

Abstract
Many cancers feature cellular hierarchies that are driven by tumor-initiating, cancer stem cells (CSCs) and rely on complex interactions with the tumor microenvironment. Standard cell culture conditions fail to recapitulate the original tumor architecture or microenvironmental gradients, and are not designed to retain the cellular heterogeneity of parental tumors. Here, we describe a three-dimensional culture system that supports the long-term growth and expansion of tumor organoids derived directly from glioblastoma specimens, including patient-derived primary cultures, xenografts, genetically engineered glioma models, or patient samples. Organoids derived from multiple regions of patient tumors retain selective tumorigenic potential. Furthermore, organoids could be established directly from brain metastases not typically amenable to in vitro culture. Once formed, tumor organoids grew for months and displayed regional heterogeneity with a rapidly dividing outer region of SOX2+, OLIG2+, and TLX+ cells surrounding a hypoxic core of primarily non-stem senescent cells and diffuse, quiescent CSCs. Notably, non-stem cells within organoids were sensitive to radiation therapy, whereas adjacent CSCs were radioresistant. Orthotopic transplantation of patient-derived organoids resulted in tumors displayed histological features, including single cell invasiveness, that were more representative of the parental tumor compared with those formed from patient-derived sphere cultures. In conclusion, we present a new ex vivo model in which phenotypically diverse stem and non-stem glioblastoma cell populations can be simultaneously cultured to explore new facets of microenvironmental influences and CSC biology.
Keywords: Glioblastoma, stem cell, organoid, microenvironment, glioma
INTRODUCTION
Glioblastoma patient prognosis is dismal with a median patient survival of 14–16 months (1). Our inability to effectively treat glioblastomas is due, in part, to their great heterogeneity on both the cellular and microenvironmental levels (2–4). Glioblastoma growth may be governed by stochastic or hierarchical models (5) and although these models are not mutually exclusive, recent studies suggest the presence of self-renewing, tumor-propagating CSCs (6–8). While CSCs remain controversial due to unresolved issues of enrichment markers, functional assays, and cellular origin, the importance of these cells has been supported by findings that CSCs are resistant to conventional therapies due to multiple mechanisms, including increased DNA repair (9).
Glioblastoma is a hierarchically organized cancer where stem-like tumor cells receive critical maintenance cues from their microenvironment. CSCs reside in perivascular niches where close proximity to the vasculature provides nutrients and oxygen (10). A second stem-like tumor cell population resides in hypoxic regions distal to the vasculature (11–13). Tumor stem, non-stem, and normal cells engage in bidirectional communication to provide instructional cues for the maintenance of cell state (14–16). Differentiated progeny and blood vessels stimulate CSC maintenance through production of cytokines (17), nitric oxide (15), Notch ligands (16), and extracellular matrix (2). CSCs are not passive recipients of microenvironmental cues, as CSCs stimulate angiogenesis through pro-angiogenic growth factor signaling (18), direct the differentiation of progeny (14), and possess lineage plasticity towards vascular pericytes (19). The CSC state is, therefore, plastic and can be influenced by the cellular microenvironment, contributing to the concept of both cell autonomous and extrinsically instructed CSCs.
Interrogating tumor cell-microenvironmental interactions is challenging. Genetically engineered mouse models are highly valuable resources but can also differ from human tumors due to species-specific distinctions and comparatively rapid evolution of the mouse tumors. For human models, the most accurate way to study tumor cell and environmental interactions is orthotopically in vivo, but this preservation of complexity also dramatically limits experimental control (20). To ask questions in vitro, researchers must select the desired culture conditions. Due to precedence and convenience, these conditions are generally not representative of tumor conditions in patients (i.e. atmospheric oxygen, neutral pH, superphysiologic glucose concentration, etc.). This choice influences and standardizes cellular responses, complicating experimental separation of instructive cues by intrinsic networks versus those generated by the culture environment. As culture selects cells to a relatively uniform state, it has been impossible to study long-term relationships of different cell populations growing together as they occur in a tumor. CSCs and non-stem tumor cells are generally maintained under incompatible conditions, complicating studies of crosstalk. Hypoxic cell culture using separate controlled incubators also precludes study of hypoxic-non-hypoxic cell interactions.
Recently described three-dimensional culture methods recapitulate features of in vivo cell growth, allowing self-organization, differentiation, and mixed heterogeneity to exist within the culture environment (21–29). Here, we describe a novel organoid culture system using patient-derived glioblastoma CSCs that recapitulates hypoxic gradients and stem cell heterogeneity found in tumors in vivo. Such gradients are not possible using current culture methods. This system will allow the study of heterogeneous cell-cell relationships including the co-culture of hypoxic and non-hypoxic CSCs.
MATERIALS AND METHODS
Human Cell and Organoid culture
Glioblastoma samples were obtained directly from patients undergoing resection in accordance with protocol #2559 approved by the Cleveland Clinic Institutional Review Board. Patient tissue samples were either finely minced prior to organoid formation, or were dissociated into single cell suspensions, red blood cells removed by brief hypotonic lysis, and counted for cell number and viability using trypan blue. All cells used in this work, with the exception of the mouse-derived RCAS-GFAP-tva/PDGFB cell culture, were patient-derived primary cultures and all specimens were verified by comparison of short tandem repeat (STR) analysis performed both immediately after isolation and periodically during the course of experimentation. Tumorspheres were maintained as xenografts and harvested for culture as previously described (9). Xenografts were dissociated (Tumor dissociation kit #130-095-929, Miltenyi, San Diego CA), magnetically sorted for CD133 (CD133 beads, Myltenyi) (IN528 and 387 cells only), and were cultured as tumorspheres in Neurobasal medium supplemented with EGF (R&D Systems, Minneapolis MN), bFGF (R&D Systems), B27 (Invitrogen, Carlsbad CA), glutamine (CCF media core), sodium pyruvate (Invitrogen), and antibiotics (Anti-anti, Invitrogen) (“NBM complete”). No cells were propagated more than 5 passages in culture after isolation.
Briefly, organoids were formed by suspending tumor cells in Matrigel (BD, San Jose) and forming 20 µl pearls on parafilm molds prior to culture. Organoids were cultured in 6-well or 10-cm plates, shaking in NBM complete media. Images of growing organoids were acquired using an EVOS FL Cell Imaging System (Invitrogen) for microscopic imaging, or a handheld Samsung Galaxy S4 for macroscopic imaging. (Please see Supplementary Methods S1 for detailed organoid procedures.)
Mice
All experiments involving mice were approved by the Cleveland Clinic Institutional Animal Care and Use Committee (IACUC). All methods and experiments were carried out in accordance with the approved relevant guidelines and regulations under IACUC protocol #2013-0935. For in vivo tumor formation studies, whole organoids were minced, dissociated (tumor dissociation kit, #130-095-929, Miltenyi), counted, and 50,000 cells were orthotopically xenografted into the right cortex of immunodeficient NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice. Mice were sacrificed upon display of overt phenotypic or neurological signs.
Immunofluorescence and imaging
Briefly, whole organoids were fixed, snap frozen in OCT, and sectioned at 10 µm before being probed with antibodies for immunofluorescence. DNA was detected using DAPI (1:10,000) or DRAQ5 (Invitrogen, 1:1000). Images were acquired using a Leica DM4000 B upright fluorescent microscope and mosaic images were assembled by hand using Adobe Photoshop. (Please see Supplementary Methods S1 for antibody lists and additional details.)
RESULTS
Establishment of glioblastoma organoids
Glioblastoma tumors contain variable regions of comparatively high oxygenation and nutrient levels in the perivascular space, changing to nutrient-poor, hypoxic, and necrotic regions distal to the vasculature (10,30). Organoids can grow beyond this size and are viable well in excess of observed diffusion limits. We reasoned that by utilizing organoid culture methods we could establish CSC cultures with mixed regions of oxygen-high, nutrient-high regions as well as chronic hypoxic and necrotic areas.
