

Abstract
Sprouty (SPRY) appears to act as a tumor suppressor in cancer, whereas we reported that SPRY2 functions as a putative oncogene in colorectal cancer (CRC) [Oncogene, 2010, 29: 5241–5253]. In general, various studies established inhibition of cell proliferation by SPRY in cancer. The mechanisms by which SPRY regulates cell proliferation in CRC are investigated. We demonstrate, for the first time, suppression of SPRY2 augmented EGF‐dependent oncogenic signaling, however, surprisingly decreased cell proliferation in colon cancer cells. Our data suggest that cell cycle inhibitor p21WAF1/CIP1 transcriptional activity being regulated by SPRY2. Indeed, suppression of SPRY2 significantly increased p21WAF1/CIP1 mRNA and protein expression as well as p21WAF1/CIP1 promoter activity. Conversely, overexpressing SPRY2 triggered a decrease in p21WAF1/CIP1 promoter activity. Concurrent down‐regulation of both SPRY1 and SPRY2 also increased p21WAF1/CIP1 expression in colon cancer cells. Increased nuclear localization of p21WAF1/CIP1 in SPRY2 downregulated colon cancer cells may explain the inhibition of cell proliferation in colon cancer cells. Underscoring the biological relevance of these findings in SPRY1 and SPRY2 mutant mouse, recombination of floxed SPRY1 and SPRY2 alleles in mouse embryonic fibroblasts (MEFs) resulted in increased expression and nuclear localization of p21WAF1/CIP1 and decreased cell proliferation. In CRC, the relationship of SPRY with p21 may provide unique strategies for cancer prevention and treatment. © 2015 The Authors. Molecular Carcinogenesis published by Wiley Periodicals, Inc.
Keywords: sprouty, colon, cancer, p21, mouse, MEF
INTRODUCTION
Sprouty (SPRY), an intracellular regulator of receptor tyrosine kinase (RTK) signaling, regulates growth, differentiation, and tumorigenesis. SPRY was initially identified as an antagonist of fibroblast growth factor (FGF) and epidermal growth factor (EGF) signaling in Drosophila 1. Four human homologs of SPRY (SPRY 1–4) have been recognized 1, 2. SPRY2 appears to be universally expressed whereas other family members show organ and tissue specificity 3. Experimental evidence demonstrates that SPRY specifically inhibits activation of extracellular regulated kinase (ERK) in response to growth factors 4, 5, 6, 7, 8. In cell and context‐dependent manner ERK activation is not always inhibited by SPRY. Surprisingly, SPRY1 and SPRY2 not only failed to suppress EGF induced Mitogen Activated Protein Kinase (MAPK) activation but enhanced its activation 9, 10, 11. Nonetheless, aberrant activation of MAPK pathway and deregulation of SPRY occurs in a variety of diseases, including neoplasia.
Expression of SPRY1 and SPRY2 is reduced in the breast, prostate, lung, and liver carcinoma suggesting a tumor suppressor role. Matched pairs of normal and cancer tissues revealed that SPRY1 and SPRY2 were consistently down‐regulated in breast cancer 12. MCF‐7 breast cancer cells proliferated faster in vitro when transfected with dominant‐negative mutant of SPRY2 and formed bigger tumors in mice. Further, low expression of SPRY2 was associated with elevated levels of EGFR2 (HER2) expression and SPRY2 was shown to act synergistically with the HER2 targeting drug trastuzumab to reduce cancer cell viability 13. Loss of SPRY2, an early event in prostate carcinogenesis, is compensated by nuclear PTEN‐mediated growth arrest. However, concomitant inactivation of PTEN and other tumor suppressor genes may lead to metastatic disease 14. Studies in non‐small cell lung cancer (NSCLC) demonstrated that SPRY2 down‐regulation contributes to tumorigenesis via ERK‐dependent and ‐independent mechanisms 15. Furthermore, loss of SPRY2 increased the tumor burden in lungs with oncogenic KRAS mutation 16 and it was suggested that tumor suppression by SPRY2 could involve targets downstream of KRAS 17. A consistent down‐regulation of SPRY2 in hepatocellular carcinoma (HCC) was also noted. SPRY2 overexpression suppressed hepatocyte growth factor (HGF)‐induced ERK and AKT‐dependent proliferation whereas loss of SPRY2 potentiated c‐Met signaling 18. Role of SPRY2 in colorectal cancer (CRC) is still unclear. We demonstrated, for the first time, increased SPRY2 protein expression in human colonic tumors 19. Contrary to our report, decreased SPRY2 mRNA transcripts were also noted in the intestinal tumors 20. However, in general, SPRY2 expression is higher in CRC tumors than in other cancers 21. In CRC, upregulation of SPRY2 in undifferentiated high‐grade tumors, at the invasive front of low‐grade tumors and in KRAS mutant tumors has been demonstrated 22, 23. In addition, transcriptional regulation of SPRY2 promoter by Wnt/β‐catenin and FOXO3a genes may suggest an oncogenic role of SPRY2 in CRC 24.
