

Abstract
Mutations in the tumor protein p53 (TP53) are the most frequently occurring genetic events in high-grade ovarian cancers, especially the prevalence of the TP53R175H mutant allele. In this study, we investigated the impact of the TP53R175H mutant allele on epithelial ovarian cancer (EOC) in vivo. We used the Pten/KrasG12D mutant mouse strain that develops serous EOC with 100% penetrance to introduce the mutant Trp53R172H allele (homolog for human TP53R175H). We demonstrate that the Trp53R172H mutation promoted EOC, but had differential effects on disease features and progression depending on the presence or absence of the wild-type (WT) TP53 allele. Heterozygous WT/Trp53R172H alleles facilitated invasion into the ovarian stroma, accelerated intraperitoneal metastasis, reduced TP53 transactivation activity, but retained responsiveness to nutlin-3a, an activator of WT TP53. Moreover, high levels of estrogen receptor alpha in these tumors enhanced the growth of both primary and metastatic tumors in response to estradiol. Ovarian tumors homozygous for Trp53R172H mutation were undifferentiated and highly metastatic, exhibited minimal TP53 transactivation activity, and expressed genes with potential regulatory functions in EOC development. Notably, heterozygous WT/Trp53R172H mice also presented mucinous cystadenocarcinomas at 12 weeks of age, recapitulating human mucinous ovarian tumors, which also exhibit heterozygous TP53 mutations (~50–60%) and KRAS mutations. Therefore, we present the first mouse model of mucinous tumor formation from ovarian cells and supporting evidence that mutant TP53 is a key regulator of EOC progression, differentiation, and responsiveness to steroid hormones.
Keywords: mutant p53, epithelial ovarian cancer, mucinous, differentiation, steroid hormone
Introduction
As the fifth leading cause of death in women, ovarian cancer is a devastating disease without effective detection and therapeutic management. The majority (>90%) of human ovarian cancers are of epithelial cell origin (1,2) and based on their histological features are categorized as serous, endometrioid, mucinous and clear cell type (3). These subtypes differ significantly in their potential sites of origin, molecular signature, prognosis and response to treatments, making epithelial ovarian cancer (EOC) not a single disease, but multiple diseases (4).
Mutations in the tumor protein TP53 (P53) are among the most common and frequent events in EOC, especially in the high-grade serous subtype (>90%) (The Cancer Genome Atlas Research Network, TCGA) (5). Furthermore, the R175H mutation of TP53 is one of the most frequently occurring (3.8%) in serous EOC (TCGA) (6). When the Trp53R172H mutation (mouse homolog for the R175H mutation in human) is expressed in cells depleted of Apc and Pten, or in cells with inactivated Rb and Brca1/2, it promotes epithelial ovarian tumor progression and metastasis (7,8). Moreover, a recent examination of TCGA database reveals that p53 gain-of-function (GOF) mutations, including the R175H mutation, in ovarian tumors of human patients are associated with a higher incidence of platinum resistance, local recurrence, and distant metastasis (9). Intriguingly and importantly, recently published studies show that TP53 mutations in human EOC tumor samples are frequently present as heterozygous (10) (and Mullany et al., Neoplasia, in press), indicating that it is critical to understand not only the biology of tumors homozygous for wild-type or mutant alleles of p53, but also the interaction between wild-type and mutant alleles in a tumor type-specific context.
Oncogenic alterations are necessary for ovarian cancer development, but strong evidence indicates that steroid hormones such as estradiol also play critical roles in promoting the timing of onset, progression and metastasis of EOC (11–13). The majority of EOC cases occur during the peri- and post-menopausal period (peak at ages 65–75), when the tumor-promoting effects of estradiol are unopposed by progesterone (1). Several recent epidemiological and clinical studies suggest that the expression of steroid hormone receptors is linked to ovarian cancer subtypes, progression, prognosis and response to hormone treatment (14–18). Thus, how oncogenic factors, TP53 and steroid hormones interact to impact EOC development is becoming an emerging area of research that is highly relevant for developing and revising clinical therapeutic strategies.
Our laboratory previously generated mice in which the Pten gene was conditionally deleted and KrasG12D was activated by Cre-recombinase driven by Amhr2 promoter (Pten/Kras mutant mice) (19,20). These mice developed ovarian surface epithelial (OSE) cell derived, low-grade serous adenocarcinomas with 100% penetrance. When Trp53 was deleted in the OSE cells of the Pten/Kras mutant mice, tumor growth was markedly reduced but the expression of estrogen receptor alpha was elevated (21), and when exposed to estradiol, the primary ovarian lesions grew rapidly and metastasized throughout the intraperitoneal cavity. These studies documented clearly and for the first time that the functional status of TRP53 can determine steroid hormone responsiveness in EOC cells.
Based on the foregoing considerations, we hypothesized that 1) specific Trp53 mutations would not only alter tumor growth and metastasis but also the responsiveness of EOC cells to steroid hormones, 2) the effect of any given mutation would also depend on whether a WT allele was present or absent, and 3) each condition would exhibit a distinct molecular signature. Specifically, because the human TP53R175H allele is frequently associated with high-grade ovarian cancer, we introduced a germ-line Trp53 R172H mutant allele into our Pten/Kras strain to generate OSE cells either heterozygous or homozygous for the Trp53R172H mutant allele. Our findings indicate that expression of heterozygous Trp53 mutant alleles drives a tumor phenotype that is distinct from that of WT Trp53 or homozygous Trp53 mutant alleles in determining ovarian tumor behavior and outcome.
Materials and Methods
Animal Studies
All animals were housed under a 14-hour light/10-hour dark schedule in the Center for Comparative Medicine at Baylor College of Medicine and provided food and water ad libitum. All animals were maintained according to the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals and approved by the Animal Care and Use Committee at Baylor college of Medicine. Mice carrying the germline Trp53R172H mutation were generated by Dr. Guillermina Lozano (MD Anderson Cancer Center, Houston, Texas) (22). These mice were bred to Ptenf/f;KrasG12D;Amhr2-Cre (PK) mice described previously to obtain PK mice expressing WT Trp53 (PKP53+/+), and either heterozygous (PKP53H/+) or homozygous mutant Trp53R172H (PKP53H/H) (19).
Laser capture microdissection and sequencing of human mucinous tumor samples
Laser capture microdissection of human mucinous tumor samples is described in detail in supplemental materials. DNA was extracted from microdissected tumor cells using a QIAamp DNA Micro Kit (Qiagen, Valencia, CA) and quantified using a NanoDrop spectrometer (NanoDrop, Rockland, DE). Mutation analysis of TP53 (Exons 5–8) was performed by Sanger Sequencing using PCR primers as described previously (23).
Injection of tumor cells into syngenic mice and hormone pellet implants
Isolation, culturing, and injection of OSE cells from genetically engineered mice are described in detail in Supplementary Materials and Methods. Genotype-confirmed mice of WT and mutant Trp53 status were implanted with one-half of a 0.36-mg estradiol pellet (0.36 mg) or with progesterone pellet (25 mg) one day prior to giving estradiol pellet (SE-121 for 17β-estradiol and P-131 for progesterone, Innovative Research of America, Sarasota, FL), and tumor progression was monitored at 1, 2 or 4 weeks after pellet implanting.
