

Abstract
Chronic inflammation in liver tissue is an underlying cause of hepatocellular carcinoma (HCC). High levels of inflammatory cytokine interleukin IL-18 in the circulation of patients with HCC correlates with poor prognosis. However, conflicting results have been reported for IL-18 in HCC development and progression. In this study, we used tissue specimens from HCC patients and clinically relevant mouse models of HCC to evaluate IL-18 expression and function. In a mouse model of liver fibrosis that recapitulates a tumor-promoting microenvironment, global deletion of the IL-18 receptor IL18R1 enhanced tumor growth and burden. Similarly, in a carcinogen-induced model of liver tumorigenesis, IL18R1 deletion increased tumor burden. Mechanistically, we found that IL-18 exerted inflammation-dependent tumor-suppressive effects largely by promoting the differentiation, activity and survival of tumor-infiltrating T cells. Finally, differences in the expression of IL-18 in tumor tissue versus non-tumor tissue was more predictive of patient outcome than overall tissue expression. Taken together, our findings resolve a long-standing contradiction regarding a tumor-suppressive role for IL-18 in established HCC and provide a mechanistic explanation for the complex relationship between its expression pattern and HCC prognosis.
Keywords: Hepatocellular carcinoma, inflammation, interleukin-18, lymphocytes, tumor microenvironment
INTRODUCTION
Liver cancer is the third most deadly cancer worldwide, and unlike many other cancers, the incidence of this disease and its most common type, hepatocellular carcinoma (HCC), is rising despite advances in surveillance and therapeutics (1,2). HCC derives from multiple etiological factors, including infection by hepatitis viruses, alcohol abuse, and metabolic syndrome (1). These etiological factors induce a state of chronic inflammation, fibrosis and cirrhosis, which in turn leads to malignant transformation of hepatocytes, HCC development, and progression (1,3–5).
Due to HCC’s well-known intimate relationship with inflammation, previous studies have demonstrated both pro- and anti-tumorigenic roles of a variety of immune cell types and mediators. Important effector cells, such as macrophages, natural killer (NK) cells, and T-cells have been shown to play varying and diverse roles in regulating the multistep tumorigenic process, often in a stage- and even etiology-dependent fashion (6–14). Cytokines, including TNFα, IL-6, IL-1α, IL-1β, TGFβ, IFNγ, and CCL22, have been reported to display robust and disparate functionalities in modulating HCC initiation and progression (7,13,15–20). Examination of HCC patient samples has also indicated a tight correlation between inflammation, disease progression, and prognosis, although these correlations may vary based on the etiology (4,14,21–24).
Interleukin 18 (IL-18) is a member of the IL-1 family of cytokines (25). Originally described as Interferon-Gamma-Inducing Factor, its canonical function is to promote production of IFNγ from a variety of immune cells, primarily Th1 CD4+ T-cells and NK cells, frequently in conjunction with IL-12 (25). IL-18 is produced by a variety of cells, including but not limited to dendritic cells, macrophages, and epithelial cells (25). Its functional production is regulated both transcriptionally and via proteolytic processing of its precursor peptide by the inflammasome (25). Clinical data have shown that IL-18 levels in serum are elevated in patients with chronic Hepatitis B, Hepatitis C, and HCC, and higher levels of circulating IL-18 are correlated with worse prognosis of HCC (26–28). However, conflicting experimental results have been reported showing either pro- or anti-tumorigenic functions of IL-18. Multiple carcinogen-induced and spontaneous tumorigenesis studies have demonstrated a pro-tumorigenic role of IL-18, while investigations utilizing implantation of tumor cells in combination with methods to enhance IL-18 expression revealed potential IL-18-induced tumor suppression (10,29,30). Thus, despite the established connection between its presence in the circulation of patients and correlated poorer prognosis, a pathological feature frequently associated with tumor promoters, the role of IL-18 in HCC development and progression is still contentious. To address this critical question, we conducted comprehensive analyses of two sets of patient samples in conjunction with clinically relevant mouse models to examine IL-18’s role in HCC. We reveal a tumor-suppressive role for IL-18 that is consistent with the complex expression patterns of this cytokine in our patient cohorts. Differing from previous reports, we demonstrate a powerful effect of endogenous IL-18 signaling modulating the accumulation and activity of both CD8+ and multiple subsets of CD4+ T-cells to effect anti-tumor activity. We further demonstrate that IL-18 signaling serves as a key mediator of complementary CD8+ T-cell and NK cell-dependent tumor suppression. Finally, we show that differences in the level of IL-18 expression between matched tumor and non-tumor tissue provided more reliable prognostic value than the overall levels of IL-18. Taken together, our findings establish that, contrary to an expected pro-tumorigenic activity due to its elevated levels in circulation, IL-18 actually exerts a stage-dependent tumor-suppressive effect on HCC progression, primarily through the anti-tumor activities of tumor-infiltrating lymphocytes.
MATERIALS AND METHODS
Patient Samples
Patient samples were obtained following informed consent according to established protocols approved by the Ethics Committees of the Eastern Hepatobiliary Surgery Hospital (Shanghai, China) and the 302 Military Hospital (Beijing, China). Details on patient collection, characterization, and survival categorization, as well as usage of the different specimens for IL-18 staining, serum quantification, and mRNA expression, are detailed in the Supplement.
Mice
All experimental procedures described were approved by the Duke University Animal Care and Use Committee. C57BL/6 (wild-type, WT) breeding mice were purchased from The Jackson Laboratory. IL18R1−/− mice backcrossed to C57BL/6 were provided by Dr. Yiping Yang (Duke University Medical Center, Durham, NC).
Reagents
For a list of antibodies, stains, and cytokines used, clone and/or catalogue numbers, and companies from which they were purchased, see Supplementary Table S1. For qPCR primers and shRNA constructs, see Supplementary Table S2. Hepa1–6 cells [Hepa1–6][ATCC®CRL-1830™] were purchased directly from ATCC which validates by short tandem repeat profiling, and passaged for fewer than 6 months after receipt, with frozen stocks of both naïve and GFP-labeled luciferase reporter-expressing Hepa1-6 cells (Hepa1-6-GFP-Luc) being thawed prior to use. Recombinant IL18BP was generated as described in (32).
Fibrosis Induction
Carbon tetrachloride (CCl4)-induced liver fibrosis: 6–7 week old male WT or IL18R1−/− mice were treated with either olive oil or CCl4 diluted 1:20 in olive oil at 600μL/kg. Mice received biweekly intraperitoneal injections for 6 weeks, and were sacrificed 2 days following the 12th injection.
Bile duct ligation-induced liver fibrosis: 12-week-old male WT or IL18R1−/− mice were anesthetized, and the common bile duct isolated and transected between two ligations. Mice were sacrificed 14 days post-surgery.
Orthotopic Implantation
Implantation occurred either 2 days after the 8th injection of CCl4 or olive oil, or immediately following bile duct ligation. Mice were anesthetized, the abdomen opened, and the liver exposed. 3×106 Hepa1-6-GFP-Luc cells were suspended in 30μL Growth Factor-Reduced Matrigel and injected into the left lobe of the liver. The peritoneum and skin were subsequently sutured shut. The 9th dose of CCl4 or olive oil occurred 2 days after surgery, and the 12th dose occurred 12 days after surgery. Mice were monitored for health, and sacrificed 14 days post-implantation for both fibrotic allograft models.
N-Nitrosodiethylamine (DEN)-Induced Carcinogenesis
Post-natal-day-15 WT or IL18R1−/− mice received a single intraperitoneal injection of DEN at 25mg/kg. Mice were sacrificed 6 or 12 months post-administration. Alternatively, starting one week post-DEN treatment, mice also received weekly doses of 500μL/kg CCl4 via intraperitoneal injection. These mice received 22 doses of CCl4, and were sacrificed 2 days after the last injection.
Sample Harvest and Preparation
Livers and spleens were harvested and either post-fixed in 10% phosphate-buffered formalin at 4°C on an orbital shaker for 1 day before washing with 70% ethanol and embedding in paraffin blocks or processed for flow cytometry/ fluorescence-activated cell sorting (FACS). Detailed tissue processing for flow cytometry, sorting, and staining protocols are in the Supplement.
Liver and Spleen as Percentage of Body Weight
Mice were euthanized, weighed, and their livers and spleens dissected and weighed. Percent of body weight was calculated as 100%*(weight of liver or spleen)/(weight of the entire mouse).