To initiate glioblastoma organoids, we modified an original procedure developed for cerebral organoids (25,26) (Supplementary Methods S1). For comparison, we grew parallel CSCs with identical media (NBM complete) and media replacement schedules either: a) in traditional serum-free sphere culture (Figure 1A–C,H), or b) as three-dimensional organoids (Figure 1D–F). Whereas standard tumorspheres reached a maximum size of approximately 300 µm within two weeks, organoids expanded prolifically to a size of approximately 3–4 millimeters after two months (Figure 1G). Establishment of viable organoids was effective from primary cultures using either whole tumorspheres or dissociated CSCs embedded in Matrigel (data not shown). After several months, isolated single organoids generated smaller, daughter spheres in culture, which could subsequently fuse to the main organoid. Although growth rates notably slowed over several months of culture, glioblastoma organoids can be stable and viable after more than a year of continuous culture without passaging (Figure 1I, J, 2A). Organoids can further be established from specimens of human origin and from genetically engineered glioma mouse models (Supplementary Figure S1A–D), allowing potential transgenic studies in organoids.
Figure 1.

Establishment of glioblastoma CSC organoids. A, Micrograph of IN528 tumorspheres, scale bar = 400 µm. B–E, Parallel IN528 tumorsphere (B, C) or organoid cultures (D, E) for 2 and 6 weeks, scale bar = 1000 µm. F, Mosaic image of multiple low-power (4×) microscope fields showing organoid growth and smaller satellite spheres. G, IN528 organoid prior to embedding and sectioning. U.S. nickel for scale. H – J, Images of 6-well plate wells containing IN528 tumorspheres (H) or organoids (I, J) at indicated time points.
Figure 2.
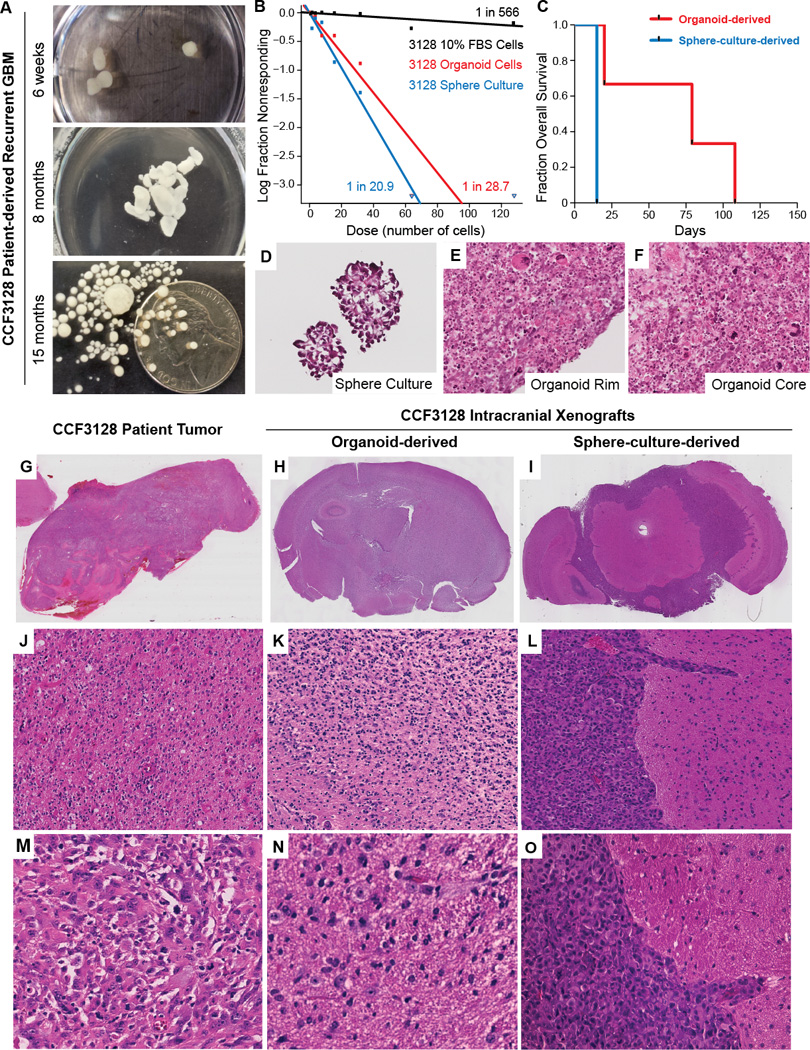
Organoid-derived glioblastoma xenografts recapitulate the diffusive phenotype of the original patient tumor. A, Longitudinal growth of CCF3128 patient-derived recurrent glioblastoma in organoid culture. B, Limiting dilution assays of organoid, tumorsphere, or FBS-differentiated CCF3128 cells. Sphere-forming cell frequencies are indicated next to each line. C, Survival plot of mice following orthotopic injection of 50,000 dissociated CCF3128 organoid or tumorsphere cells. D–F, High power micrographs of tumorsphere or organoid frozen sections (H&E, 40×). G, Low power micrograph of biopsy sample (H&E, 1×). H, Whole mount mouse brain bearing organoid derived xenograft demonstrating effacement of ventricles and asymmetric expansion of cerebral hemispheres with no clear margins of tumor (H&E, 1×). I, Whole mount mouse brain bearing tumorsphere derived xenograft showing solid growth pattern in subarachnoid space and clear margins of tumor/brain interface. (H&E, 1×). J, Intermediate power micrograph of patient biopsy specimen exhibiting a diffuse growth pattern of tumor cells with eosinophilic cytoplasm (H&E, 10×). K, Intermediate power micrograph of organoid derived xenografted tissue showing a diffuse growth pattern of tumor cells with eosinophilic cytoplasm and irregularly shaped nuclei (H&E, 10×). L, Intermediate power micrograph of tumorsphere-derived xenograft showing solid growth pattern, sharp tumor-brain interface with tumor cells growing down a the perivascular Virchow-Robin space (H&E, 10×). M, High power micrograph of patient biopsy tissue exhibiting variable amounts of eosinophilic cytoplasm with hyperchromatic irregular nuclei and pleomorphic cytoplasmic outlines (H&E, 40×). N, High power micrograph of organoid derived xenografted tissue exhibiting individual fibrillar tumor cells infiltrating into the brain substance as single cells with variable amounts of intervening brain parenchyma between the pleomorphic tumor cells (H&E, 40×). O, High power micrograph of tumorsphere-derived xenografted tissue showing solid growth pattern of basophilic tumor cells with high nuclear to cytoplasmic ratios that exhibit a sharp tumor-brain interface with infiltration along the perivascular Virchow Robin space (H&E, 40×).
Growth of tumorigenic organoids directly from primary patient samples
Avatars created from patient tumors may predict response to therapy (31). The ability to establish primary patient-derived xenografts and primary cultures from surgical specimens remains a biological, technical, and logistical barrier in tumor research. We therefore investigated whether glioblastoma organoids could be grown directly from patient tumor tissues. Fresh surgical specimens were finely minced, diluted with culture media, and mixed with Matrigel to form organoids as described above (Figure 2A). After several months of culture, we functionally quantified the stem cell population and putative tumor-initiating capabilities of these organoids using limiting dilution assays. The organoids contain sphere-forming CSCs at rates similar to, though slightly lower, than tumorspheres (Figure 2B). After 5–6 months of growth, single organoids were dissociated and orthotopically implanted into the frontal lobes of mice, which were then monitored. All recipient mice succumbed to brain tumors, with an average latency of approximately 2 months (Figure 2C), demonstrating that primary patient cells can proliferate and retain their tumorigenic potential after months of organoid culture. In comparison, the same number of tumorsphere cells established intracranial xenografts much more rapidly, with a latency of 2 weeks (Figure 2C). The conditions of tumor cell propagation prior to xenograft therefore greatly alter the growth phenotype of the subsequent xenograft tumors.