RNA extraction, real-time RT-PCR and microarray analyses
DNA expression vectors (0.20 μg DNA ; empty vector or vector expressing mutant R175H TP53) were transfected into cells using Attractene Transfection Reagent (Qiagen, Valencia, CA) according to the manufacturer’s protocol, and lysed for RNA extraction 24 hours following transfection. Total RNA extraction, reverse transcription, real-time PCR and microarray analyses were described in Supplementary Materials and Methods. The microarray data have been deposited to GEO with the accession number GSE65206.
Cell adhesion, proliferation and viability assays
To assess cell attachment to the culture dish, PKP53+/+, PKP53H/+ and PKP53H/H OSE cells at similar passages were plated at 100,000 cells/well in a 12-well plate. After culturing for 2 hours, the plates were gently swirled and pipetted three times. Unattached cells were removed; attached cells that remained on the dish were trypsinized and counted with a hemocytometer. To measure the rate of proliferation, OSE cells at similar passages were plated at 100,000 cells/well in each 12-well plate. After 36 hours of culture, when cells of all genotypes have attached well and are proliferating but not confluent, cells were trypsinized and counted using a hemocytometer. Cell viability was measured using the WST-1 assay according to manufacturer’s instruction following 72 hours of nutlin-3a treatment. The WST-1 reagent was incubated with the cells for 4 hours before its absorbance was read at 450 nm (Roche, Pleasanton, CA).
Western blot analyses
Whole cell lysate were collected using methods previous described (24). Proteins were separated using a 10% Bis-Tris gel and transferred to an immobilon-P membrane (Millipore). Primary antibodies used included ESR1 (sc-542) (1:1000), PR (sc-7208) (1:1000), and p53 (sc-6243) (1:1000) (Santa Cruz Biotechnology) and signals were detected using the Peirce ECL 2 Western Blotting Substrate (Thermo Scientific).
TP53 luciferase assay
OSE cells were transfected according to manufacturer’s instruction (P53 Cignal Reporter Assay Kit, Qiagen Sciences). Nutlin-3a (generously provided by Dr. Robert Bast, MD Anderson Cancer Center, Houston, TX) or DMSO vehicle control were added to the cultures after overnight transfection and luciferase assays were performed after 24 hours of treatment using the Dual-Glo Luciferase Assay System (Promega).
Statistical analysis
Data are represented as mean ± SEM. Comparisons between experimental groups with a single variance were analyzed using unpaired Student’s t-test, and a two-tailed p<0.05 was considered statistical significance. Comparison between experimental groups with more than one variances were analyzed using randomized two-way ANOVA. When ANOVA indicated overall significance, Student-Newman-Keuls test was used to compare individual means.
Results
Trp53 R172H mutation promotes epithelial ovarian tumor progression
Before 12 weeks of age, ovarian epithelial tumors in the PKP53+/+ and PKP53H/+ mice exhibited similar serous, papillary-like structures on the surface of the ovary that stained positively for cytokeratin 8 (KRT8), a marker for OSE cells (Fig. 1A). However, the tumors in the PKP53H/+ mice appeared more locally invasive into the ovarian stroma and more metastatic to the omentum than those in the PKP53+/+ mice of the same age (Fig. 1A). Furthermore, the PKP53H/+ mice died consistently between 12 and13 weeks of age (n=8), whereas most PKP53+/+ mice died between 16 and 40 weeks of age (Fig. 1B and 1C) (19), indicative of a more aggressive tumor phenotype in the PKP53H/+ mice (Fig. 1B). Interestingly and unexpectedly, 80% PKP53H/+ mice also developed mucinous-like tumors between 8 and 12 weeks of age, as described below in Figure 3.
Figure 1.

Mutant Trp53R172H allele promotes epithelial ovarian tumor progression. A, Representative images of tissue sections stained with H&E or KRT8 of ovaries and omentum from PKP53+/+ and PKP53H/+ mice at 12 weeks of age. Regions in frame are shown at higher magnification below. Black and red dashed lines: the surface of the ovary. Black arrow heads: ovarian bursa. Scale bar: 400 μm (top row) and 80 μm (bottom two rows). B, Survival ages and phenotypic characteristics of mutant mice with different p53 status. *Data presented here on PKP53+/+ mice are from previous published studies and new information collected from the current study (19). **PKP53H/H mice die rapidly from unknown causes. ***Female progenies with homozygous Trp53R172H/R172H mutation are born at lower than Mendelian ratio due to female-specific exencephaly, thus making it harder to obtain large sample size for the cohort. C, Kaplan-Meier analysis shows that Trp53R172H/R172H homozygous mutation is associated with the shortest term of survival, and Trp53R172H/+ heterozygous mutation is associated with significantly reduced term of survival compared to mice expressing wild type Trp53. (p<0.0002 for comparison between any two groups, log-rank test).
Figure 3.

Mucinous epithelial ovarian tumors develop in PKP53H/+ mice. A, At 12 weeks of age, 80% of PKP53H/+ mice develop mucinous structures and some of them become cystic (top panel). Regions in frame are shown at higher magnification at the right. Red arrows: OSE cells. Black arrows: secretory mucinous structures. Scale bar: 900μm, 180 μm and 90 μm. B, Representative image of H&E stained human mucinous EOC tumor (upper panel) and the corresponding sequencing results of the TP53 gene in laser capture microdissected tumor cells from the same patient. Black arrows: secretory mucinous structures. Scale bar: 200 μm. C, The RMUG-L human cell line, derived from a mucinous ovarian carcinoma, carries heterozygous WT and mutantTP53 alleles and exhibits secretory mucinous structures when innoculated into the intraperitoneal cavity of SCID mice. Scale bar: 25 μm. D, Mucinous structures in PKP53H/+ mice express MUC5AC, a marker for human mucinous ovarian carcinoma. Adjacent sections without first antibody were used as negative controls. Scale bar: 100 μm. E, Mucinous structures in PKP53H/+ mice express KRT8 but do not express FOXL2 (nuclear staining) or PAX8 (nuclear staining). Red arrowheads: mucinous structures; red arrows: epithelial structures. Scale bar: 150 μm. F, Some cystic structures in the ovary of PKP53H/+ mice contain contiguous regions that are positive for both MUC5AC and KRT8 (red arrow heads) or for KRT8 alone (red arrows) (inserts are higher magnification of regions in frames). Black arrowhead: ovarian bursa. Scale bar: 200 μm and 100 μm.
Epithelial ovarian tumor expressing homozygous Trp53R172H mutation have early onset transformation and metastasis
Papillary-like epithelial ovarian lesions also developed in homozygous PKP53H/H mice as early as 4 weeks of age and appeared to be in a more advanced stage compared to those in PKP53H/+ mice of the same age (Fig. 2A, upper panels, red arrows). Notably, a significant number of KRT8-positive metastatic tumor cells were present on the omentum of PKP53H/H mice at 4 weeks of age, while only minimal metastasis to the omentum was observed in PKP53H/+ mice (Fig. 2A, lower panels of KRT8 staining). The PKP53H/H tumor cells that metastasized to the omentum were highly proliferative as shown by KRT8 and phospho-histone H3 (p-HH3) co-staining (Fig. 2A, lower panels). Unfortunately, the PKP53H/H mice died rapidly at 5 weeks from as yet unknown causes (Fig 1B and 1C).
