

Abstract
Binge drinking induces several neurotoxic consequences including oxidative stress and neurodegeneration. Because of these effects, drugs which prevent ethanol-induced damage to the brain may be clinically beneficial.
In this study, we investigated the ethanol-mediated KLF11-MAO cell death cascade in the frontal cortex of Sprague–Dawley rats exposed to a modified Majchowicz 4-day binge ethanol model and control rats. Moreover, MAO inhibitors (MAOIs) were investigated for neuroprotective activity against binge ethanol.
Binge ethanol-treated rats demonstrated a significant increase in KLF11, both MAO isoforms, protein oxidation and caspase-3, as well as a reduction in BDNF expression in the frontal cortex compared to control rats. MAOIs prevented these binge ethanol-induced changes, suggesting a neuroprotective benefit. Neither binge ethanol nor MAOI treatment significantly affected protein expression levels of the oxidative stress enzymes, SOD2 or catalase. Furthermore, ethanol-induced antinociception was enhanced following exposure to the 4-day ethanol binge.
These results demonstrate that the KLF11-MAO pathway is activated by binge ethanol exposure and MAOIs are neuroprotective by preventing the binge ethanol-induced changes associated with this cell death cascade. This study supports KLF11-MAO as a mechanism of ethanol-induced neurotoxicity and cell death that could be targeted with MAOI drug therapy to alleviate alcohol-related brain injury. Further examination of MAOIs to reduce alcohol use disorder-related brain injury could provide pivotal insight to future pharmacotherapeutic opportunities.
Keywords: Binge ethanol, Rats, Kruppel-like factor 11 (KLF11), Transcription factor, Monoamine oxidase (MAO), Neurotoxicity, Protein oxidation, BDNF, Caspase-3
1. Introduction
Alcohol use disorders can result in devastating effects on the brain and are a major cause of global morbidity and mortality (Rehm et al., 2009). Alcohol misuse is associated with both volumetric reductions across several brain regions (Pfefferbaum et al., 1997; Agartz et al., 1999; Wrase et al., 2008; Durazzo et al., 2011; Beck et al., 2012) and loss of neuronal and glial cells (Miguel-Hidalgo et al., 2006; Duncan et al., 2015). Similar to chronic alcohol exposure, binge ethanol exposure also induces a number of neurotoxic consequences, such as edema, oxidative stress, excito-toxicity, neuroinflammation, and glial scarring which results in neurodegeneration (Crews and Nixon, 2009; Kelso et al., 2011; Collins and Neafsey, 2012). In 4-day binge models (Majchrowicz, 1975), binge drinking induces neurodegeneration even after a single binge exposure (Maynard and Leasure, 2013); however, brain damage increases with the duration of binge exposure (Collins et al., 1996; Crews et al., 2000; Obernier et al., 2002a; Hayes et al., 2013). Since reportedly 17% of Americans drink heavily (Sacks et al., 2013) and binge drinking is cited as a primary factor in predicting future abuse potential and associated neuropathology (Agartz et al., 1999; Obernier et al., 2002a), the identification of novel pharmacological targets which could curtail neurobiological damage from binge drinking is warranted.
Recently, we identified Krüppel-like factor 11 (KLF11) as an ethanol-responsive gene involved in ethanol-related brain neurodegeneration (Ou et al., 2011). KLF11 is a member of the Sp1/KLF zinc finger family of transcription factors, largely known for its role in inhibiting cell growth and inducing apoptosis (Cook et al., 1998; Fernandez-Zapico et al., 2003; Wang et al., 2007). Transforming growth factor-β1 (TGF-β1), which is induced by oxidative stress and ethanol (Chen et al., 2006), promotes the transcription of KLF11 which subsequently mediates TGF-β cell death (Cook et al., 1998; Wang et al., 2004; Buck et al., 2006). Moreover, KLF11 is a transcriptional activator of both isoforms of MAO (Ou et al., 2004; Grunewald et al., 2012), which contribute to the production of reactive oxygen species (ROS) through the catabolism of monoamines (Shih,1991; Shih et al., 1999; Duncan et al., 2012). KLF11 and MAO-B are upregulated by ethanol in vitro (Lu et al., 2008) and in chronic ethanol rodent models, as well as in the postmortem pre-frontal cortex of alcohol-dependent subjects (Ou et al., 2011, 2014; Udemgba et al., 2014; Duncan et al., 2015; Nair et al., 2015). However, it is unknown whether the KLF11-MAO pathway is also responsive to acute, high levels of ethanol exposure as seen with binge drinking.
High levels of reactive oxygen species (ROS) can damage mitochondrial DNA and induce apoptosis (Buttke and Sandstrom, 1994; Wei, 1998; Loh et al., 2006; Circu et al., 2009). Accumulation of ROS is also a critical mode of ethanol-induced cellular dysfunction (Ramachandran et al., 2003; Das and Vasudevan, 2007; Gonzalez et al., 2007; Boyadjieva and Sarkar, 2013). Oxidative stress is a devastating consequence of binge drinking and, thus, antioxidants provide substantial neuroprotection in models of binge ethanol exposure (Hamelink et al., 2005; Crews et al., 2006; Artun et al., 2010; Collins and Neafsey, 2012; Nair et al., 2015). Further, oxidative exposure of proteins due to ROS can modify their characteristics and function, such as enzymatic activity, binding of transcription factors, and increasing susceptibility to proteolytic degradation (Wolff and Dean, 1986; Davies, 1987; Davies et al., 1987). Interestingly, MAO may be a key cause of changes in levels of ROS associated with ethanol exposure. MAO-induced ROS induces DNA damage and subsequent neuronal apoptosis and neuropathology (Naoi et al., 2003; Mallajosyula et al., 2008). In fact, hydrogen peroxide alone, due to MAO catalytic activity, induces apoptosis (Naoi et al., 2003). Since MAO-induced ROS is cited as a critical source of cellular stress, drugs which inhibit its enzymatic activity may be useful therapeutics for preventing neurodegeneration.
The MAO-B inhibitors, selegiline and rasagiline, are approved by the FDA for the treatment of Parkinson’s disease and have been studied extensively in neurodegenerative rodent and cell models (Riederer et al., 2004; Youdim et al., 2014). M30, a dual, brain-selective MAOI, is currently being investigated in several neurodegenerative models related to Alzheimer’s and Huntington’s diseases (Youdim et al., 2014). Monoamine oxidase inhibitors have demonstrated an ability to reduce oxidative stress and increase neuroprotection because they inhibit amine oxidation by MAO and the subsequent formation of byproducts of hydrogen peroxide, aldehyde and ammonia (de la Cruz et al., 1996; Burke et al., 2004; Magyar and Szende, 2004; Youdim et al., 2006). In addition, N-propargylamine containing MAOIs, such as selegiline, rasagiline, and M30, have demonstrated other neuroprotective properties apart from MAO inhibition, such as increasing anti-apoptotic Bcl proteins, brain-derived and glial-derived neurotrophic factors (BDNF and GDNF), and oxidative stress scavengers, superoxide dismutase 2 (SOD2) and Catalase-1, while reducing apoptosis (Kitani et al., 1994; Carrillo et al., 2000; Youdim et al., 2003a; Avramovich-Tirosh et al., 2007; Sofic et al., 2015).
Previously, we reported that KLF11 was increased in the pre-frontal cortex (PFC) of rats and mice exposed to a chronic ethanol diet for 28 days (Ou et al., 2011, 2014), as well as in the postmortem PFC of AUD subjects (Udemgba et al., 2014). The PFC is an especially vulnerable region to the pejorative effects of ethanol exposure as several studies have highlighted anatomical and physiological aberrations in this region among chronic alcohol users (Moselhy et al., 2001; Paul et al., 2008; Beck et al., 2012). Moreover, the PFC is vastly interconnected to the limbic system and monoaminergic nuclei where insult to this region would result in widespread functional deficits in behavior and memory (Groenewegen et al., 1997; Hoover and Vertes, 2007). Therefore, in this study, we aimed to determine the response of the KLF11-MAO pathway in PFC of rats exposed to binge ethanol treatment and the efficacy of MAOIs in counteracting neurotoxicity associated with binge ethanol exposure. These data further support the KLF11/MAO pathway as a pharmacotherapeutic target with use of MAO inhibiting drugs to alleviate brain injury related to alcohol use disorder (AUD). As MAOIs are currently prescribed for several neurodegenerative diseases, there will be direct translational significance in determining their efficacy against alcohol-induced neurodegeneration.
2. Materials and methods
2.1. Animal studies
Adult male Sprague–Dawley rats (weighing 300–325 g; Charles-Rivers Laboratories, Wilmington, MA) were housed individually in a humidity- and temperature-regulated room with a 12:12-h light/dark cycle. Rats were given ad libitum access to food and water throughout the duration of ethanol feedings. The animal experiments adhered to the Ethical Guidelines on Animal Experimentation and were approved by the Institutional Animal Care and Use Committee at the University of Mississippi Medical Center.
2.2. Binge ethanol feeding and MAOI drug treatment
After acclimatization, 64 rats were randomly assigned to either the control or ethanol group. Each control or ethanol group was further subdivided into groups which received either no drug, an MAO-B inhibitor, selegiline (5 mg/kg/day) or rasagiline (5 mg/kg/ day), or an inhibitor of both MAO-A and MAO-B, M30 (5 mg/kg/ day). The doses of MAOIs administered have been well documented in studies examining the therapeutic potential for these drugs (cf. Gal et al., 2005; Saravanan et al., 2006; Eliash et al., 2009). Ethanol (Binge EtOH rats; 25% w/v) or isocalorically-substituted dextrose (Control rats) was formulated in a nutritionally complete diet (Vanilla Ensure Plus®) and administered by intragastric gavage. The gavage feeding was conducted using a stainless steel gavage secured to a syringe which was then passed through the esophagus and into the stomach to deliver the control or ethanol liquid diet. Rats were fed ethanol or control diets 3× daily (every 8 h at 7:00AM, 3:00PM, and 11:00PM) for 4 days, following a well-established 4-day binge ethanol model (Majchrowicz, 1975) which induces physical dependence on ethanol and neurodegeneration (Obernier et al., 2002a, 2002b; Crews and Braun, 2003; Kelso et al., 2011). The daily dose of ethanol (~9 g/kg/day) resulted in a mean blood alcohol content (BAC) of 95 mM, a level that is routinely found in models of binge ethanol exposure (Nixon and Crews, 2002; Obernier et al., 2002b) and among alcohol abusers who drink heavily (Majchrowicz, 1975; Henriksen et al., 1997). After the initial loading dose of 5 g/kg, administration of subsequent doses of ethanol was dependent on behavioral intoxication scores (Table 1). MAOIs were administered once daily at the second feeding of each day. Immediately following the last binge ethanol exposure, rats were sacrificed by decapitation and trunk blood was used to determine BAC using the EnzyChrom Ethanol Assay Kit according to the manufacturer’s instructions (BioAssay Systems, Hayward CA). Brains from rats were freshly dissected on ice, frozen with dry ice and stored at −80°C until further use.
