

Abstract
Current genome-editing technologies introduce double-stranded (ds) DNA breaks at a target locus as the first step to gene correction.1,2 Although most genetic diseases arise from point mutations, current approaches to point mutation correction are inefficient and typically induce an abundance of random insertions and deletions (indels) at the target locus from the cellular response to dsDNA breaks.1,2 Here we report the development of base editing, a new approach to genome editing that enables the direct, irreversible conversion of one target DNA base into another in a programmable manner, without requiring dsDNA backbone cleavage or a donor template. We engineered fusions of CRISPR/Cas9 and a cytidine deaminase enzyme that retain the ability to be programmed with a guide RNA, do not induce dsDNA breaks, and mediate the direct conversion of cytidine to uridine, thereby effecting a C→T (or G→A) substitution. The resulting “base editors” convert cytidines within a window of approximately five nucleotides (nt), and can efficiently correct a variety of point mutations relevant to human disease. In four transformed human and murine cell lines, second- and third-generation base editors that fuse uracil glycosylase inhibitor (UGI), and that use a Cas9 nickase targeting the non-edited strand, manipulate the cellular DNA repair response to favor desired base-editing outcomes, resulting in permanent correction of ∼15-75% of total cellular DNA with minimal (typically ≤ 1%) indel formation. Base editing expands the scope and efficiency of genome editing of point mutations.
Keywords: Genome editing, protein engineering, CRISPR, Cas9, genetic diseases, single-nucleotide polymorphism
The clustered regularly interspaced short palindromic repeat (CRISPR) system has been widely used to mediate genome editing in a variety of organisms and cell lines.3 CRISPR/Cas9 protein-RNA complexes localize to a target DNA sequence through base pairing with a guide RNA, and natively create a dsDNA break (DSB) at the locus specified by the guide RNA. In response to DSBs, cellular DNA repair processes mostly result in random insertions or deletions (indels) at the site of DNA cleavage through non-homologous end joining (NHEJ). In the presence of a homologous DNA template, the DNA surrounding the cleavage site can be replaced through homology-directed repair (HDR). HDR competes with NHEJ during the resolution of DSBs, and indels are generally more abundant outcomes than gene replacement. For most known genetic diseases, however, correction of a point mutation in the target locus, rather than stochastic disruption of the gene, is needed to study or address the underlying cause of the disease.4
Motivated by this need, researchers have sought to increase the efficiency of HDR and suppress NHEJ. Despite recent progress (see Supplementary Information), current strategies to correct point mutations using HDR under therapeutically relevant conditions remain inefficient (typically ∼0.1 to 5%),5,6 especially in unmodified, non-dividing cells. These observations highlight the need to develop alternative approaches to correct point mutations in genomic DNA that do not require DSBs.
We envisioned that direct conversion of one DNA base to another at a programmable target locus without requiring DSBs could increase the efficiency of gene correction relative to HDR without introducing an excess of random indels. Catalytically-dead Cas9 (dCas9), which contains Asp10Ala and His840Ala mutations that inactivate its nuclease activity, retains its ability to bind DNA in a guide RNA-programmed manner but does not cleave the DNA backbone.7 In principle, conjugation of dCas9 with an enzymatic or chemical catalyst that mediates the direct conversion of one base to another could enable RNA-programmed DNA base editing.
The deamination of cytosine (C) is catalyzed by cytidine deaminases8 and results in uracil (U), which has the base-pairing properties of thymine (T). Most known cytidine deaminases operate on RNA, and the few examples that are known to accept DNA require single-stranded (ss) DNA.9 Recent studies on the dCas9-target DNA complex reveal that at least nine nt of the displaced DNA strand are unpaired upon formation of the Cas9:guide RNA:DNA “R-loop” complex.10 Indeed, in the structure of the Cas9 R-loop complex the first 11 nt of the protospacer on the displaced DNA strand are disordered, suggesting that their movement is not highly restricted.11 It has also been speculated that Cas9 nickase-induced mutations at cytosines in the non-template strand might arise from their accessibility by cellular cytosine deaminase enzymes.12 We reasoned that a subset of this stretch of ssDNA in the R-loop might serve as an efficient substrate for a dCas9-tethered cytidine deaminase to effect direct, programmable conversion of C to U in DNA (Fig. 1a).
Figure 1. BE1 mediates specific, guide RNA-programmed C→U conversion in vitro.
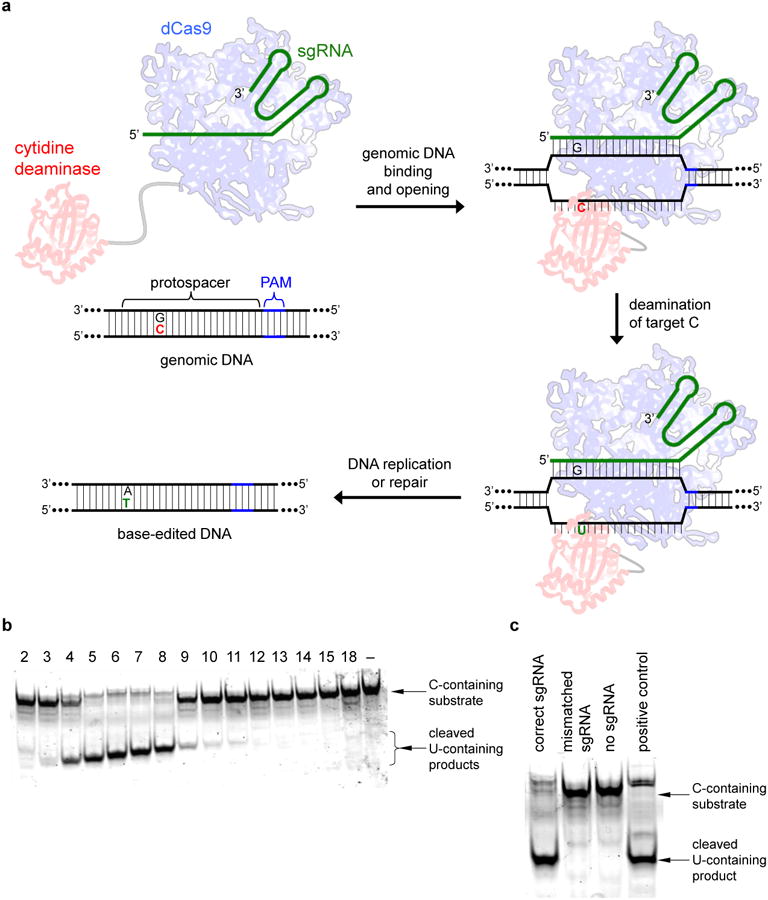
a, Base editing strategy. DNA with a target C (red) at a locus specified by a guide RNA (green) is bound by dCas9 (blue), which mediates local DNA strand separation. Cytidine deamination by a tethered APOBEC1 enzyme (red) converts the single-stranded target C→U. The resulting G:U heteroduplex can be permanently converted to an A:T bp following DNA replication or DNA repair. b, Deamination assay showing a BE1 activity window of approximately five nt. Samples were prepared as described in the Methods. Each lane is labeled according to the position of the target C within the protospacer, or with “–” if no target C is present, counting the base distal from the PAM as position 1. c, Deamination assay showing the sequence specificity and sgRNA-dependence of BE1. The DNA substrate in b was incubated with BE1 and the correct sgRNA, a mismatched sgRNA, or no sgRNA. The positive control sample used a synthetic DNA substrate with a U at position 7. For gel source data, see Supplementary Figure 1.
