

Abstract
The activation of the epithelial-to-mesenchymal transition (EMT) program is a critical step in cancer progression and metastasis, but visualization of this process at the single cell level, especially in vivo, remains challenging. We established an in vivo approach to track the fate of tumor cells based on a novel EMT-driven fluorescent color switching breast cancer mouse model and intravital two-photon laser scanning microscopy. Specifically, the MMTV-PyMT, Rosa26-RFP-GFP, and Fsp1-Cre triple transgenic mouse model was used to monitor the conversion of RFP-positive epithelial cells to GFP-positive mesenchymal cells in mammary tumors under the control of the Fsp1 (ATL1) promoter, a gate-keeper of EMT initiation. RFP-positive cells were isolated from the tumors, sorted, and transplanted into mammary fat pads of SCID mice to monitor EMT during breast tumor formation. We found that the conversion from RFP to GFP-positive and spindle-shaped cells was a gradual process, and that GFP-positive cells preferentially localized close to blood vessels, independent of tumor size. Furthermore, cells undergoing EMT expressed high levels of the HGF receptor, c-Met, and treatment of RFP-positive cells with the c-Met inhibitor, cabozantinib, suppressed the RFP-to-GFP conversion in vitro. Moreover, administration of cabozantinib to mice with palpable RFP-positive tumors resulted in a silent EMT phenotype whereby GFP-positive cells exhibited reduced motility, leading to suppressed tumor growth. In conclusion, our imaging technique provides a novel opportunity for visualizing tumor EMT at the single cell level and may help to reveal the intricacies underlying tumor dynamics and treatment responses.
Keywords: Epithelial to mesenchymal transition (EMT), intravital two-photon microscopic imaging, breast cancer
Introduction
Epithelial-mesenchymal transition (EMT) has been accepted as a critical process that allows stationary epithelial cells to gain the motile ability needed in order to migrate and invade during embryogenesis, organ development, tissue regeneration, and organ fibrosis (1,2). Extensive studies over the past decade have provided strong evidence for the activation of the EMT program in cancer progression and metastasis (3,4), mostly focusing on: 1) characterizing the function of EMT core regulators in various cancer models, and 2) revealing the clinical relevance of molecular EMT markers to prognosis (1,5,6). Consistent with these implications of EMT in cancer progression and metastasis, recent studies indicate that EMT confers stem cell-like and chemo-resistance properties to cancer cells (7,8). Although it is perceived that EMT initiates from individual cells and plays a critical role in cancer progression, a direct visualization of the EMT process at the single cell level, especially in vivo has not yet been convincingly achieved.
Most in vivo imaging methods, such as magnetic resonance imaging (MRI), positron emission tomography–computed tomography (PET-CT), or ultrasound imaging, can only reach organ-scale resolution (9). Two-photon microscopic imaging, which combines long-wavelength multi-photon fluorescence excitation and laser-scanning microscopy, enables high-resolution single cell-level and in-depth live tissue imaging with less photo-bleaching and photo-damage (10,11). We used a new EMT-driven fluorescent color switching breast cancer mouse model (12) along with two-photon microscopy to track the fate of EMT tumor cells in relation to breast cancer progression and responses to the targeted treatment in real time.
First, EMT was confirmed in vivo in the mouse breast cancer model, and the dynamic morphological changes of the EMT cells in vivo followed a similar pattern as in in vitro environments. Second, EMT initiation did not exhibit a tumor central vs. peripheral location difference; however, regardless of tumor size, there is a significant sub-population of EMT cells in close proximity to tumor stroma and blood vessels. Third, the EMT cells adjacent to blood vessels extend a long membrane extension to lean close to the vessel wall and migrate along the vessels much faster than individual EMT cells surrounded by epithelial cells. Fourth, morphological dynamics analysis identified several EMT cell types indicating different cell functions, i.e., silent EMT cells, migratory EMT cells, and fibroblast-like EMT cells. Fifth, the anti-metastatic drug Cabozantinib (XL-184), a dual inhibitor of tyrosine kinases c-Met and Flt 1/4, triggered the reversal of EMT phenotype in vitro and in vivo. Our intravital imaging approach provides a visible and direct recapitulation of tumor EMT at the single cell level and reveals cellular insights on tumor dynamics and treatment responses.
Materials and Methods
EMT mouse model and general procedures
The transgenic breast cancer mouse model was generated in the Mittal lab (12). After tumor formation, the tumor was dissected from the sacrificed mouse and pure RFP+ and GFP+ tumor cell populations were sorted using flow cytometry (BD Biosciences). For in vitro time-lapse imaging, the sorted RFP+ tumor cells were maintained in serum free DMEM medium. For in vivo transplantation, RFP+ or GFP+ cells were implanted in female CB17/Icr-Prkdc SCID mice (Charles River) in accordance with the approval and the guidelines of Institutional Animal Care and Use Committee (IACUC) of Houston Methodist Research Institute. 2×105 tumor cells were injected into the right side no. 4 mammary fat pad of the SCID mice to form in situ breast tumor. Intravital two-photon imaging was then performed at different stages of tumor development (details see below). For the drug response study, XL-184 (Cabozantinib, Selleck Chemicals) was injected intraperitoneally (30 mg/kg) daily for five days after palpable tumor formation. The primary cells from the transgenic breast cancer mouse model were authenticated by morphological observation and tested for absence of Mycoplasma contamination (MycoAlert, Lonza Rockland, Rockland, ME).
Live cell time-lapse imaging
RFP+ tumor cells were seeded in 6-well plates and given appropriate treatments. The 6-well plate was placed in the IncuCyte live cell imaging system (Essen BioScience); both bright field and GFP fluorescence channels were captured every hour for 72 hours, and 36 fields of view were taken for each well. Images were visualized as separate frames or video by the IncuCyte software. Cellular image segmentation and quantification analysis were described previously in (13).
