FEC-D arm and 95 treatment delays in the FEC-DG arm. Treatment delays were observed at a similar rate in the first three cycles applied (FEC in both arms), while in cycle 4 and particularly in cycles 5 and 6 treatment delays occurred considerably more often in the FEC-DG arm. Overall, chemotherapy cycles were delayed for more than two weeks more often in the FEC-DG arm than in the FEC-D arm (0.9 vs. 0.4 %). Table 5 outlines the reasons for the treatment delays. In the FEC-DG arm, 30.5 % of treatment delays were caused by hematological toxicity, while only 17.5 % of treatment delays were due to hematological toxicity in the FEC-D arm. 23.2 % and 25.0 % of treatment delays were caused by non-hematological toxicity in the FEC-DG and FEC-D arm, respectively.
Fig. 3.
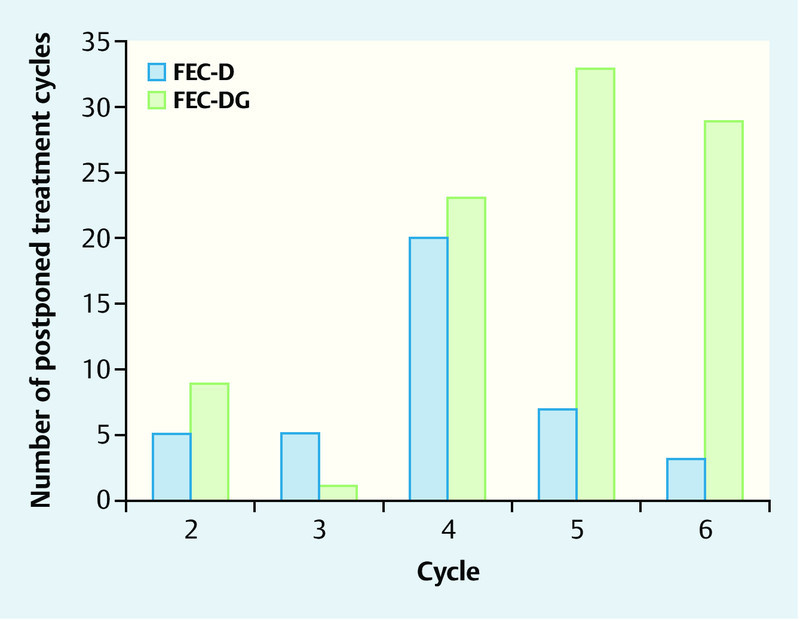
Frequency of chemotherapy treatment delays of more than two weeks for single cycles according to treatment arm.
Table 5 Reasons for chemotherapy treatment delays of more than two weeks according to treatment arm.
| Reason | Both randomization arms% (n) | FEC-Doc% (n) | FEC-DocG% (n) |
|---|---|---|---|
| Hematological toxicity | 26.7 (36) | 17.5 (7) | 30.5 (29) |
| Non-hematological toxicity | 23.7 (32) | 25.0 (10) | 23.2 (22) |
| Patient request | 2.2 (3) | 0.0 (0) | 3.2 (3) |
| Technical reasons | 8.9 (12) | 12.5 (5) | 7.4 (7) |
| Other | 38.5 (52) | 45.0 (18) | 35.8 (34) |
| Total | 100.0 (135) | 100.0 (40) | 100.0 (95) |
In the first three chemotherapy cycles administered (FEC in both arms), there were no significant differences between the two randomization arms with regard to the proportion of patients who received G-CSF support (FEC-DG: 23.4 %, FEC-D: 24.3 %, p = 0.53). In contrast, patients in the FEC-DG arm were significantly more likely to receive G-CSF support compared with patients in the FEC-D arm in chemotherapy cycles 4 to 6 (FEC-DG: 57.8 %, FEC-D: 36.3 %, p < 0.001). Likewise, patients in the two randomization arms were treated with oral antibiotics at a similar frequency in the first three chemotherapy cycles (FEC-DG: 19.0 %, FEC-D: 18.9 %; p = 0.89), while for chemotherapy cycles 4 to 6 patients in the FEC-DG arm received oral antibiotic treatment more often compared with patients in the FEC-D arm (FEC-DG: 25.5 %, FEC-D: 22.5 %; p = 0.04). During cycles 1 to 3, intravenous antibiotic therapy was applied less often, and there were no significant differences between the two randomization arms (FEC-DG: 3.0 %, FEC-D: 3.1 %; p = 0.92) or during cycles 4 to 6 (FEC-DG: 6.4 %, FEC-D: 5.3 %; p = 0.19).