Four different cytidine deaminase enzymes (hAID, hAPOBEC3G, rAPOBEC1, and pmCDA1) were evaluated for ssDNA deamination. Of the four enzymes, rAPOBEC1 showed the highest deaminase activity under the conditions tested (Extended Data Fig. 1a). Fusing rAPOBEC1 to the N-terminus, but not the C-terminus, of dCas9 preserves deaminase activity (Extended Data Fig. 1a). We expressed and purified four rAPOBEC1-dCas9 fusions with linkers of different length and composition (Extended Data Fig. 1b), and evaluated each fusion for single guide RNA (sgRNA)-programmed dsDNA deamination in vitro (Fig. 1b and Extended Data Fig. 1c-f).
We observed efficient, sequence-specific, sgRNA-dependent C to U conversion in vitro (Fig. 1c). Conversion efficiency was greatest using rAPOBEC1-dCas9 linkers over nine amino acids in length. The number of positions susceptible to deamination (the “activity window”) increases from approximately three to six nt as the linker length was extended from three to 21 amino acids (Extended Data Fig. 1c-f). The 16-residue XTEN linker13 offered a promising balance between these two characteristics, with an efficient deamination window of approximately five nt, typically from positions 4 to 8 within the protospacer, counting the end distal to the protospacer-adjacent motif (PAM) as position 1. The rAPOBEC1-XTEN-dCas9 protein served as the first-generation base editor (BE1).
We assessed the ability of BE1 in vitro to correct seven T→C mutations relevant to human disease (Extended Data Fig. 2). BE1 yielded products consistent with efficient editing of the target C, or of at least one C within the activity window when multiple Cs were present, in six of these seven targets in vitro, with an average apparent editing efficiency of 44% (Extended Data Fig. 2).
Although the preferred sequence context for APOBEC1 substrates is TC or CC,14 we anticipated that the increased effective molarity of the tethered deaminase and its ssDNA substrate upon dCas9 binding might relax this preference. To illuminate the context-dependence of BE1, we assayed its ability to edit a dsDNA 60-mer containing a single fixed C at position 7 within the protospacer, as well as all 36 single-mutant variants in which protospacer bases 1-6 and 8-13 were individually varied to each of the other three bases. High-throughput DNA sequencing (HTS) revealed 50-80% C to U conversion of substrate strands (25-40% of sequence reads from both DNA strands, one of which is not a substrate for BE1) (Fig. 2a). Editing efficiency was independent of sequence context unless the base immediately 5′ of the target C was a G, in which case editing efficiency was substantially lower (Fig. 2a). Next we assessed BE1 activity in vitro on all four NC motifs at positions 1 through 8 within the protospacer (Fig. 2b). BE1 activity followed the order TC ≥ CC ≥ AC > GC, with maximum editing efficiency achieved when the target C is at or near position 7 (See Supplementary Information). In addition, we observed that the base editor is processive, and will efficiently convert most or all Cs to Us on the same DNA strand within the 5-base activity window (Extended Data Fig. 3).
Figure 2. Effects of sequence context and target C position on base editing efficiency in vitro.

a, Effect of changing the sequence surrounding the target C on editing efficiency in vitro. The deamination yield of 80% of targeted strands (40% of total sequencing reads from both strands) for C7 in the protospacer sequence 5′-TTATTTCGTGGATTTATTTA-3′ was defined as 1.0, and the relative deamination efficiencies of substrates containing all possible single-base mutations at positions 1-6 and 8-13 are shown. b, Positional effect of each NC motif on editing efficiency in vitro. Each NC target motif was varied from positions 1 to 8 within the protospacer as indicated in the sequences shown on the right. The PAM is shown in blue. The graph shows the percentage of total DNA sequencing reads containing T at each of the numbered target C positions following incubation with BE1. Note that the maximum possible deamination yield in vitro is 50% of total sequencing reads (100% of targeted strands). Values and error bars reflect the mean and standard deviation of three (for a) or two (for b) independent biological replicates performed on different days.
While BE1 efficiently processes substrates in a test tube, in cells a tree of possible DNA repair outcomes determines the fate of the initial U:G product of base editing (Fig. 3a). We tested the ability of BE1 to convert C→T in human cells on 14 Cs in six well-studied target sites in the human genome (See Supplementary Information and Extended Data Fig. 4a).15 Although C→T editing in cells was observed for all cases, the efficiency of base editing was 0.8% to 7.7% of total DNA sequences, a large 5- to 36-fold decrease in efficiency compared to that of in vitro base editing (Fig. 3b and Extended Data Fig. 4).
Figure 3. Base editing in human cells.
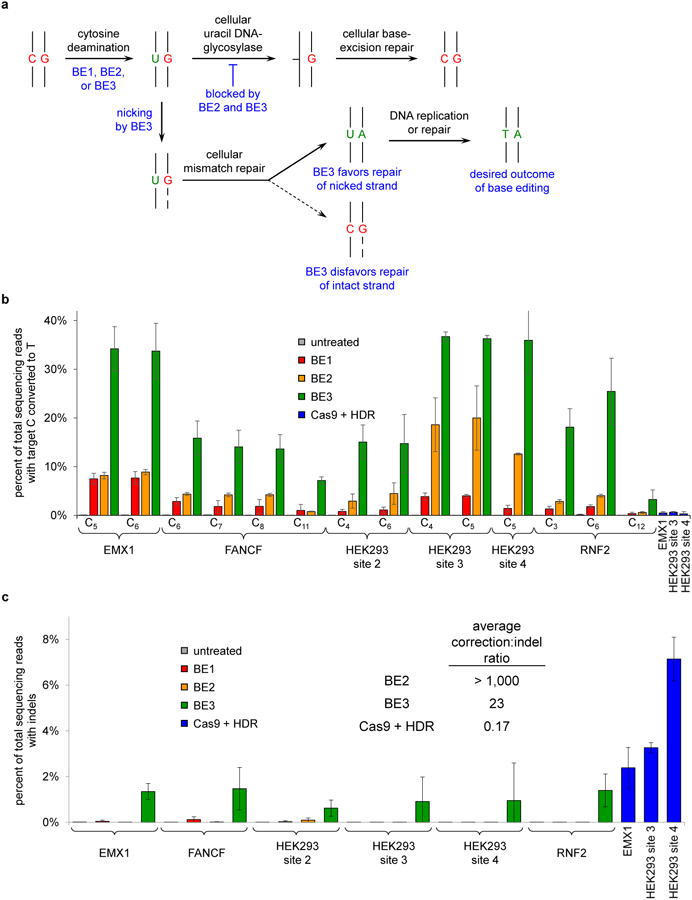
a, Possible base editing outcomes in mammalian cells. Initial editing results in a U:G mismatch. Recognition and excision of the U by uracil DNA glycosylase (UDG) initiates base excision repair (BER), which leads to reversion to the C:G starting state. BER is impeded by BE2 and BE3, which inhibit UDG. The U:G mismatch is also processed by mismatch repair (MMR), which preferentially repairs the nicked strand of a mismatch. BE3 nicks the non-edited strand containing the G, favoring resolution of the U:G mismatch to the desired U:A or T:A outcome. b, HEK293T cells were treated as described in the Methods. The percentage of total DNA sequencing reads with Ts at the target positions indicated are shown for treatment with BE1, BE2, or BE3, or for treatment with wt Cas9 with a donor HDR template. c, Frequency of indel formation (see Methods) is shown following the treatment in b. Values are listed in Supplementary Table 6. For b and c, values and error bars reflect the mean and s.d. of three independent biological replicates performed on different days.