Figure 2.

Epithelial ovarian tumors expressing homozygous Trp53R172H mutation have early onset and metastasis. A, Representative images of tissue sections stained with H&E, KRT8, or KRT8 and pHH3 on ovaries (top panels) or omentum (bottom panel) from PKP53H/H and PKP53H/+ mice at 4 weeks of age. Regions in frames are shown at higher magnification at the right. Scale bar: 400 μm, 80 μm, and 40 μm. B, Representative images of H&E and KRT8 staining on sections of tumors from intraovarian innoculated PKP53H/H cells. Region in frame is shown at higher magnification at the right. F=follicle; T=tumor; I=intestine; P=pancreas. Scale bar: 750 μm, 200 μm, 100 μm and 50 μm.
To further investigate the behavior of PKP53H/H OSE cells in vivo, we generated OSE cell lines from PKP53H/H mice and injected these cells into the ovaries of syngenic hosts. Within 2 weeks, large, undifferentiated tumors were rampant in the ovaries injected with PKP53H/H cells (Fig. 2B, upper panels). Consistent with metastatic tumors seen in PKP53H/H mice, these injected tumor cells also grew rapidly at sites throughout the intraperitoneal cavity such as the bowel mesentery, the pancreas and the omentum (Fig. 2B, lower panels). In the pancreas, these tumor cells appeared invasive.
OSE tumors expressing heterozygous Trp53R172H develop mucinous-like structures
A unique feature of the PKP53H/+ mice is that they developed large cystic-like structures within the ovary between 8 and 12 weeks of age (8/10 females) (Fig. 1B and Fig. 3A, upper panels); these structures contained serous as well as mucinous-, secretory- like cells that were not observed in PKP53+/+ or PKP53H/H mice at any age, and morphologically they resemble mucinous ovarian EOC in human patients (Fig. 3B, upper panel). KRT8 and H&E staining showed that the PKP53H/+ OSE cells (red arrows) invaded the ovarian stroma and presented various stages of differentiation into secretory mucinous-like structures (Fig. 3A, lower panels, black arrows). A previously characterized human ovarian cancer cell line (RMUG-L) that was obtained from a human mucinous ovarian carcinoma, carries heterozygous WT and mutant TP53 (c.614A>G) alleles as confirmed by us recently (23), exhibited a similar morphological and secretory phenotype when innoculated into the intraperitoneal cavity of the severely compromised immunodeficient (SCID) mice (Fig. 3C) (25,26). To confirm the mucinous tumor phenotype in the PKP53H/+ ovary, we performed immunohistochemical analyses using an antibody detecting Mucin 5AC (MUC5AC), a marker of the human ovarian mucinous-like cancer subtype (Fig. 3D) (27,28). Indeed, the secretory structures in the mouse tumors are immuno-positive for MUC5AC (Fig. 3C) and KRT8 (Fig. 3E), but not immuno-positive for FOXL2 (a marker for granulosa cells) or PAX8 (a marker for Fallopian tube secretory cells) (Fig. 3E) (29,30), suggesting that mucinous-like cells may arise from the ovarian surface epithelial cells and not from granulosa cells or Fallopian tube epithelial cells. In some cystic structures, mucinous cells that were KRT8 and MUC5AC positive were contiguous with KRT8 positive and MUC5AC negative cells, providing further evidence that MUC5AC-positive secretory cells and KRT8-positive epithelial cells may share a common precursor (Fig. 3F).
To determine the mutant status of TP53 in human mucinous EOCs, we have sequenced laser capture microdissected mucinous tumor cells from 12 human patient samples. Of these, four samples (33%) had mutations in exons 5–8 of the TP53 gene, and in all 4 cases, the wild-type allele is still expressed (Fig. 3B, lower panel; and Supplemental Fig. 1). These results support recent studies indicating that a significant number of human mucinous tumors express heterozygous mutant and wild type alleles, including the R175H mutation (Supplemental Table 1) (10,31). Specifically, supplemental data provided in the study by Mackenzie et al. (10) show that in a dataset composed of 37 laser capture microdissected human mucinous ovarian tumor samples, 63% had mutations in KRAS, 56.8% had mutations in TP53, and 36% of tumors with KRAS mutations also had mutations in TP53. Importantly, 68% of TP53 mutations in these patient samples are heterozygous, with the co-existence of a wild type and mutant allele. This observation is also consistent with the studies by Rechsteiner et al. (31), which demonstrated that at least half (57%) of human mucinous ovarian tumor samples express mutations in KRAS, and 37% of these mutations are co-expressed with mutations in TP53. To our knowledge, the PKP53H/+ mutant mouse strain is the first model that presents a mucinous-like tumor phenotype that is not derived from cells of the Müllerian duct or gut (2,32).
Epithelial ovarian tumors expressing heterozygous Trp53R172H have elevated expression of estrogen receptor alpha
To determine how expression of the Trp53R172H mutant allele affects OSE tumor responses to steroid hormones, we examined the expression and localization of estrogen receptor alpha (ESR1) and progesterone receptor (PR) in tumor cells of PKP53+/+, PKP53H/+ and PKP53H/H mice using in vivo and in vitro approaches. At 8 weeks of age (Fig. 4A and Supplemental Fig. 2), the majority of in vivo ovarian tumor cells positive for KRT8 in the PKP53+/+ mice were not ESR1-positive, whereas the majority of the KRT8-positive tumor cells in PKP53H/+ mice exhibited intense ESR1 staining. KRT8-positive tumor cells in the PKP53H/H mice exhibited low ESR1 staining. Consistent with these observations, the levels of Esr1 and Pgr mRNA are elevated in the PKP53H/+ tumor cells compared to those of the other two genotypes, and this difference was statistically different for Esr1 but not Pgr (Fig. 4B). ESR1 protein levels were also analyzed in cell lines derived from OSE cells isolated from ovaries of mice of each genotype. Western blotting confirmed that ESR1 was elevated in cells derived from the PKP53H/+ mice compared to the other two genotypes. Western blots for PR indicated that the protein levels of the PR-A isoform were higher than those of the PR-B isoform in cells of all three genotypes, and that PR-A was higher in PKP53+/+ cells compared to the other two (Fig. 4C). When OSE cells were treated with nutlin-3a, a small molecule that stabilizes active TRP53 protein, it increased Esr1 mRNA expression in PKP53+/+ and PKP53H/+ cells, but not in PKP53H/H cells (Fig. 4D), suggesting that TRP53 regulates transcription of Esr1 gene in OSE cells, and the presence of both the wild type Trp53 and mutant R172H alleles has the strongest inductive effect. These results support several previous studies that have shown direct transcriptional regulation of ESR1 by wild-type p53 using chromatin immunoprecipitation (ChIP) analyses (33,34). Under normal physiological conditions, wild type TP53 is expressed at very low levels in most cells. However, it has been shown that the expression of wild type TP53 protein increases to a very high level in the presence of mutant p53 protein (35). Thus, we speculate that the presence of heterozygous TRP53 alleles in our mouse ovarian tumor cells, as well as in relevant human tumors, elevates expression of ESR1 through wild-type p53 stabilization. In contrast to Esr1, nutlin-3a did not increase Pgr mRNA expression in any of the three cell lines (Fig. 4D). When a vector expressing the mutant R175H allele was transfected into PKP53+/+ cells, it induced increased expression of Esr1 but not Pgr mRNA (Fig. 4E), also suggesting that co-expression of wild type Trp53 and the mutant R175H alleles enhances transcription induction of the Esr1 gene. Interestingly, while many adjacent KRT8-positive epithelial tumor cells express high levels of ESR1, ESR1 expression is not present in the differentiated, secretory mucinous type tumor (Fig. 4F). This observation is consistent with variable ESR1 expression in human mucinous ovarian tumors (36), and suggests that ESR1 may be present and play a role during early stages of mucinous ovarian tumor development.