Death during the trial
Twelve deaths occurred during chemotherapy treatment. Eight deaths occurred in the FEC-DG arm, and four occurred in the FEC-D arm. The investigator and sponsor assessed causality for each case. Scores of 1 (very probable) through 5 (no relationship) were provided for each case. In the FEC-DG arm, five patients died of respiratory failure caused by fulminant pneumonia. A 69-year-old patient died of acute respiratory distress syndrome caused by pneumonia five weeks after the first cycle of DG (causality: 1), and a 60-old patient died six weeks after the third cycle of DG (causality: 2). Two 58-year-old patients died of respiratory decompensation caused by atypical pneumonia five and six weeks after the second cycle of DG (causality: 2). One 65-year-old patient died five weeks after the last cycle of DG (causality: 3). One patient succumbed to pulmonary embolism four days after the last cycle of DG. Two patients died of sudden cardiac arrest. A 67-year-old patient died after an event of generalized pain two weeks after the second cycle of DG, and a 60-year-old patient was found dead after toe amputation for gangrene two weeks after the last cycle of DG.
In the FEC-D arm, four deaths occurred; two were caused by pulmonary embolism. One 54-year-old patient suffered a spontaneous fatal pulmonary embolism 10 days after the last cycle of FEC (causality: 3), and one patient succumbed to a pulmonary embolism on the first postoperative night after cholecystectomy two weeks after the second cycle of FEC (causality: 3). The remaining two deaths (causality: 2) were a 74-year-old patient with other comorbidities, who died of cardiac decompensation during pancolitis one month after the first cycle of docetaxel, and a 70-year-old who died of cardiac complications of closely monitored type II diabetes mellitus six weeks after the second cycle of docetaxel.
Discussion
To our knowledge, no other trials have addressed the inclusion of gemcitabine in adjuvant FEC-D therapy for local high-risk breast cancer. One of the hypotheses of the SUCCESS-A trial is that reducing the docetaxel dose by 25 % and adding gemcitabine 1000 mg/m2 d1 and d8 to the chemotherapeutic regimen will lead to comparable toxicity profiles.
Comparing our toxicity results with other trials combining polychemotherapeutic regimens and gemcitabine in breast cancer patients demonstrates heterogeneous results as the various study designs differ. In the FEC-DG arm, the toxicity profile is demonstrated by leukopenia, thrombopenia, and anemia. The fact that G-CSF support and antibiotics were provided significantly more often in the FEC-DG arm might explain why no significant differences in the rates of neutropenia and febrile neutropenia were observed between the two groups. It is possible that without these preventive measures, the side effects of adding gemcitabine would have been greater. For example, in the NSABP-B38 trial 25 adjuvant chemotherapy with doxorubicin and cyclophosphamide was followed by paclitaxel and gemcitabine. G-CSF support was used in 93 % of chemotherapy cycles resulting in low rates of neutropenia (3 %). In contrast, the NSABP-B40 trial 26 reported that in the neoadjuvant docetaxel and gemcitabine arm the toxic effect with the greatest increase in frequency compared with the toxic effects of docetaxel alone was neutropenia in 34 % of the patients (NCI grade 3 and 4). Trials in the metastatic setting report increased rates of neutropenia (47.9 %) and thrombocytopenia (6.1 %) compared with our results. This may be explained by the decreased bone marrow reserve caused by prior radiation and chemotherapy or by the restricted use of G-CSF 24.
The non-hematological side effects reflect the lower taxane dose, the use of gemcitabine in the FEC-DG arm and the standard docetaxel 100 mg dose in the FEC-D arm. In the FEC-DG arm, gemcitabine caused a significant elevation of SGPT and/or SGOT, which is consistent with other trials and is well described as a limiting factor for dose escalation 27, 28. An example is the Neo-tAnGo trial 29, which included neoadjuvant gemcitabine in combination with paclitaxel before or after epirubicin/cyclophosphamide treatment where additional gemcitabine also resulted in increased rates of transaminitis. Other significant gemcitabine associated toxicities such as grade 3 or 4 nausea and infection, that were not evident when toxicities were analyzed cumulatively over all 6 cycles, were observed in cycles 4 to 6. The increased rates of neurological and musculoskeletal toxicities in the FEC-D arm reflecting the higher taxane dose are comparable with other trials administering FEC-D 30.