Expression of SPRY1 and SPRY2 is reduced in the breast, prostate, lung, and liver carcinoma suggesting a tumor suppressor role. Matched pairs of normal and cancer tissues revealed that SPRY1 and SPRY2 were consistently down‐regulated in breast cancer 12. MCF‐7 breast cancer cells proliferated faster in vitro when transfected with dominant‐negative mutant of SPRY2 and formed bigger tumors in mice. Further, low expression of SPRY2 was associated with elevated levels of EGFR2 (HER2) expression and SPRY2 was shown to act synergistically with the HER2 targeting drug trastuzumab to reduce cancer cell viability 13. Loss of SPRY2, an early event in prostate carcinogenesis, is compensated by nuclear PTEN‐mediated growth arrest. However, concomitant inactivation of PTEN and other tumor suppressor genes may lead to metastatic disease 14. Studies in non‐small cell lung cancer (NSCLC) demonstrated that SPRY2 down‐regulation contributes to tumorigenesis via ERK‐dependent and ‐independent mechanisms 15. Furthermore, loss of SPRY2 increased the tumor burden in lungs with oncogenic KRAS mutation 16 and it was suggested that tumor suppression by SPRY2 could involve targets downstream of KRAS 17. A consistent down‐regulation of SPRY2 in hepatocellular carcinoma (HCC) was also noted. SPRY2 overexpression suppressed hepatocyte growth factor (HGF)‐induced ERK and AKT‐dependent proliferation whereas loss of SPRY2 potentiated c‐Met signaling 18. Role of SPRY2 in colorectal cancer (CRC) is still unclear. We demonstrated, for the first time, increased SPRY2 protein expression in human colonic tumors 19. Contrary to our report, decreased SPRY2 mRNA transcripts were also noted in the intestinal tumors 20. However, in general, SPRY2 expression is higher in CRC tumors than in other cancers 21. In CRC, upregulation of SPRY2 in undifferentiated high‐grade tumors, at the invasive front of low‐grade tumors and in KRAS mutant tumors has been demonstrated 22, 23. In addition, transcriptional regulation of SPRY2 promoter by Wnt/β‐catenin and FOXO3a genes may suggest an oncogenic role of SPRY2 in CRC 24.
SPRY proteins are generally considered to be inhibitors of EGF and FGF signaling via Ras‐MAPK cascade. Several studies have challenged this paradigm and agonistic effect of SPRY in RTK signaling is demonstrated due to interaction of SPRY with c‐CBL that prevents c‐CBL mediated downregulation of EGFR and thus results in net increase in signaling 25. Further, in some instances, it remains unclear why SPRY2 increases EGF signaling but downregulates FGF signaling, as in both systems c‐CBL mediates growth factor receptor degradation 25. To study the effect of SPRY2 downregulation on EGFR signaling and cell proliferation in CRC, we have utilized Caco‐2 colon cancer cells, which contain high levels of endogenous EGFR, and FGFR expression. Results demonstrate that suppression of SPRY2 has no effect on EGFR expression but augments EGFR dependent MAPK activation confirming the generalized inhibitory role of SPRY2 on EGFR signaling. However, we demonstrate, for the first time, that EGF‐dependent activation of ERK, and AKT signaling cascades are insufficient to drive cancer cell proliferation in the absence of SPRY2. Suppression of SPRY2 in colon cancer cells upregulates p21WAF1/CIP1 (p21) expression. Transcriptional activation of p21 gene in SPRY2 down‐regulated colon cancer cells may account for upregulation of p21 expression and inhibition of cell proliferation. In a murine model, deletion of Spry1−/− and Spry2−/− resulted in increased p21 expression in mouse embryonic fibroblasts (MEFs) and diminished EGF‐dependent cell proliferation. Together, this study indicates that the relationship of SPRY with p21 may provide unique strategies for cancer prevention and treatment.