Histology
Tissue evaluated by histology was fixed in 10% phosphate-buffered formalin and embedded in paraffin blocks. 5μm-thick sections were stained for hematoxylin and eosin and picrosirius red using standard protocols.
Complete Blood Counts (CBCs) and Serum Evaluation
Blood collected from the inferior vena cava from anesthetized mice at sacrifice. For CBCs, blood was stored in EDTA-coated tubes and examined on an Abbot Cell-Dyn 3700. For serum evaluation, blood was incubated at room temperature for 10 minutes then centrifuged at 3000rpm for 10 minutes, following which serum was aspirated, transferred to a new tube, and stored at −80°C until assay. Alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were quantitatively evaluated by colorimetry using kits purchased from Biotron Diagnostics according to the manufacturer’s instructions. Circulating mature IL-18 was detected in the serum of mice using a mouse IL-18 ELISA kit (Medical & Biological Laboratories Co.).
qPCR Analysis of T-cells
At sacrifice, tissue was harvested and samples processed as described before. Samples were stained, and TCRβ+CD4+CD25−, TCRβ+CD4+CD25+, and TCRβ+CD8+ populations were collected. RNA was extracted from isolated cells, reverse transcribed using iScript, and samples probed for targets of interest.
Cell Differentiation and Stimulation Assays
Naïve T-cells were harvested from lymph nodes of WT or IL18R1−/− mice, sorted by commercially available untouched isolation methods, and subjected to differentiation and stimulation assays using established protocols. Information detailing these protocols can be found in the Supplement.
Statistical Analysis
Data are presented as mean±standard error of mean. Statistics were calculated using GraphPad Prism; tests are described in each figure’s legend. Significance was set at P<0.05.
RESULTS
A complex relationship between prognosis of HCC patients and expression of IL-18 signaling components
Previous studies indicated that IL-18 is frequently elevated in the circulation and liver tissue of patients with chronic liver diseases compared with healthy individuals, and a higher level is correlated with poor prognosis of HCC (26–28,31–33). To further explore this topic, we first examined the prevalence of IL-18 in the serum of patients with liver disease and found elevated levels compared with those in healthy individuals (Fig. 1A). We then determined the relationship between prognosis and expression of IL-18 in tissue specimens from a cohort of 138 HCC patients (described in Supplementary Table S3). Consistent with earlier reports, higher IL-18 expression, either intratumoral or peritumoral, was associated with poor prognosis (Figs. 1B–1C; stratification based on staining density shown in Supplementary Fig. S1), although this point was less clear with patients displaying intermediate levels of IL-18. However, when mRNA samples from a separate set of 26 chronic HBV+ patients with metastatic HCC were examined, we found significantly lower expression of both IL-18 (Fig. 1D) and its receptor IL18R1 (Fig. 1E) in primary and metastatic tumor tissue compared to matched non-tumor tissue samples. These data validate that elevated IL-18 levels in liver tissue of HCC patients could be detrimental for survival. However, in comparison with the inflamed peritumoral tissue, the expression level of IL-18 and its receptor was significantly lower within the tumor itself.
Figure 1. Elevated IL-18 correlates with poor prognosis, but expression of IL-18 and IL18R1 is often decreased within the tumor from the same patient.
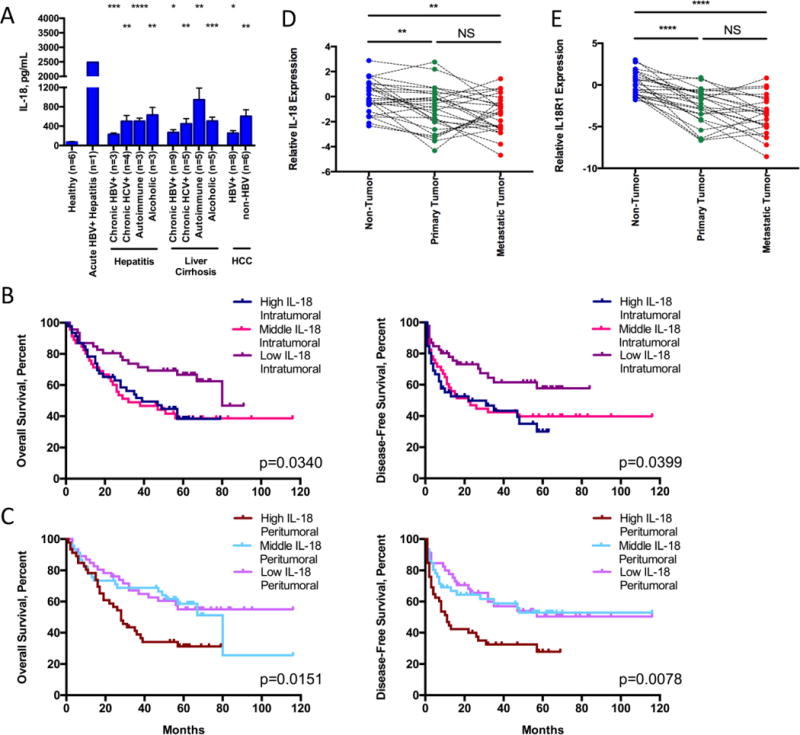
(A) Circulating plasma IL-18 measured by ELISA. (B, C) Kaplan-Meier curves for overall (left) and disease-free survival (right) of the cohort of 138 patients stratified into the highest, middle, or lowest third of patients based on IL-18 staining density either in tumor (B) or in peritumoral (C) tissue as shown in Supplementary Fig. S1. (D, E) mRNA expression of IL-18 (D) and IL18R1 (E) from matched non-, primary, and metastatic tumor tissue. Samples were normalized relative to average expression levels in all non-tumor tissue samples and displayed as log2 (fold change to average expression in non-tumor tissue). Statistics: (A) one-way ANOVA (P<0.0001), followed by unpaired t-tests comparing each group with healthy controls, n indicated under each column; (B, C) Log-rank Mantel-Cox tests, n=46 per group; (D, E) one-way ANOVA with Tukey’s multiple comparisons test, n=26 matched samples per group. NS non-significant; *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001.
IL-18 signaling exhibits suppressive effects on tumor growth
These seemingly contradictory results prompted us to address the functional relevance of IL-18 in HCC utilizing a more clinically relevant model in which the liver microenvironment resembles the diseased tissue from which a tumor develops in most HCC patients. To do this, we first established a mouse model combining carbon tetrachloride (CCl4)-induced liver fibrosis with orthotopic implantation of tumor cells (Fig. 2A and (34)). This model induced robust fibrosis in wild-type (WT) mice, as demonstrated by increases in circulating aminotransferases, hepatic stellate cell activation, and collagen deposition (Supplementary Fig. S2). Induction of inflammatory liver fibrosis enhanced tumor growth, with increased infiltration and altered immune composition in non-tumor liver and tumor compared with tumor engraftment without fibrosis induction (34). IL-18 is produced by many constituents of the tumor microenvironment (25). We therefore performed immunohistochemical staining in several HCC models, and found multiple cell populations expressed IL-18 (Supplementary Figs. S3A–S3D). We further isolated populations from the tumor and obtained similar results as shown in other pathological systems. Endothelial, myeloid, and tumor cells produced IL-18, with myeloid populations dominating production, while lymphoid cells such as CD4+ T-cells, CD8+ T-cells, and NK cells expressed higher levels of IL18R1 (Supplementary Fig. S3E). Thus, in order to examine the overall effects of IL-18 signaling in the tumor microenvironment, we employed a mouse strain with constitutive whole-body IL18R1 deletion (IL18R1−/−) (35). Consistent with a tumor-suppressive role for IL-18 signaling, orthotopically-implanted tumors grew to greater sizes in IL18R1−/− mice than in WT mice (Fig. 2B). Importantly, this enhancement in tumor burden was observed only in mice treated with CCl4, not in those treated with the vehicle control, olive oil, demonstrating a reliance on the inflammatory context for IL-18 signaling to robustly control tumor growth (Fig. 2B). Interestingly, IL18R1−/− mice displayed a slightly less severe fibrotic phenotype compared to WT animals (Supplementary Fig. S2), indicating the tumor-suppressive nature of IL-18 signaling may not be shared at other disease stages.
Figure 2. IL-18 signaling exerts a tumor-suppressive role on the growth of liver tumor cells orthotopically implanted in mice with a fibrotic liver and at late stages in tumorigenesis.