The interpretation of dose reduction and treatment delay reveals that FEC-DG is not tolerated as well as docetaxel alone due to cumulative hematological toxicity. Expectedly, in the FEC part of chemotherapy (first, second and third cycles) there were no differences in dose reduction and treatment delay between the two chemotherapy arms. As gemcitabine was administered in the next three consecutive cycles, the two randomization arms start to diverge as the frequencies of dose reduction and treatment delay increase significantly in the FEC-DG arm. Interestingly, the toxicity profile of the two arms is reflected in the reasoning for dose reduction: In the FEC-DG arm, most dose reductions were caused by hematological toxicities (45.9 %), while in the FEC-D arm they are mostly due to non-hematological toxicities (53.6 %). Surprisingly, the frequency of treatment delay for more than two weeks was more than four times as high during the fifth and sixth cycles in the FEC-DG arm compared with the FEC-D arm. Gemcitabine was given on day 1, but also on day 8 which falls into the nadir of hematological toxicity for docetaxel. This d1+d8 regimen in the study design could explain not only the numbers of dose reductions and treatment delays, but also the significantly higher administration rate of G-CSF and antibiotics in the FEC-DG arm regarding the study protocolʼs guidelines. Finally, the fact that cytostatic treatment was prematurely stopped more often in the FEC-DG arm highlights its increased toxicity profile.
More patients died during chemotherapy with FEC-DG. Notable are the five deaths caused by respiratory complications of inflammatory and/or infectious origin in the FEC-DG group. Respiratory adverse events caused by gemcitabine include dyspnea, bronchospasm and interstitial pneumonitis 24. A prospective pulmonary, cardiac and hepatic function evaluation was undertaken in the tAnGo trial 31. This trial reported temporary reductions in pulmonary function levels, being significantly greater with additive gemcitabine. Combining these facts with the higher rate of hematological toxicity registered in the FEC-DG arm and the higher predisposition for infections, the causality of the death-related events might be assumed.
Conclusion
At present gemcitabine is indicated as a first line treatment in combination with paclitaxel for patients with locally recurrent or metastatic breast cancer who have, as part of their adjuvant or neoadjuvant therapy, received anthracyclines or who have a contraindication against anthracycline therapy 24. The results of three large trials, NSABP-B38 25, NSABP-B40 26 and Neo-tAnGo 29, investigating the role of gemcitabine in the adjuvant and neoadjuvant setting, showed no benefit from the addition of gemcitabine to anthracycline and taxane-based chemotherapy treatments for early breast cancer. Our toxicity data demonstrates an increase in grade 3 and 4 hematological toxicity, in dose reductions and postponement of treatment cycles, as well as an increased need for G-CSF and antibiotic support and a higher number of fatalities in the FEC-DG arm compared with FEC-Doc alone. Although survival data from the SUCCESS A trial are still pending, our toxicity data taken in combination with the results of other trials using gemcitabine in the adjuvant setting strongly suggest that the use of gemcitabine as a component of an adjuvant breast cancer chemotherapy regimen is not a favorable therapeutic option.
Acknowledgements
This has been a collaborative project from the start. We gratefully acknowledge the thousands of patients participating in the SUCCESS-A trial, as well as all the staff members and colleagues that helped to realize this trial.
Footnotes
Conflict of Interest This translational research part of the SUCCESS trial was supported by AstraZeneca, Chugai, Lilly, Novartis, Sanofi-Aventis, Janssen Diagnostics and Veridex. W. Janni received research funding and acts as an advisor for Sanofi-Aventis, Roche, Pfizer, Novartis, Astra Zeneca, Celgene, Amgen and Janssen. B. Rack has received research funding from Sanofi-Aventis, Novartis, Lilly, Chugai, Janssen Diagnostics, and AstraZeneca. Moreover she acts as an advisor for Novartis. P. Fasching received research funding from Amgen and Novartis and has an advisory role for Amgen, Novartis, Roche, Pfizer, GSK, TEVA, Genomic Health and Nanostring. L. Schröder, H. Sommer, J. G. Koch, T. Weissenbacher, A. Schneeweiss, M. Rezai, R. Lorenz, B. Jäger, A. Schramm, L. Häberle, T. W. P. Friedl, M. W. Beckmann and C. Scholz have no conflicts of interest to declare.