MATERIALS AND METHODS
Antibodies and Molecular Reagents
Antibodies to Sprouty2 and Sprouty1 were obtained from Sigma and Origene, respectively. Antibodies for Erk, pErk, AKT, pAKT, EGFR, pEGFR, pMet, P21, and β‐actin, were obtained from Cell Signaling. Antibodies for Erb, pErb were purchased from Santa Curz. Antibody for Met was obtained from Abcam. Transfection reagent lipofectamine 3000 was purchased from Invitrogen. QRT‐PCR reagents kit was obtained from Roche (LIGHTCYCLER 480 SYBR GREEN). Human SPRY2 primers were ordered from Qiagen. Human SPRY1, and human P21, mouse SPRY2, mouse SPRY1, and mouse P21 primers were purchased from RealtimePrimer.com. Sprouty2 siRNA and non‐silencing RNA were obtained from SantaCruz. Sprouty1 siRNA and non‐silencing RNA were obtained from Origene. Duo‐Luciferase reporter assay reagents were obtained from Promega. For confocal microscopy, rabbit anti‐p21 and mouse anti‐p21 were obtained from Abcam. Secondary antibodies conjugated with fluorescent dye Alexa Fluor 488 goat anti‐rabbit, anti‐mouse, ProlongR Diamond Antifade Mountant with DAPI, and normal goat serum were obtained from Invitrogen. pGL2‐p21 promoter was purchased from Addgene.
Cell Lines and Cell Culture
Human colon cancer cell lines Caco‐2, SW480 and SW620 were purchased from the American Type Culture Collection (ATCC) and maintained in a humidified chamber with 5% CO2 at 37°C as described by the ATCC.
Spry1 and Spry2 Mutant Mice
Mice carrying Spry1flox/flox 26, Spry2flox/flox 27, and ROSA26lacZ/ROSA26lacZ 28 alleles were crossed to B6.Cg‐Tg (CAG‐cre/Esr1)5Amc/J mice (CAAG‐CreERTM, JAX stock number 004453) 29 in which a ubiquitously‐expressed Cre gene was fused to a tamoxifen‐inducible mutant of the estrogen receptor. E14.5 embryos were collected from the following genetic cross: CAAG‐CreERTM/+; Spry1flox/flox; Spry2 flox/flox X Spry1 flox/flox; Spry2 flox/flox; ROSA26lacZ/ROSA26lacZ. Head and viscera were from individual embryos. Remaining tissue was rinsed with PBS, minced, and digested with trypsin/versene by stirring with glass beads for 30 min at 37°C. Digested cells were cultured in MEF culture medium (Dulbecco's modified Eagle's medium with 10% FBS, 0.1% NEAA, 2 mM L‐glutamine, 1 mM sodium pyruvate, 50U/ml penicillin and 50 µg/ml streptomycin). Embryos and MEF lines were genotyped to determine the presence of the CAAG‐CreERTM allele and to confirm homozygosity of the Spry1flox and Spry2flox alleles. All animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC).
Cancer Cell Transfection
Caco‐2 cancer cells (0.5 × 106) were transfected by reverse transfection methods with siSPRY1, and siSPRY2 (50 nM final concentration) for 72 h using Lipofectamine 3000 as described by the manufacturer (Invitrogen, Grand Island, NY). Caco‐2 cells were also transiently transfected with 1 µg of the pHM6 empty vector or pHM6‐SPRY2 19.
RNA Isolation
Total RNA was extracted with RNA isolation kit (Exiqon, Woburn, MA) according to the manufacturer's instructions and further purified using Turbo Kit (Ambion, Carlsbad, CA). RNA purity was assessed by measuring absorptions at 260 and 280 nm and samples that had A260/A280 ratio of 1.9–2.1 were considered acceptable.
Real Time PCR
RNA was reverse transcribed with a cDNA kit (Bio‐Rad, Hercules, CA) into complementary DNA (cDNA) by utilizing specific qRT‐PCR primers in 20 μl total volume. cDNA was used as template for quantitative PCR in triplicate using SYBR green qPCR Mix (Roche, Indianapolis, IN). Amplifications were carried out in triplicate on MicroAmp optical 96‐well microliter plates (Roche). Thermal cycling conditions were as follows: 95°C for 5 min, followed by 45 cycles as follows: denaturation step at 95°C for 10 s, annealing step at 56°C for 10 s and extension step at 72°C for 10 s. GAPDH was used as an internal control for other genes. The ΔΔCT method was used to compare the relative expression levels between treatments. The final PCR results were expressed as the relative expression compared to individual control sample in each assay.
