

Abstract
Cadmium (Cd) is a highly toxic metal that affects the central nervous system. Recently we have demonstrated that inhibition of mTOR by rapamycin rescues neuronal cells from Cd-poisoning. Here we show that rapamycin inhibited Cd-induced mitochondrial ROS-dependent neuronal apoptosis. Intriguingly, rapamycin remarkably blocked phosphorylation of JNK, Erk1/2 and p38 in neuronal cells induced by Cd, which was strengthened by co-treatment with Mito-TEMPO. Inhibition of JNK and Erk1/2 by SP600125 and U0126, respectively, potentiated rapamycin’s prevention from Cd-induced apoptosis. Consistently, over-expression of dominant negative c-Jun or MKK1 also potently improved the inhibitory effect of rapamycin on Cd neurotoxicity. Furthermore, pretreatment with SP600125 or U0126, or expression of dominant negative c-Jun or MKK1 enhanced the inhibitory effects of rapamycin or Mito-TEMPO on Cd-induced ROS. Further investigation found that co-treatment with Mito-TEMPO/rapamycin more effectively rescued cells by preventing Cd inactivation of PP2A than treatment with rapamycin or Mito-TEMPO alone. Over-expression of wild-type PP2A reinforced rapamycin or Mito-TEMPO suppression of activated JNK and Erk1/2 pathways, as well as ROS production and apoptosis in neuronal cells in response to Cd. The findings indicate that rapamycin ameliorates Cd-evoked neuronal apoptosis by preventing mitochondrial ROS inactivation of PP2A, thereby suppressing activation of JNK and Erk1/2 pathways. Our results underline that rapamycin may have a potential in preventing Cd-induced oxidative stress and neurodegenerative diseases.
Keywords: Rapamycin, Cadmium, Apoptosis, MAPK, ROS, PP2A
Graphical Abstract
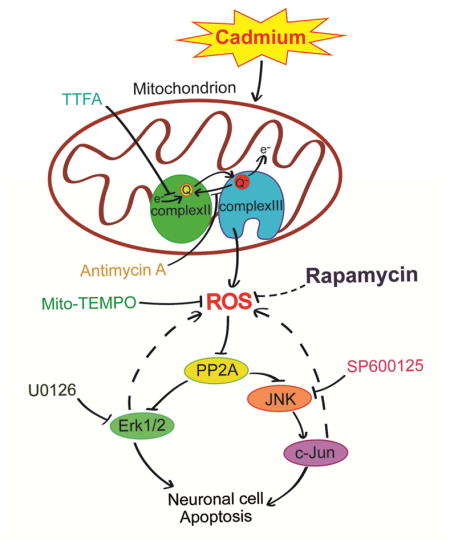
It is unclear whether and how rapamycin prevents Cd-induced neuronal apoptosis by intervening ROS-dependent activation of MAPK pathway. This study uncovers that rapamycin ameliorates Cd-evoked neuronal apoptosis by preventing mitochondrial ROS inactivation of PP2A, thereby suppressing activation of JNK and Erk1/2 pathways. The findings highlight that rapamycin may act as a potential therapeutic agent in the prevention of Cd-induced oxidative stress and neurodegenerative diseases.
1. Introduction
Cadmium (Cd) is one of the most toxic metals in the environment (water, air, and soil) (Wang and Du, 2013). Clinical data has shown that Cd, because of its long biological half-life (15–20 years), exhibits toxic effects on many organs/tissues such as blood (Kocak and Akcil, 2006), bones (Akesson et al., 2006), liver (Jomova and Valko, 2011), kidney (Johri et al., 2010), lung (Jiang et al., 2008), testis (Thompson and Bannigan, 2008), and brain (Lopez et al., 2003; Mendez-Armenta and Rios, 2007; Okuda et al., 1997). Cd can easily traverses the blood-brain barrier and accumulate in the brain, eliciting the dysfunction of the central nervous system (CNS), such as headache and vertigo, peripheral neuropathy, olfactory dysfunction, slowing of vasomotor functioning, decreased equilibrium, psychomotor speed, neurobehavioral defects in attention, and learning disabilities (Jarup et al., 1993; Pihl and Parkes, 1977; Wang and Du, 2013; Wright et al., 2006). Multiple studies have documented that Cd neurotoxicity is involved in its induction of oxidative stress, e.g., reactive oxygen species (ROS), in neuronal cells (Chen et al., 2011a; Chen et al., 2014b; Jomova and Valko, 2011; Kim et al., 2005; Mates et al., 2010). Cd exposure can initiate apoptotic cell death in distinct brain regions via oxidative stress or excessive amounts of ROS generation (Chen et al., 2014b; Goncalves et al., 2010; Watjen and Beyersmann, 2004); Excessive ROS induced by Cd can directly oxidize lipids, proteins, and nucleic acids, alter their functions, and activate related signaling pathways, thereby leading to cell dysfunction and cell death (Bertin and Averbeck, 2006; Figueiredo-Pereira et al., 1998; Genovese and Cuzzocrea, 2008; Wang and Du, 2013). Therefore, Cd-inudced ROS is thought to play a critical role in the development of neuronal cell death and consequential neurodegenerative diseases, including Parkinson’s desease (PD), Alzheimer’s disease (AD) and Huntington’s disease (HD) (Goncalves et al., 2010; Monroe and Halvorsen, 2006; Panayi et al., 2002; Wang and Du, 2013; Watjen and Beyersmann, 2004).
Mitogen-activated protein kinases (MAPKs) are evolutionarily conserved tyrosine (Tyr) and serine/threonine (Ser/Thr) protein kinases (Kyriakis and Avruch, 2012). MAPKs in mammalian cells possess at least three distinct groups, including the extracellular signal-regulated kinases ERK1/2, ERK3/4, ERK5, ERK7/8, the Jun N-terminal kinases JNK1/2/3, and the p38 MAPKs p38α/β/γ/δ (Kyriakis and Avruch, 2012). These pathways are central components of the intracellular signaling networks that control many aspects of mammalian cellular physiology, including cell proliferation/growth, differentiation, and apoptosis (Kim et al., 2008; Kyriakis and Avruch, 2012). It is well known that Ser/Thr protein phosphatase 2A (PP2A) directly dephosphorylates and inactivates Erk1/2, JNK and/or p38, involved in stress response and the regulation of cell proliferation, survival and apoptosis (Chen et al., 2008a; Chen et al., 2009; Franklin and Kraft, 1997; Han et al., 2012; Huang et al., 2004). Numerous studies have reported that Cd activates Erk1/2, JNK and/or p38 MAPK contributing to apoptosis in various types of cells, including neuronal cells (Kim et al., 2005; Rockwell et al., 2004). Our previous studies have unveiled that all the three MAPK members can be activated by Cd in neuronal cells, but Cd-induced neuronal apoptosis is only partially attributed to activation of Erk1/2 and JNK, but not p38 (Chen et al., 2008b), and pinpointed that Cd activates Erk1/2 and JNK pathways leading to apoptosis by induction of ROS and consequent inhibition of PP2A in neuronal cells (Chen et al., 2008a). Thus, we proposed that a compound that can regulate PP2A-dependent Erk1/2/JNK signaling pathways by inhibiting ROS might be useful to prevent Cd-poisoning.
Rapamycin, a macrocyclic lactone, is a potent and specific mTOR inhibitor (Zhou et al., 2010). Recent studies have shown that rapamycin acts as a neuroprotective compound in various neurological diseases (Bove et al., 2011; Erlich et al., 2007; Malagelada et al., 2010; Pan et al., 2009). Thus, we reasoned that rapamycin that can inhibit mTOR may exert preventive effect on Cd-induced neurotoxicity. As expected, inhibition of mTOR by pretreatment with rapamycin for 48 h indeed rescued neuronal cells from Cd-poisoning (Chen et al., 2008b; Xu et al., 2015). Administration of rapamycin in vivo also potently attenuated Cd-induced activation of mTOR signaling, brain damage and neuronal cell death in mice (Chen et al., 2014b). In addition, treatment with rapamycin in vitro and in vivo prevented Cd induction of ROS-dependent neuronal cell apoptosis as well (Chen et al., 2011a; Chen et al., 2014b). Interestingly, emerging evidence has implied that rapamycin may alter the activity of MAPKs including Erk1/2, JNK and/or p38 under various conditions, thus affecting cell proliferation, survival and apoptosis (Benoit et al., 2011; Hahn et al., 2005; Huang et al., 2003; Kato et al., 2013; Kawasaki et al., 2010; Shi et al., 2005). However, it is unknown whether and how rapamycin protects against Cd-induced neuronal apoptosis by preventing ROS from activation of Erk1/2, JNK and/or p38 pathways. Here, for the first time, we show that rapamycin ameliorated Cd-evoked neuronal apoptosis by preventing mitochondrial ROS inactivation of PP2A, thus suppressing activation of JNK and Erk1/2 pathways. The results improve our understanding of the molecular mechanism by which rapamycin exerts effective prevention against Cd-induced oxidative stress and neurodegenerative diseases.
2. Materials and methods
2.1. Reagents
Cadmium chloride, poly-D-lysine (PDL), 4′,6-diamidino-2-phenylindole (DAPI), antimycin A, thenoyltrifluoroacetone (TTFA), SP600125, U0126, PD169136 and protease inhibitor cocktail were purchased from Sigma (St Louis, MO, USA). 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA) was purchased from MP Biomedicals Inc (Solon, OH, USA). Rapamycin and Mito-TEMPO were from ALEXIS Biochemicals Corporation (San Diego, CA, USA). Dulbecco’s modified Eagle medium (DMEM), 0.05% Trypsin-EDTA, NEUROBASAL™ Media and B27 Supplement were purchased from Invitrogen (Grand Island, NY, USA). Horse serum and fetal bovine serum (FBS) were supplied by Hyclone (Logan, UT, USA). Annexin V-FITC/Propidium Iodide (PI) Apoptosis Detection kit was obtained from BD Biosciences (San Diego, CA, USA). Enhanced chemiluminescence reagent was from Millipore (Billerica, MA, USA). The following antibodies were used: PP2ACα (BD Biosciences, San Jose, CA, USA); phospho-Erk1/2 (Thr202/Tyr204), p38, phospho-p38 (Thr180/Tyr182), caspase-3, and PARP (Cell Signaling Technology, Beverly, MA, USA); Erk2, demethylated-PP2A, c-Jun, phospho-c-Jun (Ser63), JNK, and phospho-JNK (Thr183/Tyr185) (Santa Cruz Biotechnology, Santa Cruz, CA, USA); phospho-PP2A (Epitomics, Burlingame, CA, USA); FLAG, MKK1, and β-tubulin (Sigma); goat anti-rabbit IgG-horseradish peroxidase (HRP), goat anti-mouse IgG-HRP, and rabbit anti-goat IgG-HRP (Pierce, Rockford, IL, USA). Other chemicals were purchased from local commercial sources and were of analytical grade.