Table 1. The Majchrowicz binge ethanol feeding schedule.
64 rats were randomly assigned to control (N = 32) or ethanol groups (N = 32). Rats received either ethanol (25% w/v; EtOH rats) or isocalorically-substituted dextrose (no EtOH; Control rats) in a nutritionally complete diet (Vanilla Ensure Plus®). Binge ethanol rats (N = 32) were fed ethanol by intragastric gavage every 8 h for 4 days. Binge ethanol rats received an initial 5 g/kg loading dose, with subsequent dosages contingent on a six-point intoxication scale as follows: Normal = 5 g/kg; slightly ataxic and hypoactive = 4 g/kg; ataxic with elevated abdomen and intact righting reflex = 3 g/kg; delayed righting reflex and lack of abdominal elevation = 2 g/kg; lack of righting reflex with intact eye blink reflex = 1 g/kg; and lastly, unresponsiveness, including loss of eye blink reflex = 0 g/kg.
| Level of intoxication | Amount of ethanol administered |
|---|---|
| 0 = Normal | 5 g/kg |
| 1 = Slightly ataxic and hypoactive | 4 g/kg |
| 2 = Ataxic with elevated abdomen and intact righting reflex | 3 g/kg |
| 3 = Delayed righting reflex and lack of abdominal elevation | 2 g/kg |
| 4 = Lack of righting reflex with intact eye blink reflex | 1 g/kg |
| 5 = Unresponsive including loss of eye blink reflex | 0 g/kg |
2.3. RNA extraction and quantitative real-time RT-PCR
Total RNA was extracted from the rat frontal cortex using TRIzol reagent (Invitrogen, Carlsbad, CA). SuperScript III first-strand synthesis system (Invitrogen) was used to perform reverse transcription according to the manufacturer’s instructions. 2 μl of cDNA from each sample was used for real-time quantitative PCR using SYBR supermix kit (Bio-Rad, Hercules, CA) and iCycler MyiQ real-time PCR detection system (Bio-Rad) as described previously (Ou et al., 2011; Grunewald et al., 2012). Gene specific primers for rats were designed as follows:
MAO-A, 5′-CGTGATCGGAGGTGGCATTTC-3′ (sense) and 5′-AAA-GGCGCCCCGAAAGG-3′ (antisense); MAO-B, 5′-GGGGGCGGCATCT-CAGGT-3′ (sense) and 5′-TCAGCCGCTCAACTTCATTCACTT-3′ (anti-sense); and KLF11, 5′-TCCTGCAGGGCCGTGATGAC-3′ (sense) and 5′-GGGGAACAGGCCACCAGACTTG-3′ (antisense).
The 18S Ribosomal RNA primer was used as an internal control to normalize gene expression (Ou et al., 2004; Grunewald et al., 2012). Threshold cycle (CT) values were quantified and relative gene expression was determined using the ΔCT method (Livak and Schmittgen, 2001).
2.4. Protein extraction and western blot analysis
Brain tissue containing the frontal cortex from each rat was homogenized in RIPA buffer supplemented with Halt protease inhibitor mix (Thermo Fisher Scientific, Waltham, MA). The resulting homogenate was briefly frozen, thawed, and then centrifuged at 4 °C (12,500 rpm) for 10 min in a refrigerated microcentrifuge to pellet and eliminate cell debris. Supernatant was transferred to a new tube and stored at −20 °C until further use. Protein concentrations of each sample were assessed using the Pierce™ BCA Protein Assay Kit (Thermo Scientific). Thirty micrograms of total protein were diluted with Laemmli sample buffer, boiled for 5 min at 95 °C and cooled on ice. Samples were loaded into their respective wells and separated on a 10.5% SDS-polyacrylamide gel by electrophoresis and transferred onto a PVDF membrane using the Trans-Blot Turbo Transfer System (Bio-Rad). Membranes were blocked at room temperature for 1 h with 5% nonfat dry milk and incubated with primary antibodies for 1 h at room temperature. The primary antibodies used in this study are as follows: mouse anti-MAO-A (1:500; sc-271123; Santa Cruz Biotechnology Inc., Dallas, TX), goat anti-MAO-B (1:500; sc-18401; Santa Cruz), mouse anti-TIEG2 (KLF11; sc-136101; Santa Cruz), rabbit anti-SOD2 (1:750; sc-30080; Santa Cruz), goat anti-Catalase (1:750; sc-34281; Santa Cruz), rabbit anti-BDNF (1:500; sc-546, Santa Cruz) or rabbit anti-Caspase-3 (1:500; sc-7148; Santa Cruz). Membranes were then washed with TBS-T (Tris Buffered Saline-Tween 20) and incubated with their respective HRP-conjugated secondary antibodies, goat anti-mouse IgG, goat anti-rabbit IgG, or donkey anti-goat (sc-2020) (all 1:2000; Santa Cruz). Bands were visualized by horseradish peroxidase reaction using SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific). After primary band visualization, membranes were stripped for 20 min at room temperature in Restore Western Blot Stripping Buffer (Thermo Scientific), washed, blocked, then re-probed with mouse anti-actin (1:10,000; Millipore, Billerica, MA) and goat anti-mouse IgG (Santa Cruz) as an internal control for sample loading. Protein bands were visualized by the ChemiDoc XRS+ Imaging System (BioRad) and optical density measurements of the autoradiographic bands were quantified and normalized to those of actin using Quantity One Plus analysis software (BioRad) (Ou et al., 2011).
2.5. Protein oxidation assay
Cell lysates from the frontal cortex of each rat were assessed for levels of oxidized protein by detection of carbonylation using the Oxyblot kit (Millipore). Samples containing 15 μg of total protein were subjected to 1× dinitrophenylhydrazine (DNPH) solution for 15 min to derivatize carbonyl groups to 2,4-dinitrophenylhydrazone (DNP-hydrazone). Neutralization solution was then added to samples to halt the DNP reaction. Duplicate samples serving as negative controls were subjected to derivatization-control solution instead of DNPH solution, which was followed by addition of neutralization solution. All samples and DNP-labeled molecular weight standard ladder were then loaded into a polyacrylamide gel for electrophoresis. Following electroblotting of proteins to a PVDF membrane, membranes were blocked for 1hr. Membranes were then incubated with rabbit anti-DNP (1:150) followed by secondary incubation with goat anti-rabbit IgG for 1 h at room temperature using antibodies supplied with Oxyblot kit. Bands were visualized by chemiluminescence using Supersignal West Pico Chemiluminescent Substrate (Thermo Scientific) and densitrometric measurements were quantified using Quantity One Plus analysis software (BioRad). The carbonyl band corresponding to the molecular weight of albumin was used for assessment of protein oxidation and normalized to non-derivatized actin, following the manufacturer’s instructions and as previously described (Agarwal, 2005).
2.6. MAO catalytic activity assay
Rat frontal cortex was homogenized in assay buffer (50 mM sodium phosphate buffer). Approximately 100 μg of protein was incubated with 100 μM [14C]5-hydroxytryptamine (for MAO-A) or 10 μM [14C]-labeled phenylethylamine (for MAO-B) (PerkinElmer Inc., Waltham, MA) in assay buffer at 37 °C for 20 min. Termination of the reaction occurred with the addition of 100 μl of 6N HCl. The reaction product was then extracted with benzene/ethyl acetate (1:1) for MAO-A or ethyl acetate/toluene (1:1) for MAO-B and centrifuged at 800 ×g for 7 min. Afterwards, the organic phase from the reaction product was extracted and mixed with liquid scintillation cocktail (Beckman Coulter, Inc., Danvers, MA). The radioactivity of the reaction product was then quantified by liquid scintillation spectroscopy (Johnson et al., 2011; Ou et al., 2011; Grunewald et al., 2012).
2.7. Tail-flick test
The antinociceptive effect of binge EtOH exposure was determined using the tail-flick test. Rats were gently placed in a rat restraint chamber, while the tail was placed directly under a radiant heat source (Columbus Instruments, Columbus, OH) by a researcher blind to the experimental groups. To elicit a tail-flick response, a focused beam of light was applied to the dorsal surface of the tail at 1–1.5 cm from the tip. The tail-flick response is defined as the latency between the onset of the heat stimulus and the voluntary withdrawal of tail (Dai et al., 2008; Ou et al., 2014). Baseline response of 3–4 s was acquired by adjusting the intensity of the heat stimulus, with a cutoff time of 10 s used to avoid any injury to the animals. As previously described (Dai et al., 2008; Ou et al., 2014), the response to the pain stimulus is expressed as the percentage of maximum possible effect (%MPE), which is calculated as [(T1 − T0)/(T2 − T0)] × 100, where T0 and T1 are the tail-flick latencies before (pre-treatment) and after (post-treatment) binge ethanol exposure, respectively, and T2 represents the cutoff time.
2.8. Statistical analysis
Statistical significance in molecular assays was determined using 2-way analysis of variance (ANOVA) followed by Tukey post hoc analysis for multiple comparisons among 8 groups (ethanol or control rats receiving either no drug, selegiline, rasagiline, or M30). A 1-way ANOVA followed by Holm-Sidak post hoc analysis for multiple comparisons between control, ethanol-treated, and ethanol-treated with MAOI groups was used to compare groups used in the tail-flick test. Pearson correlation was performed between KLF11 and caspase-3 expressions. A value of p < .05 was considered statistically significant. All data are reported as mean ± SEM.
3. Results
3.1. Binge ethanol-induced upregulation of KLF11 is prevented by MAOIs
The modified Majchrowicz 4-day binge model is known to induce neurodegeneration and physical dependence on alcohol (Majchrowicz, 1975; Obernier et al., 2002b). Protein levels and mRNA were determined by western blot analysis and RT-PCR, respectively. The results indicate a significant difference in KLF11 protein (F(1,56) = 10.54; p = .002; Fig. 1B) and mRNA (F(1,56) = 23.06; p < .001; Fig. 1C) expression between the control and ethanol groups. Binge ethanol increased KLF11 protein by 67% compared to control rats (1.87 ± .29 vs 1.12 ± .13, p = .036; Fig. 1B), however MAOI treatment prevented the binge ethanol-induced increase in KLF11 expression compared to the no drug control group. Moreover, binge ethanol rats which were treated with rasagiline (1.26 ± .14 vs 1.87 ± .29, p = .049; Fig. 1B) or M30 (1.22 ± .16 vs 1.87 ± .29, p = .031; Fig. 1B) displayed less KLF11 protein compared to binge ethanol rats which received no drug by 32.6% and 36%, respectively. Similarly, binge ethanol elevated KLF11 mRNA by ~2.2-fold (2.15 ± .25 vs 1 ± .25, p = .001; Fig. 1C). In addition, rasagiline (1.48 ± .15 vs 2.15 ± .25, p = .039; Fig. 1C) and M30 (1.45 ± .12 vs 2.15 ± .25, p = .03; Fig. 1C) significantly reduced KLF11 mRNA expression by 31% and 33%, respectively, compared to rats which received no drug in the binge ethanol group. There was no significant effect found with MAOI treatment on KLF11 protein or mRNA expression within the control group. Thus, our findings show that KLF11 is upregulated by binge ethanol exposure in rats in a manner comparable to our reports in chronic ethanol models, suggesting that exposure to acute ethanol is sufficient to induce KLF11. Interestingly, each MAOI examined (selegiline, rasagiline, and M30) prevented the ethanol-induced upregulation of KLF11 compared to their control counterparts.