Cell Immunolabeling and Confocal Microscopy
Transfected cancer cells (0.5 × 104) were seeded onto collagen (10 μg/ml) coated glass coverslips. Cells were grown for additional 24 h and fixed in ice‐cold methanol for 15 min. Cells were permeabilized in 0.1% Tixton‐100 in PBS for 3 min, blocked with 10% goat serum for 1 h, incubated overnight at 4°C with rabbit anti‐p21 or mouse anti‐p21 (1:200) followed by incubation with secondary antibodies (1:400) conjugated with Alexa Fluor 488. The slides were counterstained and mounted with 4′‐6‐diamidino‐2‐phenylindoe (DAPI) and mounting medium (Vector Labs). Confocal microscopy using a Leica TCS SP8 confocal laser scanning microscope was performed as described earlier 30.
Cell Proliferation
Caco‐2 cells were transfected with non‐slencing siRNA or siRNA‐SPRY2. Transfected cells were seeded at a concentration of 5–10 × 103 cells/well in a ninety six well plate for 24 h. Cells were serum starved overnight in a 2% serum containing medium and treated with EGF (10ng/ml) for 48 h. Cells were then incubated with 100 μg of MTT per well for 4 h in a humidified 5% CO2 incubator. The MTT formazan was dissolved in DMSO, and the absorbance was measured at 490 nm in a microplate reader.
Cell Cycle Analysis
For cell cycle analysis transfected cells (1 × 105 cells/well) were plated in six well plates for 24 h. Cells were synchronized by serum deprivation and then stimulated with EGF (10 ng/ml) for the desired time points. Cells were trypsinized, fixed with 70% ethanol overnight (−20°C) and then stained with propidium iodide solution (10 µg/ml) containing RNase A (10 U/ml) at 4°C for 30 min. The distribution of cells in the different phases of cell cycle was assessed by flow cytometry (BD FACS Canto) using FlowJo software for data analysis 19.
Luciferase Promoter Assay
For the p21‐Luc reporter assay, transfected Caco‐2 cells (1.5 × 105 cells/well) were seeded in 12 well plates for 48 h. Cells were then transfected with 1 µg empty vector (pGL2‐Luc) or p21 promoter‐Luc. A Renilla luciferase vector (pRL‐TK) served as an internal control and was included in all samples. After 48 h cells were lysed and the firefly and renilla luminescence were measured sequentially using the dual luciferase assay kit (Promega, Madison, WI). Luminescence was measured by a multimode microplate reader (Turner model 9100‐002, USA). The firefly values were divided by the renilla values, and the data are expressed as the ratio of firefly to renilla luciferase activity. Independent triplicate experiments were performed for each plasmid.
Western Blotting
Total cell lysates were prepared and western blotting was performed as described previously 31. Proteins (20–40 μg) were separated by SDS–PAGE on 4–10% resolving polyacrylamide gradient gels (Bio‐Rad, Hercules, CA) and electroblotted to PVDF membranes. The membranes were incubated with specific primary antibodies, followed by 1 h incubation with appropriate peroxidase‐coupled secondary antibodies. Signals were detected using a SuperSignal chemiluminescence kit (Thermo Fisher, Rocksord, IL) and images were captured using ImageLab software (Bio‐Rad). Band intensity was quantified using the Image J software. Separate aliquots were probed for β‐actin to assess loading. Protein levels were expressed as relative expression/ β‐actin (mean ± SD).
Statistical Analysis
The results are presented as the mean ± SD of three independent experiments. An un‐paired Student's t‐test or one‐way ANOVA was used to evaluate statistically significant differences in the experimental groups and the control group. P < 0.05 was considered statistically significant.
RESULTS
Increased EGF‐Dependent Signaling in SPRY2 Knocked‐Down (KD) Cells
Growth factors regulate Ras/Raf/MAPK and PI3K‐Akt dependent pathways. SPRY2, in general, has been shown to suppress receptor tyrosine kinase (RTK)‐dependent Ras/Raf/MAPK signaling in response to growth factors. Frequently observed Ras and Raf mutations in colonic tumors, down‐stream of RTK, maintain a sustained activation of oncogenic signaling. Hence, these mutations interfere with SPRY2 functions with respect to suppression of MAPK signaling. In this investigation, Caco‐2 cell lines were preferred over other CRC cell lines as they contain wild type KRAS, BRAF, PIK3CA, and PTEN, critical genes that are mutated in CRC. Caco‐2 cells were transiently transfected with SPRY2 siRNA or non‐silencing control RNA (Nsi RNA). Following serum starvation, cells were stimulated with EGF for the indicated times and examined for activation of RTKs including EGFR, HER2, and MET by phospho‐active antibodies. Further, RTK downstream signaling was also examined by utilizing phospho‐active ERKs and AKT antibodies. EGF treatment of Nsi cells resulted in a mild activation of EGFR, HER2, and MET receptors (Figure 1). However, SPRY2 suppression led to a robust and sustained activation of RTK receptors as compared to Nsi cells. As expected, an enhanced and prolonged activation of ERKs and Akt was also observed in EGF treated SPRY2 KD cells as compared to Nsi cells (Figure 1). These results clearly implicate that RTK activation, RTK downstream signaling and SPRY2 functions are intact in Caco‐2 colon cancer cells and, in general, SPRY2 suppresses EGF dependent RTK signaling.