2.2. Cell culture
Rat pheochromocytoma (PC12) cell line (American Type Culture Collection, Manassas, VA, USA) was seeded in a 6-well or 96-well plate, or 100-mm dish coated with 0.2 μg/ml PDL and incubated as described (Chen et al., 2014a). Primary murine neurons were isolated from fetal mouse cerebral cortexes of 16–18 days of gestation in female ICR mice (being pregnant), which were handled in accordance with the guidelines of the Institutional Animal Care and Use Committee, and were in compliance with the guidelines set forth by the Guide for the Care and Use of Laboratory Animals, and then seeded in a 6-well or 96-well plate coated with 10 μg/ml PDL for experiments after 6 days of culture as described (Chen et al., 2010; Chen et al., 2014a).
2.3. Recombinant adenoviral constructs and infection of cells
The recombinant adenoviruses expressing FLAG-tagged wild-type (wt) rat PP2ACα (Ad-PP2A), FLAG-tagged dominant negative c-Jun (FLAG-Δ169) (Ad-dn-c-Jun), FLAG-tagged dominant negative MKK1 (Ad-MKK1-K97M), and the control virus expressing the green fluorescent protein (GFP) (Ad-GFP) or β-galactosidase (Ad-LacZ) were described previously (Chen et al., 2008b; Huang et al., 2003; Huang et al., 2004; Liu et al., 2010). For experiments, PC12 cells or primary neurons were grown in the growth medium and infected with the individual adenovirus for 24 h at 5 of multiplicity of infection (MOI = 5). Afterwards, cells were used for experiments. Ad-GFP served as a control. Expression of FLAG-tagged PP2A, dn-c-Jun or MKK1 was determined by Western blotting with antibodies to FLAG.
2.4. Live cell assay by trypan blue exclusion
PC12 cells, or PC12 cells infected with Ad-dn-c-Jun, Ad-MKK1-K97M, Ad-PP2A or Ad-GFP, respectively, were seeded at a density of 5 × 105 cells/well in a PDL-coated 6-well plate. Next day, cells were treated with/without Cd (20 μM) for 24 h following pre-incubation with/without rapamycin (0.2 μg/ml) for 48 h with 5 replicates of each treatment. In some cases, cells were pretreated with/without rapamycin (0.2 μg/ml) for 48 h, and then with/without Mito-TEMPO (10 μM), SP600125 (20 μM), U0126 (5 μM) or PD169136 (20 μM) for 1 h, followed by exposure to Cd (20 μM) for 24 h. Subsequently, live cells were monitored by counting viable cells using trypan blue exclusion test.
2.5. DAPI staining
PC12 cells and primary neurons, or PC12 cells infected with Ad-dn-c-Jun, Ad-MKK1-K97M, Ad-PP2A or Ad-GFP, respectively, were seeded at a density of 5 × 105 cells/well in a 6-well plate containing a PDL-coated glass coverslip per well. The next day, cells were treated with/without Cd (10 and/or 20 μM) for 24 h following pre-incubation with/without rapamycin (0.2 μg/ml) for 48 h with 5 replicates of each treatment. In some cases, cells were pretreated with/without rapamycin (0.2 μg/ml) for 48 h, and then with/without Mito-TEMPO (10 μM), SP600125 (20 μM), U0126 (5 μM) or PD169136 (20 μM) for 1 h, followed by exposure to Cd (20 μM) for 24 h. Subsequently, the apoptotic cells with fragmented and condensed nuclei were detected using DAPI staining, and imaged under a fluorescence microscope as described (Chen et al., 2008a).
2.6. Flow cytometry
PC12 cells were seeded at a density of 2 × 106 cells/dish in a PDL-coated 100-mm dish. The next day, cells were treated with/without Cd (10 and 20 μM) for 24 h following preincubation with/without rapamycin (0.2 μg/ml) for 48 h, followed by annexin-V-FITC/PI staining and analysis under a fluorescence-activated cell sorter (FACS) Vantage SE flow cytometer (Becton Dickinson, California, USA).
2.7. Cell ROS assay and ROS imaging
Detecting and imaging intracellular ROS were performed using an oxidant-sensitive probe, CM-H2DCFDA, which is a stable nonfluorescent molecule that passively diffuses into cells, where the acetate can be cleaved by intracellular esterases to produce a polar diol that is well retained within the cells (Chen et al., 2008a). In brief, PC12 and primary neurons, or PC12 cells and/or primary neurons infected with Ad-dn-c-Jun, Ad-MKK1-K97M, Ad-PP2A or and Ad-LacZ, respectively, were seeded in a 96-well plate (1 × 104 cells/well), or in a 6-well plate (5 × 105 cells/well) containing a glass coverslip per well. The next day, cells were pretreated with/without rapamycin (0.2 μg/ml) for 48 h or with/without Mito-TEMPO (10 μM) for 1 h, followed by exposure to Cd (10 and/or 20 μM) for 24 h, or to Cd (20 μM) in the presence or absence of TTFA (10 μM) or antimycin A (50 μM) for 24 h, with 5 replicates of each treatment. In some cases, cells were pretreated with/without rapamycin (0.2 μg/ml) for 48 h and then with/without Mito-TEMPO (10 μM), SP600125 (20 μM), U0126 (5 μM) or PD169136 (20 μM) for 1 h, or pretreated with/without Mito-TEMPO (10 μM) for 1 h and then with/without SP600125 (20 μM), U0126 (5 μM) or PD169136 (20 μM) for 1 h, followed by exposure to Cd (20 μM) for 24 h. The cells were then loaded with CM-H2DCFDA (10 μM) for 1 h. Subsequently, intracellular ROS fluorescence intensity was determined by excitation at 485 nm and emission at 535 nm using a Synergy™ 2 Multi-function Microplate Reader (Bio-Tek Instruments, Inc. Winooski, Vermont, USA). To image intracellular ROS, all stained specimens were rinsed three times with PBS, followed by capturing under a fluorescence microscope (Nikon 80i, Tokyo, Japan) equipped with a digital camera. For quantitative analysis of the fluorescence intensity, the integral optical density (IOD) was measured by Image-Pro Plus 6.0 software (Media Cybernetics Inc., Newburyport, MA, USA).
2.8. In vitro PP2A phosphatase assay
After treatments, cells were lysed in 50 mM Tris-HCl buffer, pH 7.0, containing 1% Nonidet P-40, 2 mM EDTA, and protease inhibitor cocktail (Sigma, 1:1000). PP2Ac was immunoprecipitated with antibodies to PP2Ac (Millipore, Temecula, CA, USA), and protein A/G agarose (Santa Cruz Biotechnology). Subsequently, the beads were washed three times with the above lysis buffer, and twice with the phosphatase assay buffer (50 mM Tris-HCl, pH 7.0, 0.1 mM CaCl2). The phosphatase activity of immunoprecipitated PP2A was assayed with a Ser/Thr Phosphatase Assay kit 1 using KRpTIRR as the substrate peptide (Millipore) following the manufacturer’s instructions. Finally, all data (from different batches of experiments) were pooled for statistical analysis as described (Liu et al., 2010).
2.9. Western blot analysis
The indicated cells, after treatments, were briefly washed with cold PBS, and then on ice, lysed in the radioimmunoprecipitation assay buffer. Subsequently, Western blotting was performed, and the blots for detected protein were semi-quantified using NIH Image J software (National Institutes of Health, Bethesda, MD, USA) as described previously (Chen et al., 2010; Chen et al., 2014a).
2.10. Statistical analysis
All data were expressed as mean ± SEM. Student’s t-test for non-paired replicates was used to identify statistically significant differences between treatment means. Group variability and interaction were compared using either one-way or two-way ANOVA followed by Bonferroni’s post-tests to compare replicate means. Significance was accepted at p < 0.05.
3. Results
3.1. Rapamycin attenuates Cd-induced mitochondrial ROS-dependent apoptosis in neuronal cells
Recently we have shown that Cd induces apoptosis of PC12 and SH-SY5Y cells by induction of ROS (Chen et al., 2008a; Chen et al., 2011a), and treatment with rapamycin in vitro and in vivo prevents Cd induction of ROS-dependent neurotoxicity (Chen et al., 2011a; Chen et al., 2014b). In line with the above findings, here we also observed that pretreatment with rapamycin (0.2 μg/ml) for 48 h attenuated the intracellular ROS production in PC12 and primary neurons induced by 24-h exposure with Cd (10 and 20 μM), as measured by an oxidant-sensitive probe, CM-H2DCFDA (Fig. 1A). To corroborate the finding, we extended the studies by imaging intracellular ROS. Imaged and quantified results revealed that treatment with Cd for 24 h evoked strong ROS fluorescence (in green) in the cells, which was potently suppressed by rapamycin pretreatment (Fig. 1B and C).
Fig. 1.

Rapamycin attenuates Cd-induced ROS in neuronal cells. PC12 cells and primary neurons were pretreated with rapamycin (Rap, 0.2 μg/ml) for 48 h, and then exposed to Cd (10 and 20 μM) for 24 h, followed by ROS assay and ROS imaging using an oxidant-sensitive probe CM-H2DCFDA. A) Rapamycin attenuated the intracellular ROS production in the cells in response to Cd. B and C) Cd-evoked strong ROS fluorescence (in green) was potently suppressed by rapamycin in the cells. Scale bar: 20 μm. Results are presented as mean ± SEM (n = 5). a p < 0.05, difference with control group; b p < 0.05, difference with 10 μM Cd group; c p < 0.05, difference with 20 μM Cd group.