Organoids recapitulate single cell tumor invasion in vivo
Although the CCF3128 tumorspheres and organoids have similar proportions of functional sphere-forming cells, xenografts derived from these sources had very different latencies. Compared to tumorspheres, organoid cultures contained marked pleomorphism in both the core and rim regions with significant variability in size, nuclear morphology, and cytologic features (Figure 2D–F). This led us to investigate the histologic variation of the resulting xenografts (Figure 2G–O). Xenografted CCF3128 tumorsphere cells exhibited a solid growth pattern (Figure 2I, L, O) as is common for glioblastoma xenografts. In contrast, glioblastoma patient tumors are often highly diffuse and infiltrative. The original CF3128 patient specimen displayed a clear single cell infiltrative phenotype, a diffuse tumor cell growth pattern, and tumor cells with eosinophilic cytoplasm (Figure 2G, J, M). This pattern was maintained in mouse orthotopic xenografts derived from CCF3128 organoid cultures (Figure 2H, K, N). Our results demonstrate that the frequent loss of a specimen’s diffuse invasive phenotype upon culture and subsequent xenograft may not be simply due to a predisposition of the patient sample or to the xenograft format per se, but can be a result of the cell expansion conditions prior to xenograft. Expansion of samples as organoids can preserve this phenotype.
Organoid creation from brain metastases
Brain metastases can be a devastating consequence of numerous cancer types; however, patient derived brain metastases are notoriously difficult to culture in vitro. We tested in parallel whether tumorsphere or organoid culture could support the growth of a patient brain metastasis sample from esophageal adenocarcinoma. As observed historically, the brain metastasis failed to grow as spheres (Supplementary Figure S2A). In contrast, niduses of cell growth became apparent in multiple organoids and expanded to comprise the complete organoid matrix within 5 weeks (Supplementary Figure S2B). Thus, organoid culture enables the growth and study of tumor specimens that would otherwise be lost.
Derivation of organoids from multiple tumor regions
In addition to cell type heterogeneity, glioblastomas demonstrate remarkable regional heterogeneity in radiographic imaging and histology (32). Selection bias in the establishment of in vitro cultures from clinical samples can therefore be a product of the sample site of the original tumor combined with how the sampled cells will propagate under given culture conditions. Organoid culture, however, may intrinsically contain microenvironmental gradients permissive for the settling and outgrowth of tumor cell populations from diverse tumor environments.
To determine whether organoid culture allows the growth of samples from varying tumor regions, we isolated samples from three spatially distinct patient tumor regions based on MRI imaging: the potentially invasive FLAIR region, the contrast enhanced tumor zone, and the inner necrotic/hypoxic core (Figure 3A). Samples from each respective region were pathologically diagnosed as infiltrating high grade astrocytoma (CW1757.1, Figure 3B), high grade astrocytoma with scant necrosis (CW1757.2, Figure 3C), and high grade astrocytoma with necrosis, gliosis, and possible treatment effect (CW1757.3, Figure 3D).
Figure 3.
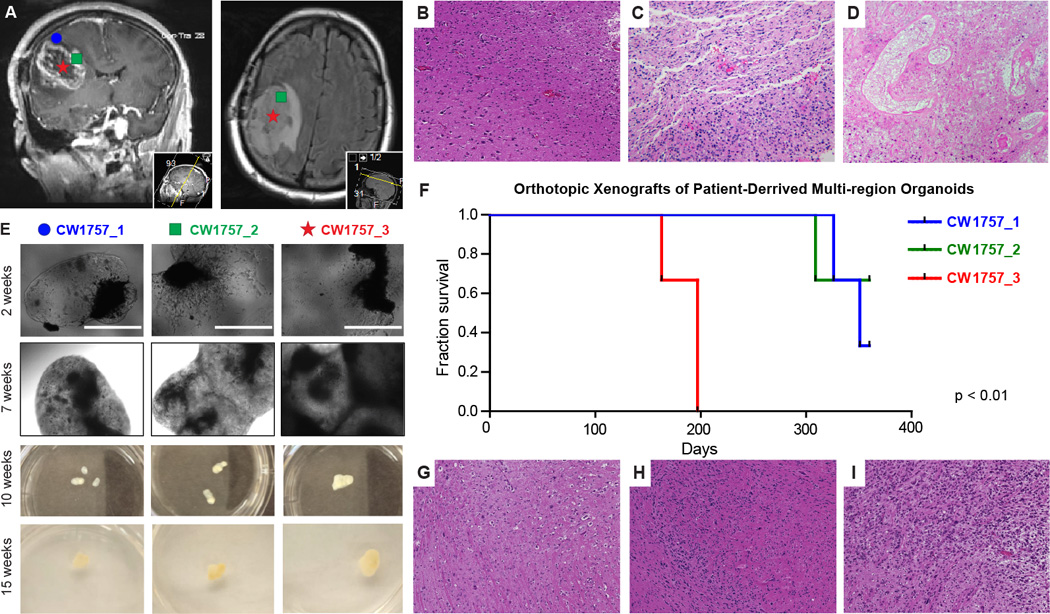
Patient-derived multi-region tumor samples. A, Based on preoperative MRI scans, surgical samples were selected from 3 distinct tumor regions for laboratory propagation. These regions were: CW1757_1 = Superficial Cortex ≥ 3mm from enhancing margin and also within hyperintense FLAIR; CW1757_2 = Enhancing margin of tumor; CW1757_3 = Tumor Center, Hypointense on T1 and non-enhancing (Typically associated with necrosis on IHC). B, High power micrograph of CW1757_1 patient derived biopsy tissue along the tumor-brain margin demonstrating mild increase in cellular density related to single cell infiltration of tumor cells (H&E, 40×). C, High power micrograph of CW1757_2 biopsy demonstrating markedly increased fibrillar tumor cellular density with variable amounts of intercellular eosinophilic brain parenchyma and no geographic necrosis (H&E, 40×). D, High power micrograph of CW1757_3 biopsy demonstrating regions of geographic necrosis (centrally) consistent with therapeutic effect rimmed by viable cells of unknown histology (H&E, 40×). E, CW1757_X specimens grown directly in organoid format for the indicated culture periods in 6-well plates, scale bar = 1000 µm. F, Kaplan-Meier survival analysis of mice bearing orthotopic xenografts from dissociated organoids originating from each tumor region in panel E. G, High power micrograph of organoid derived xenografted tumor tissue along the tumor-brain interface showing moderately increased cellular density and an indistinct tumor margin related to single cell infiltration of tumor cells into the surrounding brain, a feature also found in the biopsy tissue (B above) (H&E, 40×). H, High powered micrograph showing cellular density varying from moderate to high as detected by the variable amounts of basophilic nuclei and eosinophilic cytoplasm (H&E 40×). I, High power micrograph demonstrating a region of predominately high cellular density associated with hyperchromatic and pleomorphic basophilic nuclei, a region that differs markedly from the other regions of the tumor xenograft shown in G and H (H&E 40×).
Samples from each region were macrodissected, finely minced, and cultured as organoids. Within two weeks, cells from all three regions visibly invaded the Matrigel, filling its boundaries by 7 weeks and growing stably for months (Figure 3E). To evaluate tumor initiation capability after organoid culture, we dissociated and orthotopically xenografted organoids from each region. Although all three regions were cultured identically, cells derived from the necrotic tumor core, reinitiated tumors dramatically faster than the other two regions (Figure 3F). Cells with tumorigenic potential were maintained from all regions, but the stark contrast between region 3 and regions 1–2 demonstrates that organoids maintain functionally divergent tumor cell populations from distinct tumor regions. These organoid-derived xenografts also demonstrated marked heterogeneity and reproduced the indistinct margins and single cell infiltration present in the original patient sample (Figure 3G–I), reflecting the results from the organoid-derived CCF3128 xenografts above (Figure 2H, K, N). Organoids therefore represent a new tool for the growth and maintenance of diverse tumor populations, including infiltrative cells, in vitro.