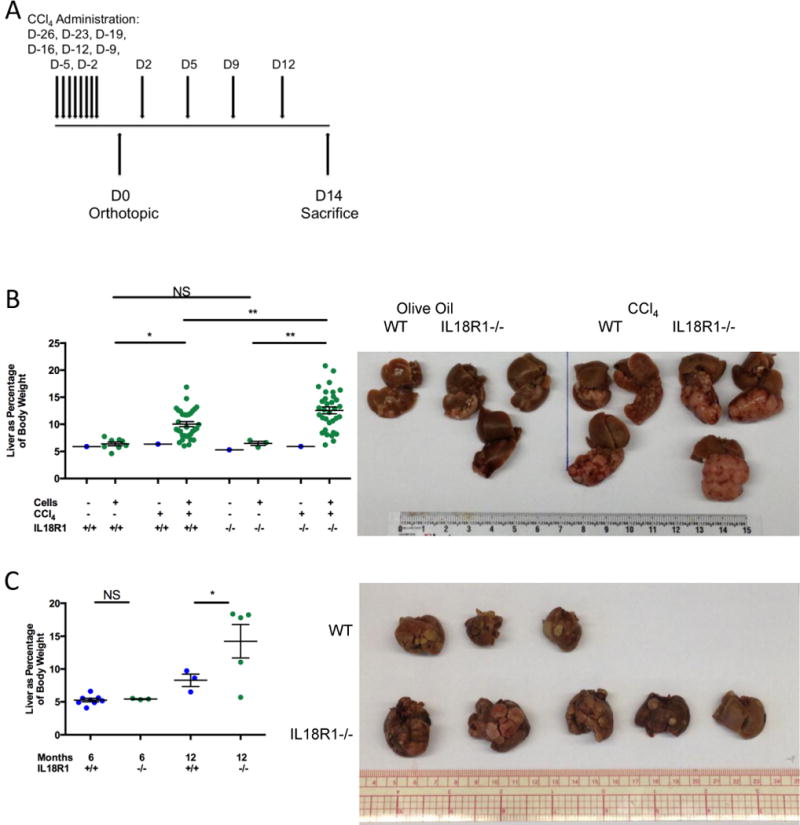
(A) Experimental schematic illustrating timing of the 12 doses of carbon tetrachloride (CCl4) administered, orthotopic implantation, and tissue harvest. (B) Tumor growth (liver as a percentage of body weight): a sham matrigel plug or Hepa1–6 cells expressing a GFP-luciferase vector (Hepa1–6-GFP-Luc) were implanted into wild-type or IL18R1-deficient C57Bl/6 mice treated with olive oil or CCl4; representative images on the right. (C) Tumor burden (liver as percentage of body weight) in wild-type or IL18R1-deficient mice 6 and 12 months after diethylnitrosamine (DEN) administration; livers harvested 12 months post-DEN treatment on right. Statistics: (B) one-way ANOVA with Tukey’s multiple comparisons test, n=3–8 for olive oil-treated mice, n=33 for CCl4-treated mice pooled over 9 experiments; (C) one-way ANOVA with Sidak’s multiple comparisons test, n=3–8 per group. NS non-significant; *P<0.05; **P<0.01.
To validate the anti-tumor effect of IL-18 signaling across multiple models, we examined liver tumorigenesis induced by a commonly-used chemical carcinogen, N-nitrosodiethylamine (DEN) (12). Importantly, this model has been shown to provoke local and systemic immune responses, particularly in later stages of tumorigenesis (12). Tumor burden at late stages in tumorigenesis was enhanced in IL18R1−/− mice compared to WT animals (Fig. 2C). Taken together, these data suggest that IL-18 signaling plays an important inflammation-dependent tumor-suppressive role.
IL-18 signaling regulates composition of tumor-infiltrating T-cell populations
IL-18 affects multiple cell types which may affect tumor growth (25). To identify which populations carried out IL-18 signaling-mediated tumor suppression, we first examined circulating immune cells. Naïve WT and IL18R1−/− mice did not show significant differences in circulating immune population proportions (Supplementary Fig. S4). In contrast, upon CCl4 treatment and orthotopic tumor cell implantation, we found increased neutrophil and decreased lymphocyte proportions in IL18R1−/− mice compared with WT animals (Supplementary Fig. S4B). This change associates with enhanced tumor burden in IL18R1−/− mice, and increased circulating neutrophil-to-lymphocyte ratios have been reported to be indicative of worse patient prognosis (36). The decreased lymphocyte counts also presents a plausible major mechanism for impaired anti-tumor function.
We then examined the immune composition of the fibrotic liver and tumor allograft. We did not find significant differences in numbers of NK cells, NKT cells, CD11b+ monocytes, Gr-1-expressing monocyte subsets, or bulk T-cells in either liver or tumor between genotypes (Supplementary Figs. S5–S7). However, we found that T-cell composition was different, specifically in the tumor. In the diseased non-tumor liver, there was a non-significant decrease in CD8+ T-cells, and a small yet significant increase in CD4+ T-cells in the IL18R1−/− mice compared to the WT animals, which was not observed in livers from naïve mice (Figs. 3A–3B; Supplementary Figs. S7A, S8A). Composition in the tumor demonstrated an accumulation of CD8+ T-cells and consequent decrease in the CD4+ T-cell proportion in WT mice compared with naïve liver tissue, which was not observed in IL18R1−/− mice (Figs. 3A, 3C; Supplementary Figs. S7B, S8B). Importantly, while there was a marginal offset in the CD4+/CD8+ T-cell ratio in the naïve spleens of WT and IL18R1−/− mice, these differences were non-significant in the spleens of tumor-bearing mice (Figs. 3D–3E; Supplementary Fig. S8C). We also confirmed this T-cell skewing in two additional models: both bile duct ligation with orthotopic tumor cell implantation and tumorigenesis combining administration of DEN with CCl4 administration yielded comparatively increased tumor-infiltrating CD4+ T-cell proportions and decreased tumor-infiltrating CD8+ T-cell proportions in IL18R1−/− mice (Supplementary Fig. S9). Together these data indicate IL18R1 deletion leads significantly reduced tumor-infiltrating CD8+ T-cells, and proportionally increased tumor-infiltrating CD4+ T-cells.
Figure 3. T-cell populations which infiltrated the tumor are skewed by loss of IL18R1.
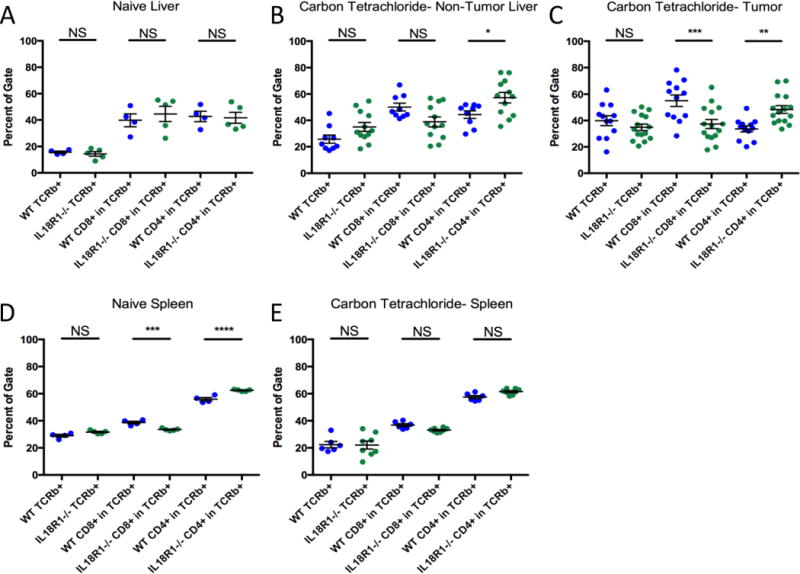
(A–E) TCRβ+, TCRβ+CD8+, and TCRβ+CD4+ cells in naïve wild-type and IL18R1-deficient mice or mice treated with CCl4 and tumor implantation were quantified in: naïve liver tissue (A), non-tumor-liver tissue from tumor-bearing mice (B), tumor tissue from tumor-bearing mice (C), naïve spleen (D), or spleen from tumor-bearing mice (E). Statistics: (A–E) one-way ANOVA with Sidak’s multiple comparisons test, n=9–12 per group for non-tumor liver samples pooled from 3 experiments, n=12–15 per group for tumor samples pooled from 4 experiments, n=6–8 per group for spleen samples pooled from 2 experiments, n=4–5 per naïve group pooled over 2 experiments. NS non-significant; *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001.