Live animal intravital two-photon microscope imaging
Image data for small (palpable tumor, 2-3 mm diameter), medium (8-10 mm diameter), and large (2 cm diameter) tumors were collected by intravital two-photon microscopy (Olympus FV1000MPE) on the anesthetized live mouse, or on ex vivo whole-mount tumor slices for montage images. Briefly, after the administration of analgesic drugs (Buprenorphine 0.1 mg/kg, Caprofen 5 mg/kg and Dexamethasone sodium phosphate 2 mg/kg), the mouse was anesthetized by Isoflurane (4% for induction and 1% for maintenance, with air flow rate at 1 L/min). A middle line incision plus a diagonal incision near the inguen was made to form a flap to expose the tumor (Fig S1a). The blood vessels on the skin especially those surrounding the tumor were protected carefully. The skin was adhered to a glass slide using tissue glue, and the exposed tumor surface was kept wet with warm PBS. A thin, glass bottom chamber was placed on top of the tumor for imaging (Fig S1b). Intravital imaging was performed on an Olympus FV1000 two-photon microscope with a 25X, 1.05 NA MPE water immersion objective; a pulsed laser system with a wavelength range of 700-1000 nm (Mai Tai Laser) was used for excitation of fluorophores. GFP and RFP were viewed simultaneously with 810 nm excitation, along with 495-460nm and 575-630nm emission filters for GFP and RFP/Texas-Red, respectively. 70 kDa Texas Red-dextran (Invitrogen) was tail vein injected to label blood vessels. To generally characterize in vivo features of the tumor cells, stacks of 508 × 508 μm at 512 × 512 pixels for the depth to 150-200 μm with 1 μm steps were taken. The same field of view was imaged every 30min for a total of 4-6 hours depending on the condition of the mouse. During the surgery and imaging, the rectal temperature of the mouse was monitored and maintained at 37 °C.
Images were reconstructed in 3D and over time using Image J. Image analysis algorithms were developed to: 1) segment tumor/lobule central and peripheral areas; 2) track tumor cell migration and membrane extension elongation; 3) segment tumor cells and blood vessels; and 4) quantify the morphological changes of the tumor cells.
Immunoblot Analysis
Cells were collected before and after treatments (TGF-β and/or XL-184), lysed, electrophoresed on SDS–polyacrylamide gels, and transferred to nitrocellulose membranes. Immunoblot analysis was performed using the following primary antibodies: rat monoclonal to E-Cadherin (Abcam, 1:1000), rabbit monoclonal to Vimentin (Abcam, 1:2000), and Goat Polyclonal to c-MET (R&D Systems, 1:1000).
Immunohistochemistry (IHC) and Immunocytochemistry (ICC)
After dissection, tumors were fixed in 4% paraformaldehyde, followed by dehydration in 30% glucose. Immunohistochemistry was performed on the tumor frozen sections. Rat anti-E-Cadherin monoclonal antibody (Abcam, 1:200) and cy5.5 conjugated second antibody were used to stain the epithelial cell marker. Tumor cells were cultured on glass-bottom chamber slides. After various treatments, the cells were fixed in 4% paraformaldehyde, followed by E-Cadherin staining.
Statistical analysis
Data are expressed as mean ± SD. For comparison of groups, we used the two-tailed t test or the Mann-Whitney rank sum test. To compare metastasis-free survival, we used the log rank test. A level of P < 0.05 was regarded as statistically significant. We did all calculations with SigmaPlot statistical software (version 11.2; Systat Software Inc. Chicago, Illinois).
Results
Imaging EMT in primary breast tumors
To demonstrate the existence of EMT in primary breast tumor and visualize the EMT process in vivo by intravital two-photon microscopy, we used a transplanted EMT-driven fluorescent color switching breast tumor model that has been established recently (12). Briefly, the tumor cells of this model are derived from MMTV-PyMT, Rosa26-RFP-GFP, and Fsp1-Cre triple transgenic mouse tumors (Fig 1a). Fsp1 is the critical gate-keeping gene of EMT initiation (14), and its early activation in this process allows for tracing the tumor cells that have undergone EMT in vivo (15).
Fig 1. Imaging demonstration of EMT presence in primary breast tumors.
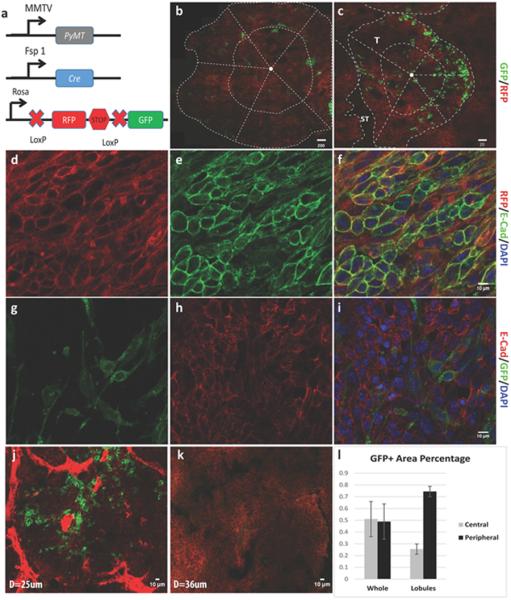
a. Scheme of the EMT-driven Cre-mediated fluorescence switching system. b. A representative montage image of the whole tumor slice. The dotted lines separate the whole slice into periphery and central parts as described in the result section and the GFP+ cells distributed sporadically in the two parts; scale bar, 200μm. c. A zoom-in lobule view of the tumor. The dotted lines also separated the periphery and central parts of the lobule. The GFP+ cells distribute in the periphery part of the lobules, adjacent to the stroma (ST) of the tumor; scale bar, 20μm. d-i. IHC staining of the primary tumor showing that E-Cadherin was co-localized well with RFP+ tumor cells, while when the tumor cells turn to GFP+, the expression of E-Cadherin was lost; scale bar, 10μm. j. A representative image showing the blood vessel-enriched stroma and the GFP+ cells distributed surrounding the blood vessels. k. A representative image showing the blood vessel-unenriched stroma where the GFP+ cells were rarely seen. l. The quantification of GFP+ cell distribution both in whole tumor slices and the tumor lobules as shown in b and c: in the whole tumor slice, the distribution of GFP+ cells at peripheral vs. central location was 49.4±18.9% vs. 50.6±18.9% of the total GFP+ cells, while in tumor lobules, more GFP+ cells located at peripheral part of the lobules (peripheral vs. central: 74.5±4.2% vs. 24.5±4.2%).