Tumor organoids generate gradients of stem cell density and hypoxia
Limitations in oxygen and nutrient availability stimulate glioblastoma self-renewal and promote maintenance of a stem-like cell state (11,33). We therefore hypothesized that organoids would have spatial differences in the populations of stem-like and differentiated cell populations present. SOX2 is a transcription factor highly associated with pluripotency (34), a Yamanaka factor capable of iPS cell reprograming (35), and a well-described marker of glioma CSCs (36,37). Glioma CSC tumorspheres almost universally express SOX2, limiting the ability to study heterogeneous populations simultaneously. We performed immunofluorescent analysis of SOX2 to visualize CSC localization in organoids from three independent patient tumor specimens. SOX2 expression was notably denser near the periphery of each organoid and transitioned to punctate staining deeper in the core (Figure 4A–C). Organoid centers often contained a mix of non-cellular areas filled with fluid or extracellular matrix, and cellular areas with a mix of both stem and non-stem cells spread throughout. Cells near the edges of the organoids were almost uniformly stem-marker positive, perhaps due to the proximity to stimulatory growth factors (EGF, bFGF). These patterns were stable at varying time points (Supplementary Figure S3A–C) including after many months (Figure 4C).
Figure 4.
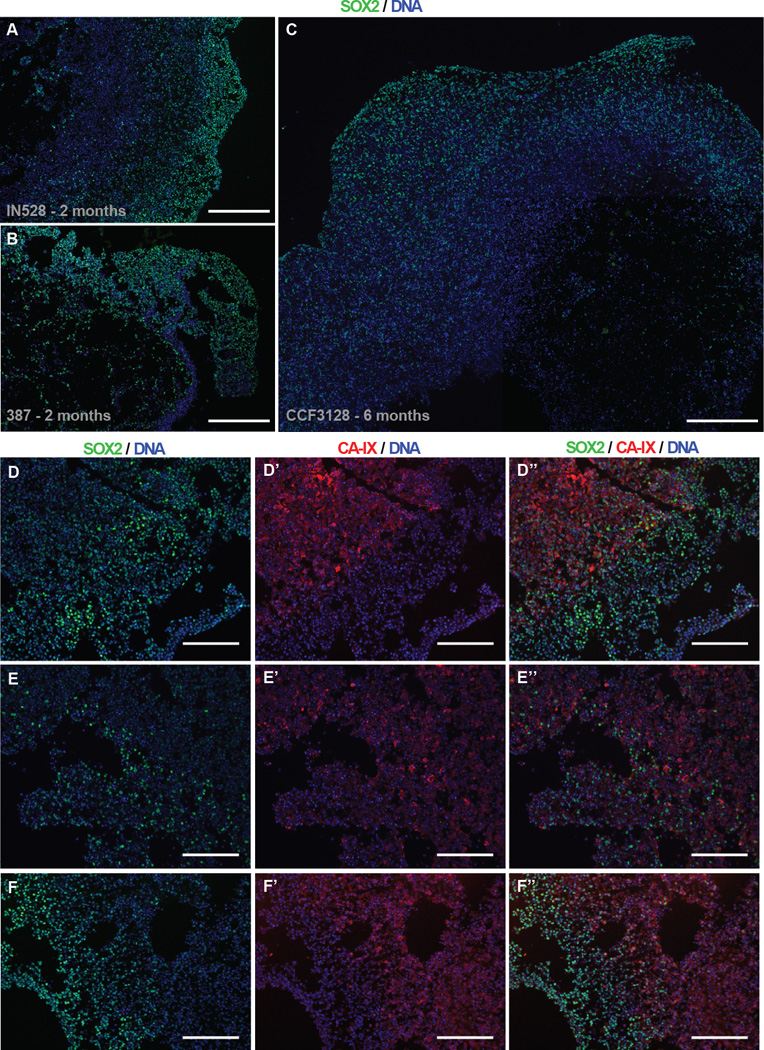
Inverse Gradients of Stem Cell Frequency and Hypoxia in Organoids. A–C, Wide field immunofluorescence imaging of nuclear SOX2 protein in IN528 (a), 387 (b), and CCF3128 (c, mosaic) organoids. Scale bars = 400 µm. D–F, Co-immunofluorescence of SOX2 and CA-IX near the edges of IN528 organoids. Scale bars = 200 µm.
We next evaluated the presence of hypoxia within organoids using antibody detection of carbonic anhydrase IX (CA-IX), a functional hypoxic marker expressed by cells in response to low oxygen conditions (38). Hypoxic gradients were observed within the organoids (Figure 4E’–G’) and strikingly, the presence of CA-IX immunofluorescence highly correlated with the spatial reduction of SOX2 expression (Figure 4E”–G”). Although the frequency of SOX2+ cells comparatively decreased crossing the hypoxic boundary, a minority of intensely SOX2+ cells remained present among negative cells throughout hypoxic areas. Thus, the influence of hypoxia upon stem state in these conditions is large, but not absolute. Furthermore, multiple molecularly distinct SOX2+ stem populations may be present within each organoid – one that requires high oxygen and growth factors to maintain its stem state (perhaps mimicking those in the perivascular niche), and one set that retains stem-like features despite resource-scarce microenvironmental conditions.
CSCs near the surface of organoids divide and die frequently whereas hypoxic CSCs are quiescent
Stem-state and hypoxia both influence cell division rates. We, therefore, evaluated the frequency of cell division across organoids using Ki-67 as a marker of proliferation. Proliferating cells were primarily localized near the organoid rim (Figure 5A). Although most activity was observed in the periphery, rare CSCs in the hypoxic core also underwent division (Figure 5B’). Although peripheral dividing cells were variably positive for SOX2 (Figure 5B”), the rare core Ki-67+ cells were typically strongly SOX2+ (Figure 5B’, arrow). The apoptotic indicator cleaved Caspase 3 was also comparatively frequent in the outer organoid rim (Figure 5C–E), suggesting that this rapidly cycling cell population also has a higher turnover rate. In contrast, CSCs in the hypoxic core rarely cycle and rarely undergo apoptosis, reminiscent of a slow-cycling stem cell population, although accepted immunologic markers have yet to be described for such cells.
Figure 5.