Figure 4.

Expression of ESR1 is elevated in OSE cells of PKP53H/+ mice. A, Representative images of immunofluorescent staining of ESR1 and KRT8 in OSE cells of ovarian tissue sections. Yellow dash lines: the surface of the ovary. F=follicles. Scale bar: 50 μm. B, Levels of Esr1 and Pgr mRNA in isolated and un-passaged OSE cells quantified by RT-PCR and normalized to Rpl19 expression. Data are represented as mean ± SEM (n=5 mice per genotype). *p<0.05. C, Representative images of Western blot for ESR1, PR-A, PR-B and beta-Actin in OSE cell lines. D, Levels of Esr1 and Pgr mRNA in OSE cells treated with DMSO or nutlin-3a (10 μM) and quantified by RT-PCR and normalized to Rpl19 expression. Data are represented as mean ± SEM (n=3 different experimental replicates). Bars without common superscript are significantly different, *p<0.05. E, Levels of Esr1 and Pgr mRNA in PKP53+/+ OSE cells expressing empty vector or vector expressing the R172H mutant allele, and quantified by RT-PCR and normalized to Rpl19 expression. Data are represented as mean ± SEM (n=3 different experimental replicates). *p<0.05.
Estradiol promotes tumor growth on the ovary and omentum in PKP53H/+ mice
Based on the elevated expression of Esr1 mRNA and protein in tumors of the PKP53H/+ mice, we next investigated the in vivo response of tumors in these mice to estradiol. Four weeks of estradiol exposure induced rampant growth and local invasion of the primary tumors within the ovarian stroma and proliferation of metastatic cells present on the omentum (Figure 5A and 5B). We also tested the effect of estradiol on PKP53H/+ tumor cells by subcutaneously injected these cells into ovariectomized syngenic hosts (Figure 5E). Estradiol significantly stimulated subcutaneous tumor growth of the PKP53H/+ tumor cells. Previously we have demonstrated that progesterone blocks effects of estradiol on tumor growth and metastasis in the PKP53−/− mice (11). However, when progesterone was given one day before estradiol to the PKP53H/+ mice, it did not block estradiol-induced tumor growth either in the primary tumor or at metastatic sites on the omentum (Figure 5C). We quantified the percentage of pHH3-positive proliferating KRT8-positive tumor cells, further confirming our observations of tumor grow rate with estradiol and progesterone treatment (Figure 5D). Thus, we conclude that estradiol promotes primary and omental metastatic tumor growth in PKP53H/+ mice, and this effect is not blocked by progesterone in the context analyzed.
Figure 5.

Estradiol promotes tumor growth on the ovary and omentum in PKP53H/+ Mice. A, Representative images of H&E and KRT8 staining of ovarian tissue sections from PKP53H/+ mice. Regions in frames are shown at higher power at the right. Scale bar: 750 μm and 150 μm. B, Immunofluorescent staining of pHH3 and KRT8 on sections of ovaries and omentum from PKP53+/+ and PKP53H/+ mice with or without in vivo estradiol treatment. Yellow dashed lines: the surface of the ovary. C, Representative images of H&E and KRT8 and pHH3 staining of ovarian tissue sections from PKP53H/+ mice implanted with progesterone pellets one day before estradiol pellets. Black arrowheads: transformed OSE cells. Dashed lines: the surface of the ovary. F: follicles. Scale bar: 100 and 50 μm. D, Quantification of the percentage of KRT8+pHH3+ cells among KRT8+ cells with visible nuclei was performed on representative images taken from different mice of each genotype (n=4). Data are represented as mean ± SEM. *p<0.05. Scale bar: 50 μm. E, Estradiol promotes the growth of subcutaneously injected PKP53H/+ OSE cells into syngenic mice (n=5). *p<0.01. D, Primary tumors on the ovary (top panels) and metastatic tumors on the omentum (bottom panel) showed extensive growth when the progesterone pellet was implanted one day before the estradiol pellet. Black arrowheads: ovarian bursa. Dashed lines: the surface of the ovary.
Expression of the Trp53R172H mutant allele alters OSE tumor differentiation and promotes their proliferation and survival
To obtain a comprehensive understanding of the molecular events occurring in the OSE tumor cells expressing Trp53R172H, we performed microarray analyses on RNA prepared using unpassaged OSE cells isolated from PKP53+/+, PKP53H/+ and PKP53H/H mice (Fig. 6A). Heatmap analyses showed that gene expression patterns are quite similar between PKP53+/+ and PKP53H/+ tumor cells, despite the marked differences in the responses of tumor cells to estradiol in these mice. PKP53H/H cells had a markedly distinct gene expression profile compared to the other two genotypes.
Figure 6.

Expression of the Trp53R172H mutant allele alters OSE tumor differentiation and promotes their proliferation and survival. A, Heatmap comparison of gene expression profiles from microarray analyses of OSE cells isolated from PKP53+/+, PKP53H/+ and PKP53H/H mice. B, RT-PCR quantification and verification of selected genes in functional categories with different levels according to the microarray analyses. Data are represented as mean ± SEM (n=5). *p<0.05. C, Expression of the Trp53R172H mutant allele alters OSE cell adhesion (top panel) and proliferation (bottom panel) in culture. This experiment was repeated 3 times and each experiment contains 4 replicates for each genotype. Data are represented as mean ± SEM. *p<0.01.
To verify the gene expression profiles, real-time qPCR analyses were performed (Fig. 6B). Genes known to be associated with OSE stem cells (Lgr5, Aldh1a1 and Aldh1a7) exhibited significantly reduced levels of mRNA in the PKP53H/H tumor cells. Three epithelial cell differentiation-regulating transcription factors, Lhx9, Amhr2, and Foxa2, were also substantially dysregulated in these cells: Lhx9 was undetectable, Amhr2 was minimal and Foxa2 was increased in the PKP53H/H tumors. The expression of several secretory factors was altered: Cxcl12 and Bdnf mRNAs increased in tumors of the PKP53H/H mice but not in the PKP53+/+, PKP53H/+ mice whereas Igfbp6 mRNA was drastically decreased in these cells. Expression of apoptosis and cell survival regulators Ddit4 and Dapk2, among many other apoptosis regulators, was diminished in PKP53H/H cells; while cell cycle regulators, such as Ccnb1, were highly expressed in these cells. Lastly, several tumoriogenic regulators in OSE tumors had differentially regulated expression in the PKP53H/H cells; Ndn was dramatically reduced whereas Ddx39, and Eya4 were significantly increased. We further analyzed cell adhesion and proliferation in the PKP53+/+, PKP53H/+ and PKP53H/H cells. As shown in Fig. 6C, cell adhesion was reduced in both PKP53H/+ and PKP53H/H cells, and the PKP53H/H cells were also significantly more proliferative than cells of the other genotypes.