Figure 1.

Silencing of SPRY2 augments EGF‐dependent signaling in Caco‐2 colon cancer cells. Caco‐2 cells were transiently transfected with non‐silencing control (Nsi) or SPRY2 siRNA (50 nM, 72 h). Cells were serum starved overnight, treated with EGF (10 ng/ml) for the indicated times and cell lysates were western blotted. Results represent a representative experiment of three independent experiments.
SPRY2 Repression Enhanced p21 Expression and Decreased Proliferation
We then hypothesized that amplified EGF‐dependent signaling in SPRY2 KD cells would result in increased cell proliferation. To our surprise, SPRY2 suppression significantly decreased basal cell proliferation in SPRY2 KD cells as compared to Nsi cells (Figure 2A). However, there was no significant difference in the rate of apoptosis (data not shown). In order to explain the inhibition of cell proliferation in SPRY2 KD cells, total RNA and protein from Nsi and SPRY2 KD cells were used to assess several cell cycle inhibitors. A significant increase in cyclin‐dependent kinase (CDK) inhibitor p21 mRNA transcripts was observed in SPRY2 KD cells as compared to Nsi cells (Figure 2B). Western immunoblotting revealed that p21 protein level is also increased in SPRY2 KD cells (Figure 2C). To assess whether p21 overexpression in SPRY2 KD cells is sustained in a physiological condition, we stimulated Nsi and SPRY2 KD cells with EGF. Results indicated that the expression of p21 remained elevated throughout the course of EGF treatment in SPRY2 KD cells (Figure 2D). These results indicate that p21 induction may account for inhibition of cell proliferation in EGF treated SPRY2 KD cells. In order to demonstrate that SPRY2 dependent p21 upregulation is not limited to only one colon cancer cell line, we utilized SW480 and SW620 colon cancer cells with different genetic backgrounds. More importantly, Caco‐2 cells that harbor wild type KRAS, BRAF, PIK3CA, and PTEN genes have mutated β‐catenin. SPRY2 suppression also increased p21 expression in SW480 and SW620 cells that contain wild type β‐catenin gene (Figure 2E).
Figure 2.
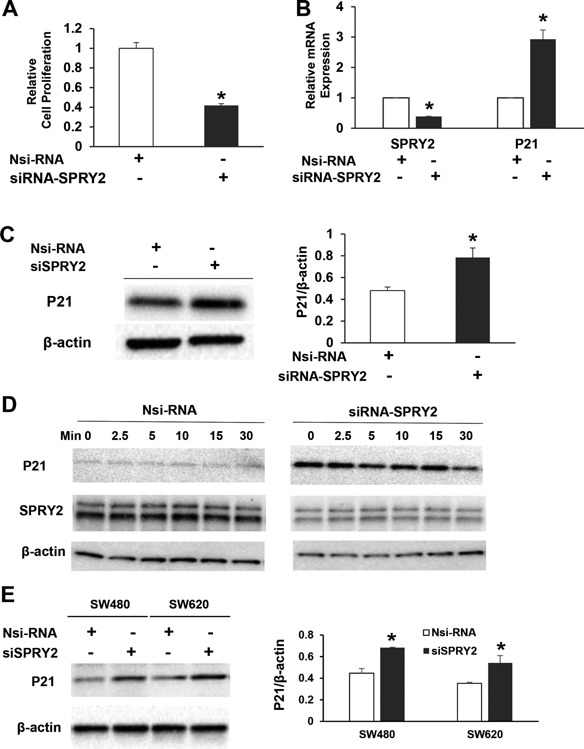
Silencing of SPRY2 upregulates p21 expression and inhibits cell proliferation. (A) Caco‐2 cells were transiently transfected with non‐silencing control (Nsi) or SPRY2 siRNA (50 nM, 72 h). Transfected cells (10 000/well) were then plated in a 96 well plate for 48 h and cell proliferation was assessed by MTT assay. Data represents means ± SD of three independent experiments, *P < 0.05, compared to Nsi cells. (B, C) Caco‐2 cells were transiently transfected with non‐silencing control (Nsi) or SPRY2 siRNA (50 nM, 72 h). p21 mRNA and protein expression was assessed by RTPCR and western blotting, respectively. Data represents means ± SD of three independent experiments, *P < 0.05, compared to Nsi cells. (D) Caco‐2 cells were transiently transfected with non‐silencing control (Nsi) or SPRY2 siRNA (50 nM, 72 h). Cells were serum starved overnight, treated with EGF (10 ng/ml) for the indicated times and cell lysates were western blotted for p21 expression. Results represent a representative experiment of three independent experiments. (E) SW480 and SW620 cells were transiently transfected with non‐silencing control (Nsi) or SPRY2 siRNA (50 nM, 72 h). p21 protein expression was assessed by western blotting. Data represents means ± SD of three independent experiments, *P < 0.05, compared to Nsi cells.