IL-18 signaling regulates T-cell differentiation, activation, proliferation, and survival
Since the most relevant difference between the WT and IL18R1−/− tumor immune composition was the CD4+/CD8+ T-cell ratio, and T-cells are well-known mediators of tumor suppression, we proceeded to determine the mechanism through which those cells affect allograft tumor growth. We found minor differences in T-cell activation and checkpoint suppression, and no difference in immune-suppressive T-regulatory cell accumulation between WT and IL18R1−/− mice (Supplementary Figs. S10–S12). Based on current literature, these differences in T-cell activation and PD-1 expression would have resulted in enhanced tumor suppression in the IL18R1−/− mice, contrary to the phenotype observed here. We then examined the functionality of tumor-infiltrating conventional CD4+ T-cells. CD4+CD25− T-cells were isolated from the tumor and subjected to qPCR analysis for cytokine, enzyme, and relevant transcription factor expression. CD4+ T-cells from IL18R1−/− mice expressed significantly less IL-2, TNFα, and IL-17a compared with WT, suggesting diminished functional potential (Fig. 4A), although GzmB expression was enhanced. IL-2 stimulates CD8+ T-cell growth and differentiation into memory cells, while TNFα mediates anti-tumor activity (37,38). Importantly, both Th1 and Th17 cells have been shown to possess anti-tumor functionality (39,40). We validated the expression profiles of these factors in tumor-infiltrating T-cells via flow cytometry (Figs. 4C–4E; Supplementary Fig. S13). While a decrease in IFNγ+IL-17a− cells among IL18R1−/− CD4+ T-cells was observed, we surprisingly found increased proportions of IFNγ−IL-17a+ cells compared with WT (Figs. 4C–4D). We also observed a trend for reduced TNFα+IL-2− (P=0.1707) and TNFα+IL-2+ (P=0.0896) cells in IL18R1−/− CD4+ T-cells compared with WT, and detected no differences in granzyme, T-bet, or Eomes production (Supplementary Fig. S13).
Figure 4. Functionality of tumor-infiltrating T-cells is altered by loss of IL18R1.
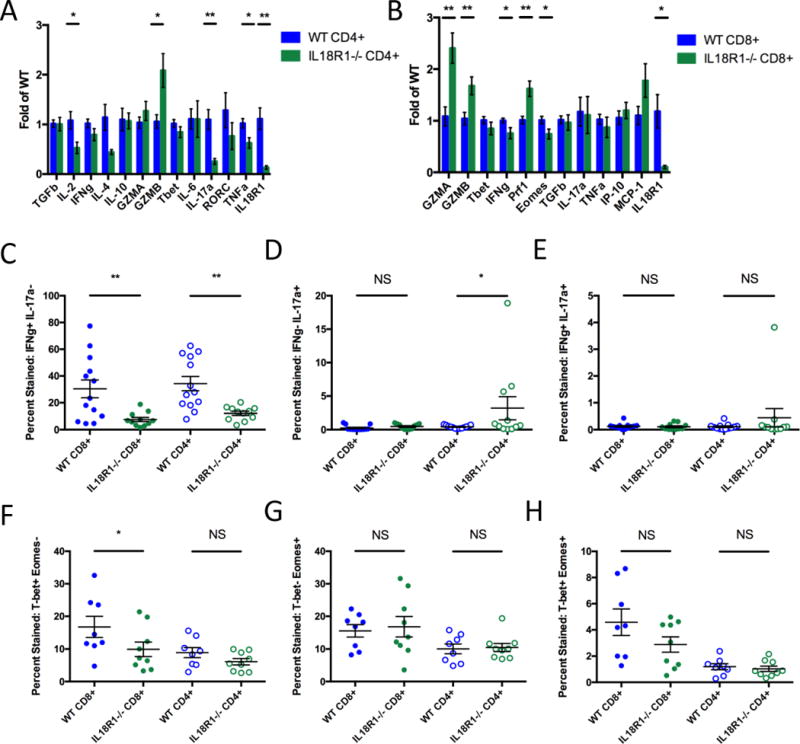
(A, B) Mice were treated with CCl4 and orthotopic tumor implantation, and TCRβ+CD4+CD25− cells (A) and TCRβ+CD8+ cells (B) were sorted from harvested tumors, mRNA extracted and reverse transcribed, and probed for expression of targets. Fold changes are calculated by normalization to the average expression level in wild-type CD4+ (A) and CD8+ T-cells (B) for the given experiment. Data pooled from two experiments, total n=7 for wild-type and n=6 for IL18R1-deficient mice. (C–H) Tumor-infiltrating TCRβ+CD4+ and TCRβ+CD8+ cells in wild-type and IL18R1-deficient mice were stained for: IFNγ and IL-17a (C-E) or T-bet and Eomes (F–H). Single- and double-stained populations were quantified as percentages of TCRβ+CD4+ and TCRβ+CD8+ cells. Statistics: (A, B) multiple t-tests, n=6–7 per group pooled over 2 experiments; (C–H) one-way ANOVA with uncorrected Fisher’s LSD test, n=11–13 per group pooled over 3 experiments for (C–E), n=8–9 per group pooled over 2 experiments for (F–H). NS non-significant; *P<0.05; **P<0.01.
We also examined the functionality of IL-18 signaling in CD8+ T-cells. Similarly, we determined the expression of relevant cytokines, enzymes, and transcription factors in tumor-infiltrating CD8+ T-cells. Expression of Prf1, GzmA, and GzmB were moderately increased in IL18R1−/− CD8+ T-cells compared to WT, while IFNγ and Eomes were mildly decreased, suggesting that on a per-cell basis, IL18R1−/− CD8+ T-cells may be slightly more potent killers than WT (Fig. 4B). Closer examination via flow cytometry also demonstrated significant differences in IFNγ production in tumor-infiltrating IL18R1−/− CD8+ T-cells compared with WT, but no differences in granzyme production (Figs. 4C–4E; Supplementary Fig. S13). We also observed a significant reduction in T-bet+Eomes− and a trend for a reduction in T-bet+Eomes+ (P=0.0510) in IL18R1−/− CD8+ T-cells compared with WT (Figs. 4F–4H).
To further test these IL-18 signaling-related T-cell-intrinsic defects, we first examined CD4+ T-cells. We differentiated naïve CD4+ T-cells from WT and IL18R1−/− mice in vitro under Th1- and Th17-inducing conditions, and then examined their IFNγ and IL-17a production in the presence or absence of IL-18. In WT CD4+ T-cells, we found efficient differentiation to both Th1 and Th17 lineages, and enhanced IFNγ production in Th1-differentiated cells upon IL-18 exposure (Figs. 5A, 5C; Supplementary Fig. S14). Th17-differentiated WT CD4+ T-cells did not respond to IL-18 exposure with enhanced IL-17a production (Fig. 5C). In contrast, IL18R1−/− CD4+ T-cells did not differentiate to Th1 as efficiently, and failed to enhance IFNγ production upon exposure to IL-18 (Fig. 5A). IL18R1−/− CD4+ T-cells also did not differentiate as efficiently to the Th17 lineage (Fig. 5C). We further assessed the proliferation and death of WT Th1 and Th17 cells in the presence of IL18BP, an antagonistic secreted decoy receptor for IL-18 (32), in the absence of exogenously-added IL-18. We found that more Th1-differentiated cells died, and Th17-differentiated cells proliferated more slowly when treated with increasing doses of IL18BP (Figs. 5B, 5D). We conclude that in Th1 cells IL-18 serves as a pro-survival factor, while in Th17 cells IL-18 modulates proliferation. Combined with the diminished production of TNFα and IL-2, these results argue for multi-faceted IL-18 modulation of inflammatory T helper cell capacities.
Figure 5. Differentiation and cytokine production of CD4+ T-cells is altered by loss of IL18R1.