Trp53 R172H mutation alters TRP53 activity in OSE tumors
To determine TRP53 activity in the PKP53+/+, PKP53H/+ and PKP53H/H cells, nutlin-3a, a small molecule stabilizer and activator of TRP53, was utilized (37). Nutlin-3a stabilized TRP53 protein in cells expressing WT Trp53 as expected (Fig. 7A) (38). Expression of the Trp53R172H stabilized TRP53 protein with or without nutlin-3a treatment in PKP53H/+ and PKP53H/H mutant cells. Enhanced stabilization of TRP53 protein was also demonstrated by TRP53 immunofluorescent staining (Fig. 7B). Despite the high levels of TRP53 protein, the activity of TRP53 signaling pathway was reduced and negligible, respectively, in PKP53H/+ and PKP53H/H mutant cells, as measured by a luciferase assay containing an inducible p53-reporter, either with or without nutlin-3a treatment (Fig. 7C).
Figure 7.
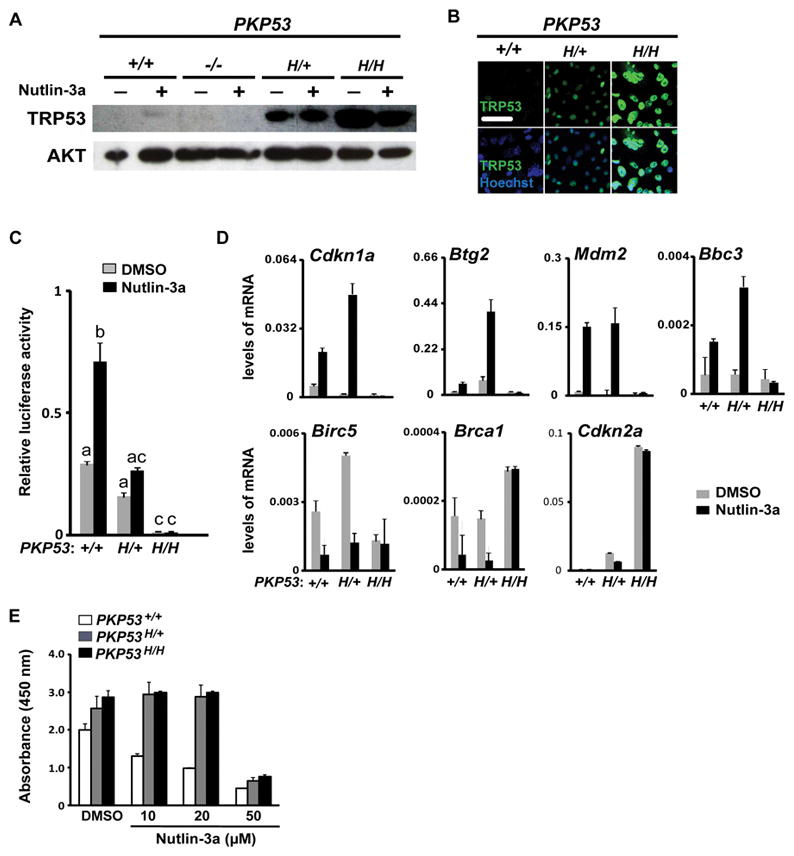
Mutant Trp53R172H alters TRP53 activity in OSE tumors. A, Representative Western blot analyses of TRP53 and AKT in OSE cells of different genotypes treated with DMSO or nutlin-3a (10 μM). B, Representative immunofluorescent staining of TRP53 in cultured OSE cells. Scale bar: 25 μm. C, Relative luciferase activity of a p53 reporter construct. Data is representative of experiments repeated 4 times and in each experiment luciferase activity was measured in triplicates. Bars without common superscript are significantly different, *p<0.05. D, Expression of the Trp53R172H mutant allele alters mRNA levels of both TRP53-induced (upper panels) and -suppressed genes (lower panels) in response to nutlin-3a treatment (n=3). E, Cell viability in response to nutlin-3a treatment (72 hr) as measured by the WST-1 assay. This experiment was repeated 3 times and each experiment contains 3 replicates for each genotype. Data are represented as mean ± SEM.
In addition, we analyzed basal and nutlin-3a induced expression or repression of known TRP53 target genes in mutant cells of each genotype (Fig. 7D). Basal levels of Cdkn1a (P21) and Mdm2 were reduced in the PKP53H/+ and PKP53H/H mutant cells compared to the PKP53+/+ cells. However, Cdkn1a, Mdm2, Btg2 and Bbc3 (Puma) were highly induced by nutlin-3a in both the PKP53+/+ and PKP53H/+ cells but not in the PKP53H/H mutant cells, indicating that the presence of one mutant allele is not sufficient to suppress the activity of the wild type allele. Likewise, expression of genes suppressed by TRP53 such as Birc5 and Brca1 were decreased by nutlin-3a treatment in the PKP53+/+ and PKP53H/+ cells but not in the PKP53H/H mutant cells. Notably, the expression of Cdkn2a (P16) was highly expressed in the PKP53H/H mutant cells as we have observed in Pten/Kras mutant cells that are null for Trp53 (21). Interestingly, despite retained transactivation activity of the wild type TRP53 in the PKP53H/+ cells, nutlin-3a treatment effectively caused death of PKP53+/+ cells in a dose-dependent manner, but this effect was abolished in PKP53H/+ and PKP53H/H cells (Fig. 7E), reflecting potential gain-of-function effect of the R172H allele in promoting tumor survival.
Discussion
Trp53 R172H mutation impacts ovarian cancer cell initiation and progression
The R175H mutation in the human TP53 gene is one of the most frequently occurring mutations in human EOC and, based on recent analyses, is frequently expressed with one wild type allele (10). These data raise a novel and critical question about the functional activity and impact of the mutant p53 allele in the presence or absence of the WT allele (Mullany, et al., Neoplasia, in press). Indeed, we demonstrate herein that tumors in the PKP53H/+ mutant mice exhibit some similarities but also some marked differences from tumors in the PKP53+/+ mice. Like the primary tumors in the PKP53+/+ mice, those in the PKP53H/+ mutant mice develop extensive papillary-like structures on the ovarian surface that stain positively for KRT8 before 8 weeks of age. However, PKP53H/+ mutant tumors 1) develop faster and are more metastatic, 2) present mucinous epithelial tumors between 8 and 12 weeks of age, 3) express higher levels of ESR1 mRNA and protein and 4) are more responsive in vivo to estradiol than the PKP53+/+ cells.