We hypothesized that the cellular DNA repair response to U:G heteroduplex DNA was responsible for the large decrease in base editing efficiency in cells (Fig. 3a). Uracil DNA glycosylase (UDG) catalyzes removal of U from DNA in cells and initiates base-excision repair (BER), with reversion of the U:G pair to a C:G pair as the most common outcome (Fig. 3a).16 Uracil DNA glycosylase inhibitor (UGI), an 83-residue protein from B. subtilis bacteriophage PBS1, potently blocks human UDG activity (IC50 = 12 pM).17 In an effort to subvert BER at the site of base editing, we fused UGI to the C-terminus of BE1 to create a second-generation base editor (BE2, APOBEC–XTEN–dCas9–UGI) and repeated editing assays on all six genomic loci. Editing efficiencies in human cells were on average 3-fold higher with BE2 than BE1, resulting in gene conversion efficiencies of up to 20% of total DNA sequenced (Fig. 3b).
Importantly, BE1 and BE2 resulted in indel formation rates ≤ 0.1% (Fig. 3c, Extended Data Table 1), consistent with the known mechanistic dependence of NHEJ on DSBs (see Supplementary Information).18 We assessed BE2-mediated base editing efficiencies on the same genomic targets in U2OS cells, and observed results similar to those in HEK293T cells (Extended Data Fig. 5). Together, these results indicate that conjugating UGI to BE1 can increase the efficiency of base editing in human cells.
Converting and protecting the substrate strand of a C:G base pair (bp) results in a maximum base editing yield of 50%. To augment base editing efficiency beyond this limit, we sought to further manipulate cellular DNA repair to induce correction of the non-edited strand containing the G. Eukaryotic mismatch repair (MMR) uses nicks present in newly synthesized DNA to direct removal and resynthesis of the newly synthesized strand (Fig. 3a).19,20 We reasoned that nicking the DNA strand containing the unedited G would simulate newly synthesized DNA, inducing MMR to preferentially resolve the U:G mismatch into desired U:A and T:A products (Fig. 3a). We therefore restored the catalytic His residue at position 840 in the Cas9 HNH domain of BE2,7 resulting in the third-generation base editor (BE3, APOBEC–XTEN–dCas9(A840H)–UGI) that nicks the non-edited strand containing a G opposite the edited U. BE3 retains the Asp10Ala mutation in Cas9 that prevents dsDNA cleavage, and also retains UGI to suppress BER.
Nicking the non-edited strand augmented base editing efficiency in human cells treated with BE3 by an additional 2- to 6-fold relative to BE2, resulting in up to 37% of total DNA sequences containing the targeted C→T conversion (Fig. 3b). Importantly, only a small frequency of indels, averaging 1.1% for the six tested loci, was observed from BE3 treatment (Fig. 3c and Supplementary Table 6). In contrast, when we treated cells with wild-type (wt) Cas9, sgRNA to target each of three loci, and a ssDNA donor template to mediate HDR, we observed C→T conversion efficiencies averaging only 0.5%, with much higher indel formation averaging 4.3% (Fig. 3c). The ratio of allele conversion to NHEJ outcomes averaged >1,000 for BE2, 23 for BE3, and 0.17 for wt Cas9 (Fig. 3c). We confirmed the permanence of base editing in human cells by monitoring editing efficiencies over multiple cell divisions in HEK293T cells at the HEK293 site 3 and 4 genomic loci (Extended Data Fig. 6 and Supplementary Information). These results collectively establish that base editing can effect much more efficient targeted single-base editing in human cells than Cas9-mediated HDR, and with much less (BE3) or almost no (BE2) indel formation.
Next we examined the off-target activity of BE1, BE2, and BE3 in human cells for five previously studied loci (see Supplementary Information and Supplementary Tables 1-5). Because the sequence preference of rAPOBEC1 is known to be independent of bases more than one nt from the target C,21 consistent with Fig. 2a, we assumed that off-target base editing arises from off-target Cas9 binding. Therefore we sequenced the top 34 known Cas9 off-target sites in human cells15, and the top 12 known dCas9 off-target binding sites (Supplementary Tables 1-5).22 We observed detectable off-target base editing at a subset of known Cas9 off-target sites (16/34 for BE1 and BE2; and 17/34 for BE3), but no detectable base editing at the known dCas9 off-target sites. All detected off-target base-editing substrates contained a C within the five-base activity window (see Supplementary Information). We also monitored C→T mutations at 3,200 cytosines surrounding the six on-target and 44 off-target loci tested and observed no evident increase in C→T conversions outside the protospacer upon BE1, BE2, or BE3 treatment compared to that of untreated cells (Extended Data Fig. 7). Taken together, these findings suggest that off-target substrates of base editors include a subset of Cas9 off-target substrates, and that base editors in human cells do not induce untargeted C→T conversion throughout the genome.
Finally, we tested the potential of base editing to correct two disease-relevant mutations in mammalian cells. The apolipoprotein E gene variant APOE4 encodes two Arg residues at amino acid positions 112 and 158, and is the largest and most common genetic risk factor for late-onset Alzheimer's disease.23 ApoE variants with Cys residues at these positions, including APOE2 (Cys112/Cys158), APOE3 (Cys112/Arg158), and APOE3r (Arg112/Cys158) have been shown or are presumed24 to confer lower Alzheimer's disease risk than APOE4. We attempted to convert APOE4 into APOE3r in immortalized mouse astrocytes in which the endogenous APOE gene was replaced by human APOE4. We delivered into these astrocytes by nucleofection DNA encoding BE3 and an appropriate sgRNA placing the target C at position 5 relative to a downstream PAM. After two days, we isolated nucleofected cells and measured editing efficiency by HTS of genomic DNA. We observed conversion of Arg158 to Cys158 in 58-75% of total DNA sequencing reads (Table 1a and Extended Data Fig. 8a). We also observed 36-50% editing of total DNA at the third position of codon 158 and 38-55% editing of total DNA at the first position of Leu159, as expected since all three of these Cs are within the base editing window. Neither of the other two C→T conversions, however, alters the amino acid sequence of the ApoE3r protein since both TGC and TGT encode Cys (all C→T changes at the third position of a codon are silent), and both CTG and TTG encode Leu.