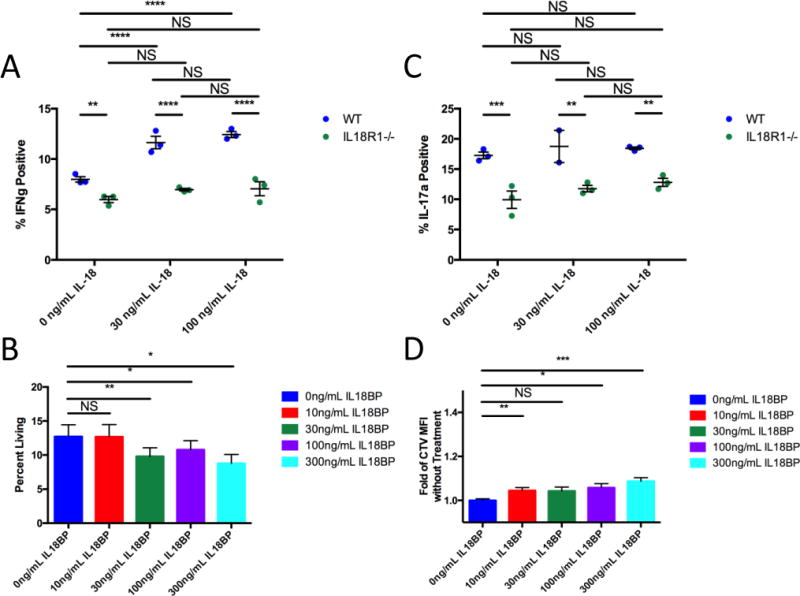
(A) Quantification of IFNγ production from wild-type and IL18R1-deficient Th1-differentiated CD4+ T-cells stimulated with IL-18. (B) Wild-type CD4+ T-cells were differentiated to Th1 and treated with IL18BP. 5 days after start of differentiation, Th1-differentiated (IFNγ+) cell survival was evaluated using a fixable Live/Dead stain. (C) Quantification of IL-17a production from wild-type and IL18R1-deficient Th17-differentiated CD4+ T-cells stimulated with IL-18. (D) Wild-type CD4+ T-cells were differentiated to Th17, dyed with CellTrace Violet, and treated with IL18BP. 5 days after start of differentiation, proliferation was evaluated by CellTrace Violet dilution. Statistics: (A, C) two-way ANOVA with uncorrected Fisher’s LSD test, n=3 per group, representative of twice-repeated experiments. (B, D) one-way ANOVA with uncorrected Fisher’s LSD test, n=12 per group pooled from 3 experiments. NS non-significant; *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001.
IL-18 signaling also had a robust effect on CD8+ T-cells: both their number and functionality were significantly reduced in IL18R1−/− tumors. Therefore, to evaluate the net effect of IL-18 signaling, we performed in vitro re-stimulation experiments (utilizing weaker T-cell antigen receptor complex stimulation than that used during initial activation) to assess effects on CD8+ T-cell proliferation and survival. Upon re-stimulation, untreated WT and IL18R1−/− CD8+ T-cells exposed to tumor conditional media (TCM) proliferate, and the majority subsequently dies (Supplementary Fig. S15). However, WT CD8+ T-cells treated with IL-18 had a higher survival rate than either untreated WT, untreated IL18R1−/−, or treated IL18R1−/− CD8+ T-cells (Fig. 6A). Treated WT CD8+ T-cells exposed to TCM also had a greater proliferation rate than both their untreated counterparts as well as IL18R1−/− CD8+ T-cells (Fig. 6B). Together, these data demonstrate that deficiency in IL-18 signaling modulates both the accumulation and functionality of both CD4+ and CD8+ tumor-infiltrating T-cells.
Figure 6. Accumulation of CD8+ T-cells is altered by loss of IL18R1; CD8+ T-cells and NK cells modulate tumor burden.
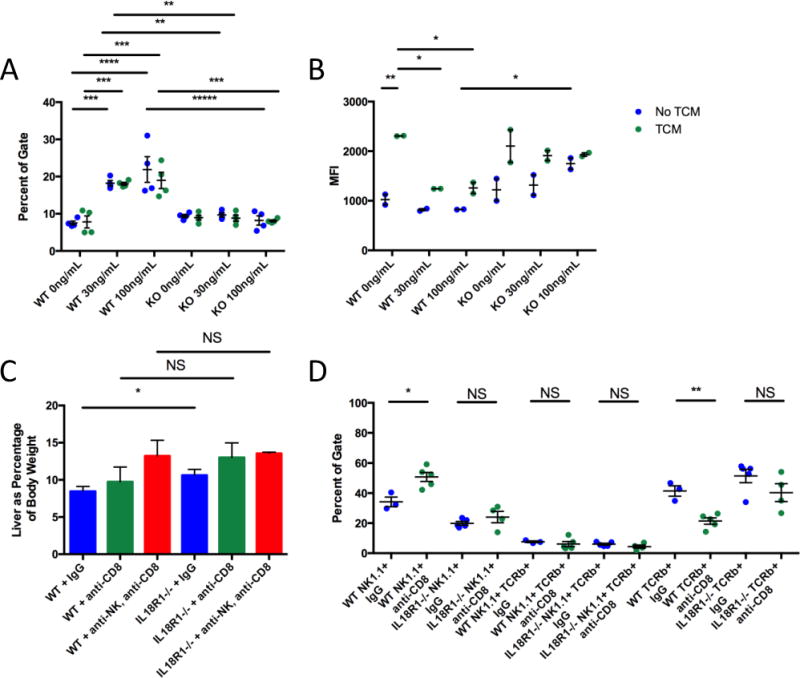
(A) Surviving wild-type and IL18R1-deficient CD8+ T-cells (defined as 7AADloCellTrace Violetlo) upon reactivation and stimulation with different doses of IL-18 with or without tumor conditional media (TCM). (B) Proliferation of CD8+ T-cells (lower mean fluorescence intensity (MFI)). (C) Tumor growth measured by liver weight as a percentage of body weight. Tumor-bearing mice were treated with IgG, anti-CD8a, or anti-NK1.1 and anti-CD8a. (D) Quantification of intratumoral NK1.1+TCRβ−, NK1.1+TCRβ+, and TCRβ+NK1.1− cells from wild-type or IL18R1-deficient C57Bl/6 mice treated with CCl4 and either IgG or anti-CD8a. Data are representative of thrice-repeated experiments. Statistics: (A) two-way ANOVA with Sidak’s multiple comparisons test, n=4 per group pooled over 2 experiments; (B) two-way ANOVA with Sidak’s multiple comparisons test, n=2 per group, representative of twice-repeated experiments; (C-D) unpaired T-tests, left to right: n=6, n=6, n=6, n=10, n=7, n=3 (C), n=3, n=5, n=5, n=5 (D). NS non-significant; *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001.
IL-18 signaling regulates lymphocyte-mediated tumor cytotoxicity in vivo
IL-18 signaling profoundly impacted T-cell differentiation, cytokine production, proliferation, and survival, so we tested if CD8+ T-cells mediated the differential anti-tumor responses between WT and IL18R1−/− mice. We ablated CD8+ T-cells in both WT and IL18R1−/− mice in our fibrotic allograft model, and examined tumor burden. Surprisingly, this did not equalize, nor did it significantly alter, tumor burden between WT and IL18R1−/− mice (Fig. 6C). Upon closer examination, we found that when CD8+ T-cells were ablated in WT mice, NK cells comprised a significantly increased portion of tumor-infiltrating cells compared with IgG-treated controls (Fig. 6D). Our earlier experiments had not shown a genotype-specific difference in intratumoral NK cell accumulation, however IL-18 markedly regulates NK cell activity (25). We then examined the functionality and maturation of tumor-infiltrating NK cells in WT and IL18R1−/− mice, and found differences in IFNγ, T-bet, and Eomes production (Supplementary Fig. S16), suggesting that their anti-tumor efficacy may be different, particularly in the absence of other cytotoxic cells. We therefore performed ablation experiments targeting NK cells. While single ablation of NK cells did not affect tumor burden (data not shown), depleting both NK cells and CD8+ T-cells equalized tumor burden between WT and IL18R1−/− mice, demonstrating complementary CD8+ T-cell- and NK cell-dependent IL-18-mediated tumor suppression (Fig. 6C).
Comparisons between IL-18 expression in tumor and non-tumor tissue provide better survival prediction
These results suggest that, rather than promoting tumor growth at late stages, IL-18 acts as a tumor suppressor, and should display reduced intratumoral expression. To probe this critical point, we re-examined the tissue specimens described earlier in Figs. 1B–1C to determine the relationship between the expression profiles of IL-18 and prognosis of those patients. In the majority of tissue samples, similar to Fig. 1D, IL-18 staining was stronger in the peritumoral tissue than within the tumor itself (Fig. 7A; Supplementary Fig. S17; quantification: Supplementary Table S4, Supplementary Figs. S1, S18). Importantly, when we quantified differences in immunostaining density between each patient’s intratumoral and peritumoral tissue, we found that this difference, particularly for those with the highest divergence, correlated more significantly to both overall and disease-free survival in patients than staining in either region alone (Fig. 7B; compare to P-values in Figs. 1B–1C). By this new measure, the ambiguity in patients with intermediate levels of IL-18 staining shown in Figs. 1B–1C was largely diminished. Thus, the differential expression of this cytokine between tumor and non-tumor tissue could be a better biomarker for predicting patient outcomes.