Fig. 1. Binge ethanol-induced upregulation of KLF11 is prevented by MAOIs.

Representative bands of KLF11 protein (A) was assessed by western blot analysis, and mRNA was assessed by RT-PCR from the frontal cortex of binge ethanol and control rats which received either no drug, selegiline, rasagiline, or M30. Binge ethanol significantly increased KLF11 protein (B) and mRNA (C), however the MAOIs, rasagiline and M30, reduced KLF11 expression in binge ethanol rats. Data presented as mean ± SEM, *p < .05, **p < .01.
3.2. Binge ethanol exposure increases expression of the monoamine oxidases
Monoamine oxidases (both type-A and -B) have been associated with alcohol dependence in humans (Carlsson et al., 1980; Porodenko and Travenko, 1991; Udemgba et al., 2014). In a chronic ethanol rodent model, we have reported an ethanol-induced increase in expression of MAO-A in mice and MAO-B in both rats and mice (Ou et al., 2011, 2014). Here, we examined whether MAO protein and mRNA expression are also affected by binge ethanol exposure or MAOI treatment in rats. For MAO-A, a significant difference was found between control and ethanol groups at the protein (F(1,56) = 12.81, p < .001) and mRNA (F(1,56) = 22.22, p < .001) expression levels. Binge ethanol elevated MAO-A protein (1.69 ± .23 vs .96 ± .06, p = .015; Fig. 2B) and mRNA (2.05 ± .27 vs 1 ± .06, p = .015; Fig. 2C) by 76% and 2-fold compared to control rats respectively. There was a modest reduction in the ethanol-induced increase in MAO-A protein and mRNA in rats receiving MAOI treatment with there being no statistical difference between control and ethanol-treated rats which received MAOI treatment.
Fig. 2. Binge ethanol exposure increases expression of monoamine oxidase.

Following 4 day binge ethanol exposure, protein and mRNA from the frontal cortex was examined for changes in monoamine oxidase by western blot analysis and RT-PCR, respectively. Representative images of protein bands are depicted in Fig. 2A. Binge ethanol significantly increased MAO-A protein (B) and mRNA (C) expression compared to control rats. Similarly, binge ethanol significantly increased MAO-B protein (D) and mRNA (E) expression compared to control rats. Data presented as mean ± SEM, *p < .05.
Similarly, a significant difference in MAO-B expression was detected between control and ethanol groups for both protein (F(1,56) = 11.87, p = .001) and mRNA (F(1,56) = 23.08, p < .001). Expression of MAOB protein was increased by 69% in binge rats (1.49 ± .14 vs .88 ± .08, p = .046; Fig. 2D), whereas binge ethanol elevated MAO-B mRNA by 1.9-fold (1.9 ± .22 vs 1 ± .07, p = .022; Fig. 2E) compared to control rats which received no ethanol. Similar to MAO-A, there was a slight drug effect on the protein or mRNA expression of MAO-B, although this was not statistically significant. This suggests some complexity of MAO transcriptional regulation in the presence of ethanol and/or MAOIs. Collectively, these results demonstrate activation of the KLF11-MAO pathway following binge ethanol exposure in rats.
3.3. MAO catalytic activity is enhanced by binge ethanol, which can be prevented by MAOI treatment
MAO enzyme activity induces a high degree of oxidative stress through its catabolism of monoamine neurotransmitters. Because we have documented increases in MAO-A and MAO-B catalytic activity in chronic ethanol rodent models (Ou et al., 2011, 2014), we examined the response of MAO catalytic activity to binge ethanol. Indeed, a significant effect was found for MAO-A catalytic activity between groups (F(1,56) = 40.36, p < .001) and within groups (F(3,56) = 11.76, p < .001). MAO-A catalytic activity was significantly reduced by rasagiline (1.62 ± .2 vs 2.29 ± .13, p = .046; Fig. 3A) and M30 (1.31 ± .14 vs 2.29 ± .13, p = .002; Fig. 3A) treatment in the control group by 29% and 43%, respectively. Binge ethanol-treated rats displayed a 41% increase in MAO-A catalytic activity compared to control rats (3.24 ± .24 vs 2.29 ± .13, p = .022; Fig. 3A). In the binge rats, rasagiline (2.45 ± .21 vs 3.24 ± .24, p = .015; Fig. 3A) and M30 (2.14 ± .21 vs 3.24 ± .24, p = .005; Fig. 3A) reduced ethanol-induced MAO-A catalytic activity by 24% and 34%, respectively. However, selegiline had no significant effect on MAO-A catalytic activity.
Fig. 3. MAO catalytic activity is enhanced by binge ethanol and MAOIs decrease MAO catalytic activity in both control and binge ethanol rats.

MAO enzymatic activity in the frontal cortex of control and binge ethanol rats receiving either no drug or a MAOI were examined using liquid scintillation spectroscopy. (A) MAO-A catalytic activity was significantly increased by binge ethanol. Rasagiline and M30 reduced MAO-A catalytic activity in both control and binge ethanol groups. (B) MAO-B catalytic activity was significantly increased by binge ethanol. All MAOIs (selegiline, rasagiline, M30) significantly decreased MAO-B activity. Data present as mean ± SEM, *p < .05, **p < .01, #p < .001.
Additionally, there was a significant difference in MAO-B catalytic activity between groups (F(1, 56) = 30.00, p < .0001) and within drug groups (F(3, 56) = 60.20, p < .0001). In control rats, a 60%, 70%, and 58% reduction in MAO-B activity was found with treatments of selegiline (.232 ± .02 vs .576 ± .06, p < .001; Fig. 3B), rasagiline (.17 ± .02 vs .576 ± .06, p < .001; Fig. 3B), and M30 (.24 ± .02 vs .576 ± .06, p < .001; Fig. 3B) compared with control rats receiving no drug. Furthermore, binge ethanol increased MAO-B activity by 34% compared to controls (.772 ± .06 vs .576 ± .06, p = .009; Fig. 3B). Similar to control rats, binge ethanol rats treated with selegiline (.365 ± .03 vs .772 ± .06, p < .001; Fig. 3B), rasagiline (.278 ± .03 vs .772 ± .06, p < .001; Fig. 3B), and M30 (.375 ± .03 vs .772 ± .06, p < .001; Fig. 3B) showed a significant reduction in MAO-B catalytic activity by 53%, 64%, and 51%, respectively, compared to binge ethanol rats which received no drug. Therefore, as in chronic ethanol models, binge ethanol exposure significantly increases MAO activity, an effect that can be prevented by MAOI administration.
3.4. Binge ethanol affects levels of oxidative stress, but not oxidative stress enzymes
Ethanol induces oxidative stress through a number of mechanisms. To determine if binge ethanol has an effect on oxidative stress and oxidative stress enzymes, and whether MAOI treatment could reverse such an effect, western blot analysis was conducted to determine protein oxidation status and levels of SOD2 and catalase (Fig. 4A). There was no statistically significant difference found for ethanol or MAOIs on the expression SOD2 protein (Fig. 4B). However, a significant group effect on catalase was found between control and ethanol-treated rats (F(1,56) = 15.69, p = .0002, Fig. 4C), indicating that catalase was increased by binge ethanol across all groups which received ethanol compared to rats that did not receive ethanol. A significant drug effect also was found for catalase protein expression between the no drug and drug groups of all rats (control and ethanol-treated) (F(3,56) = 3.344, p = .026, Fig. 4C). This suggests that both ethanol and MAOIs raise catalase protein; however, no significant difference in catalase was found when comparing individual groups. Additionally, in an effort to determine the level of oxidative stress induced by ethanol and whether MAOI treatment has any effect on oxidative stress, protein oxidation status was investigated by examining carbonyl expression on albumin proteins (Fig. 4D). There was a significant main effect between control and ethanol groups (F(1,56) = 18.14, p < .0001). Rats exposed to binge ethanol displayed increase protein oxidation levels by 2.60-fold compared to control rats (2.62 ± .512 vs 1 ± .073, p = .035; Fig. 4E). MAOIs alone did not significantly alter protein oxidation status among the control or ethanol-treated rats. However, MAOI treatment blunted the effect of binge ethanol on protein oxidation levels as there was no significant difference detected between binge ethanol rats which received MAOI treatment and control rats. These data indicate that MAOIs do not completely reverse protein carbonylation, but may mitigate oxidative stress induced by binge ethanol through inhibition of MAO catalytic activity.
Fig. 4. The effect of binge ethanol and acute MAOI treatment on the expression of oxidative stress enzymes and protein oxidation.

The frontal cortex of rats from all groups were examined for changes in oxidative stress enzymes and protein oxidation levels. Oxidative enzymes were examined by western blot analysis (A). The protein expression levels of SOD2 (B) and catalase (C) were examined in the frontal cortex of control and binge ethanol rats given no drug or a MAOI. Neither binge ethanol nor acute MAOI exposure had any significant effect on the expression of oxidative stress enzymes within individual groups. However, there was a significant group effect for increased catalase expression between control rats vs binge ethanol-exposed rats (p < .0002) and MAOI drug-treated rats vs rats given no drug (p = .026). Oxidized protein was determined by protein carbonylation status of albumin, as represented in (D). Binge ethanol rats displayed a significantly greater degree of protein oxidation compared to control rats (E), but none of the MAOIs examined had any significant effect on reducing protein oxidation levels in either control or ethanol groups. Data presented as mean ± SEM, *p < .05.
3.5. Pro-apoptotic protein Caspase-3 is increased and BDNF is reduced by binge ethanol, but MAOI treatment prevents these binge ethanol-induced effects
Because caspase-3 is increased by acute ethanol exposure in rat cerebral cortex (Han et al., 2005) and intrinsic apoptosis is reported as an ethanol-induced cell death mechanism (Lee et al., 2014), we investigated the expression of caspase-3 following binge ethanol exposure and MAOI drug treatment. Further, several studies indicate that BDNF is decreased by chronic and binge ethanol consumption, including in the PFC of rodents (Climent et al., 2002; Rueda et al., 2012; Briones and Woods, 2013). Therefore, we also examined the expression of BDNF following binge ethanol exposure and whether MAOI treatment yielded some neuroprotective effects on these markers. A significant difference between groups was found for caspase-3 (F(1,56) = 15.54, p = .0002). Caspase-3 was markedly elevated by 88% in binge ethanol rats compared to control rats (1.92 ± .4 vs 1.02 ± .1, p = .046; Fig. 5B). Treatment with MAOIs blunted the binge ethanol-induced upregulation of caspase-3 as no significant differences were found between control and binge ethanol groups treated with MAOI. Following BDNF protein analysis, a significant difference between groups was detected (F(1,56) = 17.61, p < .0001). Binge ethanol exposure in rats reduced BDNF protein expression by 55% compared to control rats (.712 ± .08 vs 1.57 ± .21, p = .03; Fig. 5C). MAOI treatment alone had no effect on BDNF levels; however, in binge ethanol-exposed rats, MAOI treatment blunted the ethanol-mediated reduction in BDNF levels. Thus, MAOIs appear to prevent the effects of binge ethanol on caspase-3 and BDNF, suggesting that acute MAOI exposure may not be an effective strategy to reverse these markers of neurodegeneration, but could be neuroprotective in preventing binge ethanol-induced changes in these markers.