Intravital two-photon imaging of 2 cm diameter primary tumors showed that the majority of stromal cells surrounding the RFP+ epithelial tumor cells were GFP positive and had characteristic fibroblast morphology (Fig S1c). Sporadic GFP+ cells were observed within the epithelial tumors, and these cells showed cuboidal or spindle morphology (Fig S1d and video S1). Due to the abundance of GFP+ stromal cells in the PyMT tumors that interfered with imaging EMT specifically, we sorted the pure RFP+ epithelial tumor cells from the triple transgenic mice and transplanted them into normal wild type mice (Fig S1e). Recently, the isolated PyMT tumor cells were proved to be a versatile cell model for studying EMT both in vitro and in vivo (16). Palpable tumors formed 7-10 days after transplantation, and the expression of GFP was detected sporadically in these tumors (Fig 1b-c, and video S2), which suggests that EMT has occurred. Further, the presence of PyMT gene in both RFP+ and GFP+ cells (Fig S1f) indicated their origin from the PyMT tumor epithelium and confirmed the epithelial to mesenchymal conversion is driven by Fsp1. The epithelial marker E-Cadherin localized well on the membranes of RFP+ cells, but when these cells turned to GFP+, they lost E-Cadherin expression (Fig 1d-i). These results indicate that the Fsp1-driven fluorescent color switching mouse model represents an effective tool to rigorously characterize the initiation of EMT in vivo.
Relation of EMT initiation to tumor sub-locations and blood vessels
To explore the location preference of EMT initiation in the tumor, we imaged the distribution of GFP+ and RFP+ cells in whole-mount tumor slices using the montage imaging function provided by the two-photon microscope. Small (2-3 mm diameter), medium (8-10 mm diameter), and large (2 cm diameter) tumors were sliced and segmented into central and peripheral areas through the midpoints of radial lines from the tumor center to the edges (Fig 1b). For the ten tumors imaged, the average distribution of GFP+ areas at peripheral vs. central location in the total GFP+ cells was 49.4±18.9% vs. 50.6±18.9%. For each tumor size category, the peripheral vs. central distribution of GFP+ areas was variable but overall there was no significant difference (Fig 1b&l; Supplementary Table 1) between the two areas.
However, we observed that many GFP+ cells localized at the border area of the tumor lobules, which is adjacent to tumor stroma (Fig 1c&l; Supplementary Table 1). In solid tumors, stroma includes connective tissue, blood vessels and inflammatory cells. To further investigate whether the GFP+ cells located in proximity to blood vessels, we differentiated the tumor stroma into two sub-groups based on the acquired Z-stack images, i.e., blood vessel-enriched stroma (could be avidly enhanced by Dextran-Texas Red) and blood vessel-unenriched stroma (could not be enhanced by Dextran-Texas Red). The results showed that among the different tumor size groups, 74.5±4.2% of the GFP+ cells located at the lobule borders adjacent to the blood vessel-enriched stroma (Fig 1j-k, Supplementary Table 1). These data indicate that no matter the tumor size, there is always a large subpopulation of EMT cells residing close to blood vessels, suggesting that interfering with tumor cell-blood vessel signaling might disrupt the fate of EMT cells.
In vitro and in vivo kinetics of EMT initiation
The sorted, pure RFP+ cells from primary tumors were cultured in DMEM with 10% fetal bovine serum. Due to the enriched EMT promoting factors in the serum including transforming growth factors (TGFs), epidermal growth factors (EGFs), and fibroblast growth factors (FGFs) (17), certain RFP+ epithelial tumor cells went through the EMT process (12). Time-lapse live-cell GFP fluorescence imaging was performed to detect GFP expression in the cells (Fig 2a-g). In a 72h time period, GFP expression emerged in about 0.5% of the cells, and the GFP signal grew gradually to reach a 4-fold increase of the fluorescence intensity within 8-14 hours (Fig 2h). Meanwhile, the shape of these cells changed from predominantly round to fusiform indicated by a ≥ 2-fold increase of the axial ratio (Fig 2i).
Fig 2. In vitro and in vivo kinetics of EMT initiation.

a-g. In vitro time-lapse imaging of EMT initiation in the cultured tumor cells. Sorted pure RFP+ tumor cells from the transgenic mice were cultured in medium with 10% FBS; GFP signal and bright field view of the cells were taken every 2 hours; the arrow indicates cell turned to GFP+ over time in one imaging field. h. The quantification of the GFP fluorescence intensity showing the increase of GFP signal over time (4.08±0.95-fold increase after 8-14h, p<0.001). i. The quantification of tumor cell axial ratio showing the increase of axial ratio after EMT (2.84±0.46 vs 1.19±0.13, p<0.001). j-l. Capture of EMT initiation in vivo. As the arrow indicates, one green tumor cell emerged during the in vivo tumor imaging. m. The quantification of GFP fluorescence intensity showing the increase of GFP signal over time (1.69±0.19-fold increase after 4-6h, p<0.001). n. The quantification of tumor cell axial ratio showing the elongation of the EMT cells after 4-6h (1.98±0.06 vs 1.11±0.05).