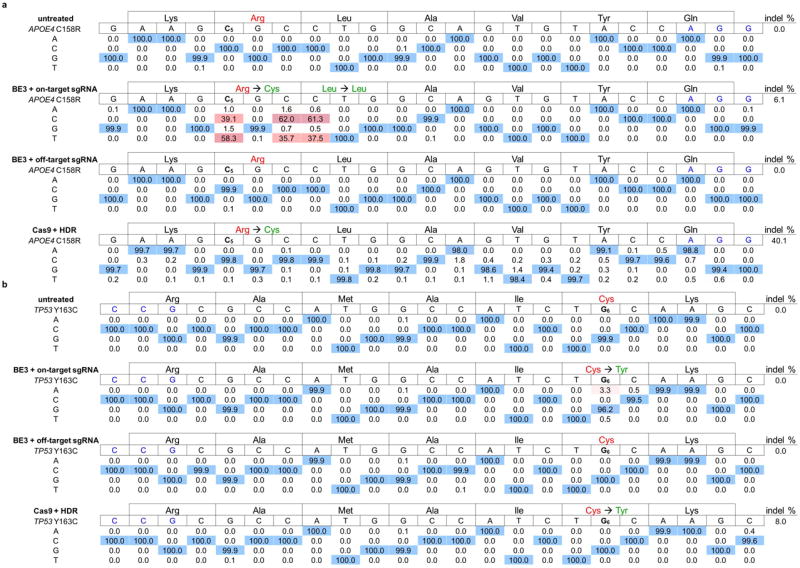
Table 1. BE3-mediated correction of two disease-relevant mutations in mammalian cells.
The sequence of the protospacer is shown to the right of the mutation, with the PAM in blue and the target base in red with a subscripted number indicating its position within the protospacer. Underneath each sequence are the percentages of total sequencing reads with the corresponding base. Cells were treated as described in the Methods. a, The Alzheimer's disease-associated APOE4 allele was converted to APOE3r in mouse astrocytes by BE3 in 74.9% of total reads. Two nearby Cs are also converted to Ts, but with no change to the predicted sequence of the resulting protein. Identical treatment of these cells with wt Cas9 and donor ssDNA results in only 0.3% correction, with 26.1% indel formation. b, The cancer-associated p53 Y163C mutation is corrected by BE3 in 7.6% of nucleofected human breast cancer cells with 0.7% indel formation. Identical treatment of these cells with wt Cas9 and donor ssDNA results in no mutation correction with 6.1% indel formation.

|
The efficiency of BE3-mediated editing of APOE4 demonstrates that a combination of suppressing BER and guiding MMR to repair the unedited strand enables base editing efficiencies to exceed the 50% maximum yield that would result from DNA replication alone. We observed no evident increase in mutations within 50 bp of either end of the protospacer compared with untreated controls (Supplementary Table 7). We observed 4.6-6.1% indels at the targeted locus following BE3 treatment. In contrast, identical treatment of astrocytes with wt Cas9 and donor ssDNA resulted in 0.1-0.3% APOE4 correction and 26-40% indels at the targeted locus, efficiencies consistent with previous reports of single-base correction using Cas9 and HDR5,6 (Table 1a and Extended Data Fig. 8a). Astrocytes treated identically but with an sgRNA targeting the VEGFA locus displayed no evidence of APOE4 base editing (Supplementary Table 6 and Extended Data Fig. 8a). These results demonstrate that base editors can mediate highly efficient and precise single-amino acid changes in the coding sequence of a protein, even when their processivity results in >1 nt change in genomic DNA.
The dominant-negative p53 mutation Tyr163Cysis strongly associated with several types ofcancer25 and can be corrected by a C→T conversion on the template strand (Extended Data Fig. 2), resulting in the translation of corrected protein even before the edited base is made permanent by DNA replication or DNA repair. We nucleofected a human breast cancer cell line homozygous for the p53 Tyr163Cys mutation (HCC1954 cells) with DNA encoding BE3 and an sgRNA programmed to correct Tyr163Cys. We observed correction of the Tyr163Cys mutation in 3.3-7.6% of nucleofected HCC1954 cells (Table 1b, Extended Data Fig. 8b, and Supplementary Table 8), with ≤ 0.7% indel formation. In contrast, treatment of cells with wt Cas9 and donor ssDNA resulted in no detectable TP53 correction (< 0.1%) with 6.1-8.0% indels at the target locus (Table 1b and Extended Data Fig. 8b). These results collectively represent the correction of disease-associated point mutations in mammalian cell lines with an efficiency and lack of other genome modification events that may not be achievable using previously described methods. An additional 300-900 clinically relevant known human genetic diseases that in principle are correctable by the base editors described in this work are shown in Extended Data Fig. 9 and Supplementary Table 8 (See Supplementary Discussion).
The development of base editing advances both the scope and effectiveness of genome editing. The base editors described here offer researchers a choice of editing with very little (< 0.1%) indel formation (BE2), or more efficient editing with ≤ 1% indel formation (BE3). That the product of base editing is, by definition, no longer a substrate likely contributes to editing efficiency by preventing subsequent product transformation, which can hamper traditional Cas9 applications. By removing the reliance on dsDNA cleavage, donor templates, and stochastic DNA repair processes that vary by cell state and cell type, base editing has the potential to expand the type of genome modifications that can be cleanly installed, the efficiency of these modifications, and the type of cells that are amenable to editing. It is likely that engineered Cas9 variants26-28 or delivery methods29 that offer improved DNA specificity or altered PAM specificities30 can provide additional base editors with improved properties. These results also suggest architectures for the fusion of other DNA-modifying enzymes, including methylases and demethylases, that may enable additional types of programmable genome and epigenome base editing.
Methods
Cloning
DNA sequences of all substrates and primers used in this paper are listed in the Supplementary Sequences. PCR was performed using VeraSeq ULtra DNA polymerase (Enzymatics), or Q5 Hot Start High-Fidelity DNA Polymerase (New England Biolabs). NBE plasmids were constructed using USER cloning (New England Biolabs). Deaminase genes were synthesized as gBlocks Gene Fragments (Integrated DNA Technologies), and Cas9 genes were obtained from previously reported plasmids.31 Deaminase and fusion genes were cloned into pCMV (mammalian codon-optimized) or pET28b (E. coli codon-optimized) backbones. sgRNA expression plasmids were constructed using site-directed mutagenesis. Briefly, the primers listed in the Supplementary Sequences were 5′ phosphorylated using T4 Polynucleotide Kinase (New England Biolabs) according to the manufacturer's instructions. Next, PCR was performed using Q5 Hot Start High-Fidelity Polymerase (New England Biolabs) with the phosphorylated primers and the plasmid pFYF1320 (EGFP sgRNA expression plasmid) as a template according to the manufacturer's instructions. PCR products were incubated with DpnI (20 U, New England Biolabs) at 37 °C for 1 h, purified on a QIAprep spin column (Qiagen), and ligated using QuickLigase (New England Biolabs) according to the manufacturer's instructions. DNA vector amplification was carried out using Mach1 competent cells (ThermoFisher Scientific).