Figure 7. Differences in IL-18 expression between tumor and non-tumor tissue strongly correlate with prognosis.
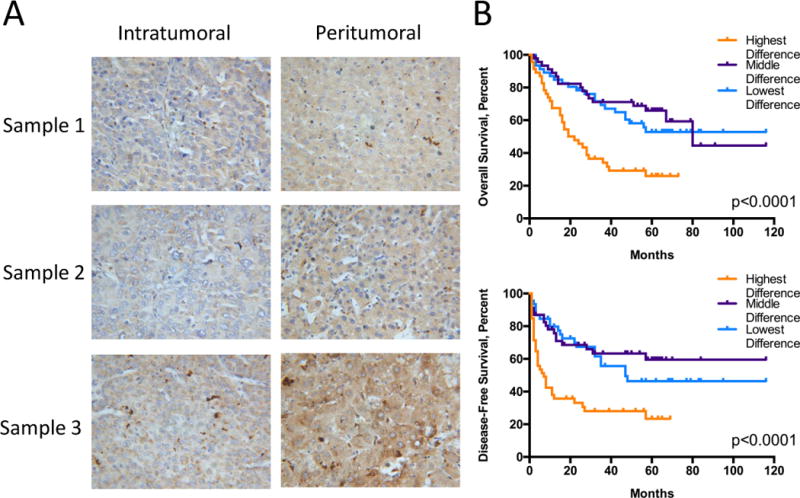
(A) Representative intratumoral and peritumoral staining (400X magnification) for IL-18 from three patients. Sample 1: middle intratumoral density, low peritumoral density, low difference in staining density; Sample 2: low intratumoral density, low peritumoral density, middle difference in staining density; Sample 3: high intratumoral density, high peritumoral density, high difference in staining density. (B) Kaplan-Meier curves for overall (top) and disease-free survival (bottom) of the 138 patients stratified into the highest, middle, or lowest third of patients based on differences in IL-18 staining density (Supplementary Table S4, Supplementary Fig. S18). Statistics: (B) Log-rank Mantel-Cox tests, n=46 per group.
DISCUSSION
Liver cancer is a devastating disease, the progression and prognosis of which are intricately intertwined with the chronically inflamed tissue in which it develops. This diseased microenvironment is drastically different from the healthy liver, including different cytokines, growth factors, activated stromal cells, extracellular matrix, and infiltrating immune cells. This altered microenvironment has been well-known to be integral to liver carcinogenesis, however studies examining the interplay between this altered environment, the tumor, and effects on the progression of the disease post-transformation have been less abundant (34). Numerous molecules have been shown to modulate this disease, but many more remain to be discovered and evaluated using more rigorous and relevant models with intact immune systems and functional microenvironments necessary for these experiments.
In this study, we demonstrated that while IL-18 is elevated in serum and non-tumor tissue of patients with HCC, its overall function in tumor progression is suppressive, modulating the accumulation and function of lymphocytes. This finding provides a novel molecular mechanism since previous alternative models had only demonstrated effects of IL-18 on NK-mediated cytotoxic activity in gene therapy studies in immunodeficient mice (10). IL-18 is a well-known regulator of Th1 functionality, affecting differentiation, proliferation, and cytokine production (25,41–44); its role in Th17 cells is more contentious (45,46). Th1 cells are critical modulators of tumor suppression in multiple cancers, including HCC (39), while the function of Th17 cells in HCC is unresolved. Increasing levels of IL-17 in the tumor have been correlated with worse prognosis, and some reports have shown significant effects of IL-17 on tumor cells (47,48). However, studies in other systems have shown that Th17 cells effect anti-tumor cytotoxicity, in part by regulating CD8+ T-cell functionality (40). Our data demonstrate IL-18 signaling-mediated effects on Th17 development and functionality in vitro and in vivo in HCC. Other systems have shown that IL-18 overexpression leads to enhanced CD8+ T-cell cytotoxicity in vitro and partial efficacy in reducing tumor burden in vivo, although not at the level of individual cells; our data mirror this and add more clarity to its effects. Cumulatively, we show multi-faceted IL-18 mediation of inflammatory T helper cell capacities, modulation of their interactions with CD8+ T-cells, and effects on the CD8+ T-cells themselves to control tumor growth. Importantly, these results were generated in the absence of IL-18 overexpression, demonstrating a robust tumor-suppressive role for endogenous IL-18.
We found that discerning endogenous IL-18’s effects on tumor growth was dependent upon an inflammatory context. These data suggest that important factors modulating tumor growth may not be easily evaluated in the absence of models mimicking clinical situation: this disease almost always presents with concurrent chronic inflammation and fibrosis. Many previous studies have utilized either immunodeficient mouse models or models without this inflamed tissue background. While the results generated from those experiments are still valuable, future studies should be designed accounting for this continually evolving inflammatory context (7,19,34,49,50).
We posit that elevated IL-18 in tissue and serum is indicative of inflammation and ongoing tissue injury, and a marker for disease progression; however its in-tumor functionality is suppressive. IL-18 expression is induced in chronic liver disease arising from a variety of etiologies, including chronic HBV, chronic HCV, and NAFLD, elevated in tissue and serum, and correlates with liver injury and fibrosis (26–28,31,33,51). Indeed, hepatotropic viruses induce expression of IL-18 in vitro, and transgenic IL-18 expression induces hepatic injury (52–54). Similar to other IL-1 family members, IL-18 is integral to the hepatocarcinogenic process (15,18,20,29,30). We observed decreased fibrosis yet increased tumor burden in IL18R1−/− mice at later stages, as well as reduced IL-18 in tumor compared with matched non-tumor tissue from patients, suggesting both late-stage tumor suppression by IL-18 and a disruption in the frequently observed correlation between fibrosis and tumor burden. Cumulatively, this describes a stage- and tissue-specific functionality for IL-18: inflammation enables carcinogenesis, however, the same ongoing inflammation in established tumors is suppressive. Indeed, tumor cells produce less IL-18 than their untransformed counterparts in distal tissue. Multiple mechanisms underlying this reduction in IL-18 production are possible, including clonal expansion, promoter methylation, transcriptional repression, and deregulation of inflammasome components, which warrant further investigation. These data emphasize that patterns of cytokine expression may differ between tumor, non-tumor tissue, and serum. Caution must be observed in choosing microenvironment-based therapeutic targets for development, taking into consideration clinical data from all relevant tissues.
Recent studies have shown that gene expression in the diseased liver correlates strongly with patient survival, and treatment targeting the non-tumor liver dramatically affects tumor progression (22,24,55). Our analyses demonstrated a stronger correlation between survival and IL-18 staining in the non-tumor liver than in the tumor. However, comparing IL-18 expression between non-tumor and tumor tissues had a stronger correlation to prognosis than either tissue alone, with the staining in the non-tumor liver determining the magnitude of this difference. Interestingly, levels of IL18R1 in either tissue alone or by comparison did not have predictive power towards patient survival (Supplementary Fig. S19), suggesting that differential expression of ligand dictates the effects of IL-18 signaling on HCC prognosis. These findings suggest that prognosis of HCC patients is determined by the combinational effects of two key factors intimately associated with IL-18 expression: reduced IL-18 signaling in the tumor compared to its surrounding non-tumor tissue, indicative of less intratumoral IL-18-induced tumor suppression; and destructive inflammatory IL-18 signaling in the diseased non-tumor liver tissue, indicative of overall tumor-independent liver function.
Supplementary Material
Acknowledgments
The authors thank members of the Duke Cancer Institute Flow Cytometry Core for aid sorting cells and members of the Wang and Li labs, particularly Jing Hu, Regina Lin, and Siqi Liu for helpful discussions and technical advice.
This work was supported by: grants CA154151 and CA164791, National Cancer Institute of the United States of America (XFW); grant 2012ZX10002-009, National Science and Technology Major Project of China, grant 30921006, National Natural Science Foundation for Creative Research Groups of China (HW); grant 81472768, National Natural Science Foundation of China (JF); grant 81222024, National Science Fund for Outstanding Young Scholars, grant 2012CB519005, National Key Basic Research Program of China (ZZ); and grant 2015CB553700-006, National Key Basic Research Program of China (PY).
Footnotes
The authors have declared that no conflict of interest exists.