To elucidate the association of Cd-induced ROS with neuronal apoptosis, annexin-V-FITC/PI staining was used, showing that rapamycin effectively diminished the relative number of apoptotic PC12 cells in response to Cd treatment (Fig. 2A and B). Subsequently, we detected the cells with nuclear fragmentation and condensation, a hallmark of apoptosis (Hao et al., 2013), using DAPI staining, exhibiting that pretreatment with rapamycin significantly reduced the percentage of cells with fragmented nuclei (arrows) in PC12 cells and primary neurons triggered by Cd exposure, compared with the control (Fig. 2C and D).
Fig. 2.
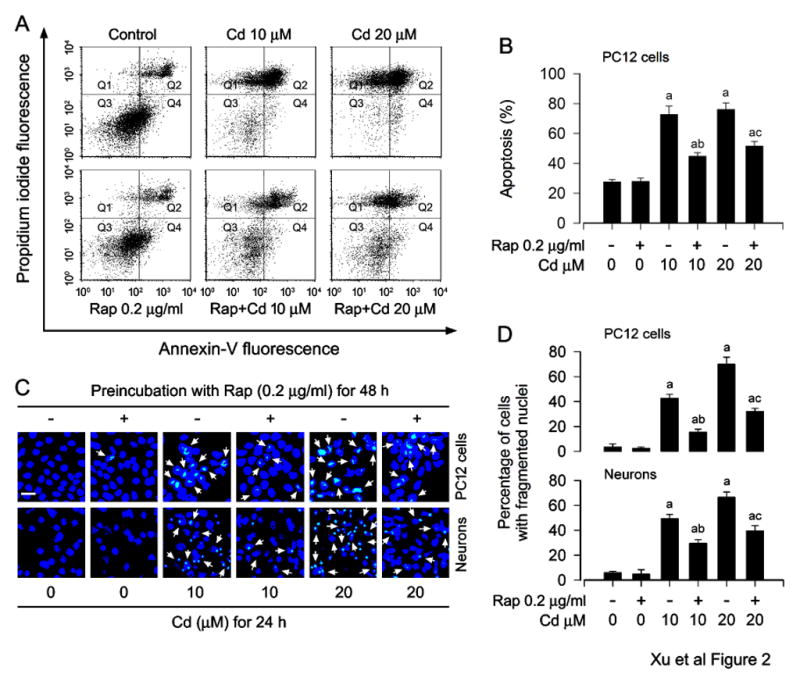
Rapamycin prevents Cd-induced apoptosis in neuronal cells. PC12 cells and/or primary neurons were pretreated with rapamycin (Rap, 0.2 μg/ml) for 48 h, and then exposed to Cd (10 and 20 μM) for 24 h. A) The percentages of live (Q3), early apoptotic (Q4), late apoptotic (Q2) and necrotic cells (Q1) were determined by FACS using annexin-V-FITC/PI staining. The results from a representative experiment are shown. B) Quantitative analysis of apoptotic PC12 cells by FACS assay. C) Apoptosis in PC12 cells and primary neurons was evaluated by nuclear fragmentation and condensation (arrows) using DAPI staining. Scale bar: 20 μm. D) The percentages of cells with fragmented nuclei were quantified, showing that rapamycin markedly attenuated Cd-induced apoptosis in the cells. Results are presented as mean ± SEM (n = 5). a p < 0.05, difference with control group; b p < 0.05, difference with 10 μM Cd group; c p < 0.05, difference with 20 μM Cd group.
Since Cd-induced ROS production has been observed in the mitochondria of anterior pituitary cells, cortical neurons and brain (Lopez et al., 2006; Poliandri et al., 2006; Wang et al., 2004), we next examined whether rapamycin prevention against Cd-induced neuronal apoptosis is dependent on mitochondrial ROS induction. For this, PC12 cells and primary neurons were pretreated with/without rapamycin (0.2 μg/ml) for 48 h, followed by exposure to Cd (20 μM) in the presence or absence of TTFA (10 μM), a mitochondrial complex II ubiquinone site inhibitor with blockage of electron supply to ubiquinol and consequential limiting the formation of ubisemiquinone (Moreno-Sanchez et al., 2013), or antimycin A (50 μM), a mitochondrial complex III inhibitor that increases the lifetime of ubisemiquinone (Lanju et al., 2014), for 24 h. We observed that co-treatment with rapamycin/TTFA inhibited Cd-evoked ROS fluorescence more potently than treatment with rapamycin or TTFA alone in the cells (Fig. 3A), whereas treatment with antimycin A alone markedly triggered ROS elevation, and there existed a further increase in ROS levels after exposure to Cd in the presence of antimycin A, which were repressed by rapamycin pretreatment (Fig. 3B). Of note, pretreatment with Mito-TEMPO (10 μM), a mitochondria-targeted antioxidant (Yeh et al., 2014), obviously inhibited Cd-induced ROS levels by 20.61% and 27.04% (Fig. 3C) as well as cell apoptosis by 19.01% and 13.88% (Fig. 3D), and pretreatment with rapamycin markedly reduced Cd-induced ROS by 27.22% and 39.71% (Fig. 3C) and cell apoptosis by 31.86% and 27.57% (Fig. 3D) compared to Cd-treated group alone in PC12 cells and primary neurons, respectively. However, Mito-TEMPO significantly strengthened the inhibitory effects of rapamycin on Cd-induced the events, exhibiting that co-treatment with Mito-TEMPO/rapamycin greatly relieved ROS production by 38.87% and 53.23% (Fig. 3C) and cell apoptosis by 44% and 38.84% (Fig. 3D) compared to Cd-treated group alone in PC12 cells and primary neurons induced by Cd, respectively. Consistently, Mito-TEMPO also diminished cleavages of caspase-3 and PARP in the cells in response to Cd, and substantially enhanced the inhibitory effect of rapamycin (Fig. 3E and F). Collectively, our findings indicate that rapamycin attenuates Cd-induced mitochondrial ROS, thereby preventing apoptosis in neuronal cells.
Fig. 3.

Rapamycin relieves Cd-induced mitochondrial ROS-dependent apoptosis in neuronal cells. PC12 and primary neurons were pretreated A and B) with/without rapamycin (0.2 μg/ml) for 48 h and then exposed to Cd (20 μM) in the presence or absence of TTFA (10 μM) or antimycin A (50 μM) for 24 h, or C-F) with/without rapamycin (0.2 μg/ml) for 48 h, Mito-TEMPO (10 μM) for 1 h and then exposed to Cd (20 μM) for 24 h, followed by A-C) ROS imaging using an oxidant-sensitive probe CM-H2DCFDA, D) cell apoptosis analysis using DAPI staining, or E) Western blotting using the indicated antibodies. The blots were probed for β-tubulin as a loading control. F) Similar results were observed in at least three independent experiments, and blots for cleaved-caspase-3, and cleaved-PARP were semi-quantified. A) Co-treatment with rapamycin/TTFA inhibited Cd-evoked ROS fluorescence more potently than treatment with rapamycin or TTFA alone in the cells. B) Treatment with antimycin A alone markedly elevated ROS and strengthened Cd-increased ROS levels, which were repressed by rapamycin pretreatment. C-F) Pretreatment with Mito-TEMPO obviously inhibited Cd-induced ROS levels and cell apoptosis, and dramatically potentiated the inhibitory effects of rapamycin on Cd-induced the events in PC12 cells and primary neurons. Results are presented as mean ± SEM (n = 3–5). a p<0.05, difference with control group; b p<0.05, difference with 20 μM Cd group; c p<0.05, difference with Cd/TTFA group, Cd/Antimycin A group, Cd/Mito-TEMPO group or Cd/Rapamycin group.
3.2. Rapamycin blocks Cd-induced activation of JNK, Erk1/2 and p38 pathways by suppressing mitochondrial ROS in neuronal cells
Numerous studies have documented that rapamycin may alter the activity of MAPKs including JNK, Erk1/2 and/or p38 under various conditions, thus affecting cell proliferation, survival and apoptosis (Benoit et al., 2011; Hahn et al., 2005; Huang et al., 2003; Kato et al., 2013; Kawasaki et al., 2010; Shi et al., 2005). Our previous studies have identified that Cd activates JNK, Erk1/2, and p38, but only JNK and Erk1/2 participate in Cd-induced apoptosis in neuronal cells (Chen et al., 2008b). Therefore, we hypothesized that rapamycin might prevent Cd-induced neuronal apoptosis by repressing MAPK pathway. To this end, PC12 and primary neurons were pretreated with rapamycin (0.2 μg/ml) for 48 h, and then exposed to Cd (10 and 20 μM) for 4 h, followed by Western blot analysis. We found that rapamycin potently suppressed Cd-induced phosphorylation of JNK, particularly protein expression and phosphorylation of c-Jun, a substrate of JNK in the cells (Fig. 4A and B). Consistently, Cd-induced phosphorylation of Erk1/2 and p38 was also effectively inhibited by rapamycin (Fig. 4A and B).
Fig. 4.

Rapamycin blocks Cd-induced activation of JNK, Erk1/2 and p38 pathways by suppressing mitochondrial ROS induction in neuronal cells. PC12 and primary neurons were pretreated A and B) with/without rapamycin (0.2 μg/ml) for 48 h, or C and D) with/without rapamycin (0.2 μg/ml) for 48 h and then Mito-TEMPO (10 μM) for 1 h, followed by exposure to Cd (10 and/or 20 μM) for 24 h. A and C) Total cell lysates were subjected to Western blot analysis using indicated antibodies. The blots were probed for β-tubulin as a loading control. B and D) Similar results were observed in at least three independent experiments, and blots for p-JNK, p-c-Jun, p-Erk1/2, and p-p38 were semi-quantified. A and B) rapamycin potently suppressed Cd-induced phosphorylation of JNK/c-Jun, Erk1/2 and p38, and C and D) co-treatment with rapamycin/Mito-TEMPO exhibited a stronger inhibitory effect on Cd-induced activation of JNK/c-Jun, Erk1/2 and p38 than treatment with rapamycin or Mito-TEMPO alone in the cells. Results are presented as mean ± SEM (n = 3). a p < 0.05, difference with control group; b p < 0.05, difference with 10 μM Cd group; c p < 0.05, difference with 20 μM Cd group; d p < 0.05, difference with Cd/Mito-TEMPO group or Cd/Rapamycin group.