Fig. 5. The effect of binge ethanol and MAOI treatment on the expression of caspase-3 and BDNF.

The protein expression levels of caspase-3 (A) and BDNF (B) were determined in the frontal cortex of control and binge ethanol rats given no drug or a MAOI by western blot analysis, as represented in (A). Binge ethanol significantly affected the expression of caspase-3 (B) and BDNF (C). MAOI treatment prevented, but did not reverse, the binge ethanol-induced change in expression of these markers. Data presented as mean ± SEM, *p < .05.
3.6. Antinociception is enhanced with binge ethanol exposure
Antinociception is a well-documented feature of chronic ethanol exposure. Although several studies have addressed nociception as a component of chronic ethanol exposure, to our knowledge, this has never been examined under conditions of binge ethanol exposure or with binge ethanol and concomitant treatment with an MAOI. Therefore, we compared the anti-nociceptive response between control rats, binge ethanol rats, and binge ethanol rats which were given an MAOI. Using the tail-flick test, EtOH-induced antinociception was measured by recording the time between applying a thermal stimulus to the tail and voluntary tail withdrawal (Dai et al., 2008). There was no statistical difference in pre-treatment baseline measurements (Fig. 6A) (F(4,35) = .812, p = .526), expressed as average tail-flick latency in seconds. However, a statistically significant effect was found among post-treatment measurements (F(4,35) = 26.49, p < .0001). Binge ethanol significantly increased tail-flick latency (98.8 ± 1.2 vs 4.9 ± 2.4 p < .001; Fig. 6B) compared to control rats. While treatment with an MAOI did not significantly affect the ethanol-induced increase in antinociception, there was a trend for rasagiline (80.54 ± 11.11 vs 98.79 ± 1.21, p = .098; Fig. 6B) and M30 (77.49 ± 12.56 vs 98.70 ± 1.21, p = .055; Fig. 6B) to attenuate the ethanol-induced increase in tail-flick latency in binge rats. These results suggest that binge ethanol exposure, in a manner similar to that of chronic ethanol exposure, induces antinociception as measured by tail-flick latency, a pain threshold response, in rats.
Fig. 6. Antinociception is increased by binge ethanol exposure.

To examine the effect of binge ethanol and MAOIs on pain threshold response, control rats, and binge ethanol rats receiving either no drug, selegiline, rasagiline, or M30 were administered the tail-flick test. (A) No differences were found in pre-treatment (before EtOH exposure) tail-flick latency. (B) Binge ethanol exposure induced antinociception compared to control rats, but there was no significant effect of MAOIs on rescuing ethanol-induced antinociception. Pre-treatment latencies are expressed as mean ± SEM seconds. Post-treatment latencies expressed as mean ± SEM % Maximal Possible Effect (MPE), #p < .001.
3.7. KLF11is positively correlated with Caspase-3 in chronic, but not binge, ethanol-treated rats
KLF11 is reported to activate caspase-3 (Gohla et al., 2008; Lomberk et al., 2012) and caspase-3 contains Sp1 binding sites, making it a putative target of KLF11 (Sudhakar et al., 2008). In effort to further investigate the relationship between KLF11 and Caspase-3, we examined protein expression of KLF11 and caspase-3 in ethanol-treated rats following either chronic or binge ethanol feedings. Data was collected from western blots performed on chronic ethanol-treated Wistar rats (Ou et al., 2011; Duncan et al., 2015) and binge ethanol-treated Sprague–Dawley rats (current study). After which, linear regression analyses were performed. No correlation was found between KLF11 and Caspase-3 protein levels (r2 = .08) in binge-treated rats. However, in chronic ethanol-treated rats, a positive correlation was found to exist between KLF11 and Caspase-3 (r2 = .45, p = .047; Fig. 7). This suggests that KLF11 in chronic, but not binge ethanol exposure, could precipitate caspase-3-mediated cell death. Under conditions of chronic ethanol exposure where blood ethanol concentration is modest (<50 mM), KLF11 could be an important regulator of caspase-3 expression. Although, in binge ethanol-treated rats, where the blood ethanol concentration is much higher (~90 mM), we find that the expression of KLF11 is not tightly coupled to Caspase-3, as further evidenced by a reduction of KLF11 expression yet no effect on caspase-3 in rats given MAOIs.
Fig. 7. KLF11 is correlated with procaspase-3 protein expression in chronic, but not binge, ethanol-treated rats.
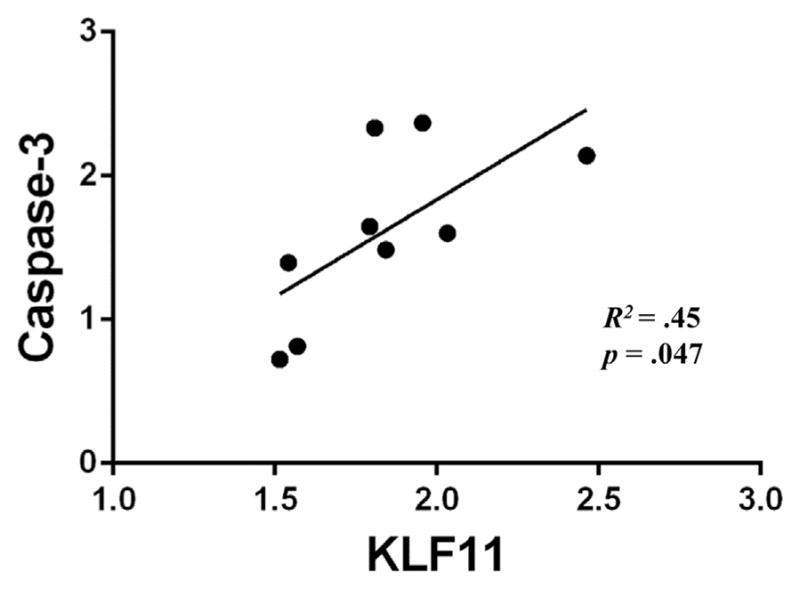
Linear regression analyses were conducted on the protein expression of KLF11 and caspase-3 in chronic and binge ethanol-treated rats. A positive correlation between KLF11 and caspase-3 protein (R2 = 0.45, p = 0.047) was found among chronic ethanol-treated rats, but not binge ethanol-treated rats (R2 = .08, p = .28).
4. Discussion
This study investigates the KLF11-MAO pathway in response to binge ethanol exposure and whether MAOIs promote neuroprotection from ethanol. The primary finding was that binge ethanol increases the KLF11-MAO pathway, protein oxidation and caspase-3, but reduces levels of BDNF. MAOIs such as rasagiline and M30 reversed the ethanol-mediated increase in MAO catalytic activity and KLF11, but did not reverse any other binge ethanol-induced changes. However, MAOIs did display some mild neuroprotective benefits by protecting against the binge ethanol-mediated increases in protein oxidation, caspase-3, and BDNF, as compared to control rats. These results are important for understanding the neuropathology associated with ethanol exposure and for identifying potential pharmacological regimens to prevent or lessen neurotoxicity from ethanol.
The 4-day binge model used in this study induces detrimental neurobiological effects, including necrosis, oxidative stress, glial scaring, a reduction in neurogenesis, and neurodegeneration (Majchrowicz, 1975; Nixon and Crews, 2002; Obernier et al., 2002a, 2002b; Crews et al., 2006; Hayes et al., 2013). Ethanol-induced cell death can occur through multiple avenues; however, one canonical mechanism involves the production and accumulation of ROS, both as a secondary effect and directly by metabolism of ethanol itself (Das and Vasudevan, 2007). Furthermore, oxidative stress is cited as a source of neurodegeneration (Srivastava et al., 2010; Chen et al., 2012) and KLF11 is reported to contribute to ROS levels by regulating the expression of oxidative stress enzymes and MAO (Fernandez-Zapico et al., 2003; Ou et al., 2004; Grunewald et al., 2012). MAO produces ROS through the deamination of mono-amine neurotransmitters, which subsequently induces cell death (Naoi et al., 2003; Fitzgerald et al., 2007). Previous reports have shown that the KLF11 cell death pathway is upregulated in response to chronic ethanol exposure in rodents (Ou et al., 2011, 2014; Duncan et al., 2015; Nair et al., 2015), as well as in postmortem brain of AUD subjects (Udemgba et al., 2014). In addition, chronic ethanol-induced increases in both MAO-A and MAO-B expression and catalytic activity have been reported in rodents and humans (Rimondini et al., 2002; Ou et al., 2014; Matthews et al., 2014; Nair et al., 2015). The present study extended these observations on the KLF11-MAO pathway from the chronic ethanol model to acute, binge-like ethanol exposure. Additionally, we evaluated the neuroprotective efficacy of propargylamine-containing MAOIs because they have demonstrated anti-apoptotic and anti-oxidative effects in rodent models of neurodegeneration (Youdim et al., 2003b, 2006; Crews et al., 2006; Avramovich-Tirosh et al., 2007; Youdim et al., 2014). The beneficial effect of MAOIs occurs not only by inhibiting MAO enzymatic activity, but also through independent propargylamine-related effects, which are known to regulate the expression of more than 50 proteins related to cellular function and viability (Youdim et al., 2014).