GJM, PY, and X-FW initiated the project. GJM, Q-JL, and X-FW designed the experiments. GJM performed the in vivo and in vitro experiments. RC aided in performing in vivo experiments. GAM and AMD helped design animal models; GAM performed bile duct ligations. JS generated IL-18BP, ZZ and F-SW collected patient serum, and BY performed IL-18 ELISA on patient serum. JF, W-HQ, and HW collected patient tissue and data, and performed IL-18 staining. GJM and X-FW wrote the manuscript, and PY, AMD, and Q-JL modified the manuscript.
References
- 1.Yang JD, Roberts LR. Hepatocellular carcinoma: a global view. Nat Rev Gastroenterol Hepatol. 2010;7(8):448–58. doi: 10.1038/nrgastro.2010.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 3.Farazi PA, DePinho RA. Hepatocellular carcinoma pathogenesis: from genes to environment. Nat Rev Cancer. 2006;6(9):674–87. doi: 10.1038/nrc1934. [DOI] [PubMed] [Google Scholar]
- 4.Kuraishy A, Karin M, Grivennikov SI. Tumor Promotion via Injury- and Death-Induced Inflammation. Immunity. 2011;35(4):467–77. doi: 10.1016/j.immuni.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thorgeirsson SS, Grisham JW. Molecular pathogenesis of human hepatocellular carcinoma. Nat Genet. 2002;31:339–46. doi: 10.1038/ng0802-339. [DOI] [PubMed] [Google Scholar]
- 6.Grimm CF, Ortmann D, Mohr L, Michalak S, Krohne TU, Meckel S, et al. Mouse α-fetoprotein–specific DNA-based immunotherapy of hepatocellular carcinoma leads to tumor regression in mice. Gastroenterology. 2000;119(4):1104–12. doi: 10.1053/gast.2000.18157. [DOI] [PubMed] [Google Scholar]
- 7.Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, et al. NF-kB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431:461–66. doi: 10.1038/nature02924. [DOI] [PubMed] [Google Scholar]
- 8.Maeda S, Kamata H, Luo J-L, Leffert H, Karin M. IKKβ Couples Hepatocyte Death to Cytokine-Driven Compensatory Proliferation that Promotes Chemical Hepatocarcinogenesis. Cell. 2005;121(7):977–90. doi: 10.1016/j.cell.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 9.Fu J, Xu D, Liu Z, Shi M, Zhao P, Fu B, et al. Increased regulatory T cells correlate with CD8 T-cell impairment and poor survival in hepatocellular carcinoma patients. Gastroenterology. 2007;132(7):2328–39. doi: 10.1053/j.gastro.2007.03.102. [DOI] [PubMed] [Google Scholar]
- 10.Tsuchiyama T, Nakamoto Y, Sakai Y, Marukawa Y, Kitahara M, Mukaida N, et al. Prolonged, NK cell-mediated antitumor effects of suicide gene therapy combined with monocyte chemoattractant protein-1 against hepatocellular carcinoma. J Immunol. 2007;178(1):574–83. doi: 10.4049/jimmunol.178.1.574. [DOI] [PubMed] [Google Scholar]
- 11.Qin L-X. Inflammatory immune responses in tumor microenvironment and metastasis of hepatocellular carcinoma. Cancer Microenviron. 2012;5(3):203–09. doi: 10.1007/s12307-012-0111-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schneider C, Teufel A, Yevsa T, Staib F, Hohmeyer A, Walenda G, et al. Adaptive immunity suppresses formation and progression of diethylnitrosamine-induced liver cancer. Gut. 2012;61(12):1733–43. doi: 10.1136/gutjnl-2011-301116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang P, Li Q-J, Feng Y, Zhang Y, Markowitz GJ, Ning S, et al. TGF-beta-miR-34a-CCL22 Signaling-Induced Treg Cell Recruitment Promotes Venous Metastases of HBV-Positive Hepatocellular Carcinoma. Cancer Cell. 2012;22(3):291–303. doi: 10.1016/j.ccr.2012.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang P, Markowitz GJ, Wang XF. The hepatitis B virus-associated tumor microenvironment in hepatocellular carcinoma. Natl Sci Rev. 2014;1(3):396–412. doi: 10.1093/nsr/nwu038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sakurai T, He G, Matsuzawa A, Yu G-Y, Maeda S, Hardiman G, et al. Hepatocyte Necrosis Induced by Oxidative Stress and IL-1α Release Mediate Carcinogen-Induced Compensatory Proliferation and Liver Tumorigenesis. Cancer Cell. 2008;14(2):156–65. doi: 10.1016/j.ccr.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tang Y, Kitisin K, Jogunoori W, Li C, Deng C-X, Mueller SC, et al. Progenitor/stem cells give rise to liver cancer due to aberrant TGF-beta and IL-6 signaling. Proc Natl Acad Sci U S A. 2008;105(7):2445–50. doi: 10.1073/pnas.0705395105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Park EJ, Lee JH, Yu G-Y, He G, Ali SR, Holzer RG, et al. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell. 2010;140(2):197–208. doi: 10.1016/j.cell.2009.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yan W, Chang Y, Liang X, Cardinal JS, Huang H, Thorne SH, et al. High-mobility group box 1 activates caspase-1 and promotes hepatocellular carcinoma invasiveness and metastases. Hepatology. 2012;55(6):1863–75. doi: 10.1002/hep.25572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.He G, Dhar D, Nakagawa H, Font-Burgada J, Ogata H, Jiang Y, et al. Identification of Liver Cancer Progenitors Whose Malignant Progression Depends on Autocrine IL-6 Signaling. Cell. 2013;155(2):384–96. doi: 10.1016/j.cell.2013.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature. 2014;498(7456):97–101. doi: 10.1038/nature12347. [DOI] [PubMed] [Google Scholar]
- 21.Ye Q-H, Qin L-X, Forgues M, He P, Kim JW, Peng AC, et al. Predicting hepatitis B virus–positive metastatic hepatocellular carcinomas using gene expression profiling and supervised machine learning. Nat Med. 2003;9(4):416–23. doi: 10.1038/nm843. [DOI] [PubMed] [Google Scholar]
- 22.Hoshida Y, Villanueva A, Kobayashi M, Peix J, Chiang DY, Camargo A, et al. Gene expression in fixed tissues and outcome in hepatocellular carcinoma. N Engl J Med. 2008;359(19):1995–2004. doi: 10.1056/NEJMoa0804525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Michelotti GA, Machado MV, Diehl AM. NAFLD, NASH and liver cancer. Nat Rev Gastroenterol Hepatol. 2013:1–10. doi: 10.1038/nrgastro.2013.183. [DOI] [PubMed] [Google Scholar]
- 24.Fuchs BC, Hoshida Y, Fujii T, Wei L, Yamada S, Lauwers GY, et al. Epidermal growth factor receptor inhibition attenuates liver fibrosis and development of hepatocellular carcinoma. Hepatology. 2014;59(4):1577–90. doi: 10.1002/hep.26898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Garlanda C, Dinarello CA, Mantovani A. The Interleukin-1 Family: Back to the Future. Immunity. 2013;39(6):1003–18. doi: 10.1016/j.immuni.2013.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng K-S, Tang H-L, Chou F-T, Chou J-W, Hsu C-H, Yu C-J, et al. Cytokine evaluation in liver cirrhosis and hepatocellular carcinoma. Hepatogastroenterology. 2009;56(93):1105–10. [PubMed] [Google Scholar]
- 27.Perrella O, Cuomo O, Sbreglia C, Monaco A, Gnarini MR, Gentile B, et al. IL-18 and interferon-gamma in HCV-related hepatocellular carcinoma: a model of interplay between immune status and cancer. J Biol Regul Homeost Agents. 2009;23(4):251–58. [PubMed] [Google Scholar]
- 28.Tangkijvanich P, Thong-Ngam D, Mahachai V, Theamboonlers A, Poovorawan Y. Role of serum interleukin-18 as a prognostic factor in patients with hepatocellular carcinoma. World J Gastroenterol. 2007;13(32):4345–49. doi: 10.3748/wjg.v13.i32.4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li S, Sun R, Chen Y, Wei H, Tian Z. TLR2 Limits Development of Hepatocellular Carcinoma by Reducing IL18-Mediated Immunosuppression. Cancer Res. 2015 doi: 10.1158/0008-5472.CAN-14-2371. [DOI] [PubMed] [Google Scholar]