To validate that rapamycin blocks activation of JNK, Erk1/2 and p38 pathways by suppressing Cd induction of mitochondrial ROS in neuronal cells, PC12 cells and primary neurons were pretreated with/without rapamycin (0.2 μg/ml) for 48 h, and then treated with/without Mito-TEMPO (10 μM) for 1 h, followed by exposure to Cd (20 μM) for 4 h. As shown in Fig. 4C and D, Mito-TEMPO or rapamycin obviously suppressed Cd-induced phosphorylation of JNK/c-Jun, Erk1/2 and p38 in the cells. Especially, co-treatment with rapamycin/Mito-TEMPO exhibited a stronger inhibitory effect on Cd-induced activation of JNK/c-Jun, Erk1/2 and p38 in the cells (Fig. 4C and D). This was consistent with the findings of rapamycin and/or Mito-TEMPO inhibition of Cd-increased neuronal apoptosis and cleavages of caspase-3 and PARP observed by DAPI staining and Western blotting, respectively (Fig. 3D–F). The results suggest that rapamycin blocks Cd-induced mitochondrial ROS, attenuating activation of the MAPK pathways and apoptosis in neuronal cells.
3.3. Rapamycin prevents Cd-induced neuronal apoptosis only in part by blocking both JNK and Erk1/2 pathways
To pinpoint how JNK, Erk1/2 and p38 are involved in rapamycin protection against Cd-induced neuronal cell apoptosis, SP600125 (JNK inhibitor), U0126 (MEK1/2 inhibitor) and PD169136 (p38 inhibitor) were employed. When PC12 cells and primary neurons were treated with rapamycin (0.2 μg/ml) for 48 h alone, or with SP600125 (20 μM), U0126 (5 μM), or PD169136 (20 μM) for 1 h alone, or co-treated with rapamycin (0.2 μg/ml) for 48 h in the presence or absence of SP600125 (20 μM), U0126 (5 μM), or PD169136 (20 μM) for 1 h, and then exposed to Cd (20 μM) for 4 h or 24 h, we found that SP600125 or U0126 alone remarkably attenuated Cd-induced activation of JNK/c-Jun or Erk1/2, and caspase-3 in the cells, respectively (Fig. 5A and B). Furthermore, co-treatment with rapamycin/SP600125 or rapamycin/U0126 exhibited a stronger inhibitory effect on Cd-induced JNK or Erk1/2, and caspase-3 activation (Fig. 5A and B). In line with this, co-treatment with rapamycin/SP600125 or rapamycin/U0126 also rescued cells from Cd-induced apoptotic cell death more potently than rapamycin, SP600125 or U0126 alone, as determined by live and apoptotic cells using trypan blue exclusive and DAPI staining, respectively (Fig. 5C–E). In addition, consistent with our previous findings (Chen et al., 2008b), although PD169136 (20 μM) blocked Cd-induced phosphorylation of p38, co-treatment with rapamycin/PD169136 failed to enhance the protective effect of rapamycin on Cd-induced cytotoxicity (data not shown). Taken together, our data suggest that rapamycin may prevent Cd-induced neuronal apoptosis partially by blocking JNK and Erk1/2 pathways.
Fig. 5.

Rapamycin inhibits Cd-induced neuronal apoptosis by blocking JNK and Erk1/2 pathways. PC12 cells and primary neurons were pretreated with/without rapamycin (0.2 μg/ml) for 48 h, and then SP600125 (20 μM) or U0126 (5 μM) for 1 h, followed by exposure to Cd (20 μM) for 4 h (for Western blotting) or 24 h (for live cell analysis, DAPI staining). A) Cell lysates were subjected to Western blot analysis using indicated antibodies, showing that inhibitors of JNK (SP600125) and Erk1/2 (U0126) strengthened the inhibitory activity of rapamycin. The blots were probed for β-tubulin as a loading control. B) Similar results were observed in at least three independent experiments, and blots for p-JNK, p-c-Jun, p-Erk1/2, and cleaved-caspase-3 were semi-quantified. C) Live cells were detected by counting viable cells using trypan blue exclusion and D and E) the percentages of apoptotic cells with fragmented nuclei were quantified by DAPI staining, showing that pharmacological inhibition of JNK and Erk1/2 enhanced rapamycin prevention of Cd-induced cell viability reduction and apoptosis, respectively. Results are presented as mean ± SEM (n = 3–5). a p < 0.05, difference with control group; b p < 0.05, difference with 20 μM Cd group; c p < 0.05, difference with Cd/SP600125 group, Cd/U0126 group or Cd/Rapamycin group.
To confirm the above findings that rapamycin inhibits Cd-induced neuronal cell apoptosis by blocking activation of JNK and Erk1/2 cascades, we carried out the experiments for gene over-expression or gene silencing. PC12 cells, infected with Ad-dn-c-Jun, Ad-MKK1-K97M and Ad-GFP (as control), were pretreated with rapamycin (0.2 μg/ml) for 48 h, and then exposed to Cd (20 μM) for 4 h or 24 h. Western blot analysis revealed that ectopic expression of dn-c-Jun and MKK1-K97M, but not GFP, obviously blocked Cd-induced phosphorylation of c-Jun and Erk1/2 (Fig. 6A and B). Consistently, Cd-induced cleavage of caspase-3 was apparently attenuated by expression of dn-c-Jun or MKK1-K97M (Fig. 6A and B). Furthermore, our assay for live and apoptotic cells exhibited that expression of dn-c-Jun or MKK1-K97M also partially rescued PC12 cells from apoptosis triggered by Cd exposure (Fig. 6C and D). Of importance, addition of rapamycin showed more inhibitory effect on Cd-induced activation of c-Jun and Erk1/2, as well as apoptosis in Ad-dn-c-Jun group or Ad-MKK1-K97M group than in Ad-GFP group (Fig. 6A–D). In addition, infection of PC12 cells with lentiviral shRNAs to Erk1/2, c-Jun and p38, respectively, down-regulated expression of these proteins by ~ 90%. Similarly, silencing Erk1/2 and c-Jun, but not p38, strengthened rapamycin’s prevention of Cd-induced neuronal apoptosis (data not shown). Thus, our results demonstrate that rapamycin inhibits Cd-induced cell apoptosis in neuronal cells, at least in part, through blocking JNK and Erk1/2 cascades.
Fig. 6.

Expression of dominant negative c-Jun or dominant negative MKK1 reinforces rapamycin inhibition of Cd-induced neuronal apoptosis. PC12 cells, infected with Ad-dn-c-Jun, Ad-MKK1-K97M and Ad-GFP (as control), respectively, were pretreated with/without rapamycin (0.2 μg/ml) for 48 h, and then exposed to Cd (20 μM) for 4 h (for Western blotting) or 24 h (for live cell analysis, DAPI staining). A) Cell lysates were subjected to Western blot analysis using indicated antibodies. The blots were probed for β-tubulin as a loading control. B) Similar results were observed in at least three independent experiments, and blots for p-c-Jun, p-Erk1/2, and cleaved-caspase-3 were semi-quantified. C) Live cells were detected by counting viable cells using trypan blue exclusion. D) The percentages of apoptotic cells with fragmented nuclei were quantified by DAPI staining. Results are presented as mean ± SEM (n = 3–5). a p < 0.05, difference with control group; b p < 0.05, difference with 20 μM Cd group; c p < 0.05, Ad-dn-c-Jun group or Ad-MKK1-K97M group versus Ad-GFP group.
3.4. Pharmacological inhibition of JNK or Erk1/2, or expression of dominant negative c-Jun or MKK1 strengthens rapamycin’s inhibition of Cd-induced mitochondrial ROS in neuronal cells
To dissect whether JNK and Erk1/2 positively mediates Cd-induced mitochondrial ROS and the role of rapamycin in blocking Cd-induced mitochondrial ROS activation of JNK and Erk1/2 pathways, PC12 cells and primary neurons were pretreated with/without rapamycin (0.2 μg/ml) for 48 h or Mito-TEMPO (10 μM) for 1 h, and then with/without SP600125 (20 μM) or U0126 (5 μM) for 1 h, followed by exposure to Cd (20 μM) for 24 h. We noticed that treatment with SP600125 or U0126 alone substantially blocked Cd-induced ROS production (Fig. 7A and B), in line with our previous findings (Chen et al., 2008a). Of importance, there existed more inhibitory effects on Cd-induced ROS in the cells co-treated with rapamycin/SP600125 or rapamycin/U0126, or with Mito-TEMPO/SP600125 or Mito-TEMPO/U0126 than in the ones treated with rapamycin, Mito-TEMPO, SP600125 or U0126 alone (Fig. 7A and B). In addition, we also observed that inhibition of p38 by PD169136 (20 μM) did not block Cd-elicited ROS, and also failed to enhance the inhibitory effect of rapamycin or Mito-TEMPO on Cd-induced ROS (Fig. 7A and B).
Fig. 7.

Pharmacological inhibition of JNK or Erk1/2, or expression of dominant negative c-Jun or MKK1 strengthens rapamycin’s inhibition of Cd-induced mitochondrial ROS in neuronal cells. A and B) PC12 and primary neurons were pretreated with/without rapamycin (0.2 μg/ml) for 48 h or with/without Mito-TEMPO (10 μM) for 1 h, and then SP600125 (20 μM), U0126 (5 μM) or PD169136 (20 μM) for 1 h, followed by exposure to Cd (20 μM) for 24 h. C and D) PC12 cells and primary neurons, infected with Ad-dn-c-Jun, Ad-MKK1-K97M or Ad-LacZ (as control), were exposed to Cd (20 μM) for 24 h post pre-incubation with/without rapamycin (0.2 μg/ml) for 48 h or Mito-TEMPO (10 μM) for 1 h. A and B) ROS imaging using an oxidant-sensitive probe CM-H2DCFDA was quantified, showing more inhibitory effects on Cd-induced ROS in the cells co-treated with rapamycin/SP600125 or rapamycin/U0126, or with Mito-TEMPO/SP600125 or Mito-TEMPO/U0126 than in the ones treated with rapamycin, Mito-TEMPO, SP600125 or U0126 alone. C and D) Expression of dn-c-Jun and MKK1-K97M, but not LacZ, partially prevented the cells from ROS overproduction induced by Cd, and expression of dn-c-Jun and MKK1-K97M strengthened the inhibitory effects of rapamycin or Mito-TEMPO on Cd-induced ROS in the cells. Results are presented as mean ± SEM (n = 5). a p < 0.05, difference with control group; b p < 0.05, difference with 20 μM Cd group; c p < 0.05, – SP600125 group versus + SP600125 group or – U0126 group versus + U0126 group; d p < 0.05, Ad-dn-c-Jun group or Ad-MKK1-K97M group versus Ad-LacZ group.