Suppression of p21 Reversed Inhibition of Proliferation in SPRY2 KD Cells
To establish the significance of upregulated p21 in the inhibition of cell proliferation, we investigated whether p21 downregulation will reverse the inhibitory effect of SPRY2 downregulation on cell proliferation. Caco‐2 cells were transfected with either SPRY2 siRNA alone or a mixture of SPRY2 and p21 siRNA for concomitant suppression of SPRY2 and p21. As expected, SPRY2 suppression resulted in inhibition of basal and EGF‐stimulated cell proliferation (Figure 3). However, concurrent suppression of both p21 and SPRY2 in Caco‐2 cells reversed the inhibition on cell proliferation that was triggered by SPRY2 suppression alone (Figure 3). These results, therefore, suggest that p21 expression is required for inhibition of cell proliferation in SPRY2 KD cells.
Figure 3.
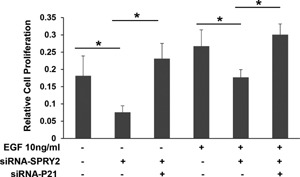
Silencing of p21 reverts the effect of SPRY2 downregulation on EGF‐induced cell proliferation Caco‐2 cells were transiently transfected with non‐silencing control (Nsi) or SPRY2 siRNA (50 nM, 72 h) or a mixture of SPRY2 siRNA and p21 siRNA (50 nM each, 72 h). Transfected cells (10 000/well) were plated in a ninety six well plate, serum starved overnight, treated with EGF (10 ng/ml) or vehicle for 48 h and cell proliferation was assessed by MTT assay. Data represents means ± SD of three independent experiments, *P < 0.05 (one way ANOVA).
Suppression of SPRY2 Delayed Cell Cycle Transition
To assess whether decreased cell proliferation reflected a change in cell cycle, we measured cell cycle phase distribution (Figure 4). Caco‐2 cells were transfected either with Nsi‐RNA or siRNA‐SPRY2 and serum starved for 24 h for cell synchronization. SPRY2 suppression noticeably increased cell population in G0/G1 phase when compared to Nsi‐RNA treated cells (At 0 h, 51% vs 61%). Serum starved cells were then treated with EGF and cell cycle phase distribution was measured at 4 and 16 h. After 4 h of EGF stimulation the cell populations in different phases of cell cycle were comparable in control and SPRY2 siRNA treated cells (Figure 4A and B). At a later time point, however, in Nsi‐RNA cells a rapid decrease in G0/G1 with concomitant increase in S and G2/M phases was observed (Figure 4A). In contrast, we observed a lingering and gradual decrease in G0/G1 in SPRY2 siRNA treated cells (Figure 4B). Data indicate that following EGF stimulation Nsi‐RNA treated control cells cycled faster as compared to SPRY2 downregulated cells. These results, therefore, demonstrate that delayed cell cycle transition and slow release from G1 phase could be responsible for decreased cell proliferation in SPRY2 downregulated cells.
Figure 4.

Silencing of SPRY2 delays cell cycle transition. Caco‐2 cells were transiently transfected with non‐silencing control (Nsi) or SPRY2 siRNA (50 nM, 72 h). Transfected cells were plated in six well plates for 24 h. Cells were synchronized by serum deprivation and then stimulated with EGF for indicated times. The percentage of cells in G0/G1, S and G2/M phases of cell cycle are shown within each histogram. Representative results from three independent experiments are shown.