EMT initiation was also evaluated in vivo. About ninety 3D image stacks and images of more than 25,000 cells were acquired from 15 mice with 508×508 μm field of view by intravital two-photon microscopy, and each field was imaged for at least 4-6 hours. The overall patterns of in vivo EMT initiation followed the similar dynamics as the in vitro scenario, i.e., the gradual increase of GFP signals, and the shapes of these GFP+ cells become more fusiform (Fig 2j-n). In addition, within the 4-6 hours imaging acquisition time window, we were only able to capture the faint dynamic RFP-to-GFP color switching process on thirty cells (0.12% of the total 25,000 cells), suggesting a wider time window of EMT initiation in individual tumor cells during tumor development. Compared with the in vitro EMT, this result also indicates the complicated impact of the in vivo tumor microenvironment on the EMT process.
Characterization of the migration and invasion of EMT cells in vivo
The migration and invasion features of tumor cells are mostly reflected by their morphological changes. To characterize these features of the EMT cells, we performed higher magnification (40X objective) intravital imaging of the GFP+ cells in the small size breast tumors of the transplanted mice. Quantification data were obtained from ~800 GFP+ cells and ~1200 adjacent RFP+ cells from 10 tumors. Not surprisingly, the GFP+ cells showed an elongated shape with a bigger axial ratio than that of the RFP+ cells (GFP+: 2.17±0.27; RFP+: 1.28±0.26; p=0.003) (Fig 3a). In addition, we found that more GFP+ tumor cells protruded membrane extensions longer than 3 μm (GFP+: 3-5μm 46±4%, >5μm 4.67±1.53%; vs RFP+: 3-5μm 15±4.35%, >5μm 0.33±0.57%; p<0.001 Fig 3b), while the direction of the long membrane extensions was orientated toward blood vessels or tumor stroma (Fig 3c).
Fig 3. In vivo migration of EMT tumor cells.

a. The quantification of tumor cell axial ratio showing the elongated GFP+ tumor cells after EMT compared with original RFP+ epithelial tumor cells (2.17±0.27 vs 1.28±0.26; p=0.003). b. The quantification of length distribution of membrane extensions in GFP+ and RFP+ cells (in RFP+ cells, 59.67±2.52% of them have no extensions, 25±3% have extensions <3μm, 15±4.36% have extensions 3-5μm, only 0.33±0.58% of them have extensions longer than 5μm. In GFP+ cells, the percentage of each corresponding groups were 11.25±3%, 34.33±3.05%, 46±4% and 4.67±1.53 respectively). c. The main direction of the long membrane extensions was orientated toward blood vessels or tumor stroma as indicated by white arrows. d-f. Dynamic in vivo cell migration of the GFP+ cells which were located in the middle of tumor cell clusters and adjacent to the blood vessels (g-i). The migration speed differed between the two groups: 0.993±0.06 and 6.06±2.98 μm/hour, respectively. j-l. GFP+ tumor cells migrated in clusters along the blood vessel. The upper right insert pictures in d-i are the maximum intensity projection of the selected GFP+ cells in the Z-stack. Long dotted lines in g-l mark the blood vessels.
Dynamic in vivo cell migration of the GFP+ cells was recorded (Fig 3d-i). The migration kinetics of GFP+ cells varied depending on their locations. The individual GFP+ cells surrounded by clusters of RFP+ epithelial cells moved slowly, 0.993±0.06 μm/hour (Fig 3d-f). GFP+ cells adjacent to blood vessels extended membrane extensions to lean close to the vessel wall and migrated along the vessels much faster, 6.06±2.98 μm/hour (Fig 3g-i). We also observed that small clusters of GFP+ cells move together towards the blood vessel or tumor stroma (Fig 3j-l), in agreement with previous reports of multicellular streaming that is critical to tumor metastasis (18). These morphological observations confirm that the EMT process endows tumor cells with more motile characteristics and that blood vessels play an important role, impacting the migration of EMT tumor cells.
Single cell morphological dynamics of EMT cells in vivo
During EMT, tumor cells lose cell polarity and cell-cell conjunction which are necessary to maintain the epithelial phenotype (4). A fraction of these cells retain some polarity and gain fibroblast-like features forming a small part of the cancer-associated fibroblast population (19-21). These cells attach to the extracellular matrix (ECM) tightly, but do not have high migratory ability. Conversely, the other part of the tumor cells that have undergone EMT lose all polarity, taking on an amoeba-like shape. These cells have loosened ECM attachment, allowing for rapid migration (Fig 4a-f).
Fig 4. Single cell morphological dynamics of EMT cells in vivo.
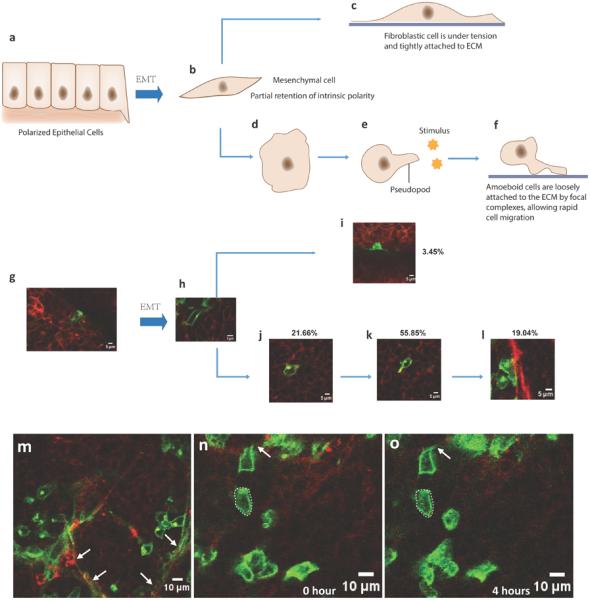
a-f. A sketch of the cell morphological dynamics after EMT in the tumor. After EMT, some of tumor cells kept part of their polarity and will transform to fibroblast cells and attach to extracellular matrix firmly (c) while other tumor cells will lose their polarity and transform into ‘amoeba’-like cells; they will be attracted by stimuli and can migrate towards the stimuli by extending membrane extensions (d-f). g-l. Representative images captured by intravital multi-photon microscopy and the percentage statistics of each cell stages after EMT which corresponds with the steps shown in a-f. m. A representative in vivo image of the big tumor showing more fibroblast-like GFP+ cells (indicated by arrows) at the tumor stromal area. n-o. Representative in vivo images of the small tumor showing the silent GFP+ cells (in the dotted line) and its close-by migratory EMT cells with an elongated membrane extension (arrow).