Following the 4-day binge exposure to ethanol, rats displayed an increase in expression of both mRNA and protein of KLF11, MAO-A, and MAO-B. These results suggest that indeed, the KLF11-MAO pathway is activated by binge exposure to ethanol in rats. Interestingly, MAOI treatment prevented the ethanol-induced increase of this pathway compared to control rats treated only with ethanol. Further, rasagiline and M30, but not selegiline, significantly reduced the ethanol-mediated upregulation of KLF11. This result is consistent with our report that MAOI treatment during chronic ethanol exposure reduces the ethanol-responsive increase in KLF11 expression (Lu et al., 2008). Moreover, concomitant antioxidant administration during chronic ethanol exposure in rats significantly decreases the ethanol-mediated induction of KLF11 (Nair et al., 2015). Thus, one possible mechanism of KLF11 induction by ethanol appears to occur through oxidative stress. Our data indicates that inhibiting both MAO isoforms via rasagiline or M30, but not solely MAO-B inhibition by selegiline, is necessary to reduce the induction of KLF11. In fact, the non-selective MAO inhibitor, pargyline, is documented to reduce TGF-β1 expression (Chaava et al., 2001), highlighting the importance of dual MAO inhibition in attenuating the activation of this ROS-inducible signaling cascade which targets KLF11 (Buck et al., 2006; Chen et al., 2006). Further, inhibiting MAO-A may interplay upon other cell death signaling cascades which involve KLF11, since MAO-A is reported as a pro-apoptotic factor apart from it’s role in monoamine catabolism (Ou et al., 2006). Differences in pharamacokenetics could explain also why rasagiline and M30, but not selegiline, significantly reduced KLF11. This is because selegiline is metabolized to the neurotoxic methamphetamine, while the rasagiline metabolite, aminoindan, and iron-chelating functions of M30 have demonstrated neuroprotective activity independent of MAO inhibition (Youdim et al., 2001, 2014; Ou et al., 2009). In addition, none of the MAOIs used in this study were capable of significantly diminishing the increase in MAO expression brought on by binge ethanol. Because rasagiline and M30 significantly reduced KLF11 in binge rats, but not MAO, other MAO-related transcription factors may be involved in the regulation of MAO during ethanol exposure, particularly in the presence of an MAOI. Moreover, MAOIs may elicit compensatory cellular mechanisms in an effort to upregulate MAO expression and thereby overwhelm MAO inhibition. In a KLF11 knockout (KO) mouse model, increases in expression of both MAO-A and MAO-B mRNA persisted following ethanol treatment, which supports the notion that other regulatory factors are involved in regulation of MAO by ethanol (Ou et al., 2014) in the absence or repression of KLF11. Thus, it appears that there is added complexity in MAO regulation under exposure to ethanol.
In a comparable manner to MAO protein expression, an increase was detected in catalytic activity of both MAO-A and MAO-B following exposure to binge ethanol. We have previously documented increases in MAO catalytic activity in chronic ethanol rodent studies (Ou et al., 2011, 2014). Similarly, the present study indicates that MAO catalytic activity is elevated by exposure to binge ethanol and may serve as at least one source of ethanol-induced oxidative stress. As expected, MAOI administration significantly reduced MAO catalytic activity in control rats and these drugs also blunted the binge ethanol induced increase in catalytic activity. At the doses used in this study (5 mg/kg/day), all three MAOIs (selegiline, rasagiline, M30) significantly reduced MAOB catalytic activity. However, only rasagiline and M30 significantly inhibited MAOA activity. The potent MAOB inhibitor rasagiline is documented to lose specificity for MAOB at higher concentrations (3 mg/kg) (Speiser et al., 1999), an effect we observed in this study at 5 mg/kg. These inhibitory effects seen on MAO catalytic activity here are similar to other studies investigating MAOIs at the doses studied (Paterson et al., 1991; Speiser et al., 1999; Youdim and Tipton, 2002; Gal et al., 2005). Therefore, MAOI treatment may at least partially reduce oxidative stress associated with exposure to binge ethanol. Interestingly, since only rasagiline and M30 reduced KLF11 expression in binge ethanol rats, inhibition of both MAO-A and MAO-B appear to contribute to reversing KLF11 expression brought on by ethanol. The precise mechanism whereby rasagiline and M30 reduce KLF11 expression in binge ethanol rats is not known, although the added inhibition of MAO-A by these compounds may lower levels of ROS. For example, MAO-A is involved in cell death signaling pathways apart from its role in oxidative stress (De Zutter and Davis, 2001; Ou et al., 2006), suggesting that there may be some downstream effects of MAO-A which interact with KLF11 signaling. The propargylamine moiety of the MAOIs used in this study have also demonstrated anti-apoptotic effects aside from their inhibition of MAO (Youdim et al., 2006, 2014). In addition, metabolic products of some MAOIs, such as rasagiline, yield neuroprotective metabolites (Youdim et al., 2006).
In an effort to further investigate oxidative stress induced by ethanol, we examined levels of protein oxidation, superoxide dismutase 2 (SOD2), and catalase in binge rats with or without MAOI treatment. Protein oxidation by ethanol has been documented, including in rodents exposed to binge ethanol (Nogales et al., 2014). Like MAO enzymatic activity, protein oxidation was strikingly elevated by binge ethanol exposure. Although MAOIs did not significantly reverse the binge ethanol-induced increase in protein oxidation, they did prevent a significant rise in protein oxidation compared to control rats, suggesting some mild protection against binge ethanol-related protein oxidation. Therefore, MAO catalytic activity may not be solely responsible for ethanol-induced oxidative stress production, but may only contribute partially to ROS levels. Moreover, neither binge ethanol nor MAOIs had any significant effect on the protein expression of SOD2 or catalase. Several studies have reported changes in expression/activity of SOD2 and catalase following ethanol exposure, while others have not (Carmiel-Haggai et al., 2003; Artun et al., 2010; Nogales et al., 2014). However, in the brain, catalase is largely responsible for the metabolism of ethanol to acetylaldehyde (Ledesma et al., 2014), which explains why we observed a group effect in binge ethanol-treated rats for increased catalase expression compared to control rats, regardless of MAOI administration. Interestingly, MAOIs increase SOD2 and catalase in several rodent models (Carrillo et al., 1992; Kitani et al., 1994; Carrillo et al., 2000; Youdim et al., 2014). However, these studies used chronic and subchronic exposure to MAOIs, suggesting that a period of acute exposure, such as 4 days, may not be sufficient to significantly affect SOD2 and catalase expression. Pretreating rats with MAOIs which continues during exposure to binge ethanol may be more beneficial in suppressing ROS levels in response to binge ethanol.
Binge ethanol is reported to induce caspase-3 and decrease BDNF (Carmiel-Haggai et al., 2003; Briones and Woods, 2013). Moreover, ethanol not only reduces BDNF levels, it also inhibits BDNF-mediated signaling (Li et al., 2004), which could influence pro-apoptotic routes of cell death and converge with KLF11-mediated cell death signaling. Conversely, MAOIs reduce apoptosis and increase BDNF in cell culture and in rodents (Kontkanen and Castren, 1999; Mizuta et al., 2000; Youdim et al., 2014; Bar-Am et al., 2015). In the current study, binge ethanol exposure nearly doubled caspase-3 levels and reduced BDNF by half, strongly suggesting that cell death processes are enhanced by binge ethanol. Although caspase-3 and BDNF levels of binge ethanol-treated rats given MAOIs were not significantly different from levels of controls, a strong trend still suggested that MAOIs were not entirely successful in reversing the effect of binge ethanol on caspase-3 and BDNF. As with SOD2 and catalase, chronic treatment with MAOIs may be necessary to observe a more robust effect in reversing the detrimental effects of binge ethanol on rat brain. Alternatively, the high level of intoxication induced by binge ethanol feeding in this study may supersede the full neuroprotective potential of these drugs. It would be worthwhile to examine the pharmacotherapeutic potential of MAOIs in a chronic ethanol model, where rodents would receive a much longer exposure period to the drugs and where the ethanol exposure pattern is more relevant to chronic alcohol use observed in AUD.
To further categorize the relationship between KLF11 and caspase-3, we performed linear regression analyses on protein levels of KLF11 and caspase-3 in both binge and chronic ethanol-treated rats. The positive correlation between KLF11 and caspase-3 suggest that under conditions of chronic ethanol exposure where blood ethanol concentration is modest (<50 mM), KLF11 could be an important regulator of caspase-3 expression. Although, in binge ethanol-treated rats where the blood ethanol concentration is much higher (~90 mM), we find that the expression of KLF11 is not tightly coupled to Caspase-3, as further evidenced by a reduction of KLF11 expression yet no effect on caspase-3 in rats given MAOIs. Future studies to more closely examine the relationship of ethanol-induced KLF11 on caspase-3 expression are warranted.
Finally, an antinociceptive effect of binge ethanol exposure is reported here, which has also been widely documented in chronic ethanol models (Egli et al., 2012; Ou et al., 2014). There was a trend for a modest, non-significant reduction in pain threshold among rasagiline- and M30-treated, but not selegiline-treated rats exposed to binge ethanol. We previously reported that knock out of KLF11 in mice rescued pain perception that was diminished by ethanol, signifying that KLF11 is involved in ethanol-mediated antinociception (Ou et al., 2014). Further examination of MAOIs and KLF11 to restore ethanol-induced antinociception is needed. KLF11 may be involved in ethanol-induced neuropathy and nerve damage, resulting in decreased sensation and detection of pain.
This study provides evidence that the KLF11-MAO pathway is responsive to exposure to binge ethanol. Interestingly, MAOI treatment was able to partially or fully prevent the ethanol-induced changes in KLF11, MAO, protein oxidation, caspase-3, and BDNF, suggesting modestly enhanced neuroprotection from the effects of binge ethanol exposure in rats. Additional investigation of MAOIs using different administration regimens are needed to assess their neuroprotective efficacy in models of both chronic and binge exposure to ethanol. The potential of MAOIs to attenuate brain injury in response to acute or chronic exposure to ethanol or ethanol poisoning is of particular translational interest. Overall, this study supports the KLF11-MAO pathway as a novel ethanol-inducible signaling cascade which could mediate molecular events leading to cellular aberrations and neural cell death. In addition, treatment with propargylamine MAOIs may represent a novel pharmacotherapeutic strategy to alleviate alcohol-related neurotoxicity and neurodegeneration.
Acknowledgments
The authors appreciate the support provided by the Laboratory Animal Facilities at the University of Mississippi Medical Center. This study was supported by Public Health Service Grants R01 AA020103, P30 GM103328, The Brain & Behavior Research Foundation (NARSAD), an Alcohol Research Resource Award Grant (R24 AA015512-02) and a grant from the Intramural Research Support Program from the University of Mississippi Medical Center.