SPRY2 Regulates p21 Gene Transcription
To further explore the relationship between SPRY2 and p21, we then asked whether SPRY2 could regulate p21 expression. Induction of p21 mRNA in SPRY2 KD cells suggested a possible role of SPRY2 in the transcriptional regulation of this gene. A major player in cell cycle control p21 is mainly regulated at transcriptional level 32. To determine whether SPRY2 regulates p21 by transcriptional mechanisms, we then examined the effect of SPRY2 downregulation on upregulation on the p21‐luciferase reporter activity. Caco‐2 cells were transiently transfected with SPRY2 cDNA expression plasmid or SPRY2 siRNA. As a control, cells were transfected either with empty vector or non‐silencing control RNA. SPRY2 overexpression or suppression was confirmed by RTPCR and western blotting (data not shown). Cells were then transiently transfected with a p21 promoter luciferase reporter construct containing 2.4 kb of upstream promoter sequence of the p21 gene. SPRY2 overexpression significantly decreased p21 promoter activity (Figure 5A) whereas SPRY2 suppression resulted in increased p21 promoter activity (Figure 5B). These results indicate that p21 promoter activity is inversely related to SPRY2 expression in Caco‐2 colon cancer cells. However, it remains unclear whether SPRY2 regulates p21 gene transcription through direct or indirect effects on its promoter. Further experiments are required to address this issue.
Figure 5.
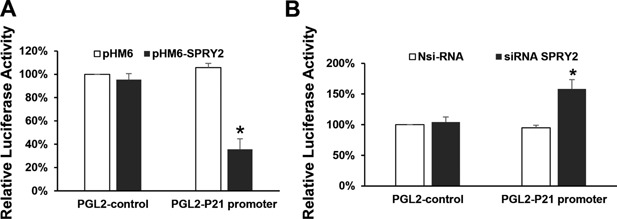
Transcriptional regulation of p21 promoter by SPRY2. Caco‐2 cells were seeded in a 12 well plate for 24 h. Cells were then transiently transfected with (A) empty vector pHM6 or pHM6‐SPRY2 (1 μg) (B) Nsi RNA or siRNA‐SPRY2 (50 nM) for 72 h. Cells were then transfected with luciferase reporter construct pGL2‐Luc containing 2.4 kb of p21 native promoter or empty vector (pGL2) for 48 h. As an internal standard a Renilla luciferase vector (pRL‐TK) was included in all samples. After 48 h, cell lysates were prepared and the Firefly and Renilla luminescence were measured sequentially using the dual luciferase assay kit. Data represents means ± SD of three independent experiments, *P < 0.05, compared to control vector(s) or non‐silencing RNA treated cells.
Increased Nuclear Localization of p21 in SPRY2 KD Colon Cancer Cells
Localization of p21 is of particular importance considering that nuclear accumulation of p21 is associated with growth inhibition whereas oncogenic activities of p21 are frequently associated with its cytoplasmic accumulation 33. The subcellular localization of p21 in Nsi and SPRY2 KD cells was studied to examine if there is a p21 nuclear shuttling after SPRY2 suppression. Confocal immunofluorescence microscopy established a significant upregulation and redistribution of p21 in SPRY2 KD cells when compared to Nsi cells. In Nsi control cells, p21 was present both in the cytoplasm as well as nucleus. A weak and diffuse cytoplasmic and nuclear p21 distribution was noted in Nsi control cells, however, strong immunoreactivity of p21 in the nuclei of SPRY2 KD cells indicate a tendency of p21 to accumulate in nucleus following SPRY2 suppression (Figure 6).
Figure 6.
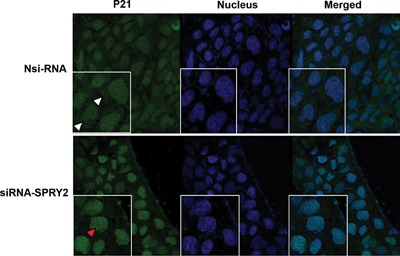
Increased nuclear localization of p21 in SPRY2 downregulated cells. Representative confocal images showing expression of p21 in Nsi RNA or SPRY2 siRNA (50 nM, 72 h) treated Caco‐2 cells. Cells were fixed with cold methanol, permeabilized, and blocked with normal goat serum. Sequentially, the cells were incubated with rabbit anti‐p21 and Alexa Fluor 488‐conjugated goat anti‐rabbit immunoglobulin. The cells were then stained with DAPI to label the nuclei. Images were obtained using a Leica TCS SP8 confocal laser scanning microscope. Cytoplasmic (white arrow head) and nuclear (red arrow head) localization of p21 is shown. All images were collected using identical microscope settings and magnification. The panels are representative of at least three independent experiments in which similar patterns were obtained for the indicated cells.