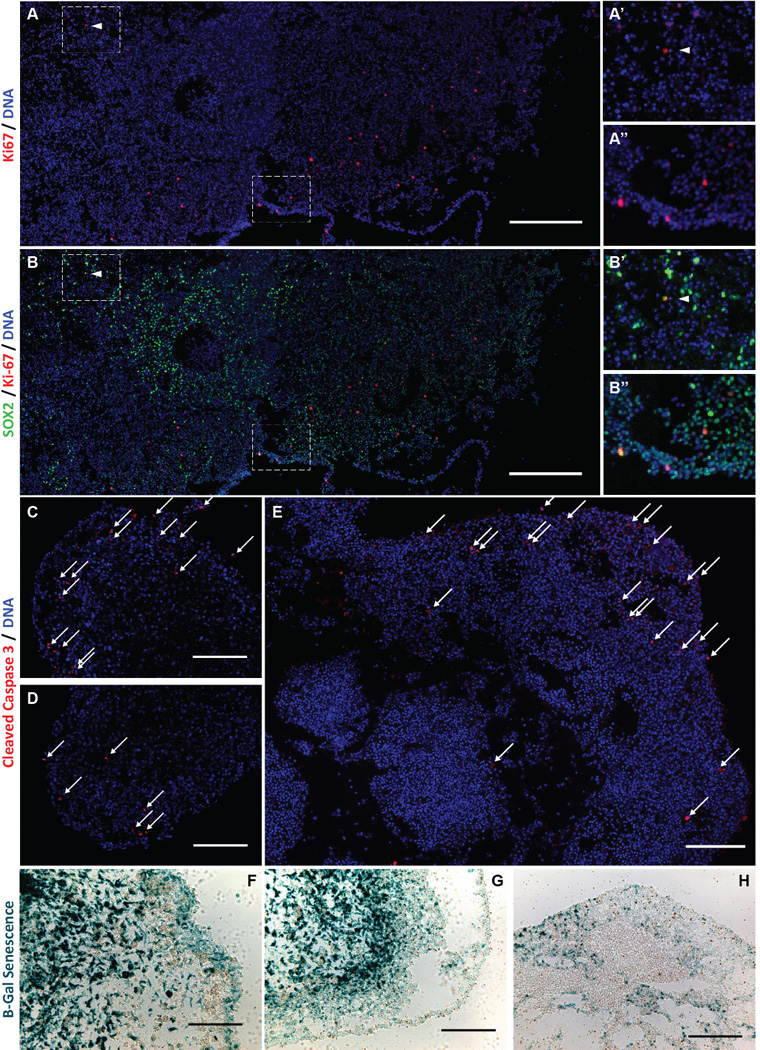
Spatial and phenotypic cellular heterogeneity in organoids. A, B, Immunofluorescence mosaic imaging of Ki-67 protein and SOX2 in IN528 organoids, scale bars = 400µ. Insets (A’–B”) are magnified regions of the mosaic span as indicated by dashed boxes. C–E, Immunofluorescence imaging specific to cleaved Caspase 3 protein in IN528 (C, D) and 387 (E) organoids. White arrows indicate positive cells, scale bars = 200µm. F–H, Light micrographs of X-Gal detection of Senesence-associated β-galactosidase in IN528 (F, G) and 387 (H) organoids. Scale bars = 100 µm (F), 200 µm (G), 400 µm (H).
In complement to proliferative rates, we also investigated cellular senescence within organoids. Although no reliable protein markers for senescence have been described in glioblastoma, the presence of beta-galactosidase (β-gal) activity (pH 6.5) is an indicator of senescence (39). X-gal staining marked β-gal positive senescent cells present throughout tumor organoids (Figure 5F–H). Senescent cells were most frequent throughout the inner hypoxic regions, but also present at a moderate frequency throughout the proliferative rim including at the outer edge. These results suggest that a cell’s choice to become senescent is not simply a response to microenvironmental gradients but can also be made amidst proliferative stimuli. Thus, senescent cells can be maintained in organoid culture for months, enabling their study amidst proliferating cells that would overwhelm them in standard culture methods.
CSCs in organoids are radioresistant whereas adjacent non-stem tumor cells are radiosensitive
Radiation therapy is a standard of care for glioblastoma patients, but CSCs are more resistant to radiation than their non-stem progeny (9). Glioblastoma organoids contain adjacent stem and non-stem cells and maintain tumor-like microenvironmental gradients. We therefore investigated whether heterogeneous radiation sensitivity is present throughout glioblastoma organoids. After irradiation, Caspase 3 cleavage was significantly increased in glioblastoma cells around the organoid rim, but not within the organoid core (Figure 6A, C, F). These apoptotic cells were also almost exclusively SOX2 negative (non-stem) (Figure 6D, G). These findings functionally validate the ability of SOX2+ cells within the organoid to display a common CSC phenotype – radioresistance. They also demonstrate the potential for organoids to be used as a screening tool to identify sensitizers to standard of care therapies.
Figure 6.
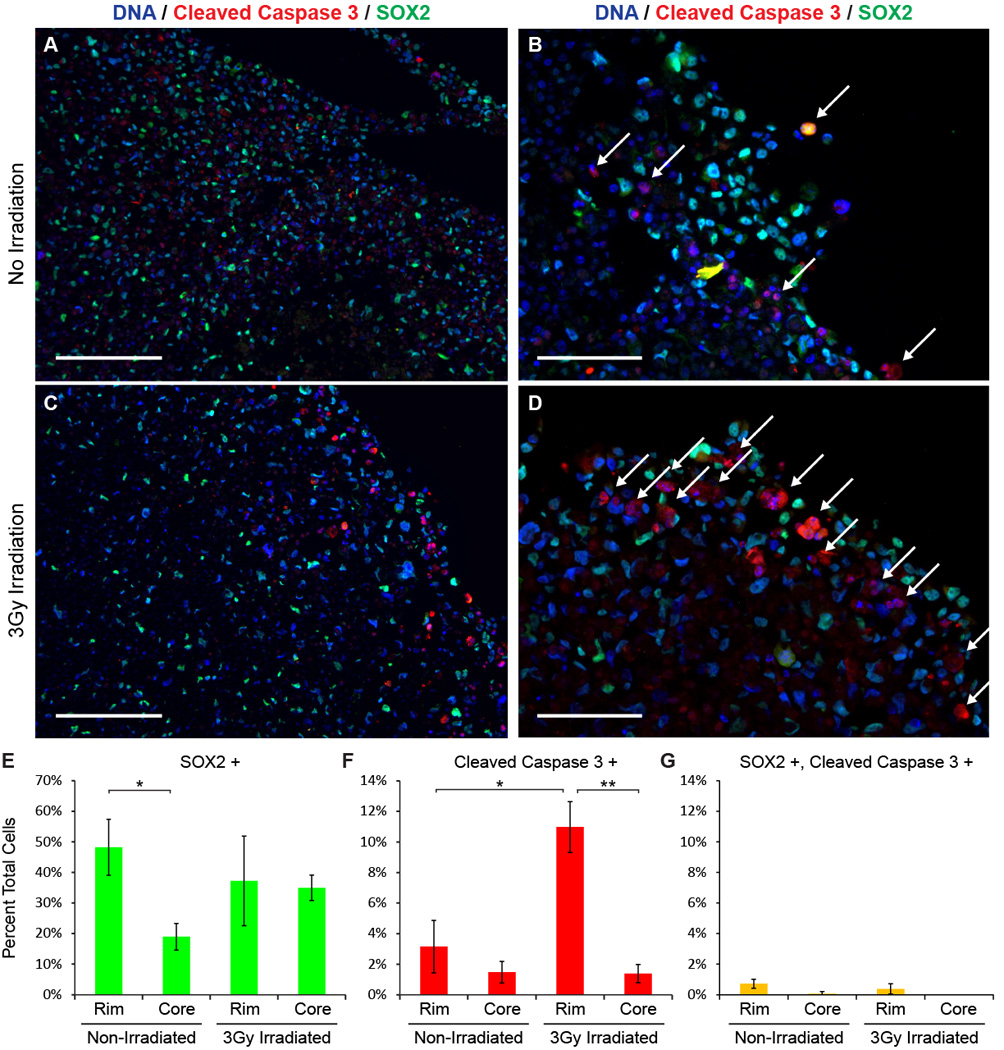
Organoid rim non-CSCs are radiosensitive. A–D, Immunofluorescence imaging of cleaved Caspase 3 protein and SOX2 protein in IN528 organoids 96 hours after 3 Gy irradiation. White arrows indicate cleaved Caspase 3 positive cells. Scale bars = 200 µm (A, C) or 100 µm (B, D). E–G, Total cells and cells positive for SOX2 (E), cleaved Caspase 3 (F), and both (G) were blindly counted from 3 non-overlapping high power fields within the indicated organoid regions. Student's t-Test; *, p < 0.02; **, p < .001.