In vitro deaminase assay on ssDNA
Sequences of all ssDNA substrates are listed in the Supplementary Sequences. All Cy3-labelled substrates were obtained from Integrated DNA Technologies (IDT). Deaminases were expressed in vitro using the TNT T7 Quick Coupled Transcription/Translation Kit (Promega) according to the manufacturer's instructions using 1 μg of plasmid. Following protein expression, 5 μL of lysate was combined with 35 μL of ssDNA (1.8 μM) and USER enzyme (1 unit) in CutSmart buffer (New England Biolabs) (50 mM potassium acetate, 29 mM Tris-acetate, 10 mM magnesium acetate, 100 ug/mL BSA, pH 7.9) and incubated at 37 °C for 2 h. Cleaved U-containing substrates were resolved from full-length unmodified substrates on a 10% TBE-urea gel (Bio-Rad).
Expression and purification of His6-rAPOBEC1-linker-dCas9 fusions
E. coli BL21 STAR (DE3)-competent cells (ThermoFisher Scientific) were transformed with plasmids encoding pET28b-His6-rAPOBEC-linker-dCas9 with GGS, (GGS)3, XTEN, or (GGS)7 linkers. The resulting expression strains were grown overnight in Luria-Bertani (LB) broth containing 100 μg/mL of kanamycin at 37 °C. The cells were diluted 1:100 into the same growth medium and grown at 37 °C to OD600 = ∼0.6. The culture was cooled to 4 °C over a period of 2 h, and isopropyl -β-d-1- thiogalactopyranoside (IPTG) was added at 0.5 mM to induce protein expression. After ∼16 h, the cells were collected by centrifugation at 4,000 g and resuspended in lysis buffer (50 mM tris(hydroxymethyl)-aminomethane (Tris)-HCl[pH 7.0], 1 M NaCl, 20% glycerol, 10 mM tris(2-carboxyethyl)phosphine (TCEP, Soltec Ventures)). The cells were lysed by sonication (20 s pulse-on, 20 s pulse-off for 8 min total at 6 W output) and the lysate supernatant was isolated following centrifugation at 25,000 g for 15 min. The lysate was incubated with His-Pur nickel-nitriloacetic acid (nickel-NTA) resin (ThermoFisher Scientific) at 4 °C for 1 h to capture the His-tagged fusion protein. The resin was transferred to a column and washed with 40 mL of lysis buffer. The His-tagged fusion protein was eluted in lysis buffer supplemented with 285 mM imidazole, and concentrated by ultrafiltration (Amicon-Millipore, 100-kDa molecular weight cut-off) to 1 mL total volume. The protein was diluted to 20 mL in low-salt purification buffer containing 50 mM tris(hydroxymethyl)-aminomethane (Tris)-HCl [pH 7.0], 0.1 M NaCl, 20% glycerol, 10 mM TCEP and loaded onto SP Sepharose Fast Flow resin (GE Life Sciences). The resin was washed with 40 mL of this low-salt buffer, and the protein eluted with 5 mL of activity buffer containing 50 mM tris(hydroxymethyl)-aminomethane (Tris)-HCl [pH 7.0], 0.5 M NaCl, 20% glycerol, 10 mM TCEP. The eluted proteins were quantified by SDS-PAGE.
In vitro transcription of sgRNAs
Linear DNA fragments containing the T7 promoter followed by the 20-bp sgRNA target sequence were transcribed in vitro using the primers listed in the Supplementary Sequences with the TranscriptAid T7 High Yield Transcription Kit (ThermoFisher Scientific) according to the manufacturer's instructions. sgRNA products were purified using the MEGAclear Kit (ThermoFisher Scientific) according to the manufacturer's instructions and quantified by UV absorbance.
Preparation of Cy3-conjugated dsDNA substrates
Sequences of 80-nt unlabeled strands are listed in the Supplementary Sequences and were ordered as PAGE-purified oligonucleotides from IDT. The 25-nt Cy3-labeled primer listed in the Supplementary Sequences is complementary to the 3′ end of each 80-nt substrate. This primer was ordered as an HPLC-purified oligonucleotide from IDT. To generate the Cy3-labeled dsDNA substrates, the 80-nt strands (5 μL of a 100 μM solution) were combined with the Cy3-labeled primer (5 μL of a 100 μM solution) in NEBuffer 2 (38.25 μL of a 50 mM NaCl, 10 mM Tris-HCl, 10 mM MgCl2, 1 mM DTT, pH 7.9 solution, New England Biolabs) with dNTPs (0.75 μL of a 100 mM solution) and heated to 95 °C for 5 min, followed by a gradual cooling to 45 °C at a rate of 0.1 °C/s. After this annealing period, Klenow exo– (5 U, New England Biolabs) was added and the reaction was incubated at 37 °C for 1 h. The solution was diluted with Buffer PB (250 μL, Qiagen) and isopropanol (50 μL) and purified on a QIAprep spin column (Qiagen), eluting with 50 μL of Tris buffer.
Deaminase assay on dsDNA
The purified fusion protein (20 μL of 1.9 μM in activity buffer) was combined with 1 equivalent of appropriate sgRNA and incubated at ambient temperature for 5 min. The Cy3-labeled dsDNA substrate was added to final concentration of 125 nM and the resulting solution was incubated at 37 °C for 2 h. The dsDNA was separated from the fusion by the addition of Buffer PB (100 μL, Qiagen) and isopropanol (25 μL) and purified on aEconoSpin micro spin column (Epoch Life Science), eluting with 20 μL of CutSmart buffer (New England Biolabs). USER enzyme (1 U, New England Biolabs) was added to the purified, edited dsDNA and incubated at 37 °C for 1 h. The Cy3-labeled strand was fully denatured from its complement by combining 5 μL of the reaction solution with 15 μL of a DMSO-based loading buffer (5 mM Tris, 0.5 mM EDTA, 12.5% glycerol, 0.02% bromophenol blue, 0.02% xylene cyan, 80% DMSO). The full-length C-containing substrate was separated from any cleaved, U-containing edited substrates on a 10% TBE-urea gel (Bio-Rad) and imaged on a GE Amersham Typhoon imager.