In line with the tumor-promoting effects of the Trp53R172H allele in our mouse model of EOC, homozygous PKP53H/H OSE cells exhibit early lesion onset and undergo more advanced papillary transformation and prominent metastasis onto the omentum as early as 4 weeks of age (39). When we injected PKP53H/H tumor OSE cells into the ovaries of syngenic host mice, they grew rapidly in the ovary and intraperitoneal cavity and invaded the pancreas. Thus homozygous mutant alleles exert a potent effect on the initiation, early progression and metastasis of ovarian tumor in the same genetic background. Gene profiles of the PKP53H/H EOC cells further document that these cells are markedly distinct from those in the PKP53+/+ or PKP53H/+ mice, as discussed below.
Trp53 R172H mutation alters epithelial ovarian tumor differentiation
Particularly striking, and of high clinical relevance, was the emergence of KRT8-positive mucinous-like tumors in the PKP53H/+ mice that invade the ovary, and present a variety of differentiation stages by 8–12 weeks of age. Thus, the Trp53R172H mutant allele promotes, in a subset of cells, a phenotype that is distinct from the serous, papillary-like structures that develop earlier. To our knowledge, the PKP53H/+ mice may potentially provide the first mouse model of mucinous EOC subtype arising from OSE cells, a site far removed from the presumed cervical/gut origin of mucinous tumors in women (3). Among human patients, the mucinous subtype is considered relatively uncommon but often has worse outcomes and is more resistant to conventional chemotherapy (36,40). Tumor development in the PKP53H/+ mice resembles features of human mucinous ovarian cancer in several aspects: 1) mutation in KRAS is a notable feature of the human ovarian mucinous subtype (in ~50% cases) (10,31,41–43), 2) concurrence of KRAS and TP53 mutations occurs in human mucinous ovarian cancer samples (36%) (10,31), 3) a significant number of human mucinous tumors are heterozygous for mutant TP53 allele and a wild type allele (Fig 3B and Supplemental Table 1) (10), and 4) mutations in the PI3K/PTEN pathway are also a prominent feature of human mucinous ovarian cancer (10). That OSE cells may have the potential to trans-differentiate into mucinous-like, secretory tumor cells highlights 1) the plasticity of these ovarian surface epithelial cells and 2) the importance of the interactions among p53 status, cell-of-origin and the ovarian microenvironment in determining ovarian tumor cell fate decisions.
TRP53 and steroid hormone receptor status impact tumor prognosis and responsiveness to steroid hormones
We show that tumors in the PKP53H/+ mice express elevated levels of ESR1 in vivo and in vitro, and undergo marked proliferation in the primary tumor and at metastatic sites on the omentum in response to estradiol treatment in vivo. Although PGR(A and B) are present in these cells, progesterone treatment prior to estradiol does not block estradiol-induced tumor growth. These results differ from those described previously in the same mutant background (11), where estradiol potently stimulated proliferation and metastasis of Trp53 null PKP53−/− tumors and these responses to estradiol were completely blocked by progesterone treatment. According to these observations, TP53 status and steroid hormone receptor status could be considered together for predicting tumor prognosis and responsiveness to steroid hormones in ovarian cancer patients.
Heterozygous state with co-expression of both wild-type and Trp53 R172H mutation represents a distinct tumor phenotype
A number of reports indicate that the Trp53 R172H mutation exerts dominant negative (DN) effects on the WT Trp53 allele (44,45). However, our data indicate that this mutant has other effects on the WT allele in the PKP53H/+ tumor cells that may be context specific. First, as mentioned above, tumor morphology appeared to be quite similar between PKP53H/+ mice and PKP53+/+ mice, at least until later stages of tumor development when mucinous tumors arise in the PKP53H/+ mice. Second, whereas microarray gene expression profiles are similar (but not identical) between OSE cells of these two genotypes, they are dramatically distinct from the gene expression profiles in the PKP53H/H cells. Third, in the presence of the R172H allele, p53 signaling seems to be active based on both luciferase TRP53 activity reporter assays and induction of P53 target genes upon nutlin-3a treatment. Collectively, our data and other studies provide evidence that it is possible that some ovarian cancer patients with heterozygous mutant TP53 expression will respond to therapeutic interventions which activate WT p53 signaling (46) (and Mullany et al., Neoplasia, in press).
The vast number of genes that exhibit increased and decreased expression in the PKP53H/H tumor cells compared to PKP53H/+ and PKP53+/+ cells suggest that the homozygous mutants exert profound changes in the molecular events controlling these cells, some of which may mediate GOF mechanisms to ensure tumor survival in the absence of wild type TRP53. The uniqueness of the gene profiles in the PKP53H/H tumor cells is further supported by the fact that they are also quite distinct from those in the p53 null cells of the Pten/Kras background (21). One example is Eya4, the overexpression of which has been reported in many different cancers (47–50). In comparison to cells expressing wild-type Trp53, the level of Eya4 mRNA is dramatically elevated in PKP53H/H cells, but reduced in PKP53 null OSE cells (21). Eya4 and other differentially regulated genes in OSE cells expressing homozygous R172H mutant alleles versus null Trp53 alleles may explain the distinctly different behaviors of these two OSE tumors. In particular, while epithelial ovarian tumors in the PKP53H/H homozygous mice exhibit early onset and metastasis, epithelial ovarian tumors in the PKP53−/− homozygous mice exhibit slow growth and minimal transformation (21). Thus, the status of p53 in tumor cells impacts tumor cell behavior, growth morphology, and metastatic potential.
Supplementary Material
Acknowledgments
Financial support: This work is supported by NIH-CA-181808 (JSR), NIH-HD-16229 (JSR), U54-HD07945 (Project 2, JSR) of SCCPIR Specialized Cooperative Centers Program in Reproduction and Infertility Research (Bert W O’Malley, BWO), and NIH-T32-HD-1765-36 (BWO). K-K W is supported in part by grants from the National Institutes of Health, including The University of Texas MD Anderson Cancer Center Specialized Program of Research Excellence in Ovarian Cancer (P50 CA08369), CA133057, and the Blanton-Davis Ovarian Cancer Research Program.
The authors thank Dr. Markus Rechsteiner for sharing data and for their insights in personal communications. The authors also thank Dr. Victor Kostyuk for statistical analyses. This project was supported by the Integrated Microscopy Core at Baylor College of Medicine with funding from the NIH (HD007495, DK56338, and CA125123), the Dan L. Duncan Cancer Center, and the John S. Dunn Gulf Coast Consortium for Chemical Genomics. This project was also supported by the Pathology and Histology Core at Baylor College of Medicine with funding from the NIH (NCI P30-CA125123). We also thank the Genomic and RNA profiling Core at Baylor College of Medicine for performing the microarray analyses.
Footnotes
The authors do not have any conflicts of interest to disclose.
Author Contributions
Y.A.R. and J.S.R. provided the conception and design for the studies. Y.A.R., L.K.M., and K.K.W. developed methodology. Z.L. analyzed the microarray data. A.J.H. provided pathological diagnosis. Y.A.R. performed all experiments and acquisition of data.