To further clarify which type of EMT cells dominate in the tumors, we tracked individual GFP+ cells in early-stage tumors (2 mm diameter) and evaluated their morphological dynamics after EMT (Fig 4g-i). The GFP+ fibroblasts were recognized by their spindle-like shape and long, thin linear processes tightly attached to the stromal component of the tumor (Fig 4m, indicated by arrows). After counting the GFP+ cells in the tumors, these fibroblast-like EMT cells constituted only a tiny part of the post-EMT cells (3.45%). More than half of the cells (55.85%) exhibited tropism, and iterative elongation and contraction of the membrane extensions along one main direction (Fig 4n-o, indicated by arrows). In addition, the membrane extensions of the nearby cells protruded in similar main directions (Fig 3g-i), which indicated that they extended the membrane extensions toward same stimuli. 19.04% of post-EMT cells migrated into the adjacent tumor stroma. These cells attached loosely to the ECM (no obvious flat contact surface to the stroma) and had shorter contact surface than the fibroblast-like GFP+ cells, suggesting that they migrated more easily than the fibroblast-like GFP+ cells. These data further indicate that most EMT cells are actively motile, and that a portion of the EMT cells might be transformed into cancer related fibroblasts. Besides the migratory and fibroblast-like GFP+ cells, 21.66% of the GFP+ cells kept the amoeboid appearance without membrane extensions, and they kept static without obvious shape changes during the 4-6 hours imaging period (Fig 4n-o, indicated by dotted lines). Since these cells are mostly located surrounding the migratory EMT cells, they are most likely temporary silent EMT cells.
When GFP+ cells in mid-size and large tumors were imaged, we found more clusters than sporadic GFP+ cells at the tumor stromal area (Fig S2a-b, video S3). These could be attributed to the cell proliferation and the migration assembly of GFP+ cells. At these stages, the morphological analysis revealed that 60-80% of these cells appeared fibroblast-like in shape with long contact surface to the tumor stroma. Although the morphology of these cells did not appear to favor cell migration, we observed a dramatically rapid tumor growth phase after the initial slow growth phases when pure RFP+ cells were injected into mammary fat pad. This suggests that the fibroblast-like GFP+ cells might play a promotional role in tumor growth (19,20).
Imaging and visualization of drug effects on EMT
As presented above, the post-EMT GFP+ cells were either actively motile or able to promote tumor growth as fibroblast-like cells. Our results also point out that blood vessels are crucial to the spatial location and temporal migration of the EMT cells, suggesting that targeting blood vessel related factors might be beneficial in impeding EMT occurrence and restraining EMT cells’ behaviors. Blood vessels supply nutrients and growth factors that allow tumors to grow (22). A panel of growth factors, including TGF-β, EGF, FGF, hepatocyte growth factor (HGF), Insulin-like growth factor 1 (IGF-1), and platelet-derived growth factor (PDGF), was investigated in inducing the EMT-like morphological changes of the PyMT tumor-derived EMT cells; of these TGF-β and HGF provoked obvious loss of cell-cell contacts, which was not observed with other growth factors (16). Furthermore, we found that the HGF receptor c-Met (Fig 5a-c), but not other growth factor receptors (Supplementary Table 2), was highly expressed highly in EMT cells, and the high expression of c-Met in clinical breast tumors correlated with early metastasis (Fig S3a,d). In addition, the vascular endothelial growth factor receptor (VEGFR) coding genes Flt1 and Flt 4 were also elevated in patients with metastatic cancer (Fig S3b-c, e-f). We thus chose Cabozantinib (XL-184), a potent c-Met inhibitor, as well as an inhibitor of Flt1/4, to test whether our intravital EMT imaging platform could be useful in visualizing and evaluating the drug effects on EMT at the single cell level.
Fig 5. Imaging and quantification of Cabozantinib (XL-184) effects on EMT in vitro.

a-c. Western blot analysis of E-Cadherin and c-Met expression in the XL-184-treated RFP+ tumor cells. d-f. GFP imaging merged with bright field view of the XL-184-treated RFP+ tumor cells. g. GFP+ cells counting of the XL-184-treated RFP+ tumor cells. The TGF- β induced increase of GFP+ tumor cells was suppressed by XL-184 (p<0.001). h. Elongation of cell axial ratio induced by TGF-β was completely reversed by XL-184. i-k. Immunocytochemistry staining of E-Cadherin in the XL-184-treated tumor cells.
In vitro, treatment with Cabozantinib (XL-184) at 1 μM for 3 days impeded the RFP-to-GRP color switch of the cultured pure RFP+ cells and even inhibited more than 50% of the EMT occurrence induced by TGF-β (10ng/ml) (Fig 5d-f, g). Furthermore, morphological analysis of the residual GFP+ cells indicated that these cells retained more silent EMT phenotype with no membrane extensions and have the cell axis ratio of 2.83±0.15 in comparing with 6.01±0.86 for the GFP+ cells induced by TGF-β (p<0.001, Fig 5df, h), accompanied by the suppressed c-Met expression and elevated E-Cadherin expression (Fig 5a-c, i-k).