Next, PC12 cells and primary neurons were infected with Ad-dn-c-Jun, Ad-MKK1-K97M and Ad-LacZ (as control), and then exposed to Cd (20 μM) for 24 h post pre-incubation with/without rapamycin (0.2 μg/ml) for 48 h or Mito-TEMPO (10 μM) for 1 h, respectively. The results showed that expression of dn-c-Jun and MKK1-K97M, but not LacZ, partially prevented the cells from ROS overproduction induced by Cd (Fig. 7C and D). Of note, expression of dn-c-Jun and MKK1-K97M strengthened the inhibitory effects of rapamycin or Mito-TEMPO on Cd-induced ROS in the cells (Fig. 7C and D). Collectively, the findings support the notion that rapamycin plays a crucial role in blocking Cd-induced mitochondrial ROS activation of JNK and Erk1/2 pathways, which, in turn, also positively mediates Cd-induced mitochondrial ROS.
3.5. Rapamycin ameliorates Cd-induced neuronal apoptosis by preventing mitochondrial ROS inactivation of PP2A, thereby repressing activation of JNK and Erk1/2 pathways
It has been reported that PP2A negatively regulate Erk1/2, JNK and/or p38, involved in stress response and the regulation of cell proliferation, survival and apoptosis (Chen et al., 2008a; Han et al., 2012; Junttila et al., 2008). Our previous research has demonstrated that Cd activates Erk1/2 and JNK pathways leading to apoptosis by inhibition of PP2A in neuronal cells (Chen et al., 2008a), and the current study has found that rapamycin inhibited Cd-induced mitochondrial ROS-dependent activation of Erk1/2 and JNK pathways, preventing neuronal apoptosis (Fig. 4–7). Therefore, we postulated that rapamycin might block Cd activation of JNK, Erk1/2 and p38 pathways by preventing Cd-induced mitochondrial ROS from inhibiting PP2A. To this end, PC12 cells and primary neurons were pretreated with/without rapamycin (0.2 μg/ml) for 48 h, and then treated with/without Mito-TEMPO (10 μM) for 1 h, followed by exposure to Cd (20 μM) for 4 h. As shown in Fig. 8A and B, rapamycin, Mito-TEMPO and/or Cd did not apparently alter cellular protein levels of PP2Ac. However, rapamycin or Mito-TEMPO suppressed the basal or Cd-increased expression of demethylated- and phospho-PP2A (Tyr307) (Fig. 8A and B), two events that are related to decreased activity of PP2A (Janssens and Goris, 2001). Especially, co-treatment with rapamycin/Mito-TEMPO exhibited a stronger inhibitory effect on Cd-induced demethylated- and phospho-PP2A in the cells (Fig. 8A and B), indicating rapamycin’s activation of PP2A by preventing Cd-induced mitochondrial ROS. This is further supported by the finding that co-treatment with rapamycin/Mito-TEMPO prevented Cd from inhibiting PP2A activity more effectively than treatment with rapamycin or Mito-TEMPO alone in PC12 cells and primary neurons (Fig. 8C), as determined by the in vitro Ser/Thr phosphatase assay.
Fig. 8.

Rapamycin ameliorates Cd-induced neuronal apoptosis by preventing mitochondrial ROS inactivation of PP2A, thereby suppressing activation of JNK and Erk1/2 pathways. PC12 and primary neurons, or PC12 cells infected with Ad-PP2A or Ad-GFP/Ad-LacZ (as control), respectively, were pretreated with/without rapamycin (0.2 μg/ml) for 48 h and then with/without Mito-TEMPO (10 μM) for 1 h, followed by exposure to Cd (20 μM) for 4 h (for Western blotting) or 24 h (for PP2A phosphatase assay, live cell analysis, DAPI staining, ROS imaging). A and E) Cell lysates were subjected to Western blot analysis using indicated antibodies. The blots were probed for β-tubulin as a loading control. B and F) Similar results were observed in at least three independent experiments, and blots for p-Erk1/2, p-JNK, p-c-Jun, p-p38, and cleaved-caspase-3 were semi-quantified. C and D) PP2A in cell lysates was immunoprecipitated with antibodies to PP2Ac plus protein A/G agarose beads, followed by in vitro phosphatase assay using Ser/Thr Phosphatase Assay Kit 1 (Millipore). G) Live cells were detected by counting viable cells using trypan blue exclusion. I) The percentages of apoptotic cells with fragmented nuclei were quantified by DAPI staining. H) ROS imaging using an oxidant-sensitive probe CM-H2DCFDA was quantified. Results are presented as mean ± SEM (n = 3–5). a p < 0.05, difference with control group; b p < 0.05, difference with 20 μM Cd group; c p < 0.05, difference with Cd/Mito-TEMPO group or Cd/Rapamycin group; d p < 0.05, Ad-PP2A group versus Ad-GFP group or Ad-LacZ group.
To substantiate the roles of PP2A in rapamycin inhibition of Cd-induced mitochondrial ROS-dependent activation of MAPKs and neuronal cell apoptosis, PC12 cells, infected respectively with Ad-PP2A and Ad-GFP/Ad-LacZ (as control), were pretreated with/without rapamycin (0.2 μg/ml) for 48 h or Mito-TEMPO (10 μM) for 1 h, and then exposed to Cd (20 μM) for 4 h or 24 h. As demonstrated in Fig. 8D, over-expression of PP2A, but not GFP, greatly elevated the phosphatase activity of PP2A in the cells, and rendered significant resistance to Cd-reduced PP2A activity. Over-expression of PP2A further reinforced rapamycin’s or Mito-TEMPO’s prevention of Cd inactivation of PP2A (Fig. 8D). By Western blot analysis, we observed that over-expression of FLAG-tagged PP2A partially prevented Cd-induced activation of Erk1/2, JNK/c-Jun and caspase-3, but not p38, and potentiated the inhibitory effect of rapamycin or Mito-TEMPO on the events (Fig. 8E and F). Using trypan blue exclusion and DAPI staining, we found that over-expression of PP2A alone partially prevented Cd-induced live cell reduction (Fig. 8G) and apoptosis (Fig. 8I) in PC12 cells. However, addition of rapamycin or Mito-TEMPO rendered more significant resistance to Cd-induced cell death in Ad-PP2A-infected group than in Ad-GFP-infected group (Fig. 8G and I). Furthermore, interestingly, we also noted that over-expression of PP2A did not obviously alter the intracellular ROS levels, but substantially attenuated Cd-induced ROS, and especially enhanced the inhibitory effect of rapamycin or Mito-TEMPO on Cd-induced ROS in PC12 cells (Fig. 8H), as determined by imaging intracellular ROS using CM-H2DCFDA. Taken together, our data strongly suggest that rapamycin ameliorates Cd-induced neuronal apoptosis by preventing mitochondrial ROS inactivation of PP2A, thus blocking activation of JNK and Erk1/2 cascades.
4. Discussion
Cadmium, a toxic environmental and industrial pollutant, is known to easily penetrate the blood-brain barrier and accumulate in the brain, thus evoking ROS overproduction and consequent neuronal cell death of CNS (Bertin and Averbeck, 2006; Figueiredo-Pereira et al., 1998; Genovese and Cuzzocrea, 2008; Wang and Du, 2013). Especially, Cd-induced ROS in human neurodegenerative diseases, such as PD, AD and HD, has received more attentions as an etiological factor (Goncalves et al., 2010; Monroe and Halvorsen, 2006; Panayi et al., 2002; Wang and Du, 2013; Watjen and Beyersmann, 2004). It is of great importance to find effective interventions for prevention and treatment of Cd-induced oxidative damage and disorders of CNS. Rapamycin is not only a lipophilic macrolide antibiotic but also a specific mTOR inhibitor (Zhou et al., 2010). A series of recent studies have shown that rapamycin is very effective in both treatment of various neurological diseases and extension of lifespan, as a therapeutic or neurotrophic compound (Bove et al., 2011; Ehninger et al., 2014; Erlich et al., 2007; Harrison et al., 2009; Malagelada et al., 2010; Pan et al., 2008; Pan et al., 2009; Selman et al., 2009). Rapamycin can directly alter molecular abnormalities in neuronal cells related to calcium signaling and mTOR signaling pathways (Chen et al., 2011b; Ruan et al., 2008; Xu et al., 2015). Our recent studies have clarified that inhibition of mTOR signaling by rapamycin rescues neuronal cells from Cd-poisoning (Chen et al., 2008b; Xu et al., 2015). We have also identified that treatment with rapamycin in vitro and in vivo attenuates Cd-induced ROS activation of mTOR contributing to neuronal apoptosis (Chen et al., 2011a; Chen et al., 2014b). However, it is not clear whether and how rapamycin protects against Cd-induced neuronal apoptosis by preventing ROS from activation of MAPK pathways. Here we provide evidence that rapamycin blocked Cd-induced activation of JNK, Erk1/2 and p38 pathways by suppressing induction of mitochondrial ROS in neuronal cells. Further, we found that rapamycin ameliorates Cd-induced neuronal apoptosis by preventing mitochondrial ROS inactivation of PP2A, thereby suppressing activation of JNK and Erk1/2 pathways.
Mitochondria play a crucial role in cellular redox homeostasis and apoptosis induction (Cheng et al., 2012; Koopman et al., 2010). The mitochondrion is not only the energy factory but also the main generator of ROS for cells. Under pathological conditions, excessive ROS in the mitochondria are formed and reduce mitochondrial biogenesis (Lu et al., 2012; Seo et al., 2010), which, in turn, further stimulates ROS generation and reduces mitochondrial respiration (Woo and Shadel, 2011). The dysfunctional mitochondria commit the cell to undergo apoptosis by producing excessive ROS (Kim et al., 2008). It has been reported that Cd induces ROS production in the mitochondria of anterior pituitary cells, cortical neurons and brain (Lopez et al., 2006; Poliandri et al., 2006; Wang et al., 2004). Therefore, in this study we firstly focused on investigating Cd-induced mitochondrial ROS-dependent apoptosis and rapamycin’s effects on the event in neuronal cells. Using TTFA, antimycin A, or Mito-TEMPO, we unveiled that rapamycin targeted inhibition of Cd-induced mitochondrial ROS production, thereby relieving robust cleavages of caspase-3 and PARP, as well as cell apoptosis in PC12 cells and primary neurons (Fig. 3), as detected by ROS imaging, DAPI staining and Western blot analysis. Based on the entire data from our observations and other literatures, to our knowledge, this is the first report showing that rapamycin prevents mitochondrial ROS-dependent apoptosis in neuronal cells induced by Cd. However, further study will be needed to delineate underlying signaling mechanisms of rapamycin’s inhibition of Cd-induced mitochondria ROS-dependent apoptosis.