Supporting Information
References
- 1.Early Breast Cancer Trialistʼs Collaborative Group (EBCTCG) . Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet. 2005;365:1687–1717. doi: 10.1016/S0140-6736(05)66544-0. [DOI] [PubMed] [Google Scholar]
- 2.Andergassen U, Kasprowicz N S, Hepp P. et al. Participation in the SUCCESS-A trial improves intensity and quality of care for patients with primary breast cancer. Geburtsh Frauenheilk. 2013;73:63–69. doi: 10.1055/s-0032-1328147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eisemann N, Waldmann A, Katalinic A. Epidemiology of breast cancer – current figures and trends. Geburtsh Frauenheilk. 2013;73:130–135. doi: 10.1055/s-0032-1328075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maass N, Schütz F, Fasching P A. et al. Breast cancer update 2014 – focus on the patient and the tumour. Geburtsh Frauenheilk. 2015;75:170–182. doi: 10.1055/s-0035-1545704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Petrelli F, Borgonovo K, Cabiddu M. et al. Mortality, leukemic risk, and cardiovascular toxicity of adjuvant anthracycline and taxane chemotherapy in breast cancer: a meta-analysis. Breast Cancer Res Treat. 2012;135:335–346. doi: 10.1007/s10549-012-2121-6. [DOI] [PubMed] [Google Scholar]
- 6.Levine M N, Bramwell V H, Pritchard K I. et al. Randomized trial of intensive cyclophosphamide, epirubicin, and fluorouracil chemotherapy compared with cyclophosphamide, methotrexate, and fluorouracil in premenopausal women with node-positive breast cancer. National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 1998;16:2651–2658. doi: 10.1200/JCO.1998.16.8.2651. [DOI] [PubMed] [Google Scholar]
- 7.Mamounas E P, Bryant J, Lembersky B. et al. Paclitaxel after doxorubicin plus cyclophosphamide as adjuvant chemotherapy for node-positive breast cancer: results from NSABP B-28. J Clin Oncol. 2005;23:3686–3696. doi: 10.1200/JCO.2005.10.517. [DOI] [PubMed] [Google Scholar]
- 8.Henderson I C, Berry D A, Demetri G D. et al. Improved outcomes from adding sequential Paclitaxel but not from escalating Doxorubicin dose in an adjuvant chemotherapy regimen for patients with node-positive primary breast cancer. J Clin Oncol. 2003;21:976–983. doi: 10.1200/JCO.2003.02.063. [DOI] [PubMed] [Google Scholar]
- 9.Hershman D L, Shao T. Anthracycline cardiotoxicity after breast cancer treatment. Oncology (Williston Park) 2009;23:227–234. [PubMed] [Google Scholar]
- 10.Sandström M, Lindman H, Nygren P. et al. Model describing the relationship between pharmacokinetics and hematologic toxicity of the epirubicin-docetaxel regimen in breast cancer patients. J Clin Oncol. 2005;23:413–421. doi: 10.1200/JCO.2005.09.161. [DOI] [PubMed] [Google Scholar]
- 11.Poirier E, Desbiens C, Poirier B. et al. Comparison of serious adverse events between the original and a generic docetaxel in breast cancer patients. Ann Pharmacother. 2014;48:447–455. doi: 10.1177/1060028013514941. [DOI] [PubMed] [Google Scholar]
- 12.Markman M. Managing taxane toxicities. Support Care Cancer. 2003;11:144–147. doi: 10.1007/s00520-002-0405-9. [DOI] [PubMed] [Google Scholar]
- 13.Carmichael J, Possinger K, Phillip P. et al. Advanced breast cancer: a phase II trial with gemcitabine. J Clin Oncol. 1995;13:2731–2736. doi: 10.1200/JCO.1995.13.11.2731. [DOI] [PubMed] [Google Scholar]
- 14.Akrivakis K, Schmid P, Flath B. et al. Prolonged infusion of gemcitabine in stage IV breast cancer: a phase I study. Anticancer Drugs. 1999;10:525–531. doi: 10.1097/00001813-199907000-00003. [DOI] [PubMed] [Google Scholar]
- 15.OʼShaughnessy J, Schwartzberg L S, Danso M A. et al. A randomized phase III study of iniparib (BSI-201) in combination with gemcitabine/carboplatin (G/C) in metastatic triple-negative breast cancer (TNBC) J Clin Oncol. 2011;29:Abstr. 1007. [Google Scholar]
- 16.Heinemann V. Gemcitabine in metastatic breast cancer. Expert Rev Anticancer Ther. 2004;5:37–44. doi: 10.1586/14737140.5.3.429. [DOI] [PubMed] [Google Scholar]
- 17.Colomer R, Llombart-Cussac A, Tusquets I. et al. Biweekly gemcitabine plus vinorelbine in first-line metastatic breast cancer: efficacy and correlation with HER2 extracellular domain. Clin Transl Oncol. 2006;8:896–902. doi: 10.1007/s12094-006-0153-2. [DOI] [PubMed] [Google Scholar]