In vivo, when palpable tumor formed in the RFP+ cells implanted mice, 30mg/kg Cabozantinib (XL-184) was administered once daily for 5 days through intraperitoneal injection (23). Tumor growth was completely suppressed during the drug treatment, and the tumors grew consistently slower than the tumors in the vehicle group even after stopping the treatment (Fig 6a). At the end of 5-day treatment, primary tumor intravital imaging was performed on all mice. Because EMT may occur even before formation of palpable tumor and our EMT driven color switch is not reversible, there were still clusters of GFP+ cells found in the tumors. Strikingly, the GFP+ cells in the Cabozantinib (XL-184)–treated tumors were quite different from those in the vehicle-treated tumors, i.e., most of them were attached together in clusters and had similar axial ratios as the surrounding RFP+ tumor cells (Fig 6b). In addition, they rarely had membrane extensions and were static (Fig 6c), which complied with the silent EMT cell type. Further IHC staining demonstrated the epithelial phenotype of these clustered GFP+ cells (Fig 6d-f). Treatment with Cabozantinib (XL-184) has shown great efficacy in inhibiting metastasis on a breast cancer mouse model (24) and multiple cancer patients (25). Although the critical role of c-Met in EMT has been well characterized (26) and Cabozantinib (XL-184) inhibition of tumor progression has been extensively studied (27), our imaging data provided a visible and direct recapitulation at the single cell level for the first time. It proves the potential of the color-switching animal model system and intravital imaging platform in evaluating the drug's effects on EMT.
Fig 6. Imaging and quantification of Cabozantinib (XL-184) effects on EMT in vivo.
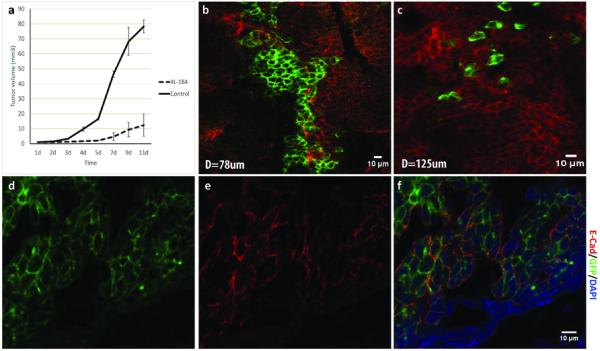
a. Tumor growth curve of the XL-184 treated mice (30mg/kg, once daily for 5 days, IP) and the PBS control group. b. A representative intravital multi-photon microscopy image of the XL-184 treated tumor. The GPF+ tumor cells were in clusters and had similar axial ratios as the surrounding RFP+ tumor cells, which was different from the sporadically distributed GFP+ cells in the control group (c). d-f. Immunohistochemistry staining of E-Cadherin in the XL-184 treated tumor. The E-Cadherin collocated with the clustered GFP+ tumor cells, indicating the epithelial phenotype of the clustered GFP+ cells.
Discussion
In this work, we report the visualization of tumor EMT at the single cell level by intravital two-photon microscopy on a newly developed EMT driven color switching breast cancer model. To our best knowledge, only a few strategies have been attempted to image the tumor EMT process in live cells; for example, the use of a fluorescence-based alternative splicing receptor (fibroblast growth factor receptor 2) (28-30), and microRNA-200a sequence-based molecular beacon (31). However, these techniques are affected by multiple variables and limited in their ability to monitor the rapid and dynamic EMT process in vivo. The combination of intravital two-photon microscopy and the MMTV-PyMT/Rosa26-RFP-GFP/Fsp1-Cre mouse tumor model allows the tracking of individual EMT cells in the tumor directly for hours, which has been a major challenge of EMT research over the past decade (3). One-time histological experiments provide snapshots but cannot track longitudinal phenomena, and most in vivo imaging techniques have resolutions insufficient to study single cells (9,32). Our platform offers several advantages, as it is not only minimally invasive but also sensitive to single cells, assesses the EMT occurrence throughout the whole tumor at all stages, differentiates between silent, migratory and fibroblast-like EMT cells, and makes it possible to examine the effects of drugs on the EMT process. In this way, we are able to comprehensively characterize the previously unknown EMT dynamics in tumor development.
Using the color-switching model and reported intravital microscopy imaging platform, we were able to capture the in vivo EMT occurrence in early tumor development. Furthermore, a large sub-population of the EMT cells resides in close proximity to stroma and blood vessels, and more than half of these cells exhibited tropism towards or along the blood vessels. These data may implicate an early tumor cell dissemination mediated by EMT-endowed motile characteristics. Recent clinical and experimental studies indicate that systemic tumor cell spread initiates in the early stages and small tumors in breast (33), and epidemiological analysis of more than 12,000 breast cancer patients also shows that metastasis may be initiated 5–7 years before diagnosis of the primary tumor (34). Furthermore, gene expression studies reveal that patients with poor prognosis can be identified before manifestation of metastasis (35), and a core EMT gene-expression signature has been identified in association with poor prognosis in aggressive breast cancer subtypes (36).
In addition, many other studies strongly support the role of EMT in the early steps of the metastatic cascade, i.e., EMT program not only allows carcinoma cells to dissociate from each other but also provides them the ability to degrade m for single-cell invasion to initiate the metastatic cascade (3). Since the linkage between EMT and early metastasis has shown up prominently, the model and imaging platform we developed can facilitate the design and evaluation of effective therapies targeting cancer metastasis. For example, molecular targets on those early EMT cells can be identified, and drug effects on the early EMT process can be evaluated as we showed with Cabozantinib (XL-184) as a prove-of-concept example.
Through quantitative image analysis, we were able to define the heterogeneous sub-populations of EMT cells at different tumor stages, i.e., silent EMT cells, fibroblast-like EMT cells, and migratory EMT cells. The migratory EMT cells were characterized by losing cell polarity, acting like amoeba, and migrating towards stimuli, which may mainly contribute to metastasis (21). The fibroblast-like EMT cells, which may develop to cancer associated fibroblasts, kept part of the polarity and attached tightly to the ECM without much movement (20). We examined a progressive increase of such fibroblast-like EMT cells across the small, middle, and large tumors, and in parallel, the tumor growth showed a two-phase curve with one slow growth phase and one rapid growth phase. These correlation results suggest that the fibroblast-like EMT cells may promote the tumor progression.