CSCs in organoids heterogeneously co-express stem cell markers SOX2, OLIG2, and TLX
The perivascular and hypoxic CSC niches in brain tumors display distinct molecular regulation (30). Our findings suggest that there may be molecularly and functionally distinct SOX2+ CSC populations within organoids. We therefore investigated the co-expression of additional stem cell markers potentially important in CSC biology. The orphan nuclear receptor tailless (TLX) is a mediator of self-renewal and long term proliferation of embryonic and neural stem cells (NSCs) and may be critical in the initiation of glioma (40,41). In a genetically engineered mouse model, TLX plays an essential role in gliomagenesis and may mark a quiescent CSC population, distinct from the SOX2+ cells, which are proliferative (42,43). Based on these findings, we investigated whether TLX was differentially expressed within potential slow-cycling CSCs of the hypoxic core. Surprisingly, rather than marking rare hypoxic cells, TLX immunofluorescence was nearly universal among SOX2-positive cells (Figure 7A–A’”). TLX and SOX2 staining overwhelmingly coincided in the proliferative rim, hypoxic core, and in CSC nests among differentiated tumor cells (Figure 7A’–7A’”). This suggests that TLX function may be distinct in human gliomas compared to previous mouse models. The correspondence between TLX and SOX2 expression in CSCs parallels other work in adult NSCs showing a transcriptional network where SOX2 directly regulates TLX transcription and TLX and SOX2 form transcription complex at TLX-controlled genes(44). In light of these data, our staining pattern suggests that the SOX2 and TLX regulatory networks in our CSCs may have similarities to TLX regulation in adult NSCs.
Figure 7.
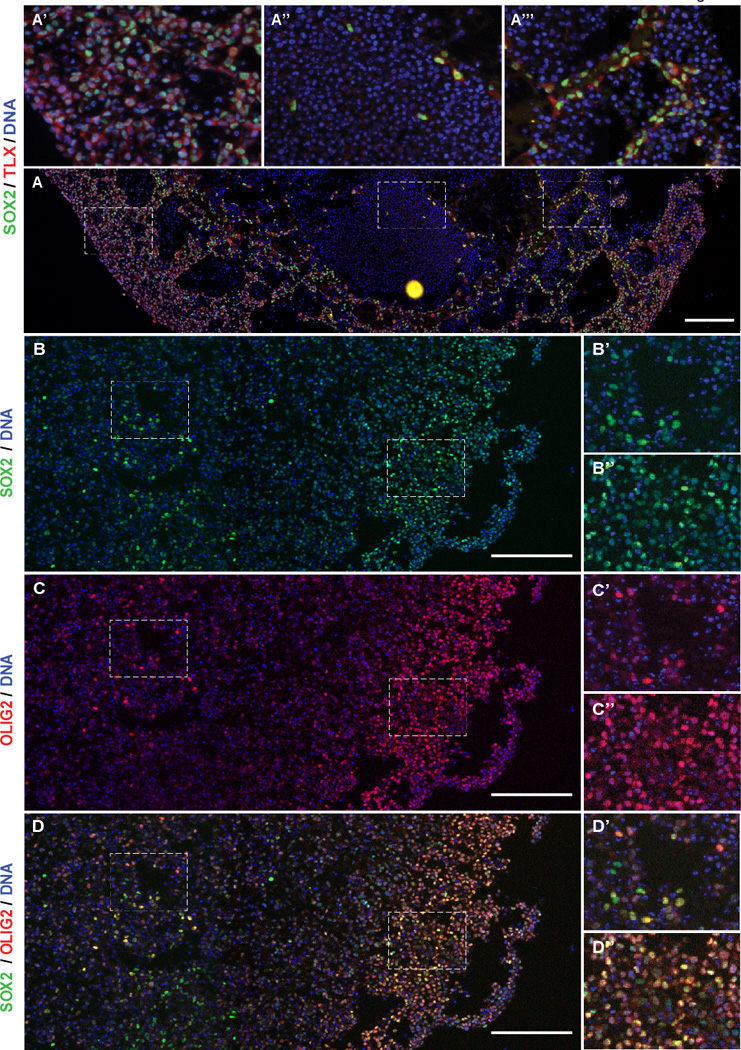
Partially overlapping stem cell marker expression in glioblastoma organoids. A, Immunofluorescence mosaic imaging of SOX2 and TLX protein in 387 organoids. Scale bars = 200 µm. Insets (a’–a’”) are magnified regions of the mosaic span as indicated by dashed boxes. B–D, Immunofluorescence mosaic imaging of SOX2 and OLIG2 protein expression in IN528 organoids. Insets (B’–D”) are magnified regions of the mosaic span as indicated by dashed boxes. Scale bars = 200 µm.
The basic helix-loop-helix transcription factor OLIG2 controls replication competence in NSCs and glioblastoma, is preferentially expressed in CSCs, is required for glioblastoma initiation in mouse models, and is one of four factors capable of reprograming glioma cells to a CSC state (36,45,46). CSC tumorspheres are almost universally positive for SOX2 and for OLIG2 (Supplementary Figure S4A–D). We investigated whether OLIG2 is co-expressed with SOX2 in organoids and found that such co-expression is common in both inner and outer organoid regions, but to varying degrees (Figure 7B–D, Supplementary Figure S4E). In the CSC-rich proliferative rim, the vast majority of cells co-expressed OLIG2 and SOX2 (Figure 7D”). SOX2/OLIG2 double-positive cells were still frequent amongst CSCs in the hypoxic core, but strongly single-positive cells were frequently present as well (Figure 7D’). Once away from the proliferative rim, this heterogeneity extends deep within the organoids (Supplementary Figure S5). These findings suggest that separate organoid regions may harbor distinct but overlapping CSC subpopulations.
DISCUSSION
Three-dimensional organoid culture systems permit a complex structure to develop, mimicking organ development (21,26,29). While some cancers display spatial orientation relative to the stem cell niche, gliomas generate neo-niches that lack coherent organization. We investigated the spatial distribution of CSCs within organoids and observed, similar to patient tumors, little evidence of self-organization or higher-level patterning. Small stem cell nests were present in some organoid cultures (Figure 7A’”, Supplementary Figure S4E), but gradients of stem cell markers appear primarily driven by resource diffusion with SOX2-positive cells densely located near the organoid periphery with the highest exposure to oxygen, nutrients, and media growth factors. This region could be viewed as the in vitro equivalent of the perivascular niche, whereas the organoid interior is a mimic of the resource-poor hypoxic niche in vivo. This analogy is supported by the inverse relationship between SOX2+ frequency and regions staining for the functional hypoxic marker CA-IX (Figure 4D–F). Interestingly, this gradient is not linear. Although markedly fewer cells are SOX2+ within hypoxic regions, the punctate distribution of strongly SOX2+ cells remained throughout the organoid interior (Supplementary Figure S5). This inverse relationship of markers from rim to center combined with clear nuclear localization of the SOX2 transcription factor shows that these results are not a technical artifact such as “edge effect” or incomplete fixation. This biological relationship suggests a cellular choice driven by a threshold rather than a simple reflection of resource diffusion.
We further investigated the relationship between SOX2 and expression of another key glioma stem cell gene, OLIG2 (36,45,46). Overlap between SOX2 and OLIG2 expression is particularly prominent in the proliferative rim of glioblastoma organoids whereas CSCs in the core are more heterogeneous in their expression or co-expression of these CSC markers (Figure 7B–D). This suggests that tumor organoids may harbor distinct molecular subpopulations within the stem cell hierarchy and that the distribution of these cells may be influenced by their microenvironment. We see similar stem cell behavior in tumors in vivo where CSCs are enriched within perivascular niches yet also reside in hypoxic niches far from the vasculature. The microenvironmental gradients present in organoids allow similar heterogeneity of CSCs growing simultaneously. The molecular profiles of these CSC subtypes as well as their influence upon each other and their non-stem glioblastoma cell neighbors in vivo or in organoid culture have yet to be determined.