MAPK pathways are central components of the intracellular signaling networks that control many cellular events, including cell proliferation/growth, differentiation, and apoptosis (Kim et al., 2008; Kyriakis and Avruch, 2012). Numerous studies have described that MAPKs are involved in molecular mechanisms for the action of Cd, showing that Cd activates Erk1/2, JNK and/or p38 MAPK, causing apoptosis in various types of cells, such as macrophages (Misra et al. 2002), renal cells (mesangial or glomerular) (Hirano et al. 2005), tumor cell lineages (Lee et al. 2005), and neuronal cells (Kim et al., 2005; Rockwell et al., 2004). This discrepancy might be due to differences in cell type. In our previous studies, we have observed that all the three MAPK members can be activated by Cd, but only Erk1/2 and JNK participate in Cd-induced apoptosis in neuronal cells (Chen et al., 2008b). We have also revealed that Cd induction of ROS activates Erk1/2 and JNK pathways leading to apoptosis in neuronal cells (Chen et al., 2008a). However, of note, some data have documented that rapamycin affects the activity of JNK, Erk1/2 and p38 under different conditions (Benoit et al., 2011; Hahn et al., 2005; Huang et al., 2003; Kato et al., 2013; Kawasaki et al., 2010; Shi et al., 2005). This prompted us to ask whether and how rapamycin blocks Cd-induced mitochondrial ROS activation of JNK, Erk1/2 and p38 pathways, involved in rapamycin protection against Cd neurotoxicity. In the present study, we observed that rapamycin obviously inhibited Cd-induced phosphorylation of JNK, Erk1/2 and p38, including protein expression and phosphorylation of c-Jun, the substrate of JNK, in PC12 cells and primary neurons. Intriguingly, rapamycin’s inhibition of Cd-activated MAPKs was strengthened by co-treatment with Mito-TEMPO, implying that rapamycin prevents Cd-induced a pernicious interaction between mitochondrial ROS and the MAPK pathways in neuronal cells.
In this study, using pharmacological JNK inhibitor SP600125, Erk1/2 inhibitor U0126 and p38 inhibitor PD169136, we found that SP600125 or U0126 (Fig. 5), but not PD169136 (data not shown), potentiated rapamycin’s prevention of Cd-induced cell viability reduction and apoptosis in PC12 cells and primary neurons, as determined by live and apoptotic cells using trypan blue exclusive and DAPI staining. To corroborate the above findings, we extended our studies using genetic manipulation of c-Jun, MKK1, Erk1/2 or p38 activity, showing that ectopic expression of dominant negative c-Jun or MKK1 (Fig. 6), silencing Erk1/2 and c-Jun, but not p38 (data not shown), also potently enhanced the inhibitory effect of rapamycin on Cd neurotoxicity. It is worth mentioning that p38 inhibitor PD169136 and silencing p38 had no effect on rapamycin prevention of Cd-induced apoptosis in neuronal cells, indicating that rapamycin exerts its rescue against neuronal apoptosis independently of p38, although rapamycin also suppressed Cd-evoked activation of p38 pathway. Furthermore, we noticed that pretreatment with SP600125 or U0126, but not with PD169136, or over-expression of dominant negative c-Jun or MKK1 enhanced the inhibitory effects of rapamycin or Mito-TEMPO on Cd-induced ROS (Fig. 7A–D), suggesting that activated JNK or Erk1/2 may feedback positively mediate Cd-induced mitochondrial ROS. Taken together, our data support the notion that rapamycin plays an important role in blocking Cd-induced mitochondrial ROS, which prevents activation of JNK and Erk1/2 pathways, thereby rescuing against neuronal apoptosis. However, it remains to be determined whether rapamycin controls ROS homeostasis by intervening ROS-generating and -eliminating systems in neuronal cells in response to Cd. Understanding the underlying mechanisms may be helpful to uncover why pharmacological inhibition of JNK or Erk1/2, or expression of dominant negative c-Jun or MKK1 strengthens rapamycin’s inhibition of Cd-induced mitochondrial ROS in neuronal cells.
It is well known that PP2A negatively regulates Erk1/2, JNK and/or p38, in response to stress response (Chen et al., 2008a; Han et al., 2012; Junttila et al., 2008). Our previous studies have pinpointed that Cd activates Erk1/2 and JNK pathways leading to apoptosis by inducing ROS inhibition of PP2A in neuronal cells (Chen et al., 2008a), and the present study has unveiled that rapamycin blocked Cd induction of mitochondrial ROS, thus suppressing activation of Erk1/2 and JNK pathways and neuronal apoptosis (Fig. 4–7). In addition, as PP2A has been linked to rapamycin’s sensitivity (Liu et al., 2010), this led us to investigate whether rapamycin blocks Cd activation of JNK, Erk1/2 and/or p38 pathways by preventing Cd-induced mitochondrial ROS from inactivation of PP2A. In this study, we did not observe that rapamycin and/or Mito-TEMPO altered cellular protein expression of the catalytic subunit (PP2Ac) (Fig. 8A and B). However, we found that Cd-induced robust expression of demethylated-PP2Ac and phospho-PP2Ac (Tyr307) was substantially diminished by rapamycin and/or Mito-TEMPO in PC12 cells and primary neurons, and a stronger inhibitory effect on Cd-induced events in the cells co-treated with rapamycin/Mito-TEMPO was observed (Fig. 8A and B). This is further evidenced by the in vitro Ser/Thr phosphatase assay (Fig. 8C). These data indicate that rapamycin activates the phosphatase activity of PP2A, at least in part, by preventing Cd-induced mitochondrial ROS-mediated elevation of demethylation and phosphorylation of PP2Ac, two events responsible for PP2A inactivation (Janssens and Goris, 2001). To corroborate the above findings, genetic recue studies for PP2A were carried out. We found that over-expression of wild-type PP2A significantly strengthened rapamycin or Mito-TEMPO suppression of activated JNK and Erk1/2, but not p38, as well as ROS production and apoptosis in neuronal cells in response to Cd (Fig. 8D–H). Collectively, these results reveal that inhibition of ROS by rapamycin is required for rapamycin’s prevention of inactivation of PP2A and activation of Erk1/2 and JNK in neuronal cells triggered by Cd. The findings enhance our understanding of rapamycin’s preventive effect on Cd-induced oxidative damage or apoptosis in neuronal cells.
In conclusion, we have identified that rapamycin inhibited Cd-induced mitochondrial ROS-dependent neuronal apoptosis. Mechanistically, rapamycin prevented Cd-evoked mitochondrial ROS inactivation of PP2A, thus suppressing activation of JNK and Erk1/2 pathways, and rescuing neuronal apoptosis. Our findings underline that rapamycin may be exploited for the prevention of Cd-induced oxidative stress and consequent neurodegenerative disorders.
Highlights.
Rapamycin attenuates Cd-induced mitochondrial ROS-dependent neuronal apoptosis.
Rapamycin ameliorates Cd neurotoxicity by preventing mitochondrial ROS-mediated PP2A-JNK/Erk1/2 pathways.
Rapamycin can prevent Cd-induced oxidative stress and neurodegeneration.
Acknowledgments
This work was supported in part by the grants from National Natural Science Fundation of China (No. 30971486, 81271416; L.C.), NIH (CA115414; S.H.), the Scientific Research Foundation of the State Education Ministry of China (SEMR20091341, L.C.), the Project for the Priority Academic Program Development and the Natural Science Foundation of Jiangsu Higher Education Institutions of China (10KJA180027; L. C.), American Cancer Society (RSG-08-135-01-CNE; S.H.), Louisiana Board of Regents (NSF-2009-PFUND-144; S.H.), and Innovative Research Program of Jiangsu College Graduate of China (KYLX15_0733; C.X.).