In addition, we found a significant percentage of EMT cells were silent subtype (~20%), however, we did not observe any changeovers between the silent and migratory EMT cells within the current imaging period (~6h). We observed that most of the migratory and silent EMT cells reside within a restricted distance, but all are far from the clusters of fibroblast-like EMT cells. We postulate that the migratory and silent type of EMT cells might either have paracrine interplays between them or have autocrine signaling via secreted factors to maintain their equilibration in the tumor niche that gives rise to metastasis, and the fibroblast-like EMT cells might have intrinsic molecular characteristics that predispose them to promote tumor growth. In our study, HGF and its receptor c-Met were identified prominently in the EMT cells. Treatment with Cabozantinib (XL-184) in vivo pushed the emergence of more silent EMT cells rather than migratory and fibroblast-like EMT cells. Cabozantinib (XL-184) has shown having great efficacy in inhibiting metastasis and reducing tumor growth (24), indicating that HGF and c-Met signaling are critical in both processes. More importantly, this proof-of-concept study of Cabozantinib (XL-184) on the EMT heterogeneity manifests the potential of the single-cell intravital EMT imaging platform in discovering and evaluating therapies targeting cancer metastasis.
Imaging primary tumor EMT may not elucidate metastasis comprehensively, as mesenchymalepithelial transition (MET) may be required in distant organs to establish epithelial metastatic colonization (3,5). However, the technological platform of EMT color switching model and intravital two-photon microscopy imaging presented in this paper provides a powerful tool to characterize the single cell MET, i.e., the spatiotemporal localization of the metastatic cells using the irreversible color of “M” cells, the delineation of single cell morphological dynamics and the heterogeneity of MET cells, and the evaluation of drug effects on MET in in vivo tumor micro-environments.
Supplementary Material
Acknowledgments
All microscopic imaging studies were conducted at Houston Methodist Research Institute's Advanced Cellular and Tissue Microscope Core Facility.
Grant support
This research was funded by NIH U54 CA149196 and a John S. Dunn Research Foundation grant (S.T.C. Wong), and an NIH U54 CA149196 pilot project grant (H. Zhao).
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosure.
Authors’ Contributions
Conception and design: Z. Zhao, H. Zhao, S.T. Wong
Development of methodology: Z. Zhao, X.P. Zhu, K. Cui, J. Mancuso, R. Federley, H. Zhao
Acquisition of data (provided animals, provided facilities, etc.): Z. Zhao, X.P. Zhu, K. Cui, V. Mittal, D.C. Gao, G.J. Teng, S.T. Wong
Analysis and interpretation of data (e.g., statistical analysis): Z. Zhao, X.P. Zhu, K. Cui, H. Zhao, S. T. Wong
Writing, review, and/or revision of the manuscript: Z. Zhao, J. Mancuso, R. Federley, H. Zhao, S.T. Wong
Administrative, technical, or material support: G.J. Teng, S.T. Wong
Study supervision: H. Zhao, S.T. Wong
Reference
- 1.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119(6):1420–28. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871–90. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 3.Tsai JH, Yang J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev. 2013;27(20):2192–206. doi: 10.1101/gad.225334.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nieto MA. Epithelial plasticity: a common theme in embryonic and cancer cells. Science. 2013;342(6159):1234850. doi: 10.1126/science.1234850. [DOI] [PubMed] [Google Scholar]
- 5.Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2(6):442–54. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 6.Fidler IJ. The pathogenesis of cancer metastasis: the 'seed and soil' hypothesis revisited. Nat Rev Cancer. 2003;3(6):453–8. doi: 10.1038/nrc1098. [DOI] [PubMed] [Google Scholar]
- 7.Santisteban M, Reiman JM, Asiedu NK, Behrens MD, Nassar A, Kalli KR, et al. Immune-Induced Epithelial to Mesenchymal Transition In vivo Generates Breast Cancer Stem Cells. Cancer Res. 2009;69(7):2887–95. doi: 10.1158/0008-5472.CAN-08-3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xie M, Zhang L, He CS, Xu F, Liu JL, Hu ZH, et al. Activation of notch-1 enhances epithelial mesenchymal transition in gefitinib-acquired resistant lung cancer cells. J Cell Biochem. 2012;113(5):1501–13. doi: 10.1002/jcb.24019. [DOI] [PubMed] [Google Scholar]
- 9.Wessels JT, Busse AC, Mahrt J, Dullin C, Grabbe E, Mueller GA. In vivo imaging in experimental preclinical tumor research--a review. Cytometry A. 2007;71(8):542–9. doi: 10.1002/cyto.a.20419. [DOI] [PubMed] [Google Scholar]
- 10.Niesner RA, Hauser AE. Recent advances in dynamic intravital multi-photon microscopy. Cytometry A. 2011;79(10):789–98. doi: 10.1002/cyto.a.21140. [DOI] [PubMed] [Google Scholar]
- 11.Tanaka K, Toiyama Y, Inoue Y, Uchida K, Araki T, Mohri Y, et al. Intravital imaging of gastrointestinal diseases in preclinical models using two-photon laser scanning microscopy. Surg Today. 2013;43(2):123–29. doi: 10.1007/s00595-012-0283-9. [DOI] [PubMed] [Google Scholar]
- 12.Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong ST, et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature. 2015;527(7579):472–6. doi: 10.1038/nature15748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yin Z, Sadok A, Sailem H, McCarthy A, Xia XF, Li FH, et al. A screen for morphological complexity identifies regulators of switch-like transitions between discrete cell shapes. Nat Cell Biol. 2013;15(7):860. doi: 10.1038/ncb2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xue CS, Plieth D, Venkov C, Xu C, Neilson EG. The gatekeeper effect of epithelial-mesenchymal transition regulates the frequency of breast cancer metastasis. Cancer Res. 2003;63(12):3386–94. [PubMed] [Google Scholar]