Preparation of in vitro-edited dsDNA for high-throughput sequencing (HTS)
The oligonucleotides listed in the Supplementary Sequences were obtained from IDT. Complementary sequences were combined (5 μL of a 100 μM solution) in Tris buffer and annealed by heating to 95 °C for 5 min, followed by a gradual cooling to 45 °C at a rate of 0.1 °C/s to generate 60-bp dsDNA substrates. Purified fusion protein (20 μL of 1.9 μM in activity buffer) was combined with 1 equivalent of appropriate sgRNA and incubated at ambient temperature for 5 min. The 60-mer dsDNA substrate was added to final concentration of 125 nM and the resulting solution was incubated at 37 °C for 2 h. The dsDNA was separated from the fusion by the addition of Buffer PB (100 μL, Qiagen) and isopropanol (25 μL) and purified on a EconoSpin micro spin column (Epoch Life Science), eluting with 20 μL of Tris buffer. The resulting edited DNA (1 μL was used as a template) was amplified by PCR using the HTS primer pairs specified in the Supplementary Sequences and VeraSeq Ultra (Enzymatics) according to the manufacturer's instructions with 13 cycles of amplification. PCR reaction products were purified using RapidTips (Diffinity Genomics), and the purified DNA was amplified by PCR with primers containing sequencing adapters, purified, and sequenced on a MiSeq high-throughput DNA sequencer (Illumina) as previously described.32
Cell culture
HEK293T (ATCC CRL-3216) and U2OS (ATCC-HTB-96) were maintained in Dulbecco's Modified Eagle's Medium plus GlutaMax (ThermoFisher) supplemented with 10% (v/v) fetal bovine serum (FBS), at 37 °C with 5% CO2. HCC1954 cells (ATCC CRL-2338) were maintained in RPMI-1640 medium (ThermoFisher Scientific) supplemented as described above. Immortalized rat astrocytes containing the ApoE4 isoform of the APOE gene (Taconic Biosciences) were cultured in Dulbecco's Modified Eagle's Medium plus GlutaMax (ThermoFisher Scientific) supplemented with 10% (v/v) fetal bovine serum (FBS) and 200 μg/mL Geneticin (ThermoFisher Scientific).
Transfections
HEK293T cells were seeded on 48-well collagen-coated BioCoat plates (Corning) and transfected at approximately 85% confluency. Briefly, 750 ng of BE and 250 ng of sgRNA expression plasmids were transfected using 1.5 μl of Lipofectamine 2000 (ThermoFisher Scientific) per well according to the manufacturer's protocol.
Astrocytes, U2OS, HCC1954 and HEK293T cells were transfected using appropriate Amaxa Nucleofector™ II programs according to manufacturer's instructions (basic glial cell, V, V, and V kits using programs T-020, X-001, X-005, and Q-001 for astrocytes, U2OS, HCC1954, and HEK293T cells, respectively). 40 ng of iRFP670 (Addgene plasmid 45457)33 was added to the nucleofection solution to assess nucleofection efficiencies in these cell lines. Astrocytes and HCC1954 cells were filtered through a 40 μm strainer (Fisher Scientific) after harvesting, and the nucleofected cells were collected on a Beckman Coulter MoFlo XDP Cell Sorter using the iRFP signal (abs 643 nm, em 670 nm). The U2OS and HEK293T cells were used without enrichment of nucleofected cells.
High-throughput DNA sequencing of genomic DNA samples
Transfected cells were harvested after 3 d and the genomic DNA was isolated using the Agencourt DNAdvance Genomic DNA Isolation Kit (Beckman Coulter) according to the manufacturer's instructions. On-target and off-target genomic regions of interest were amplified by PCR with flanking HTS primer pairs listed in the Supplementary Sequences. PCR amplification was carried out with Phusion high-fidelity DNA polymerase (ThermoFisher) according to the manufacturer's instructions using 5 ng of genomic DNA as a template. Cycle numbers were determined separately for each primer pair as to ensure the reaction was stopped in the linear range of amplification (30, 28, 28, 28, 32, and 32 cycles for EMX1, FANCF, HEK293 site 2, HEK293 site 3, HEK293 site 4, and RNF2 primers, respectively). PCR products were purified using RapidTips (Diffinity Genomics). Purified DNA was amplified by PCR with primers containing sequencing adaptors. The products were gel-purified and quantified using the Quant-iT™ PicoGreen dsDNA Assay Kit (ThermoFisher) and KAPA Library Quantification Kit-Illumina (KAPA Biosystems). Samples were sequenced on an Illumina MiSeq as previously described.32
Data analysis
Sequencing reads were automatically demultiplexed using MiSeq Reporter (Illumina), and individual FASTQ files were analyzed with a custom Matlab script provided in the Supplementary Notes. Each read was pairwise aligned to the appropriate reference sequence using the Smith-Waterman algorithm. Base calls with a Q-score below 31 were replaced with N's and were thus excluded in calculating nucleotide frequencies. This treatment yields an expected MiSeq base-calling error rate of approximately 1 in 1,000. Aligned sequences in which the read and reference sequence contained no gaps were stored in an alignment table from which base frequencies could be tabulated for each locus.
Indel frequencies were quantified with a custom Matlab script shown in the Supplementary Notes using previously described criteria.29 Sequencing reads were scanned for exact matches to two 10-bp sequences that flank both sides of a window in which indels might occur. If no exact matches were located, the read was excluded from analysis. If the length of this indel window exactly matched the reference sequence the read was classified as not containing an indel. If the indel window was two or more bases longer or shorter than the reference sequence, then the sequencing read was classified as an insertion or deletion, respectively.
Extended Data
Extended Data Figure 1. Effects of deaminase, linker length, and linker composition on base editing.

a, Gel-based deaminase assay showing activity of rAPOBEC1, pmCDA1, hAID, hAPOBEC3G, rAPOBEC1-GGS-dCas9, rAPOBEC1-(GGS)3-dCas9, and dCas9-(GGS)3-rAPOBEC1 on ssDNA. Enzymes were expressed in a mammalian cell lysate-derived in vitro transcription-translation system and incubated with 1.8 μM dye-conjugated ssDNA and USER enzyme (uracil DNA glycosylase and endonuclease VIII) at 37 °C for 2 h. The resulting DNA was resolved on a denaturing polyacrylamide gel and imaged. The positive control is a sequence with a U synthetically incorporated at the same position as the target C. b, Coomassie-stained denaturing PAGE of the expressed and purified proteins used in (c), (d), (e), and (f). c-f, Gel-based deaminase assay showing the deamination window of base editors with deaminase–Cas9 linkers of GGS (c), (GGS)3 (d), XTEN (e), or (GGS)7 (f). Following incubation of 1.85 μM deaminase-dCas9 fusions complexed with sgRNA with 125 nM dsDNA substrates at 37 °C for 2 h, the dye-conjugated DNA was isolated and incubated with USER enzyme at 37 °C for 1 h to cleave the DNA backbone at the site of any Us. The resulting DNA was resolved on a denaturing polyacrylamide gel, and the dye-conjugated strand was imaged. Each lane is numbered according to the position of the target C within the protospacer, or with – if no target C is present. 8U is a positive control sequence with a U synthetically incorporated at position 8. For gel source data, see Supplementary Figure 1.
Extended Data Figure 2. BE1 is capable of correcting disease-relevant mutations in vitro.

a, Protospacer and PAM sequences (red) of seven disease-relevant mutations. The disease-associated target C in each case is indicated with a subscripted number reflecting its position within the protospacer. For all mutations except both APOE4 SNPs, the target C resides in the template (non-coding) strand. b, Deaminase assay showing each dsDNA 80-mer oligonucleotide before (–) and after (+) incubation with BE1, DNA isolation, and incubation with USER enzymes to cleave DNA at positions containing U. Positive control lanes from incubation of synthetic oligonucleotides containing U at various positions within the protospacer with USER enzymes are shown with the corresponding number indicating the position of the U. Editing efficiencies were quantitated by dividing the intensity of the cleaved product band by that of the entire lane for each sample. For gel source data, see Supplementary Figure 1.
Extended Data Figure 3. Processivity of BE1.