Abbreviations
- AD
Alzheimer disease
- Cd
cadmium
- CM-H2DCFDA
5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate
- DAPI
4′, 6-diamidino-2-phenylindole
- DMEM
Dulbecco’s Modified Eagle’s Medium
- Erk1/2
extracellular signal-regulated kinase 1/2
- FBS
fetal bovine serum
- HD
Huntington’s disease
- JNK
c-Jun N-terminal kinase
- MAPK
mitogen-activated protein kinase
- MKK
mitogen-activated protein kinase kinase
- mTOR
mammalian target of rapamycin
- PBS
phosphate buffered saline
- PD
Parkinson disease
- PDL
poly-D-lysine
- PI
propidium Iodide
- PP2A
protein phosphatases 2A
- ROS
reactive oxygen species
- Ser/Thr
serine/threonine
- TTFA
thenoyltrifluoroacetone
- Tyr
tyrosine
Footnotes
Conflict of interest
The authors declare that they have no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akesson A, Bjellerup P, Lundh T, Lidfeldt J, Nerbrand C, Samsioe G, Skerfving S, Vahter M. Cadmium-induced effects on bone in a population-based study of women. Environ Health Perspect. 2006;114(6):830–834. doi: 10.1289/ehp.8763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoit M, Dormond-Meuwly A, Demartines N, Dormond O. Targeting the JNK signaling pathway potentiates the antiproliferative efficacy of rapamycin in LS174T colon cancer cells. J Surg Res. 2011;167(2):e193–198. doi: 10.1016/j.jss.2011.01.015. [DOI] [PubMed] [Google Scholar]
- Bertin G, Averbeck D. Cadmium: cellular effects, modifications of biomolecules, modulation of DNA repair and genotoxic consequences (a review) Biochimie. 2006;88(11):1549–1559. doi: 10.1016/j.biochi.2006.10.001. [DOI] [PubMed] [Google Scholar]
- Bove J, Martinez-Vicente M, Vila M. Fighting neurodegeneration with rapamycin: mechanistic insights. Nat Rev Neurosci. 2011;12(8):437–452. doi: 10.1038/nrn3068. [DOI] [PubMed] [Google Scholar]
- Chen L, Liu L, Huang S. Cadmium activates the mitogen-activated protein kinase (MAPK) pathway via induction of reactive oxygen species and inhibition of protein phosphatases 2A and 5. Free Radic Biol Med. 2008a;45(7):1035–1044. doi: 10.1016/j.freeradbiomed.2008.07.011. [DOI] [PubMed] [Google Scholar]
- Chen L, Liu L, Luo Y, Huang S. MAPK and mTOR pathways are involved in cadmium-induced neuronal apoptosis. J Neurochem. 2008b;105(1):251–261. doi: 10.1111/j.1471-4159.2007.05133.x. [DOI] [PubMed] [Google Scholar]
- Chen L, Liu L, Yin J, Luo Y, Huang S. Hydrogen peroxide-induced neuronal apoptosis is associated with inhibition of protein phosphatase 2A and 5, leading to activation of MAPK pathway. Int J Biochem Cell Biol. 2009;41(6):1284–1295. doi: 10.1016/j.biocel.2008.10.029. [DOI] [PubMed] [Google Scholar]
- Chen L, Xu B, Liu L, Luo Y, Yin J, Zhou H, Chen W, Shen T, Han X, Huang S. Hydrogen peroxide inhibits mTOR signaling by activation of AMPKalpha leading to apoptosis of neuronal cells. Lab Invest. 2010;90(5):762–773. doi: 10.1038/labinvest.2010.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Xu B, Liu L, Luo Y, Zhou H, Chen W, Shen T, Han X, Kontos CD, Huang S. Cadmium induction of reactive oxygen species activates the mTOR pathway, leading to neuronal cell death. Free Radic Biol Med. 2011a;50(5):624–632. doi: 10.1016/j.freeradbiomed.2010.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Gu C, Xu C, Zhang J, Xu Y, Ren Q, Guo M, Huang S, Chen L. Celastrol prevents cadmium-induced neuronal cell death via targeting JNK and PTEN-Akt/mTOR network. J Neurochem. 2014a;128(2):256–266. doi: 10.1111/jnc.12474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Ren Q, Zhang J, Ye Y, Zhang Z, Xu Y, Guo M, Ji H, Xu C, Gu C, Gao W, Huang S, Chen L. N-acetyl-L-cysteine protects against cadmium-induced neuronal apoptosis by inhibiting ROS-dependent activation of Akt/mTOR pathway in mouse brain. Neuropathol Appl Neurobiol. 2014b;40(6):759–777. doi: 10.1111/nan.12103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Xu Y, Xu B, Guo M, Zhang Z, Liu L, Ma H, Chen Z, Luo Y, Huang S, Chen L. CaMKII is involved in cadmium activation of MAPK and mTOR pathways leading to neuronal cell death. J Neurochem. 2011b;119(5):1108–1118. doi: 10.1111/j.1471-4159.2011.07493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng G, Kong RH, Zhang LM, Zhang JN. Mitochondria in traumatic brain injury and mitochondrial-targeted multipotential therapeutic strategies. Br J Pharmacol. 2012;167(4):699–719. doi: 10.1111/j.1476-5381.2012.02025.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehninger D, Neff F, Xie K. Longevity, aging and rapamycin. Cell Mol Life Sci. 2014;71(22):4325–4346. doi: 10.1007/s00018-014-1677-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlich S, Alexandrovich A, Shohami E, Pinkas-Kramarski R. Rapamycin is a neuroprotective treatment for traumatic brain injury. Neurobiol Dis. 2007;26(1):86–93. doi: 10.1016/j.nbd.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Figueiredo-Pereira ME, Yakushin S, Cohen G. Disruption of the Intracellular Sulfhydryl Homeostasis by Cadmium-induced Oxidative Stress Leads to Protein Thiolation and Ubiquitination in Neuronal Cells. J Biol Chem. 1998;273(21):12703–12709. doi: 10.1074/jbc.273.21.12703. [DOI] [PubMed] [Google Scholar]
- Franklin CC, Kraft AS. Conditional expression of the mitogen-activated protein kinase (MAPK) phosphatase MKP-1 preferentially inhibits p38 MAPK and stress-activated protein kinase in U937 cells. J Biol Chem. 1997;272(27):16917–16923. doi: 10.1074/jbc.272.27.16917. [DOI] [PubMed] [Google Scholar]
- Genovese T, Cuzzocrea S. Role of free radicals and poly(ADP-ribose)polymerase-1 in the development of spinal cord injury: new potential therapeutic targets. Curr Med Chem. 2008;15(5):477–487. doi: 10.2174/092986708783503177. [DOI] [PubMed] [Google Scholar]
- Goncalves JF, Fiorenza AM, Spanevello RM, Mazzanti CM, Bochi GV, Antes FG, Stefanello N, Rubin MA, Dressler VL, Morsch VM, Schetinger MR. N-acetylcysteine prevents memory deficits, the decrease in acetylcholinesterase activity and oxidative stress in rats exposed to cadmium. Chem Biol Interact. 2010;186(1):53–60. doi: 10.1016/j.cbi.2010.04.011. [DOI] [PubMed] [Google Scholar]
- Hahn M, Li W, Yu C, Rahmani M, Dent P, Grant S. Rapamycin and UCN-01 synergistically induce apoptosis in human leukemia cells through a process that is regulated by the Raf-1/MEK/ERK, Akt, and JNK signal transduction pathways. Mol Cancer Ther. 2005;4(3):457–470. doi: 10.1158/1535-7163.MCT-04-0137. [DOI] [PubMed] [Google Scholar]
- Han X, Xu B, Beevers CS, Odaka Y, Chen L, Liu L, Luo Y, Zhou H, Chen W, Shen T, Huang S. Curcumin inhibits protein phosphatases 2A and 5, leading to activation of mitogen-activated protein kinases and death in tumor cells. Carcinogenesis. 2012;33(4):868–875. doi: 10.1093/carcin/bgs029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao B, Cheng S, Clancy CJ, Nguyen MH. Caspofungin kills Candida albicans by causing both cellular apoptosis and necrosis. Antimicrob Agents Chemother. 2013;57(1):326–332. doi: 10.1128/AAC.01366-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460(7253):392–395. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, Shu L, Dilling MB, Easton J, Harwood FC, Ichijo H, Houghton PJ. Sustained activation of the JNK cascade and rapamycin-induced apoptosis are suppressed by p53/p21(Cip1) Mol Cell. 2003;11(6):1491–1501. doi: 10.1016/s1097-2765(03)00180-1. [DOI] [PubMed] [Google Scholar]
- Huang S, Shu L, Easton J, Harwood FC, Germain GS, Ichijo H, Houghton PJ. Inhibition of mammalian target of rapamycin activates apoptosis signal-regulating kinase 1 signaling by suppressing protein phosphatase 5 activity. J Biol Chem. 2004;279(35):36490–36496. doi: 10.1074/jbc.M401208200. [DOI] [PubMed] [Google Scholar]
- Janssens V, Goris J. Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem J. 2001;3539(3):417–439. doi: 10.1042/0264-6021:3530417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarup L, Persson B, Edling C, Elinder CG. Renal function impairment in workers previously exposed to cadmium. Nephron. 1993;64(1):75–81. doi: 10.1159/000187282. [DOI] [PubMed] [Google Scholar]
- Jiang G, Xu L, Song S, Zhu C, Wu Q, Zhang L, Wu L. Effects of long-term low-dose cadmium exposure on genomic DNA methylation in human embryo lung fibroblast cells. Toxicology. 2008;244(1):49–55. doi: 10.1016/j.tox.2007.10.028. [DOI] [PubMed] [Google Scholar]
- Johri N, Jacquillet G, Unwin R. Heavy metal poisoning: the effects of cadmium on the kidney. Biometals. 2010;23(5):783–792. doi: 10.1007/s10534-010-9328-y. [DOI] [PubMed] [Google Scholar]
- Jomova K, Valko M. Advances in metal-induced oxidative stress and human disease. Toxicology. 2011;283(2–3):65–87. doi: 10.1016/j.tox.2011.03.001. [DOI] [PubMed] [Google Scholar]
- Junttila MR, Li SP, Westermarck J. Phosphatase-mediated crosstalk between MAPK signaling pathways in the regulation of cell survival. FASEB J. 2008;22(4):954–965. doi: 10.1096/fj.06-7859rev. [DOI] [PubMed] [Google Scholar]
- Kato H, Katoh R, Kitamura M. Dual regulation of cadmium-induced apoptosis by mTORC1 through selective induction of IRE1 branches in unfolded protein response. PLoS One. 2013;8(5):e64344. doi: 10.1371/journal.pone.0064344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki Y, Harashima S, Sasaki M, Mukai E, Nakamura Y, Harada N, Toyoda K, Hamasaki A, Yamane S, Yamada C, Yamada Y, Seino Y, Inagaki N. Exendin-4 protects pancreatic beta cells from the cytotoxic effect of rapamycin by inhibiting JNK and p38 phosphorylation. Horm Metab Res. 2010;42(5):311–317. doi: 10.1055/s-0030-1249035. [DOI] [PubMed] [Google Scholar]