References
- 1.Romero I, Bast RC., Jr Minireview: human ovarian cancer: biology, current management, and paths to personalizing therapy. Endocrinology. 2012;153(4):1593–602. doi: 10.1210/en.2011-2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kurman RJ, Shih Ie M. Molecular pathogenesis and extraovarian origin of epithelial ovarian cancer--shifting the paradigm. Human pathology. 2011;42(7):918–31. doi: 10.1016/j.humpath.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cho KR, Shih Ie M. Ovarian cancer. Annual review of pathology. 2009;4:287–313. doi: 10.1146/annurev.pathol.4.110807.092246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schuler S, Ponnath M, Engel J, Ortmann O. Ovarian epithelial tumors and reproductive factors: a systematic review. Archives of gynecology and obstetrics. 2013;287(6):1187–204. doi: 10.1007/s00404-013-2784-1. [DOI] [PubMed] [Google Scholar]
- 5.Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609–15. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brachova P, Thiel KW, Leslie KK. The consequence of oncomorphic TP53 mutations in ovarian cancer. International journal of molecular sciences. 2013;14(9):19257–75. doi: 10.3390/ijms140919257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Szabova L, Yin C, Bupp S, Guerin TM, Schlomer JJ, Householder DB, et al. Perturbation of Rb, p53, and Brca1 or Brca2 cooperate in inducing metastatic serous epithelial ovarian cancer. Cancer research. 2012;72(16):4141–53. doi: 10.1158/0008-5472.CAN-11-3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu R, Baker SJ, Hu TC, Norman KM, Fearon ER, Cho KR. Type I to type II ovarian carcinoma progression: mutant Trp53 or Pik3ca confers a more aggressive tumor phenotype in a mouse model of ovarian cancer. The American journal of pathology. 2013;182(4):1391–9. doi: 10.1016/j.ajpath.2012.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kang HJ, Chun SM, Kim KR, Sohn I, Sung CO. Clinical relevance of gain-of-function mutations of p53 in high-grade serous ovarian carcinoma. PloS one. 2013;8(8):e72609. doi: 10.1371/journal.pone.0072609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mackenzie R, Kommoss S, Winterhoff BJ, Kipp BR, Garcia JJ, Voss J, et al. Targeted deep sequencing of mucinous ovarian tumors reveals multiple overlapping RAS-pathway activating mutations in borderline and cancerous neoplasms. BMC cancer. 2015;15:415. doi: 10.1186/s12885-015-1421-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mullany LK, Liu Z, Wong KK, Deneke V, Ren YA, Herron A, et al. Tumor repressor protein 53 and steroid hormones provide a new paradigm for ovarian cancer metastases. Mol Endocrinol. 2014;28(1):127–37. doi: 10.1210/me.2013-1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laviolette LA, Garson K, Macdonald EA, Senterman MK, Courville K, Crane CA, et al. 17beta-estradiol accelerates tumor onset and decreases survival in a transgenic mouse model of ovarian cancer. Endocrinology. 2010;151(3):929–38. doi: 10.1210/en.2009-0602. [DOI] [PubMed] [Google Scholar]
- 13.Trevino LS, Giles JR, Wang W, Urick ME, Johnson PA. Gene expression profiling reveals differentially expressed genes in ovarian cancer of the hen: support for oviductal origin? Hormones & cancer. 2010;1(4):177–86. doi: 10.1007/s12672-010-0024-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Stefano I, Zannoni GF, Prisco MG, Fagotti A, Tortorella L, Vizzielli G, et al. Cytoplasmic expression of estrogen receptor beta (ERbeta) predicts poor clinical outcome in advanced serous ovarian cancer. Gynecologic oncology. 2011;122(3):573–9. doi: 10.1016/j.ygyno.2011.05.025. [DOI] [PubMed] [Google Scholar]
- 15.Zhao D, Zhang F, Zhang W, He J, Zhao Y, Sun J. Prognostic role of hormone receptors in ovarian cancer: a systematic review and meta-analysis. International journal of gynecological cancer : official journal of the International Gynecological Cancer Society. 2013;23(1):25–33. doi: 10.1097/IGC.0b013e3182788466. [DOI] [PubMed] [Google Scholar]
- 16.Sieh W, Köbel M, Longacre TA, Bowtell DD, deFazio A, Goodman MT, et al. Hormone-receptor expression and ovarian cancer survival: an Ovarian Tumor Tissue Analysis consortium study. The Lancet Oncology. 2013;14(9):853–62. doi: 10.1016/S1470-2045(13)70253-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chan KK, Leung TH, Chan DW, Wei N, Lau GT, Liu SS, et al. Targeting estrogen receptor subtypes (ERalpha and ERbeta) with selective ER modulators in ovarian cancer. The Journal of endocrinology. 2014;221(2):325–36. doi: 10.1530/JOE-13-0500. [DOI] [PubMed] [Google Scholar]
- 18.Murdoch WJ, Van Kirk EA, Isaak DD, Shen Y. Progesterone facilitates cisplatin toxicity in epithelial ovarian cancer cells and xenografts. Gynecologic oncology. 2008;110(2):251–5. doi: 10.1016/j.ygyno.2008.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fan HY, Liu Z, Paquet M, Wang J, Lydon JP, DeMayo FJ, et al. Cell type-specific targeted mutations of Kras and Pten document proliferation arrest in granulosa cells versus oncogenic insult to ovarian surface epithelial cells. Cancer research. 2009;69(16):6463–72. doi: 10.1158/0008-5472.CAN-08-3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mullany LK, Fan HY, Liu Z, White LD, Marshall A, Gunaratne P, et al. Molecular and functional characteristics of ovarian surface epithelial cells transformed by KrasG12D and loss of Pten in a mouse model in vivo. Oncogene. 2011;30(32):3522–36. doi: 10.1038/onc.2011.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mullany LK, Liu Z, King ER, Wong KK, Richards JS. Wild-type tumor repressor protein 53 (Trp53) promotes ovarian cancer cell survival. Endocrinology. 2012;153(4):1638–48. doi: 10.1210/en.2011-2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM, et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell. 2004;119(6):861–72. doi: 10.1016/j.cell.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 23.Crane EK, Kwan SY, Izaguirre DI, Tsang YT, Mullany LK, Zu Z, et al. Nutlin-3a: A Potential Therapeutic Opportunity for TP53 Wild-Type Ovarian Carcinomas. PloS one. 2015;10(8):e0135101. doi: 10.1371/journal.pone.0135101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu S, Han SJ, Smith CL. Cooperative activation of gene expression by agonists and antagonists mediated by estrogen receptor heteroligand dimer complexes. Molecular pharmacology. 2013;83(5):1066–77. doi: 10.1124/mol.112.084228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matsuo K, Nishimura M, Bottsford-Miller JN, Huang J, Komurov K, Armaiz-Pena GN, et al. Targeting SRC in mucinous ovarian carcinoma. Clin Cancer Res. 2011;17(16):5367–78. doi: 10.1158/1078-0432.CCR-10-3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu T, Hu W, Dalton HJ, Choi HJ, Huang J, Kang Y, et al. Targeting SRC and tubulin in mucinous ovarian carcinoma. Clin Cancer Res. 2013;19(23):6532–43. doi: 10.1158/1078-0432.CCR-13-1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang J, El-Bahrawy M. Expression profile of mucins (MUC1, MUC2, MUC5AC, and MUC6) in ovarian mucinous tumours: changes in expression from benign to malignant tumours. Histopathology. 2014 doi: 10.1111/his.12578. [DOI] [PubMed] [Google Scholar]