- 18.Spielmann M, Llombart-Cussac A, Kalla S. et al. Single-agent gemcitabine is active in previously treated metastatic breast cancer. Oncology. 2001;60:303–307. doi: 10.1159/000058524. [DOI] [PubMed] [Google Scholar]
- 19.Heinemann V, Stemmler H J, Wohlrab A. et al. High efficacy of gemcitabine and cisplatin in patients with predominantly anthracycline- and taxane-pretreated metastatic breast cancer. Cancer Chemother Pharmacol. 2006;57:640–646. doi: 10.1007/s00280-005-0093-5. [DOI] [PubMed] [Google Scholar]
- 20.Nagourney R A, Flam M, Link J. et al. Carboplatin plus gemcitabine repeating doublet therapy in recurrent breast cancer. Clin Breast Cancer. 2008;8:432–435. doi: 10.3816/CBC.2008.n.052. [DOI] [PubMed] [Google Scholar]
- 21.Allouache D, Gawande S R, Tubiana-Hulin M. et al. First-line therapy with gemcitabine and paclitaxel in locally, recurrent or metastatic breast cancer: a phase II study. BMC Cancer. 2005;5:151. doi: 10.1186/1471-2407-5-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dranitsaris G, Beegle N, Kalberer T. et al. A comparison of toxicity and health care resource use between eribulin, capecitabine, gemcitabine, and vinorelbine in patients with metastatic breast cancer treated in a community oncology setting. J Oncol Pharm Pract. 2015;21:170–177. doi: 10.1177/1078155214525369. [DOI] [PubMed] [Google Scholar]
- 23.Maas K W, van der Lee I, Bolt K. et al. Lung function changes and pulmonary complications in patients with stage III non-small cell lung cancer treated with gemcitabine/cisplatin as part of combined modality treatment. Lung Cancer. 2003;41:345–351. doi: 10.1016/s0169-5002(03)00237-x. [DOI] [PubMed] [Google Scholar]
- 24.Albain K S, Nag S M, Calderillo-Ruiz G. et al. Gemcitabine plus Paclitaxel versus Paclitaxel monotherapy in patients with metastatic breast cancer and prior anthracycline treatment. J Clin Oncol. 2008;26:3950–3957. doi: 10.1200/JCO.2007.11.9362. [DOI] [PubMed] [Google Scholar]
- 25.Swain S M, Tang G, Geyer C E. et al. Definitive results of a phase III adjuvant trial comparing three chemotherapy regimens in women with operable, node-positive breast cancer: the NSABP B-38 trial. J Clin Oncol. 2013;31:3197–3204. doi: 10.1200/JCO.2012.48.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bear H D, Tang G, Rastogi P. et al. Bevacizumab added to neoadjuvant chemotherapy for breast cancer. N Engl J Med. 2012;366:310–320. doi: 10.1056/NEJMoa1111097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fossella F V, Lippman S M, Shin D M. et al. Maximum-tolerated dose defined for single-agent gemcitabine: a phase I dose-escalation study in chemotherapy-naive patients with advanced non-small-cell lung cancer. J Clin Oncol. 1997;15:310–316. doi: 10.1200/JCO.1997.15.1.310. [DOI] [PubMed] [Google Scholar]
- 28.Seidman A D, Brufsky A, Ansari R H. et al. Phase III trial of gemcitabine plus docetaxel versus capecitabine plus docetaxel with planned crossover to the alternate single agent in metastatic breast cancer. Ann Oncol. 2011;22:1094–1101. doi: 10.1093/annonc/mdq578. [DOI] [PubMed] [Google Scholar]
- 29.Earl H M, Vallier A L, Hiller L. et al. Effects of the addition of gemcitabine, and paclitaxel-first sequencing, in neoadjuvant sequential epirubicin, cyclophosphamide, and paclitaxel for women with high-risk early breast cancer (Neo-tAnGo): an open-label, 2×2 factorial randomised phase 3 trial. Lancet Oncol. 2014;15:201–212. doi: 10.1016/S1470-2045(13)70554-0. [DOI] [PubMed] [Google Scholar]
- 30.Roché H, Fumoleau P, Spielmann M. et al. Sequential adjuvant epirubicin-based and docetaxel chemotherapy for node-positive breast cancer patients: the FNCLCC PACS01 Trial. J Clin Oncol. 2006;24:5664–5671. doi: 10.1200/JCO.2006.07.3916. [DOI] [PubMed] [Google Scholar]
- 31.Wardley A M, Hiller L, Howard H C. et al. tAnGo: a randomised phase III trial of gemcitabine in paclitaxel-containing, epirubicin/cyclophosphamide-based, adjuvant chemotherapy for early breast cancer: a prospective pulmonary, cardiac and hepatic function evaluation. Br J Cancer. 2008;99:597–603. doi: 10.1038/sj.bjc.6604538. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.