- Kim SD, Moon CK, Eun SY, Ryu PD, Jo SA. Identification of ASK1, MKK4, JNK, c-Jun, and caspase-3 as a signaling cascade involved in cadmium-induced neuronal cell apoptosis. Biochem Biophys Res Commun. 2005;328(1):326–334. doi: 10.1016/j.bbrc.2004.11.173. [DOI] [PubMed] [Google Scholar]
- Kim SJ, Jeong HJ, Myung NY, Kim MC, Lee JH, So HS, Park RK, Kim HM, Um JY, Hong SH. The protective mechanism of antioxidants in cadmium-induced ototoxicity in vitro and in vivo. Environ Health Perspect. 2008;116(7):854–862. doi: 10.1289/ehp.10467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocak M, Akcil E. The effects of chronic cadmium toxicity on the hemostatic system. Pathophysiol Haemost Thromb. 2006;35(6):411–416. doi: 10.1159/000102047. [DOI] [PubMed] [Google Scholar]
- Koopman WJ, Nijtmans LG, Dieteren CE, Roestenberg P, Valsecchi F, Smeitink JA, Willems PH. Mammalian mitochondrial complex I: biogenesis, regulation, and reactive oxygen species generation. Antioxid Redox Signal. 2010;12(12):1431–1470. doi: 10.1089/ars.2009.2743. [DOI] [PubMed] [Google Scholar]
- Kyriakis JM, Avruch J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: a 10-year update. Physiol Rev. 2012;92(2):689–737. doi: 10.1152/physrev.00028.2011. [DOI] [PubMed] [Google Scholar]
- Lanju X, Jing X, Shichang L, Zhuo Y. Induction of apoptosis by antimycin A in differentiated PC12 cell line. J Appl Toxicol. 2014;34(6):651–657. doi: 10.1002/jat.2890. [DOI] [PubMed] [Google Scholar]
- Liu L, Chen L, Luo Y, Chen W, Zhou H, Xu B, Han X, Shen T, Huang S. Rapamycin inhibits IGF-1 stimulated cell motility through PP2A pathway. PLoS One. 2010;5(5):e10578. doi: 10.1371/journal.pone.0010578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez E, Arce C, Oset-Gasque MJ, Canadas S, Gonzalez MP. Cadmium induces reactive oxygen species generation and lipid peroxidation in cortical neurons in culture. Free Radic Biol Med. 2006;40(6):940–951. doi: 10.1016/j.freeradbiomed.2005.10.062. [DOI] [PubMed] [Google Scholar]
- Lopez E, Figueroa S, Oset-Gasque MJ, Gonzalez MP. Apoptosis and necrosis: two distinct events induced by cadmium in cortical neurons in culture. Br J Pharmacol. 2003;138(5):901–911. doi: 10.1038/sj.bjp.0705111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Wu DM, Zheng YL, Hu B, Cheng W, Zhang ZF. Purple sweet potato color attenuates domoic acid-induced cognitive deficits by promoting estrogen receptor-alpha-mediated mitochondrial biogenesis signaling in mice. Free Radic Biol Med. 2012;52:646–659. doi: 10.1016/j.freeradbiomed.2011.11.016. [DOI] [PubMed] [Google Scholar]
- Malagelada C, Jin ZH, Jackson-Lewis V, Przedborski S, Greene LA. Rapamycin protects against neuron death in in vitro and in vivo models of Parkinson’s disease. J Neurosci. 2010;30(3):1166–1175. doi: 10.1523/JNEUROSCI.3944-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mates JM, Segura JA, Alonso FJ, Marquez J. Roles of dioxins and heavy metals in cancer and neurological diseases using ROS-mediated mechanisms. Free Radic Biol Med. 2010;49(9):1328–1341. doi: 10.1016/j.freeradbiomed.2010.07.028. [DOI] [PubMed] [Google Scholar]
- Mendez-Armenta M, Rios C. Cadmium neurotoxicity. Environ Toxicol Pharmacol. 2007;23(3):350–358. doi: 10.1016/j.etap.2006.11.009. [DOI] [PubMed] [Google Scholar]
- Monroe RK, Halvorsen SW. Cadmium blocks receptor-mediated Jak/STAT signaling in neurons by oxidative stress. Free Radic Biol Med. 2006;41(3):493–502. doi: 10.1016/j.freeradbiomed.2006.04.023. [DOI] [PubMed] [Google Scholar]
- Moreno-Sanchez R, Hernandez-Esquivel L, Rivero-Segura NA, Marin-Hernandez A, Neuzil J, Ralph SJ, Rodriguez-Enriquez S. Reactive oxygen species are generated by the respiratory complex II--evidence for lack of contribution of the reverse electron flow in complex I. FEBS J. 2013;280(3):927–938. doi: 10.1111/febs.12086. [DOI] [PubMed] [Google Scholar]
- Okuda B, Iwamoto Y, Tachibana H, Sugita M. Parkinsonism after acute cadmium poisoning. Clin Neurol Neurosurg. 1997;99(4):263–265. doi: 10.1016/s0303-8467(97)00090-5. [DOI] [PubMed] [Google Scholar]
- Pan T, Kondo S, Zhu W, Xie W, Jankovic J, Le W. Neuroprotection of rapamycin in lactacystin-induced neurodegeneration via autophagy enhancement. Neurobiol Dis. 2008;32(1):16–25. doi: 10.1016/j.nbd.2008.06.003. [DOI] [PubMed] [Google Scholar]
- Pan T, Rawal P, Wu Y, Xie W, Jankovic J, Le W. Rapamycin protects against rotenone-induced apoptosis through autophagy induction. Neuroscience. 2009;164(2):541–551. doi: 10.1016/j.neuroscience.2009.08.014. [DOI] [PubMed] [Google Scholar]
- Panayi AE, Spyrou NM, Iversen BS, White MA, Part P. Determination of cadmium and zinc in Alzheimer’s brain tissue using inductively coupled plasma mass spectrometry. J Neurol Sci. 2002;195(1):1–10. doi: 10.1016/s0022-510x(01)00672-4. [DOI] [PubMed] [Google Scholar]
- Pihl RO, Parkes M. Hair element content in learning disabled children. Science. 1977;198(4313):204–206. doi: 10.1126/science.905825. [DOI] [PubMed] [Google Scholar]
- Poliandri AH, Machiavelli LI, Quinteros AF, Cabilla JP, Duvilanski BH. Nitric oxide protects the mitochondria of anterior pituitary cells and prevents cadmium-induced cell death by reducing oxidative stress. Free Radic Biol Med. 2006;40(4):679–688. doi: 10.1016/j.freeradbiomed.2005.09.021. [DOI] [PubMed] [Google Scholar]
- Rockwell P, Martinez J, Papa L, Gomes E. Redox regulates COX-2 upregulation and cell death in the neuronal response to cadmium. Cell Signal. 2004;16(3):343–353. doi: 10.1016/j.cellsig.2003.08.006. [DOI] [PubMed] [Google Scholar]
- Ruan B, Pong K, Jow F, Bowlby M, Crozier RA, Liu D, Liang S, Chen Y, Mercado ML, Feng X, Bennett F, von Schack D, McDonald L, Zaleska MM, Wood A, Reinhart PH, Magolda RL, Skotnicki J, Pangalos MN, Koehn FE, Carter GT, Abou-Gharbia M, Graziani EI. Binding of rapamycin analogs to calcium channels and FKBP52 contributes to their neuroprotective activities. Proc Natl Acad Sci USA. 2008;105(1):33–38. doi: 10.1073/pnas.0710424105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selman C, Tullet JM, Wieser D, Irvine E, Lingard SJ, Choudhury AI, Claret M, Al-Qassab H, Carmignac D, Ramadani F, Woods A, Robinson IC, Schuster E, Batterham RL, Kozma SC, Thomas G, Carling D, Okkenhaug K, Thornton JM, Partridge L, Gems D, Withers DJ. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science. 2009;326(5949):140–144. doi: 10.1126/science.1177221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo AY, Joseph AM, Dutta D, Hwang JC, Aris JP, Leeuwenburgh C. New insights into the role of mitochondria in aging: mitochondrial dynamics and more. J Cell Sci. 2010;123(15):2533–2542. doi: 10.1242/jcs.070490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Sharma A, Wu H, Lichtenstein A, Gera J. Cyclin D1 and c-myc internal ribosome entry site (IRES)-dependent translation is regulated by AKT activity and enhanced by rapamycin through a p38 MAPK- and ERK-dependent pathway. J Biol Chem. 2005;280(12):10964–10973. doi: 10.1074/jbc.M407874200. [DOI] [PubMed] [Google Scholar]
- Thompson J, Bannigan J. Cadmium: toxic effects on the reproductive system and the embryo. Reprod Toxicol. 2008;25(3):304–315. doi: 10.1016/j.reprotox.2008.02.001. [DOI] [PubMed] [Google Scholar]
- Wang B, Du Y. Cadmium and its neurotoxic effects. Oxid Med Cell Longev. 2013;2013:898034. doi: 10.1155/2013/898034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Fang J, Leonard SS, Rao KM. Cadmium inhibits the electron transfer chain and induces reactive oxygen species. Free Radic Biol Med. 2004;36(11):1434–1443. doi: 10.1016/j.freeradbiomed.2004.03.010. [DOI] [PubMed] [Google Scholar]
- Watjen W, Beyersmann D. Cadmium-induced apoptosis in C6 glioma cells: influence of oxidative stress. Biometals. 2004;17(1):65–78. doi: 10.1023/a:1024405119018. [DOI] [PubMed] [Google Scholar]
- Woo DK, Shadel GS. Mitochondrial stress signals revise an old aging theory. Cell. 2011;144(1):11–12. doi: 10.1016/j.cell.2010.12.023. [DOI] [PubMed] [Google Scholar]
- Wright RO, Amarasiriwardena C, Woolf AD, Jim R, Bellinger DC. Neuropsychological correlates of hair arsenic, manganese, and cadmium levels in school-age children residing near a hazardous waste site. Neurotoxicology. 2006;27(2):210–216. doi: 10.1016/j.neuro.2005.10.001. [DOI] [PubMed] [Google Scholar]
- Xu C, Liu C, Liu L, Zhang R, Zhang H, Chen S, Luo Y, Chen L, Huang S. Rapamycin prevents cadmium-induced neuronal cell death via targeting both mTORC1 and mTORC2 pathways. Neuropharmacology. 2015;97:35–45. doi: 10.1016/j.neuropharm.2015.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh YT, Yeh H, Su SH, Lin JS, Lee KJ, Shyu HW, Chen ZF, Huang SY, Su SJ. Phenethyl isothiocyanate induces DNA damage-associated G2/M arrest and subsequent apoptosis in oral cancer cells with varying p53 mutations. Free Radic Biol Med. 2014;74:1–13. doi: 10.1016/j.freeradbiomed.2014.06.008. [DOI] [PubMed] [Google Scholar]
- Zhou H, Luo Y, Huang S. Updates of mTOR inhibitors. Anticancer Agents Med Chem. 2010;10(7):571–581. doi: 10.2174/187152010793498663. [DOI] [PMC free article] [PubMed] [Google Scholar]