The protospacer and PAM (red) of a 60-mer DNA oligonucleotide containing eight consecutive Cs is shown at the top. The oligonucleotide (125 nM) was incubated with BE1 (2 μM) for 2 h at 37 °C. The DNA was isolated and analyzed by high-throughput sequencing. Shown are the percent of total reads for the most frequent nine sequences observed. The vast majority of edited strands (>93%) have more than one C converted to T.
Extended Data Figure 4. BE1 base editing efficiencies are dramatically decreased in mammalian cells.

a, Protospacer (black and red) and PAM (blue) sequences of the six mammalian cell genomic loci targeted by base editors. Target Cs are indicated in red with subscripted numbers corresponding to their positions within the protospacer. b, Synthetic 80-mers with sequences matching six different genomic sites were incubated with BE1 then analyzed for base editing by HTS. For each site, the sequence of the protospacer is indicated to the right of the name of the site, with the PAM highlighted in blue. Underneath each sequence are the percentages of total DNA sequencing reads with the corresponding base. We considered a target C as “editable” if the in vitro conversion efficiency is >10%. Note that maximum yields are 50% of total DNA sequencing reads since the non-targeted strand is unaffected by BE1. Values are shown from a single experiment. c, HEK293T cells were transfected with plasmids expressing BE1 and an appropriate sgRNA. Three days after transfection, genomic DNA was extracted and analyzed by HTS at the six loci. Cellular C to T conversion percentages, defined as the percentage of total DNA sequencing reads with Ts at the target positions indicated, are shown for BE1 at all six genomic loci. Values and error bars of all data from HEK293T cells reflect the mean and standard deviation of three independent biological replicates performed on different days.
Extended Data Figure 5. Base editing efficiencies of BE2 in U2OS and HEK293T cells.

Cellular C to T conversion percentages by BE2 are shown for each of the six targeted genomic loci in HEK293T cells and U2OS cells. HEK293T cells were transfected using lipofectamine 2000, and U2OS cells were nucleofected. Three days after plasmid delivery, genomic DNA was extracted and analyzed for base editing at the six genomic loci by HTS. Values and error bars reflect the mean and standard deviation of at least two biological experiments done on different days.
Extended Data Figure 6. Base editing persists over multiple cell divisions.

Cellular C to T conversion percentages by BE2 and BE3 are shown for HEK293 sites 3 and 4 in HEK293T cells before and after passaging the cells. HEK293T cells were nucleofected with plasmids expressing BE2 or BE3 and an sgRNA targeting HEK293 site 3 or 4. Three days after nucleofection, the cells were harvested and split in half. One half was subjected to HTS analysis, and the other half was allowed to propagate for approximately five cell divisions, then harvested and subjected to HTS analysis. Values and error bars reflect the mean and standard deviation of at least two biological experiments.
Extended Data Figure 7. Non-target C/G mutation rates.

Shown here are the C to T and G to A mutation rates at 2,500 distinct cytosines and guanines surrounding the six on-target and 34 off-target loci tested, representing a total of 14,700,000 sequence reads derived from approximately 1.8×106 cells. a, Cellular non-target C to T and G to A conversion percentages by BE1, BE2, and BE3 are plotted individually against their positions relative to a protospacer for all 2,500 cytosines/guanines. The side of the protospacer distal to the PAM is designated with positive numbers, while the side that includes the PAM is designated with negative numbers. b, Average non-target cellular C to T and G to A conversion percentages by BE1, BE2, and BE3 are shown, as well as the highest and lowest individual conversion percentages.
Extended Data Figure 8. Additional data sets of BE3-mediated correction of two disease-relevant mutations in mammalian cells.
For each site, the sequence of the protospacer is indicated to the right of the name of the mutation, with the PAM highlighted in blue and the base responsible for the mutation indicated in red bold with a subscripted number corresponding to its position within the protospacer. The amino acid sequence above each disease-associated allele is shown, together with the corrected amino acid sequence following base editing in green. Underneath each sequence are the percentages of total sequencing reads with the corresponding base. Cells were nucleofected with plasmids encoding BE3 and an appropriate sgRNA. Two days after nucleofection, genomic DNA was extracted from the nucleofected cells and analyzed by HTS to assess pathogenic mutation correction. a, The Alzheimer's disease-associated APOE4 allele is converted to APOE3r in mouse astrocytes by BE3 in 58.3% of total reads only when treated with the correct sgRNA. Two nearby Cs are also converted to Ts, but with no change to the predicted sequence of the resulting protein. Identical treatment of these cells with wt Cas9 and donor ssDNA results in 0.2% correction, with 26.7% indel formation. b, The cancer-associated p53 Y163C mutation is corrected by BE3 in 3.3% of nucleofected human breast cancer cells only when treated with the correct sgRNA. Identical treatment of these cells with wt Cas9 and donor ssDNA results in no detectable mutation correction with 8.0% indel formation.
Extended Data Figure 9. Genetic variants from ClinVar that in principle can be corrected by base editingg.

The NCBI ClinVar database of human genetic variations and their corresponding phenotypes (see main text ref. 4) was searched for genetic diseases that can be corrected by current base editing technologies. The results were filtered by imposing the successive restrictions listed on the left. The x-axis shows the number of occurrences satisfying that restriction and all above restrictions on a logarithmic scale.
Extended Data Table 1. Indel formation following treatment of HEK293T cells with BE1, BE2, BE3, or wt Cas9 + assDNA template for HDR.
Indel frequencies were calculated as described in the Methods following treatment of HEK293T cells with BE1, BE2, and BE3 for all six genomic loci, or with wt Cas9 and a ssDNA template for HDR at three of the six sites (EMX1, HEK293 site 3, and HEK293 site 4). Values reflect the mean and standard deviation of at least three independent biological replicates performed on different days.
| EMX1 indel (%) | FANCF indel (%) | HEK293 site 2 indel (%) | HEK293 site 3 indel (%) | HEK293 site 4 indel (%) | RNF2 indel (%) | |
|---|---|---|---|---|---|---|
|
|
||||||
| untreated | 0.01±0.00 | 0.00±0.00 | 0.00±0.00 | 0.00±0.00 | 0.00±0.00 | 0.00±0.00 |
| BE1 | 0.04±0.05 | 0.11±0.13 | 0.02±0.04 | 0.00±0.00 | 0.00±0.00 | 0.00±0.00 |
| BE2 | 0.01±0.00 | 0.01±0.01 | 0.09±0.09 | 0.00±0.00 | 0.00±0.00 | 0.00±0.00 |
| BE3 | 1.34±0.35 | 1.47±0.93 | 0.62±0.35 | 0.91±1.07 | 0.95±1.64 | 1.39±0.72 |
| Cas9 + HDR | 2.38±0.89 | 3.26±0.22 | 7.14±0.96 | |||
Supplementary Material
Acknowledgments
This work was supported by U.S. National Institutes of Health (NIH) R01 EB022376 (formerly R01 GM065400), F-Prime Biomedical Research Initiative (A28161), and the Howard Hughes Medical Institute. A.C.K. is a Ruth L. Kirchstein National Research Service Awards Postdoctoral Fellow (F32 GM 112366-2). Y.B.K. holds a Natural Sciences and Engineering Research Council of Canada Postgraduate Scholarship (NSERC PGS-D). M.S.P. is an NSF Graduate Research Fellow and was supported by the Harvard Biophysics NIH training grant T32 GM008313. J.A.Z. was a Ruth L. Kirschstein National Research Service Award Postdoctoral Fellow (F32 GM 106601-2). We thank Brad Hyman and Eloise Hudry for providing immortalized mouse astrocytes containing APOE4.