References
- Agartz I, Momenan R, Rawlings RR, Kerich MJ, Hommer DW. Hippocampal volume in patients with alcohol dependence. Arch Gen Psychiatry. 1999;56:356–363. doi: 10.1001/archpsyc.56.4.356. [DOI] [PubMed] [Google Scholar]
- Agarwal R. Smoking, oxidative stress and inflammation: impact on resting energy expenditure in diabetic nephropathy. BMC Nephrol. 2005;6:13. doi: 10.1186/1471-2369-6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artun BC, Kusku-Kiraz Z, Gulluoglu M, Cevikbas U, Kocak-Toker N, Uysal M. The effect of carnosine pretreatment on oxidative stress and hepatotoxicity in binge ethanol administered rats. Hum Exp Toxicol. 2010;29:659–665. doi: 10.1177/0960327109359460. [DOI] [PubMed] [Google Scholar]
- Avramovich-Tirosh Y, Amit T, Bar-Am O, Zheng H, Fridkin M, Youdim MB. Therapeutic targets and potential of the novel brain- permeable multi-functional iron chelator-monoamine oxidase inhibitor drug, M-30, for the treatment of Alzheimer’s disease. J Neurochem. 2007;100:490–502. doi: 10.1111/j.1471-4159.2006.04258.x. [DOI] [PubMed] [Google Scholar]
- Bar-Am O, Amit T, Youdim MB, Weinreb O. Neuroprotective and neurorestorative potential of propargylamine derivatives in ageing: focus on mitochondrial targets. J Neural Transm. 2015 doi: 10.1007/s00702-015-1395-3. [DOI] [PubMed] [Google Scholar]
- Beck A, Wustenberg T, Genauck A, Wrase J, Schlagenhauf F, Smolka MN, Mann K, Heinz A. Effect of brain structure, brain function, and brain connectivity on relapse in alcohol-dependent patients. Arch Gen Psychiatry. 2012;69:842–852. doi: 10.1001/archgenpsychiatry.2011.2026. [DOI] [PubMed] [Google Scholar]
- Boyadjieva NI, Sarkar DK. Microglia play a role in ethanol-induced oxidative stress and apoptosis in developing hypothalamic neurons. Alcohol Clin Exp Res. 2013;37:252–262. doi: 10.1111/j.1530-0277.2012.01889.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briones TL, Woods J. Chronic binge-like alcohol consumption in adolescence causes depression-like symptoms possibly mediated by the effects of BDNF on neurogenesis. Neuroscience. 2013;254:324–334. doi: 10.1016/j.neuroscience.2013.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck A, Buchholz M, Wagner M, Adler G, Gress T, Ellenrieder V. The tumor suppressor KLF11 mediates a novel mechanism in transforming growth factor beta-induced growth inhibition that is inactivated in pancreatic cancer. Mol Cancer Res MCR. 2006;4:861–872. doi: 10.1158/1541-7786.MCR-06-0081. [DOI] [PubMed] [Google Scholar]
- Burke WJ, Li SW, Chung HD, Ruggiero DA, Kristal BS, Johnson EM, Lampe P, Kumar VB, Franko M, Williams EA, Zahm DS. Neurotoxicity of MAO metabolites of catecholamine neurotransmitters: role in neurodegenerative diseases. Neurotoxicology. 2004;25:101–115. doi: 10.1016/S0161-813X(03)00090-1. [DOI] [PubMed] [Google Scholar]
- Buttke TM, Sandstrom PA. Oxidative stress as a mediator of apoptosis. Immunol Today. 1994;15:7–10. doi: 10.1016/0167-5699(94)90018-3. [DOI] [PubMed] [Google Scholar]
- Carlsson A, Adolfsson R, Aquilonius SM, Gottfries CG, Oreland L, Svennerholm L, Winblad B. Biogenic amines in human brain in normal aging, senile dementia, and chronic alcoholism. Adv Biochem Psychopharmacol. 1980;23:295–304. [PubMed] [Google Scholar]
- Carmiel-Haggai M, Cederbaum AI, Nieto N. Binge ethanol exposure increases liver injury in obese rats. Gastroenterology. 2003;125:1818–1833. doi: 10.1053/j.gastro.2003.09.019. [DOI] [PubMed] [Google Scholar]
- Carrillo MC, Kanai S, Sato Y, Ivy GO, Kitani K. Sequential changes in activities of superoxide dismutase and catalase in brain regions and liver during (−)deprenyl infusion in male rats. Biochem Pharmacol. 1992;44:2185–2189. doi: 10.1016/0006-2952(92)90345-j. [DOI] [PubMed] [Google Scholar]
- Carrillo MC, Minami C, Kitani K, Maruyama W, Ohashi K, Yamamoto T, Naoi M, Kanai S, Youdim MB. Enhancing effect of rasagiline on superoxide dismutase and catalase activities in the dopaminergic system in the rat. Life Sci. 2000;67:577–585. doi: 10.1016/s0024-3205(00)00643-3. [DOI] [PubMed] [Google Scholar]
- Chaava R, Alfarano C, Guilbeau-Frugier C, Coatrieux C, Kesteman AS, Parini A, Fares N, Gue M, Schanstra JP, Bascands JL. Pargyline reduces renal damage associated with ischaemia-reperfusion and cyclosporin. Nephrol Dial Transpl. 2001;26(2):489–498. doi: 10.1093/ndt/gfq445. [DOI] [PubMed] [Google Scholar]
- Chen CP, Kuhn P, Chaturvedi K, Boyadjieva N, Sarkar DK. Ethanol induces apoptotic death of developing beta-endorphin neurons via suppression of cyclic adenosine monophosphate production and activation of transforming growth factor-beta1-linked apoptotic signaling. Mol Pharmacol. 2006;69:706–717. doi: 10.1124/mol.105.017004. [DOI] [PubMed] [Google Scholar]
- Chen G, Ke Z, Xu M, Liao M, Wang X, Qi Y, Zhang T, Frank JA, Bower KA, Shi X, Luo J. Autophagy is a protective response to ethanol neurotoxicity. Autophagy. 2012;8:1577–1589. doi: 10.4161/auto.21376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Circu ML, Moyer MP, Harrison L, Aw TY. Contribution of glutathione status to oxidant-induced mitochondrial DNA damage in colonic epithelial cells. Free Radic Biol Med. 2009;47:1190–1198. doi: 10.1016/j.freeradbiomed.2009.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Climent E, Pascual M, Renau-Piqueras J, Guerri C. Ethanol exposure enhances cell death in the developing cerebral cortex: role of brain-derived neurotrophic factor and its signaling pathways. J Neurosci Res. 2002;68:213–225. doi: 10.1002/jnr.10208. [DOI] [PubMed] [Google Scholar]
- Collins MA, Corso TD, Neafsey EJ. Neuronal degeneration in rat cerebrocortical and olfactory regions during subchronic “binge” intoxication with ethanol: possible explanation for olfactory deficits in alcoholics. Alcohol Clin Exp Res. 1996;20:284–292. doi: 10.1111/j.1530-0277.1996.tb01641.x. [DOI] [PubMed] [Google Scholar]
- Collins MA, Neafsey EJ. Neuroinflammatory pathways in binge alcohol-induced neuronal degeneration: oxidative stress cascade involving aquaporin, brain edema, and phospholipase A2 activation. Neurotox Res. 2012;21:70–78. doi: 10.1007/s12640-011-9276-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook T, Gebelein B, Mesa K, Mladek A, Urrutia R. Molecular cloning and characterization of TIEG2 reveals a new subfamily of transforming growth factor-beta-inducible Sp1-like zinc finger-encoding genes involved in the regulation of cell growth. J Biol Chem. 1998;273:25929–25936. doi: 10.1074/jbc.273.40.25929. [DOI] [PubMed] [Google Scholar]
- Crews F, Nixon K, Kim D, Joseph J, Shukitt-Hale B, Qin L, Zou J. BHT blocks NF-kappaB activation and ethanol-induced brain damage. Alcohol Clin Exp Res. 2006;30:1938–1949. doi: 10.1111/j.1530-0277.2006.00239.x. [DOI] [PubMed] [Google Scholar]
- Crews FT, Braun CJ. Binge ethanol treatment causes greater brain damage in alcohol-preferring P rats than in alcohol-nonpreferring NP rats. Alcohol Clin Exp Res. 2003;27:1075–1082. doi: 10.1097/01.ALC.0000075826.35688.0D. [DOI] [PubMed] [Google Scholar]
- Crews FT, Braun CJ, Hoplight B, Switzer RC, 3rd, Knapp DJ. Binge ethanol consumption causes differential brain damage in young adolescent rats compared with adult rats. Alcohol Clin Exp Res. 2000;24:1712–1723. [PubMed] [Google Scholar]
- Crews FT, Nixon K. Mechanisms of neurodegeneration and regeneration in alcoholism. Alcohol Alcohol. 2009;44:115–127. doi: 10.1093/alcalc/agn079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai X, Brunson CD, Rockhold RW, Loh HH, Ho IK, Ma T. Gender differences in the antinociceptive effect of tramadol, alone or in combination with gabapentin, in mice. J Biomed Sci. 2008;15:645–651. doi: 10.1007/s11373-008-9252-0. [DOI] [PubMed] [Google Scholar]
- Das SK, Vasudevan DM. Alcohol-induced oxidative stress. Life Sci. 2007;81:177–187. doi: 10.1016/j.lfs.2007.05.005. [DOI] [PubMed] [Google Scholar]
- Davies KJ. Protein damage and degradation by oxygen radicals. I General aspects J Biol Chem. 1987;262:9895–9901. [PubMed] [Google Scholar]
- Davies KJ, Lin SW, Pacifici RE. Protein damage and degradation by oxygen radicals. IV Degradation of denatured protein. J Biol Chem. 1987;262:9914–9920. [PubMed] [Google Scholar]
- de la Cruz CP, Revilla E, Steffen V, Rodriguez-Gomez JA, Cano J, Machado A. Protection of the aged substantia nigra of the rat against oxidative damage by (−)-deprenyl. Br J Pharmacol. 1996;117:1756–1760. doi: 10.1111/j.1476-5381.1996.tb15350.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Zutter GS, Davis RJ. Pro-apoptotic gene expression mediated by the p38 mitogen-activated protein kinase signal transduction pathway. Proc Natl Acad Sci U S A. 2001;98:6168–6173. doi: 10.1073/pnas.111027698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan J, Johnson S, Ou XM. Monoamine oxidases in major depressive disorder and alcoholism. Drug Discov Ther. 2012;6:112–122. [PubMed] [Google Scholar]
- Duncan J, Wang N, Zhang X, Johnson S, Harris S, Zheng B, Zhang Q, Rajkowska G, Miguel-Hidalgo JJ, Sittman D, Ou XM, Stockmeier CA, Wang JM. Chronic social stress and ethanol increase expression of KLF11, a cell death mediator, in rat brain. Neurotox Res. 2015;28(1):18–31. doi: 10.1007/s12640-015-9524-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durazzo TC, Tosun D, Buckley S, Gazdzinski S, Mon A, Fryer SL, Meyerhoff DJ. Cortical thickness, surface area, and volume of the brain reward system in alcohol dependence: relationships to relapse and extended abstinence. Alcohol Clin Exp Res. 2011;35:1187–1200. doi: 10.1111/j.1530-0277.2011.01452.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egli M, Koob GF, Edwards S. Alcohol dependence as a chronic pain disorder. Neurosci Biobehav Rev. 2012;36:2179–2192. doi: 10.1016/j.neubiorev.2012.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliash S, Dror V, Cohen S, Rehavi M. Neuroprotection by rasagiline in thiamine deficient rats. Brain Res. 2009;1256:138–148. doi: 10.1016/j.brainres.2008.11.097. [DOI] [PubMed] [Google Scholar]