- 30.Terakura D, Shimizu M, Iwasa J, Baba A, Kochi T, Ohno T, et al. Preventive effects of branched-chain amino acid supplementation on the spontaneous development of hepatic preneoplastic lesions in C57BL/KsJ-db/db obese mice. Carcinogenesis. 2012;33(12):2499–506. doi: 10.1093/carcin/bgs303. [DOI] [PubMed] [Google Scholar]
- 31.Chia CS, Ban K, Ithnin H, Singh H, Krishnan R, Mokhtar S, et al. Expression of interleukin-18, interferon-gamma and interleukin-10 in hepatocellular carcinoma. Immunol Lett. 2002;84(3):163–72. doi: 10.1016/s0165-2478(02)00176-1. [DOI] [PubMed] [Google Scholar]
- 32.Novick D, Schwartsburd B, Pinkus R, Suissa D, Belzer I, Sthoeger Z, et al. A NOVEL IL-18BP ELISA SHOWS ELEVATED SERUM IL-18BP IN SEPSIS AND EXTENSIVE DECREASE OF FREE IL-18. Cytokine. 2001;14(6):334–42. doi: 10.1006/cyto.2001.0914. [DOI] [PubMed] [Google Scholar]
- 33.Zhang Y, Li Y, Ma Y, Liu S, She Y, Zhao P, et al. Dual effects of interleukin-18: inhibiting hepatitis B virus replication in HepG2.2.15 cells and promoting hepatoma cells metastasis. Am J Physiol Gastrointest Liver Physiol. 2011;301(3):G565–G73. doi: 10.1152/ajpgi.00058.2011. [DOI] [PubMed] [Google Scholar]
- 34.Markowitz GJ, Michelotti GA, Diehl AM, Wang X-F. Inflammatory models drastically alter tumor growth and the immune microenvironment in hepatocellular carcinoma. Science Bulletin. 2015;60(8):762–72. doi: 10.1007/s11434-015-0772-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hoshino K, Tsutsui H, Kawai T, Takeda K, Nakanishi K, Takeda Y, et al. Cutting edge: generation of IL-18 receptor-deficient mice: evidence for IL-1 receptor-related protein as an essential IL-18 binding receptor. J Immunol. 1999;162(9):5041–44. [PubMed] [Google Scholar]
- 36.Kinoshita A, Onoda H, Imai N, Iwaku A, Oishi M, Fushiya N, et al. Comparison of the prognostic value of inflammation-based prognostic scores in patients with hepatocellular carcinoma. Br J Cancer. 2012;107(6):988–93. doi: 10.1038/bjc.2012.354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Calzascia T, Pellegrini M, Hall H, Sabbagh L, Ono N, Elford AR, et al. TNF-α is critical for antitumor but not antiviral T cell immunity in mice. J Clin Invest. 2007 doi: 10.1172/JCI32567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rosenberg SA. IL-2: The First Effective Immunotherapy for Human Cancer. J Immunol. 2014;192(12):5451–58. doi: 10.4049/jimmunol.1490019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Budhu A, Forgues M, Ye Q-H, Jia H-L, He P, Zanetti KA, et al. Prediction of venous metastases, recurrence, and prognosis in hepatocellular carcinoma based on a unique immune response signature of the liver microenvironment. Cancer Cell. 2006;10(2):99–111. doi: 10.1016/j.ccr.2006.06.016. [DOI] [PubMed] [Google Scholar]
- 40.Martin-Orozco N, Muranski P, Chung Y, Yang XO, Yamazaki T, Lu S, et al. T Helper 17 Cells Promote Cytotoxic T Cell Activation in Tumor Immunity. Immunity. 2009;31(5):787–98. doi: 10.1016/j.immuni.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Greene CM, Meachery G, Taggart CC, Rooney CP, Coakley R, O’Neill SJ, et al. Role of IL-18 in CD4+ T Lymphocyte Activation in Sarcoidosis. J Immunol. 2000;165(8):4718–24. doi: 10.4049/jimmunol.165.8.4718. [DOI] [PubMed] [Google Scholar]
- 42.Kohno K, Kataoka J, Ohtsuki T, Suemoto Y, Okamoto I, Usui M, et al. IFN-gamma-inducing factor (IGIF) is a costimulatory factor on the activation of Th1 but not Th2 cells and exerts its effect independently of IL-12. J Immunol. 1997;158(4):1541–50. [PubMed] [Google Scholar]
- 43.Micallef MJ, Ohtsuki T, Kohno K, Tanabe F, Ushio S, Namba M, et al. Interferon-gamma-inducing factor enhances T helper 1 cytokine production by stimulated human T cells: synergism with interleukin-12 for interferon-gamma production. Eur J Immunol. 1996;26(7):1647–51. doi: 10.1002/eji.1830260736. [DOI] [PubMed] [Google Scholar]
- 44.Puren AJ, Fantuzzi G, Gu Y, Su MS, Dinarello CA. Interleukin-18 (IFNgamma-inducing factor) induces IL-8 and IL-1beta via TNFalpha production from non-CD14+ human blood mononuclear cells. J Clin Invest. 1998;101(3):711–21. doi: 10.1172/JCI1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lalor SJ, Dungan LS, Sutton CE, Basdeo SA, Fletcher JM, Mills KHG. Caspase-1-Processed Cytokines IL-1 and IL-18 Promote IL-17 Production by and CD4 T Cells That Mediate Autoimmunity. J Immunol. 2011;186(10):5738–48. doi: 10.4049/jimmunol.1003597. [DOI] [PubMed] [Google Scholar]
- 46.Nakae S, Iwakura Y, Suto H, Galli SJ. Phenotypic differences between Th1 and Th17 cells and negative regulation of Th1 cell differentiation by IL-17. J Leukoc Biol. 2007;81(5):1258–68. doi: 10.1189/jlb.1006610. [DOI] [PubMed] [Google Scholar]
- 47.Gu F-M, Li Q-L, Gao Q, Jiang J-H, Zhu K, Huang X-Y, et al. IL-17 induces AKT-dependent IL-6/JAK2/STAT3 activation and tumor progression in hepatocellular carcinoma. Mol Cancer. 2011;10(1):150. doi: 10.1186/1476-4598-10-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liao R, Sun J, Wu H, Yi Y, Wang J-X, He H-W, et al. High expression of IL-17 and IL-17RE associate with poor prognosis of hepatocellular carcinoma. J Exp Clin Cancer Res. 2013;32(1):1–1. doi: 10.1186/1756-9966-32-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dapito DH, Mencin A, Gwak G-Y, Pradere J-P, Jang M-K, Mederacke I, et al. Promotion of Hepatocellular Carcinoma by the Intestinal Microbiota and TLR4. Cancer Cell. 2012;21(4):504–16. doi: 10.1016/j.ccr.2012.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang M-C, Chang C-P, Lei H-Y. Induction of liver fibrosis in a murine hepatoma model by thioacetamide is associated with enhanced tumor growth and suppressed antitumor immunity. Lab Invest. 2010;90(12):1782–93. doi: 10.1038/labinvest.2010.139. [DOI] [PubMed] [Google Scholar]
- 51.Negash AA, Gale M. Hepatitis regulation by the inflammasome signaling pathway. Immunol Rev. 2015;265(1):143–55. doi: 10.1111/imr.12279. [DOI] [PubMed] [Google Scholar]
- 52.Finotto S, Siebler J, Hausding M, Schipp M, Wirtz S, Klein S, et al. Severe hepatic injury in interleukin 18 (IL-18) transgenic mice: a key role for IL-18 in regulating hepatocyte apoptosis in vivo. Gut. 2004;53(3):392–400. doi: 10.1136/gut.2003.018572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee M-O, Choi Y-H, Shin E-C, Kang H-J, Kim Y-M, Jeong S-Y, et al. Hepatitis B virus X protein induced expression of interleukin 18 (IL-18): a potential mechanism for liver injury caused by hepatitis B virus (HBV) infection. J Hepatol. 2002;37(3):380–86. doi: 10.1016/s0168-8278(02)00181-2. [DOI] [PubMed] [Google Scholar]
- 54.Shrivastava S, Mukherjee A, Ray R, Ray RB. Hepatitis C virus induces interleukin-1beta (IL-1beta)/IL-18 in circulatory and resident liver macrophages. J Virol. 2013;87(22):12284–90. doi: 10.1128/JVI.01962-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Philips GM, Chan IS, Swiderska M, Schroder VT, Guy C, Karaca GF, et al. Hedgehog Signaling Antagonist Promotes Regression of Both Liver Fibrosis and Hepatocellular Carcinoma in a Murine Model of Primary Liver Cancer. PLoS One. 2011;6(9):e23943. doi: 10.1371/journal.pone.0023943. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.