- 28.Cheng W, Liu J, Yoshida H, Rosen D, Naora H. Lineage infidelity of epithelial ovarian cancers is controlled by HOX genes that specify regional identity in the reproductive tract. Nature medicine. 2005;11(5):531–7. doi: 10.1038/nm1230. [DOI] [PubMed] [Google Scholar]
- 29.Mork L, Maatouk DM, McMahon JA, Guo JJ, Zhang P, McMahon AP, et al. Temporal differences in granulosa cell specification in the ovary reflect distinct follicle fates in mice. Biology of reproduction. 2012;86(2):37. doi: 10.1095/biolreprod.111.095208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perets R, Wyant GA, Muto KW, Bijron JG, Poole BB, Chin KT, et al. Transformation of the fallopian tube secretory epithelium leads to high-grade serous ovarian cancer in Brca;Tp53;Pten models. Cancer cell. 2013;24(6):751–65. doi: 10.1016/j.ccr.2013.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rechsteiner M, Zimmermann AK, Wild PJ, Caduff R, von Teichman A, Fink D, et al. TP53 mutations are common in all subtypes of epithelial ovarian cancer and occur concomitantly with KRAS mutations in the mucinous type. Experimental and molecular pathology. 2013;95(2):235–41. doi: 10.1016/j.yexmp.2013.08.004. [DOI] [PubMed] [Google Scholar]
- 32.Shan W, Mercado-Uribe I, Zhang J, Rosen D, Zhang S, Wei J, et al. Mucinous adenocarcinoma developed from human fallopian tube epithelial cells through defined genetic modifications. Cell Cycle. 2012;11(11):2107–13. doi: 10.4161/cc.20544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rasti M, Arabsolghar R, Khatooni Z, Mostafavi-Pour Z. p53 Binds to estrogen receptor 1 promoter in human breast cancer cells. Pathology oncology research : POR. 2012;18(2):169–75. doi: 10.1007/s12253-011-9423-6. [DOI] [PubMed] [Google Scholar]
- 34.Shirley SH, Rundhaug JE, Tian J, Cullinan-Ammann N, Lambertz I, Conti CJ, et al. Transcriptional regulation of estrogen receptor-alpha by p53 in human breast cancer cells. Cancer research. 2009;69(8):3405–14. doi: 10.1158/0008-5472.CAN-08-3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Junk DJ, Vrba L, Watts GS, Oshiro MM, Martinez JD, Futscher BW. Different Mutant/Wild-Type p53 Combinations Cause a Spectrum of Increased Invasive Potential in Nonmalignant Immortalized Human Mammary Epithelial Cells. Neoplasia. 2008;10(5):450–61. doi: 10.1593/neo.08120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brown J, Frumovitz M. Mucinous tumors of the ovary: current thoughts on diagnosis and management. Current oncology reports. 2014;16(6):389. doi: 10.1007/s11912-014-0389-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303(5659):844–8. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 38.Tovar C, Rosinski J, Filipovic Z, Higgins B, Kolinsky K, Hilton H, et al. Small-molecule MDM2 antagonists reveal aberrant p53 signaling in cancer: implications for therapy. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(6):1888–93. doi: 10.1073/pnas.0507493103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kenny HA, Chiang CY, White EA, Schryver EM, Habis M, Romero IL, et al. Mesothelial cells promote early ovarian cancer metastasis through fibronectin secretion. The Journal of clinical investigation. 2014;124(10):4614–28. doi: 10.1172/JCI74778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ledermann JA, Luvero D, Shafer A, O’Connor D, Mangili G, Friedlander M, et al. Gynecologic Cancer InterGroup (GCIG) consensus review for mucinous ovarian carcinoma. International journal of gynecological cancer : official journal of the International Gynecological Cancer Society. 2014;24(9 Suppl 3):S14–9. doi: 10.1097/IGC.0000000000000296. [DOI] [PubMed] [Google Scholar]
- 41.Gemignani ML, Schlaerth AC, Bogomolniy F, Barakat RR, Lin O, Soslow R, et al. Role of KRAS and BRAF gene mutations in mucinous ovarian carcinoma. Gynecologic oncology. 2003;90(2):378–81. doi: 10.1016/s0090-8258(03)00264-6. [DOI] [PubMed] [Google Scholar]
- 42.Mayr D, Hirschmann A, Lohrs U, Diebold J. KRAS and BRAF mutations in ovarian tumors: a comprehensive study of invasive carcinomas, borderline tumors and extraovarian implants. Gynecologic oncology. 2006;103(3):883–7. doi: 10.1016/j.ygyno.2006.05.029. [DOI] [PubMed] [Google Scholar]
- 43.Auner V, Kriegshauser G, Tong D, Horvat R, Reinthaller A, Mustea A, et al. KRAS mutation analysis in ovarian samples using a high sensitivity biochip assay. BMC cancer. 2009;9:111. doi: 10.1186/1471-2407-9-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Silva JL, De Moura Gallo CV, Costa DC, Rangel LP. Prion-like aggregation of mutant p53 in cancer. Trends in biochemical sciences. 2014;39(6):260–7. doi: 10.1016/j.tibs.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 45.Willis A, Jung EJ, Wakefield T, Chen X. Mutant p53 exerts a dominant negative effect by preventing wild-type p53 from binding to the promoter of its target genes. Oncogene. 2004;23(13):2330–8. doi: 10.1038/sj.onc.1207396. [DOI] [PubMed] [Google Scholar]
- 46.Wang Y, Suh YA, Fuller MY, Jackson JG, Xiong S, Terzian T, et al. Restoring expression of wild-type p53 suppresses tumor growth but does not cause tumor regression in mice with a p53 missense mutation. The Journal of clinical investigation. 2011;121(3):893–904. doi: 10.1172/JCI44504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miller SJ, Lan ZD, Hardiman A, Wu J, Kordich JJ, Patmore DM, et al. Inhibition of Eyes Absent Homolog 4 expression induces malignant peripheral nerve sheath tumor necrosis. Oncogene. 2010;29(3):368–79. doi: 10.1038/onc.2009.360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Depreux FF, Darrow K, Conner DA, Eavey RD, Liberman MC, Seidman CE, et al. Eya4-deficient mice are a model for heritable otitis media. The Journal of clinical investigation. 2008;118(2):651–8. doi: 10.1172/JCI32899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li H, Diao TY, Zhou ZY, Yang FY, Ma Q, Li QH. Relationship between the expression of hTERT and EYA4 mRNA in peripheral blood mononuclear cells with the progressive stages of carcinogenesis of the esophagus. Journal of experimental & clinical cancer research : CR. 2009;28:145. doi: 10.1186/1756-9966-28-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tadjuidje E, Hegde RS. The Eyes Absent proteins in development and disease. Cellular and molecular life sciences : CMLS. 2013;70(11):1897–913. doi: 10.1007/s00018-012-1144-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.