- Fernandez-Zapico ME, Mladek A, Ellenrieder V, Folch-Puy E, Miller L, Urrutia R. An mSin3A interaction domain links the transcriptional activity of KLF11 with its role in growth regulation. EMBO J. 2003;22:4748–4758. doi: 10.1093/emboj/cdg470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald JC, Ufer C, Billett EE. A link between monoamine oxidase-A and apoptosis in serum deprived human SH-SY5Y neuroblastoma cells. J Neural Transm. 2007;114:807–810. doi: 10.1007/s00702-007-0692-x. [DOI] [PubMed] [Google Scholar]
- Gal S, Zheng H, Fridkin M, Youdim MB. Novel multifunctional neuroprotective iron chelator-monoamine oxidase inhibitor drugs for neurodegenerative diseases. In vivo selective brain monoamine oxidase inhibition and prevention of MPTP-induced striatal dopamine depletion. J Neurochem. 2005;95:79–88. doi: 10.1111/j.1471-4159.2005.03341.x. [DOI] [PubMed] [Google Scholar]
- Gohla G, Krieglstein K, Spittau B. Tieg3/Klf11 inducesapoptosis in OLI-neu cells and enhances the TGF-beta signaling pathway by transcriptional repression of Smad7. J Cell Biochem. 2008;104(3):850–861. doi: 10.1002/jcb.21669. [DOI] [PubMed] [Google Scholar]
- Gonzalez A, Pariente JA, Salido GM. Ethanol stimulates ROS generation by mitochondria through Ca2+ mobilization and increases GFAP content in rat hippocampal astrocytes. Brain Res. 2007;1178:28–37. doi: 10.1016/j.brainres.2007.08.040. [DOI] [PubMed] [Google Scholar]
- Groenewegen HJ, Wright CI, Uylings HBM. The anatomical relationships of the prefrontal cortex with limbic structures and the basal ganglia. J Psychopharmacol. 1997;11(2):99–106. doi: 10.1177/026988119701100202. [DOI] [PubMed] [Google Scholar]
- Grunewald M, Johnson S, Lu D, Wang Z, Lomberk G, Albert PR, Stockmeier CA, Meyer JH, Urrutia R, Miczek KA, Austin MC, Wang J, Paul IA, Woolverton WL, Seo S, Sittman DB, Ou XM. Mechanistic role for a novel glucocorticoid-KLF11 (TIEG2) protein pathway in stress-induced monoamine oxidase A expression. J Biol Chem. 2012;287:24195–24206. doi: 10.1074/jbc.M112.373936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamelink C, Hampson A, Wink DA, Eiden LE, Eskay RL. Comparison of cannabidiol, antioxidants, and diuretics in reversing binge ethanol-induced neurotoxicity. J Pharmacol Exp Ther. 2005;314:780–788. doi: 10.1124/jpet.105.085779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JY, Joo Y, Kim YS, Lee YK, Kim HJ, Cho GJ, Choi WS, Kang SS. Ethanol induces cell death by activating caspase-3 in the rat cerebral cortex. Mol Cells. 2005;20:189–195. [PubMed] [Google Scholar]
- Hayes DM, Deeny MA, Shaner CA, Nixon K. Determining the threshold for alcohol-induced brain damage: new evidence with gliosis markers. Alcohol Clin Exp Res. 2013;37:425–434. doi: 10.1111/j.1530-0277.2012.01955.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriksen JH, Gronbaek M, Moller S, Bendtsen F, Becker U. Carbohydrate deficient transferrin (CDT) in alcoholic cirrhosis: a kinetic study. J Hepatol. 1997;26:287–292. doi: 10.1016/s0168-8278(97)80043-8. [DOI] [PubMed] [Google Scholar]
- Hoover WB, Vertes RP. Anatomical analysis of afferent projections to the medial prefrontal cortex in the rat. Brain Struct Funct. 2007;212:149–179. doi: 10.1007/s00429-007-0150-4. [DOI] [PubMed] [Google Scholar]
- Johnson S, Stockmeier CA, Meyer JH, Austin MC, Albert PR, Wang J, May WL, Rajkowska G, Overholser JC, Jurjus G, Dieter L, Johnson C, Sittman DB, Ou XM. The reduction of R1, a novel repressor protein for monoamine oxidase A, in major depressive disorder. Neuropsychopharmacol Off Publ Am Coll Neuropsychopharmacol. 2011;36:2139–2148. doi: 10.1038/npp.2011.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelso ML, Liput DJ, Eaves DW, Nixon K. Upregulated vimentin suggests new areas of neurodegeneration in a model of an alcohol use disorder. Neuroscience. 2011;197:381–393. doi: 10.1016/j.neuroscience.2011.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitani K, Kanai S, Carrillo MC, Ivy GO. (−)Deprenyl increases the life span as well as activities of superoxide dismutase and catalase but not of glutathione peroxidase in selective brain regions in Fischer rats. Ann N Y Acad Sci. 1994;717:60–71. doi: 10.1111/j.1749-6632.1994.tb12073.x. [DOI] [PubMed] [Google Scholar]
- Kontkanen O, Castren E. Trophic effects of selegiline on cultured dopaminergic neurons. Brain Res. 1999;829:190–192. doi: 10.1016/s0006-8993(99)01363-3. [DOI] [PubMed] [Google Scholar]
- Ledesma JC, Miguel M, Pascual M, Guerri C, Aragon CM. Induction of brain cytochrome P450 2E1 boosts the locomotor-stimulating effects of ethanol in mice. Neuropharmacology. 2014;85:36–44. doi: 10.1016/j.neuropharm.2014.05.018. [DOI] [PubMed] [Google Scholar]
- Lee M, Song BJ, Kwon Y. Ethanol mediates cell cycle arrest and apoptosis in SK-N-SH neuroblastoma cells. J Cancer Prev. 2014;19:39–46. doi: 10.15430/jcp.2014.19.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Li Z, Ding M, Thiele CJ, Luo J. Ethanol inhibits brain-derived neurotrophic factor-mediated intracellular signaling and activator protein-1 activation in cerebellar granule neurons. Neuroscience. 2004;126(1):149–162. doi: 10.1016/j.neuroscience.2004.03.028. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Loh KP, Huang SH, De Silva R, Tan BK, Zhu YZ. Oxidative stress: apoptosis in neuronal injury. Curr Alzheimer Res. 2006;3:327–337. doi: 10.2174/156720506778249515. [DOI] [PubMed] [Google Scholar]
- Lomberk G, Mathison AJ, Grzenda A, Seo S, DeMars CJ, Rizvi S, Bonilla-Velez J, Calvo E, Fernandez-Zapico ME, Iovanna J, Buttar NS, Urrutia R. Sequence-specific recruitment of heterochromatin protein 1 via interaction with Kruppel-like factor 11, a human transcription factor involved in tumor suppression and metabolic diseases. J Biol Chem. 2012;287:13026–13039. doi: 10.1074/jbc.M112.342634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu D, Johnson C, Johnson S, Tazik S, Ou XM. The neuroprotective effect of antidepressant drug via inhibition of TIEG2-MAO B mediated cell death. Drug Discov Ther. 2008;2:289–295. [PMC free article] [PubMed] [Google Scholar]
- Magyar K, Szende B. (−)-Deprenyl, a selective MAO-B inhibitor, with apoptotic and anti-apoptotic properties. Neurotoxicology. 2004;25:233–242. doi: 10.1016/S0161-813X(03)00102-5. [DOI] [PubMed] [Google Scholar]
- Majchrowicz E. Induction of physical dependence upon ethanol and the associated behavioral changes in rats. Psychopharmacologia. 1975;43:245–254. doi: 10.1007/BF00429258. [DOI] [PubMed] [Google Scholar]
- Mallajosyula JK, Kaur D, Chinta SJ, Rajagopalan S, Rane A, Nicholls DG, Di Monte DA, Macarthur H, Andersen JK. MAO-B elevation in mouse brain astrocytes results in Parkinson’s pathology. PLoS One. 2008;3:e1616. doi: 10.1371/journal.pone.0001616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews BA, Kish SJ, Xu X, Boileau I, Rusjan PM, Wilson AA, DiGiacomo D, Houle S, Meyer JH. Greater monoamine oxidase a binding in alcohol dependence. Biol Psychiatry. 2014;75:756–764. doi: 10.1016/j.biopsych.2013.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard ME, Leasure JL. Exercise enhances hippocampal recovery following binge ethanol exposure. PLoS One. 2013;8:e76644. doi: 10.1371/journal.pone.0076644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miguel-Hidalgo JJ, Overholser JC, Meltzer HY, Stockmeier CA, Rajkowska G. Reduced glial and neuronal packing density in the orbitofrontal cortex in alcohol dependence and its relationship with suicide and duration of alcohol dependence. Alcohol Clin Exp Res. 2006;30:1845–1855. doi: 10.1111/j.1530-0277.2006.00221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuta I, Ohta M, Ohta K, Nishimura M, Mizuta E, Hayashi K, Kuno S. Selegiline and desmethylselegiline stimulate NGF, BDNF, and GDNF synthesis in cultured mouse astrocytes. Biochem Biophys Res Commun. 2000;279:751–755. doi: 10.1006/bbrc.2000.4037. [DOI] [PubMed] [Google Scholar]
- Moselhy HF, Georgiou G, Kahn A. Frontal lobe changes in alcoholism: a review of the literature. Alcohol Alcohol. 2001;36:357–368. doi: 10.1093/alcalc/36.5.357. [DOI] [PubMed] [Google Scholar]
- Nair SS, Prathibha P, Syam Das S, Kavitha S, Indira M. All trans retinoic acid (ATRA) mediated modulation of N-methyl D-aspartate receptor (NMDAR) and Kruppel like factor 11 (KLF11) expressions in the mitigation of ethanol induced alterations in the brain. Neurochem Int. 2015;83–83:41–47. doi: 10.1016/j.neuint.2015.02.007. [DOI] [PubMed] [Google Scholar]
- Naoi M, Maruyama W, Youdim MB, Yu P, Boulton AA. Anti-apoptotic function of propargylamine inhibitors of type-B monoamine oxidase. Inflammopharmacology. 2003;11:175–181. doi: 10.1163/156856003765764344. [DOI] [PubMed] [Google Scholar]
- Nixon K, Crews FT. Binge ethanol exposure decreases neurogenesis in adult rat hippocampus. J Neurochem. 2002;83:1087–1093. doi: 10.1046/j.1471-4159.2002.01214.x. [DOI] [PubMed] [Google Scholar]
- Nogales F, Rua RM, Ojeda ML, Murillo ML, Carreras O. Oral or intraperitoneal binge drinking and oxidative balance in adolescent rats. Chem Res Toxicol. 2014;27:1926–1933. doi: 10.1021/tx5002628. [DOI] [PubMed] [Google Scholar]