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature
Author contributions: A.C.K. and Y.B.K. designed the research, performed experiments, analyzed data, and wrote the manuscript. M.S.P. assisted with the data analysis. J.A.Z. assisted with the preparation of materials and the design of experiments. D.R.L designed and supervised the research and wrote the manuscript. All of the authors contributed to editing the manuscript.
Author Information: Plasmids encoding BE1, BE2, and BE3 are available from Addgene (plasmids 73018, 73019, 73020, 73021). HTS data have been deposited in the NCBI Sequence Read Archive database under Bioproject accession code PRJNA316460.
The authors declare competing financial interests: A.C.K. and D.R.L. have filed a provisional patent application on this work. D.R.L. is a consultant and co-founder of Editas Medicine, a company that seeks to develop genome-editing therapeutics.
References
- 1.Cox DB, Platt RJ, Zhang F. Therapeutic genome editing: prospects and challenges. Nature medicine. 2015;21:121–131. doi: 10.1038/nm.3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hilton IB, Gersbach CA. Enabling functional genomics with genome engineering. Genome research. 2015;25:1442–1455. doi: 10.1101/gr.190124.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sander JD, Joung JK. CRISPR-Cas systems for editing, regulating and targeting genomes. Nature biotechnology. 2014;32:347–355. doi: 10.1038/nbt.2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Landrum MJ, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic acids research. 2015 doi: 10.1093/nar/gkv1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cong L, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ran FA, et al. Genome engineering using the CRISPR-Cas9 system. Nat Protocols. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jinek M, et al. A Programmable Dual-RNA–Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Conticello SG. The AID/APOBEC family of nucleic acid mutators. Genome biology. 2008;9:229. doi: 10.1186/gb-2008-9-6-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harris RS, Petersen-Mahrt SK, Neuberger MS. RNA Editing Enzyme APOBEC1 and Some of Its Homologs Can Act as DNA Mutators. Molecular Cell. 2002;10:1247–1253. doi: 10.1016/s1097-2765(02)00742-6. [DOI] [PubMed] [Google Scholar]
- 10.Jore MM, et al. Structural basis for CRISPR RNA-guided DNA recognition by Cascade. Nature structural & molecular biology. 2011;18:529–536. doi: 10.1038/nsmb.2019. [DOI] [PubMed] [Google Scholar]
- 11.Jiang F, et al. Structures of a CRISPR-Cas9 R-loop complex primed for DNA cleavage. Science. 2016 doi: 10.1126/science.aad8282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsai SQ, et al. Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat Biotech. 2014;32:569–576. doi: 10.1038/nbt.2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schellenberger V, et al. A recombinant polypeptide extends the in vivo half-life of peptides and proteins in a tunable manner. Nature biotechnology. 2009;27:1186–1190. doi: 10.1038/nbt.1588. [DOI] [PubMed] [Google Scholar]
- 14.Saraconi G, Severi F, Sala C, Mattiuz G, Conticello SG. The RNA editing enzyme APOBEC1 induces somatic mutations and a compatible mutational signature is present in esophageal adenocarcinomas. Genome biology. 2014;15:417. doi: 10.1186/s13059-014-0417-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsai SQ, et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nature biotechnology. 2015;33:187–197. doi: 10.1038/nbt.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kunz C, Saito Y, Schar P. DNA Repair in mammalian cells: Mismatched repair: variations on a theme. Cell Mol Life Sci. 2009;66:1021–1038. doi: 10.1007/s00018-009-8739-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mol CD, et al. Crystal structure of human uracil-DNA glycosylase in complex with a protein inhibitor: protein mimicry of DNA. Cell. 1995;82:701–708. doi: 10.1016/0092-8674(95)90467-0. [DOI] [PubMed] [Google Scholar]
- 18.Lieber MR, Ma Y, Pannicke U, Schwarz K. Mechanism and regulation of human non-homologous DNA end-joining. Nat Rev Mol Cell Biol. 2003;4:712–720. doi: 10.1038/nrm1202. [DOI] [PubMed] [Google Scholar]
- 19.Heller RC, Marians KJ. Replisome assembly and the direct restart of stalled replication forks. Nat Rev Mol Cell Biol. 2006;7:932–943. doi: 10.1038/nrm2058. [DOI] [PubMed] [Google Scholar]
- 20.Pluciennik A, et al. PCNA function in the activation and strand direction of MutLα endonuclease in mismatch repair. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:16066–16071. doi: 10.1073/pnas.1010662107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beale RC, et al. Comparison of the differential context-dependence of DNA deamination by APOBEC enzymes: correlation with mutation spectra in vivo. Journal of molecular biology. 2004;337:585–596. doi: 10.1016/j.jmb.2004.01.046. [DOI] [PubMed] [Google Scholar]
- 22.Kuscu C, Arslan S, Singh R, Thorpe J, Adli M. Genome-wide analysis reveals characteristics of off-target sites bound by the Cas9 endonuclease. Nature biotechnology. 2014;32:677–683. doi: 10.1038/nbt.2916. [DOI] [PubMed] [Google Scholar]
- 23.Kim J, Basak JM, Holtzman DM. The role of apolipoprotein E in Alzheimer's disease. Neuron. 2009;63:287–303. doi: 10.1016/j.neuron.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seripa D, et al. The missing ApoE allele. Annals of human genetics. 2007;71:496–500. doi: 10.1111/j.1469-1809.2006.00344.x. [DOI] [PubMed] [Google Scholar]
- 25.Stephens PJ, et al. The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012;486:400–404. doi: 10.1038/nature11017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Slaymaker IM, et al. Rationally engineered Cas9 nucleases with improved specificity. Science. 2015 doi: 10.1126/science.aad5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Davis KM, Pattanayak V, Thompson DB, Zuris JA, Liu DR. Small molecule-triggered Cas9 protein with improved genome-editing specificity. Nature chemical biology. 2015;11:316–318. doi: 10.1038/nchembio.1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kleinstiver BP, et al. High-fidelity CRISPR–Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 2016;529:490–495. doi: 10.1038/nature16526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zuris JA, et al. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nature biotechnology. 2015;33:73–80. doi: 10.1038/nbt.3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kleinstiver BP, et al. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature. 2015;523:481–485. doi: 10.1038/nature14592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mali P, et al. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat Biotech. 2013;31838:833. doi: 10.1038/nbt.2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pattanayak V, et al. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat Biotech. 2013;31843:839. doi: 10.1038/nbt.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shcherbakova DM, Verkhusha VV. Near-infrared fluorescent proteins for multicolor in vivo imaging. Nat Meth. 2013;10:751–754. doi: 10.1038/nmeth.2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.