- 15.Okada H, Danoff TM, Kalluri R, Neilson EG. Early role of Fsp1 in epithelial-mesenchymal transformation. American Journal of Physiology-Renal Physiology. 1997;273(4):F563–F74. doi: 10.1152/ajprenal.1997.273.4.F563. [DOI] [PubMed] [Google Scholar]
- 16.Waldmeier L, Meyer-Schaller N, Diepenbruck M, Christofori G. Py2T murine breast cancer cells, a versatile model of TGFbeta-induced EMT in vitro and in vivo. PLoS ONE. 2012;7(11):e48651. doi: 10.1371/journal.pone.0048651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dumont N, Wilson MB, Crawford YG, Reynolds PA, Sigaroudinia M, Tlsty TD. Sustained induction of epithelial to mesenchymal transition activates DNA methylation of genes silenced in basal-like breast cancers. Proc Natl Acad Sci U S A. 2008;105(39):14867–72. doi: 10.1073/pnas.0807146105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Patsialou A, Bravo-Cordero JJ, Wang Y, Entenberg D, Liu H, Clarke M, et al. Intravital multiphoton imaging reveals multicellular streaming as a crucial component of in vivo cell migration in human breast tumors. Intravital. 2013;2(2):e25294. doi: 10.4161/intv.25294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cirri P, Chiarugi P. Cancer associated fibroblasts: the dark side of the coin. Am J Cancer Res. 2011;1(4):482–97. [PMC free article] [PubMed] [Google Scholar]
- 20.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nature Reviews Cancer. 2006;6(5):392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- 21.Condeelis J, Segall JE. Intravital imaging of cell movement in tumours. Nat Rev Cancer. 2003;3(12):921–30. doi: 10.1038/nrc1231. [DOI] [PubMed] [Google Scholar]
- 22.Vannucci L. Stroma as an Active Player in the Development of the Tumor Microenvironment. Cancer Microenviron. 2014:1–8. doi: 10.1007/s12307-014-0150-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.You WK, Sennino B, Williamson CW, Falcon B, Hashizume H, Yao LC, et al. VEGF and c-Met Blockade Amplify Angiogenesis Inhibition in Pancreatic Islet Cancer. Cancer Res. 2011;71(14):4758–68. doi: 10.1158/0008-5472.CAN-10-2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yakes FM, Chen J, Tan J, Yamaguchi K, Shi Y, Yu P, et al. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol Cancer Ther. 2011;10(12):2298–308. doi: 10.1158/1535-7163.MCT-11-0264. [DOI] [PubMed] [Google Scholar]
- 25.Castellone MD, Carlomagno F, Salvatore G, Santoro M. Receptor tyrosine kinase inhibitors in thyroid cancer. Best Practice & Research Clinical Endocrinology & Metabolism. 2008;22(6):1023–38. doi: 10.1016/j.beem.2008.09.012. [DOI] [PubMed] [Google Scholar]
- 26.Mazzone M, Comoglio PM. The Met pathway: master switch and drug target in cancer progression. FASEB J. 2006;20(10):1611–21. doi: 10.1096/fj.06-5947rev. [DOI] [PubMed] [Google Scholar]
- 27.Sennino B, Ishiguro-Oonuma T, Wei Y, Naylor RM, Williamson CW, Bhagwandin V, et al. Suppression of Tumor Invasion and Metastasis by Concurrent Inhibition of c-Met and VEGF Signaling in Pancreatic Neuroendocrine Tumors. Cancer Discovery. 2012;2(3):270–87. doi: 10.1158/2159-8290.CD-11-0240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oltean S, Sorg BS, Albrecht T, Bonano VI, Brazas RM, Dewhirst MW, et al. Alternative inclusion of fibroblast growth factor receptor 2 exon IIIc in Dunning prostate tumors reveals unexpected epithelial mesenchymal plasticity. Proc Natl Acad Sci U S A. 2006;103(38):14116–21. doi: 10.1073/pnas.0603090103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oltean S, Febbo PG, Garcia-Blanco MA. Dunning rat prostate adenocarcinomas and alternative splicing reporters: powerful tools to study epithelial plasticity in prostate tumors in vivo. Clin Exp Metastasis. 2008;25(6):611–19. doi: 10.1007/s10585-008-9186-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Somarelli JA, Schaeffer D, Bosma R, Bonano VI, Sohn JW, Kemeny G, et al. Fluorescence-based alternative splicing reporters for the study of epithelial plasticity in vivo. Rna-a Publication of the Rna Society. 2013;19(1):116–27. doi: 10.1261/rna.035097.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Choi Y, Kim HS, Woo J, Hwang EH, Cho KW, Kim S, et al. Real-Time Imaging of the Epithelial- Mesenchymal Transition Using microRNA-200a Sequence-Based Molecular Beacon-Conjugated Magnetic Nanoparticles. PLoS ONE. 2014:9(7). doi: 10.1371/journal.pone.0102164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nature Reviews Cancer. 2002;2(8):563–72. doi: 10.1038/nrc865. [DOI] [PubMed] [Google Scholar]
- 33.Husemann Y, Geigl JB, Schubert F, Musiani P, Meyer M, Burghart E, et al. Systemic spread is an early step in breast cancer. Cancer Cell. 2008;13(1):58–68. doi: 10.1016/j.ccr.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 34.Engel J, Eckel R, Kerr J, Schmidt M, Furstenberger G, Richter R, et al. The process of metastasisation for breast cancer. Eur J Cancer. 2003;39(12):1794–806. doi: 10.1016/s0959-8049(03)00422-2. [DOI] [PubMed] [Google Scholar]
- 35.van't Veer LJ, Dai HY, van de Vijver MJ, He YDD, Hart AAM, Mao M, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415(6871):530–36. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- 36.Taube JH, Herschkowitz JI, Komurov K, Zhou AY, Gupta S, Yang J, et al. Core epithelial-to mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc Natl Acad Sci U S A. 2010;107(35):15449–54. doi: 10.1073/pnas.1004900107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.