NSCs are long-lived and quiescent with the proliferative burden arising from more lineage committed transit-amplifying cells. In contrast, in most cell culture methods including neural stem cell and glioblastoma stem cell cultures, rapidly proliferating populations outcompete slow cycling, quiescent, or senescent cells. The influence of these populations in scientific experiments is thus limited. In organoids, the vast majority of proliferative activity (as determined by Ki-67 staining) was localized to the peripheral rim (Figure 5A). This is not surprising as this region contains the highest levels of growth factors, oxygen, and nutrients, all of which are required for proliferative activity. These cells are variably positive for the stem cell marker SOX2, suggesting the proliferation of both stem-like and more differentiated (such as transit-amplifying) cell populations in this region (Figure 5B”). A rare population of SOX2+ cells in the hypoxic core also showed clear Ki-67 positivity (Figure 5B’). Furthermore, staining for cleaved Caspase 3 revealed relatively high apoptotic rates in the organoid periphery compared to the core (Figure 5C–E). This suggests that the proliferative edge is a region of high cell turnover surrounding a more stable core, rather than the presence of a conveyor-belt-like effect from edge proliferation to core death.
Cytotoxic cancer therapies may be less effective in slower cycling or non-proliferative cancer cells. The above finding may indicate a long-lived population of SOX2+ cells in the core which have retained the ability to divide but do so very infrequently, as would be expected for a slow-cycling or quiescent stem cell. The presence of senescent glioblastoma cells, in concert with the potential presence of quiescent CSCs in tumor organoids suggests that organoids may reflect greater complexity of heterogeneous tumor cell populations compared to other culture techniques. For instance, we have demonstrated radiosensitive non-stem cells and radioresistant CSCs growing adjacently within the organoid rim (Figure 6). This preserved heterogeneity underscores a potential utility of organoids derived directly from patients for screening assays, the study of therapies and therapeutic resistance, and in the development of patient avatar models.
We have shown the striking ability of organoid-derived xenografts from multiple patient samples and tumor regions to recapitulate the single-cell infiltrative phenotype of the original patient tumor (Figure 2H, K, N; Figure 3 G–I) whereas tumorsphere-derived xenografts produce a solid mass (Figure 2I, L, O). This finding suggests that organoid culture may preserve a patient tumor cell population capable of reproducing this infiltrative phenotype. Organoid culture may therefore be essential to recapitulate and study this deadly component of glioblastoma. Infiltrative mouse xenografts derived from organoids and solid xenograft tumors derived from tumorspheres may also have different responses to candidate therapeutics. Parallel organoid- and tumorsphere-derived xenograft animals may therefore complement each other in preclinical trials.
Organoid growth has a different spectrum of benefits and drawbacks compared to tumorspheres or to xenografts (Supplemental Table T1). Organoids are more time consuming at the time of establishment than tumorspheres and may be impractical for high-throughput screening efforts. Compared to tumorspheres, organoids have low and regionally variable cellular growth rates. Also in contrast to traditional culture methods, the cellular heterogeneity present in organoids could obscure molecular responses to experimental perturbations if the organoid is lysed in bulk prior to analysis (i.e., for immunoblot or qPCR assay).
The cellular and microenvironmental heterogeneity within tumor organoids enables the simultaneous culture of functionally and phenotypically diverse stem and non-stem glioblastoma cell populations. Neither in vivo xenograft propagation nor any currently described culture systems can similarly pair such cellular diversity and microenvironmental gradients with the experimental control of in vitro culture. Organoid culture may allow the study of stem and non-stem glioblastoma cell populations in the same culture, the investigation of interactions between CSCs in proliferative and hypoxic niches, and the further dissection of subpopulations within the glioblastoma stem cell hierarchy. Future studies utilizing organoids may empower medium-throughput drug screening with the ability to detect mixed responses that are often discovered only in clinical trials. Combinatorial studies in organoid format may circumvent the feasibility barrier posed by in vivo studies. The creation of organoids may permit more reliable models for precision medicine, even from tumor types such as like cerebral metastases that are challenging to perpetuate. Finally, genetically engineered organoids have been developed to interrogate the genetic initiation and progression of tumors in the gastrointestinal system (47). Our studies suggest such models are feasible in brain tumors as well.
Supplementary Material
Acknowledgments
Financial Support: This work was supported by The National Institutes of Health grants CA189647 (C.G.H.), CA154130, CA171652, CA169117, NS087913, NS089272, CA197718 (J.N.R), Research Programs Committees of Cleveland Clinic (J.N.R), and James S. McDonnell Foundation (J.N.R). A.E.S. is supported by the Peter D. Cristal Chair in Neurosurgical Oncology as well as the Kimble endowment for Neurosurgical Oncology. S.C.M. is supported by a CIHR Banting Fellowship.
We would like to thank our funding sources and members of the Rich Lab for their support. We thank the Cleveland Clinic Imaging, Histology, and Flow Cytometry Cores and Biological Resources Unit for their assistance and technical expertise. We thank the laboratory of Justin Lathia for the gift of the RCAS-GFAP-tva/PDGFB cell culture.
Footnotes
Competing financial interests: The authors declare no competing financial interests.
REFERENCES
- 1.Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. The lancet oncology. 2009;10(5):459–466. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 2.Lathia JD, Heddleston JM, Venere M, Rich JN. Deadly teamwork: neural cancer stem cells and the tumor microenvironment. Cell stem cell. 2011;8(5):482–485. doi: 10.1016/j.stem.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meyer M, Reimand J, Lan X, Head R, Zhu X, Kushida M, et al. Single cell-derived clonal analysis of human glioblastoma links functional and genomic heterogeneity. Proceedings of the National Academy of Sciences of the United States of America. 2015;112(3):851–856. doi: 10.1073/pnas.1320611111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science (New York, NY. 2014;344(6190):1396–1401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vescovi AL, Galli R, Reynolds BA. Brain tumour stem cells. Nature reviews. 2006;6(6):425–436. doi: 10.1038/nrc1889. [DOI] [PubMed] [Google Scholar]
- 6.Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, De Vitis S, et al. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer research. 2004;64(19):7011–7021. doi: 10.1158/0008-5472.CAN-04-1364. [DOI] [PubMed] [Google Scholar]
- 7.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al. Identification of human brain tumour initiating cells. Nature. 2004;432(7015):396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 8.Hemmati HD, Nakano I, Lazareff JA, Masterman-Smith M, Geschwind DH, Bronner-Fraser M, et al. Cancerous stem cells can arise from pediatric brain tumors. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(25):15178–15183. doi: 10.1073/pnas.2036535100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444(7120):756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 10.Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B, et al. A perivascular niche for brain tumor stem cells. Cancer cell. 2007;11(1):69–82. doi: 10.1016/j.ccr.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 11.Li Z, Bao S, Wu Q, Wang H, Eyler C, Sathornsumetee S, et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer cell. 2009;15(6):501–513. doi: 10.1016/j.ccr.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seidel S, Garvalov BK, Wirta V, von Stechow L, Schanzer A, Meletis K, et al. A hypoxic niche regulates glioblastoma stem cells through hypoxia inducible factor 2 alpha. Brain. 2010;133(Pt 4):983–995. doi: 10.1093/brain/awq042. [DOI] [PubMed] [Google Scholar]