- Obernier JA, Bouldin TW, Crews FT. Binge ethanol exposure in adult rats causes necrotic cell death. Alcohol Clin Exp Res. 2002a;26:547–557. [PubMed] [Google Scholar]
- Obernier JA, White AM, Swartzwelder HS, Crews FT. Cognitive deficits and CNS damage after a 4-day binge ethanol exposure in rats. Pharmacol Biochem Behav. 2002b;72:521–532. doi: 10.1016/s0091-3057(02)00715-3. [DOI] [PubMed] [Google Scholar]
- Ou XM, Chen K, Shih JC. Dual functions of transcription factors, transforming growth factor-beta-inducible early gene (TIEG)2 and Sp3, are mediated by CACCC element and Sp1 sites of human monoamine oxidase (MAO) B gene. J Biol Chem. 2004;279:21021–21028. doi: 10.1074/jbc.M312638200. [DOI] [PubMed] [Google Scholar]
- Ou XM, Chen K, Shih JC. Monoamine oxidase A and repressor R1 are involved in apoptotic signaling pathway. Proc Natl Acad Sci U S A. 2006;103:10923–10928. doi: 10.1073/pnas.0601515103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou XM, Lu D, Johnson C, Chen K, Youdim MBH, Rajkowska G, Shih JC. Glyerceraldehyde-3 phosphate dehydrogenase-monoamine oxidase B-mediated cell death-induced by ethanol is prevented by rasagiline and 1R-amino-indan. Neurotox Res. 2009;16:148–159. doi: 10.1007/s12640-009-9064-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou XM, Johnson C, Lu D, Johnson S, Paul IA, Austin MC, Iyo AH, Miguel-Hidalgo JJ, Luo J, Bell RL, Grunewald M, Wang J, Sittman DB. Ethanol increases TIEG2-MAO B cell death cascade in the prefrontal cortex of ethanol-preferring rats. Neurotox Res. 2011;19:511–518. doi: 10.1007/s12640-010-9164-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou XM, Udemgba C, Wang N, Dai X, Lomberk G, Seo S, Urrutia R, Wang J, Duncan J, Harris S, Fairbanks CA, Zhang X. Diabetes-causing gene, kruppel-like factor 11, modulates the antinociceptive response of chronic ethanol intake. Alcohol Clin Exp Res. 2014;38:401–408. doi: 10.1111/acer.12258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson IA, Juorio AV, Berry MD, Zhu MY. Inhibition of monoamine oxidase-B by (−)-deprenyl potentiates neuronal responses to dopamine agonists but does not inhibit dopamine catabolism in the rat striatum. J Pharmacol Exp Ther. 1991;258:1019–1026. [PubMed] [Google Scholar]
- Paul CA, Au R, Fredman L, Massaro JM, Seshadri S, Decarli C, Wolf PA. Association of alcohol consumption with brain volume in the Framingham study. Arch Neurol. 2008;65:1363–1367. doi: 10.1001/archneur.65.10.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfefferbaum A, Sullivan EV, Mathalon DH, Lim KO. Frontal lobe volume loss observed with magnetic resonance imaging in older chronic alcoholics. Alcohol Clin Exp Res. 1997;21:521–529. doi: 10.1111/j.1530-0277.1997.tb03798.x. [DOI] [PubMed] [Google Scholar]
- Porodenko VA, Travenko EN. Biochemical research on monoamine oxidases and their significance in the diagnosis of alcoholic intoxication. Sud med ekspert. 1991;34:45–46. [PubMed] [Google Scholar]
- Ramachandran V, Watts LT, Maffi SK, Chen J, Schenker S, Henderson G. Ethanol-induced oxidative stress precedes mitochondrially mediated apoptotic death of cultured fetal cortical neurons. J Neurosci Res. 2003;74:577–588. doi: 10.1002/jnr.10767. [DOI] [PubMed] [Google Scholar]
- Rehm J, Mathers C, Popova S, Thavorncharoensap M, Teerawattananon Y, Patra J. Global burden of disease and injury and economic cost attributable to alcohol use and alcohol-use disorders. Lancet. 2009;373:2223–2233. doi: 10.1016/S0140-6736(09)60746-7. [DOI] [PubMed] [Google Scholar]
- Riederer P, Lachenmayer L, Laux G. Clinical applications of MAO-inhibitors. Curr Med Chem. 2004;11:2033–2043. doi: 10.2174/0929867043364775. [DOI] [PubMed] [Google Scholar]
- Rimondini R, Arlinde C, Sommer W, Heilig M. Long-lasting increase in voluntary ethanol consumption and transcriptional regulation in the rat brain after intermittent exposure to alcohol. FASEB J Off Publ Fed Am Soc Exp Biol. 2002;16:27–35. doi: 10.1096/fj.01-0593com. [DOI] [PubMed] [Google Scholar]
- Rueda AV, Teixeira AM, Yonamine M, Camarini R. Environmental enrichment blocks ethanol-induced locomotor sensitization and decreases BDNF levels in the prefrontal cortex in mice. Addict Biol. 2012;17:736–745. doi: 10.1111/j.1369-1600.2011.00408.x. [DOI] [PubMed] [Google Scholar]
- Sacks JJ, Roeber J, Bouchery EE, Gonzales K, Chaloupka FJ, Brewer RD. State costs of excessive alcohol consumption, 2006. Am J Prev Med. 2013;45:474–485. doi: 10.1016/j.amepre.2013.06.004. [DOI] [PubMed] [Google Scholar]
- Saravanan KS, Sindhu KM, Senthilkumar KS, Mohanakumar KP. L-deprenyl protects against rotenone-induced, oxidative stress-mediated dopaminergic neurodegeneration in rats. Neurochem Int. 2006;49:28–40. doi: 10.1016/j.neuint.2005.12.016. [DOI] [PubMed] [Google Scholar]
- Shih JC. Molecular basis of human MAO A and B. Neuropsychopharmacol. Off Publ Am Coll Neuropsychopharmacol. 1991;4:1–7. [PubMed] [Google Scholar]
- Shih JC, Chen K, Ridd MJ. Monoamine oxidase: from genes to behavior. Annu Rev Neurosci. 1999;22:197–217. doi: 10.1146/annurev.neuro.22.1.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofic E, Salkovic-Petrisic M, Tahirovic I, Sapcanin A, Mandel S, Youdim M, Riederer P. Brain catalase in the streptozotocin-rat model of sporadic Alzheimer’s disease treated with the iron chelator-monoamine oxidase inhibitor, M30. J Neural Transm. 2015;122:559–564. doi: 10.1007/s00702-014-1307-y. [DOI] [PubMed] [Google Scholar]
- Speiser Z, Mayk A, Eliash S, Cohen S. Studies with rasagiline, a MAO-B inhibitor, in experimental focal ischemia in the rat. J Neural Transm. 1999;106:593–606. doi: 10.1007/s007020050182. [DOI] [PubMed] [Google Scholar]
- Srivastava V, Buzas B, Momenan R, Oroszi G, Pulay AJ, Enoch MA, Hommer DW, Goldman D. Association of SOD2, a mitochondrial antioxidant enzyme, with gray matter volume shrinkage in alcoholics. Neuropsychopharmacol Off Publ Am Coll Neuropsychopharmacol. 2010;35:1120–1128. doi: 10.1038/npp.2009.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhakar C, Jain N, Swarup G. Sp1-like sequences mediate human caspase-3 promoter activation by p73 and cisplatin. FEBS J. 2008;275(9):2200–2213. doi: 10.1111/j.1742-4658.2008.06373.x. [DOI] [PubMed] [Google Scholar]
- Udemgba C, Johnson S, Stockmeier CA, Luo J, Albert PR, Wang J, May WL, Rajkowska G, Harris S, Sittman DB, Ou XM. The expression of KLF11 (TIEG2), a monoamine oxidase B transcriptional activator in the prefrontal cortex of human alcohol dependence. Alcohol Clin Exp Res. 2014;38:144–151. doi: 10.1111/acer.12229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Peters B, Klussmann S, Bender H, Herb A, Krieglstein K. Gene structure and evolution of Tieg3, a new member of the Tieg family of proteins. Gene. 2004;325:25–34. doi: 10.1016/j.gene.2003.09.045. [DOI] [PubMed] [Google Scholar]
- Wang Z, Spittau B, Behrendt M, Peters B, Krieglstein K. Human TIEG2/ KLF11 induces oligodendroglial cell death by downregulation of Bcl-XL expression. J Neural Transm. 2007;114:867–875. doi: 10.1007/s00702-007-0635-6. [DOI] [PubMed] [Google Scholar]
- Wei YH. Oxidative stress and mitochondrial DNA mutations in human aging. Proc Soc Exp Biol Med Soc Exp Biol Med. 1998;217:53–63. doi: 10.3181/00379727-217-44205. [DOI] [PubMed] [Google Scholar]
- Wolff SP, Dean RT. Fragmentation of proteins by free radicals and its effect on their susceptibility to enzymic hydrolysis. Biochem J. 1986;234:399–403. doi: 10.1042/bj2340399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrase J, Makris N, Braus DF, Mann K, Smolka MN, Kennedy DN, Caviness VS, Hodge SM, Tang L, Albaugh M, Ziegler DA, Davis OC, Kissling C, Schumann G, Breiter HC, Heinz A. Amygdala volume associated with alcohol abuse relapse and craving. Am J Psychiatry. 2008;165:1179–1184. doi: 10.1176/appi.ajp.2008.07121877. [DOI] [PubMed] [Google Scholar]
- Youdim MB, Amit T, Falach-Yogev M, Bar Am O, Maruyama W, Naoi M. The essentiality of Bcl-2, PKC and proteasome-ubiquitin complex activations in the neuroprotective-antiapoptotic action of the anti-Parkinson drug, rasagiline. Biochem Pharmacol. 2003a;66:1635–1641. doi: 10.1016/s0006-2952(03)00535-5. [DOI] [PubMed] [Google Scholar]
- Youdim MB, Edmondson D, Tipton KF. The therapeutic potential of monoamine oxidase inhibitors. Nat Rev Neurosci. 2006;7:295–309. doi: 10.1038/nrn1883. [DOI] [PubMed] [Google Scholar]
- Youdim MB, Kupershmidt L, Amit T, Weinreb O. Promises of novel multi-target neuroprotective and neurorestorative drugs for Parkinson’s disease. Park Relat Disord. 2014;20(Suppl 1):S132–S136. doi: 10.1016/S1353-8020(13)70032-4. [DOI] [PubMed] [Google Scholar]
- Youdim MB, Tipton KF. Rat striatal monoamine oxidase-B inhibition by l-deprenyl and rasagiline: its relationship to 2-phenylethylamine-induced stereotypy and Parkinson’s disease. Park Relat Disord. 2002;8:247–253. doi: 10.1016/s1353-8020(01)00011-6. [DOI] [PubMed] [Google Scholar]
- Youdim MBH, Amit T, Falach-Yogev M, Am OB, Maruyama W, Naoi M. The essentiality of Bcl-2, PKC and proteasome–ubiquitin complex activations in the neuroprotective-antiapoptotic action of the anti-Parkinson drug, rasagiline. Biochem Pharmacol. 2003b;66:1635–1641. doi: 10.1016/s0006-2952(03)00535-5. [DOI] [PubMed] [Google Scholar]
- Youdim MB, Gross A, Finberg JP. [N-propargyl-1R(+)-aminoindan], a selective and potent inhibitor of mitochondrial monoamine oxidase B. Br J Pharmacol. 2001;132:500–506. doi: 10.1038/sj.bjp.0703826. [DOI] [PMC free article] [PubMed] [Google Scholar]