- 13.Bar EE, Lin A, Mahairaki V, Matsui W, Eberhart CG. Hypoxia increases the expression of stem-cell markers and promotes clonogenicity in glioblastoma neurospheres. Am J Pathol. 2010;177(3):1491–1502. doi: 10.2353/ajpath.2010.091021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yan K, Wu Q, Yan DH, Lee CH, Rahim N, Tritschler I, et al. Glioma cancer stem cells secrete Gremlin1 to promote their maintenance within the tumor hierarchy. Genes Dev. 2014;28(10):1085–1100. doi: 10.1101/gad.235515.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Charles N, Ozawa T, Squatrito M, Bleau AM, Brennan CW, Hambardzumyan D, et al. Perivascular nitric oxide activates notch signaling and promotes stem-like character in PDGF-induced glioma cells. Cell stem cell. 2010;6(2):141–152. doi: 10.1016/j.stem.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhu TS, Costello MA, Talsma CE, Flack CG, Crowley JG, Hamm LL, et al. Endothelial cells create a stem cell niche in glioblastoma by providing NOTCH ligands that nurture self-renewal of cancer stem-like cells. Cancer research. 2011;71(18):6061–6072. doi: 10.1158/0008-5472.CAN-10-4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang H, Lathia JD, Wu Q, Wang J, Li Z, Heddleston JM, et al. Targeting interleukin 6 signaling suppresses glioma stem cell survival and tumor growth. Stem cells (Dayton, Ohio) 2009;27(10):2393–2404. doi: 10.1002/stem.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bao S, Wu Q, Sathornsumetee S, Hao Y, Li Z, Hjelmeland AB, et al. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer research. 2006;66(16):7843–7848. doi: 10.1158/0008-5472.CAN-06-1010. [DOI] [PubMed] [Google Scholar]
- 19.Cheng L, Huang Z, Zhou W, Wu Q, Donnola S, Liu JK, et al. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell. 2013;153(1):139–152. doi: 10.1016/j.cell.2013.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Becher OJ, Holland EC. Genetically engineered models have advantages over xenografts for preclinical studies. Cancer research. 2006;66(7):3355–3358. doi: 10.1158/0008-5472.CAN-05-3827. discussion 58-9. [DOI] [PubMed] [Google Scholar]
- 21.Shamir ER, Ewald AJ. Three-dimensional organotypic culture: experimental models of mammalian biology and disease. Nat Rev Mol Cell Biol. 2014;15(10):647–664. doi: 10.1038/nrm3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boj SF, Hwang CI, Baker LA, Chio II, Engle DD, Corbo V, et al. Organoid models of human and mouse ductal pancreatic cancer. Cell. 2015;160(1–2):324–338. doi: 10.1016/j.cell.2014.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McCracken KW, Cata EM, Crawford CM, Sinagoga KL, Schumacher M, Rockich BE, et al. Modelling human development and disease in pluripotent stem-cell-derived gastric organoids. Nature. 2014;516(7531):400–404. doi: 10.1038/nature13863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smith SJ, Wilson M, Ward JH, Rahman CV, Peet AC, Macarthur DC, et al. Recapitulation of tumor heterogeneity and molecular signatures in a 3D brain cancer model with decreased sensitivity to histone deacetylase inhibition. PLoS One. 2012;7(12):e52335. doi: 10.1371/journal.pone.0052335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lancaster MA, Knoblich JA. Generation of cerebral organoids from human pluripotent stem cells. Nat Protoc. 2014;9(10):2329–2340. doi: 10.1038/nprot.2014.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lancaster MA, Renner M, Martin CA, Wenzel D, Bicknell LS, Hurles ME, et al. Cerebral organoids model human brain development and microcephaly. Nature. 2013;501(7467):373–379. doi: 10.1038/nature12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gao D, Vela I, Sboner A, Iaquinta PJ, Karthaus WR, Gopalan A, et al. Organoid cultures derived from patients with advanced prostate cancer. Cell. 2014;159(1):176–187. doi: 10.1016/j.cell.2014.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karthaus WR, Iaquinta PJ, Drost J, Gracanin A, van Boxtel R, Wongvipat J, et al. Identification of multipotent luminal progenitor cells in human prostate organoid cultures. Cell. 2014;159(1):163–175. doi: 10.1016/j.cell.2014.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459(7244):262–265. doi: 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- 30.Mohyeldin A, Garzon-Muvdi T, Quinones-Hinojosa A. Oxygen in stem cell biology: a critical component of the stem cell niche. Cell stem cell. 2010;7(2):150–161. doi: 10.1016/j.stem.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 31.Joo KM, Kim J, Jin J, Kim M, Seol HJ, Muradov J, et al. Patient-specific orthotopic glioblastoma xenograft models recapitulate the histopathology and biology of human glioblastomas in situ. Cell Rep. 2013;3(1):260–273. doi: 10.1016/j.celrep.2012.12.013. [DOI] [PubMed] [Google Scholar]
- 32.Cloughesy TF, Cavenee WK, Mischel PS. Glioblastoma: from molecular pathology to targeted treatment. Annu Rev Pathol. 2014;9:1–25. doi: 10.1146/annurev-pathol-011110-130324. [DOI] [PubMed] [Google Scholar]
- 33.Flavahan WA, Wu Q, Hitomi M, Rahim N, Kim Y, Sloan AE, et al. Brain tumor initiating cells adapt to restricted nutrition through preferential glucose uptake. Nat Neurosci. 2013;16(10):1373–1382. doi: 10.1038/nn.3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Young RA. Control of the embryonic stem cell state. Cell. 2011;144(6):940–954. doi: 10.1016/j.cell.2011.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 36.Suva ML, Rheinbay E, Gillespie SM, Patel AP, Wakimoto H, Rabkin SD, et al. Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell. 2014;157(3):580–594. doi: 10.1016/j.cell.2014.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de la Rocha AM, Sampron N, Alonso MM, Matheu A. Role of SOX family of transcription factors in central nervous system tumors. Am J Cancer Res. 2014;4(4):312–324. [PMC free article] [PubMed] [Google Scholar]
- 38.Potter C, Harris AL. Hypoxia inducible carbonic anhydrase IX, marker of tumour hypoxia, survival pathway and therapy target. Cell cycle (Georgetown, Tex. 2004;3(2):164–167. [PubMed] [Google Scholar]
- 39.Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(20):9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Qu Q, Sun G, Li W, Yang S, Ye P, Zhao C, et al. Orphan nuclear receptor TLX activates Wnt/beta-catenin signalling to stimulate neural stem cell proliferation and self-renewal. Nat Cell Biol. 2010;12(1):31–40. doi: 10.1038/ncb2001. sup pp 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zou Y, Niu W, Qin S, Downes M, Burns DK, Zhang CL. The nuclear receptor TLX is required for gliomagenesis within the adult neurogenic niche. Mol Cell Biol. 2012;32(23):4811–4820. doi: 10.1128/MCB.01122-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu HK, Wang Y, Belz T, Bock D, Takacs A, Radlwimmer B, et al. The nuclear receptor tailless induces long-term neural stem cell expansion and brain tumor initiation. Genes Dev. 2010;24(7):683–695. doi: 10.1101/gad.560310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhu Z, Khan MA, Weiler M, Blaes J, Jestaedt L, Geibert M, et al. Targeting self-renewal in high-grade brain tumors leads to loss of brain tumor stem cells and prolonged survival. Cell stem cell. 2014;15(2):185–198. doi: 10.1016/j.stem.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 44.Shimozaki K, Zhang CL, Suh H, Denli AM, Evans RM, Gage FH. SRY-box-containing gene 2 regulation of nuclear receptor tailless (Tlx) transcription in adult neural stem cells. J Biol Chem. 2012;287(8):5969–5978. doi: 10.1074/jbc.M111.290403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ligon KL, Huillard E, Mehta S, Kesari S, Liu H, Alberta JA, et al. Olig2-regulated lineage-restricted pathway controls replication competence in neural stem cells and malignant glioma. Neuron. 2007;53(4):503–517. doi: 10.1016/j.neuron.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mehta S, Huillard E, Kesari S, Maire CL, Golebiowski D, Harrington EP, et al. The central nervous system-restricted transcription factor Olig2 opposes p53 responses to genotoxic damage in neural progenitors and malignant glioma. Cancer cell. 2011;19(3):359–371. doi: 10.1016/j.ccr.2011.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Matano M, Date S, Shimokawa M, Takano A, Fujii M, Ohta Y, et al. Modeling colorectal cancer using CRISPR-Cas9-mediated engineering of human intestinal organoids. Nature medicine. 2015;21(3):256–262. doi: 10.1038/nm.3802. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.