Concurrent Suppression of SPRY1 and SPRY2 Upregulated p21 Expression
SPRY1 has also been implicated with tumorigenesis in many human cancers 34. SPRY1 and SPRY2 share a unique highly conserved COOH‐terminal cysteine‐rich domain. However, the NH2‐terminal portion of SPRY protein is less conserved and exhibits only 25–37% identity among the different family members. These sequence differences could be responsible for the functional divergence among the SPRY proteins. We raised the question whether suppression of both SPRY1 and SPRY2 in colon cancer cells would exhibit a diverse effect on p21 expression. Concurrent suppression of SPRY1 and SPRY2 by siRNA also increased p21 mRNA (Figure 7A) and protein expression in Caco‐2 cells (Figure 7B).
Figure 7.
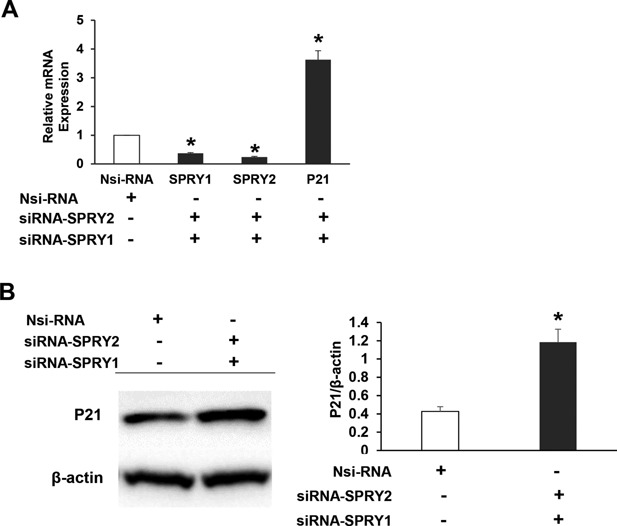
Concurrent SPRY1 and SPRY2 suppression in Caco‐2 cells upregulates p21 expression. Caco‐2 cells were transiently transfected with non‐silencing control (Nsi) or a mixture of SPRY1 and SPRY2 siRNA (50 nM each, 72 h). (A) SPRY1, SPRY2, and p21 mRNA, and (B) p21 protein expression was assessed by RTPCR and western blotting, respectively. Nsi arbitrary value 1 represents mRNA expression levels of SPRY1, SPRY2 and p21 in non‐targeted siRNA transfected cells. Data represents means ± SD of three independent experiments, *P < 0.05, compared to non‐silencing RNA treated cells.
Spry1−/−Spry2−/− MEFs Exhibited Increased Nuclear p21 Expression and Decreased Proliferation
We utilized Spry1 and Spry2 floxed mouse to evaluate biological significance of the loss of Spry1 and Spry2 in MEFs. CAAG‐CreERTM/+; Spry1flox/flox; Spry2 flox/flox; ROSA26lacZ/+ MEF lines [here onwards referred as Spry1f/f; Spry2f/f] were cultured and treated with 1 μM tamoxifen in MEF culture medium at 37°C for 48 h to generate CAAG‐CreERTM/+; Spry1−/−; Spry2 −/−; ROSA26lacZ/+ double mutant cells (here onwards referred as Spry1−/−; Spry2−/−). Addition of tamoxifen resulted in deletion of Spry1 and Spry2 that was confirmed by qRT PCR and lacZ expression. A complete recombination and greater than 90% reduction in Spry1 and Spry2 transcripts was noted after 48 h tamoxifen treatment. MEFs were further incubated without tamoxifen for the indicated time periods (48–120 h) and p21 mRNA contents were assessed. Spry1−/−; Spry2−/− MEFs demonstrated a significant increase in p21 mRNA transcripts (Figure 8A). Studies were extended to assess the effect of EGF on MEF proliferation. EGF treatment significantly increased proliferation in both Spry1f/f; Spry2f/f and Spry1−/−; Spry2−/− cells (Figure 8B). However, EGF‐treated Spry1−/−; Spry2−/− MEFs exhibited reduced proliferation (32%, P < 0.05) when compared to EGF‐treated Spry1f/f; Spry2f/f counterparts (Figure 8B). Furthermore, confocal immunofluorescence microscopy demonstrated a significant upregulation and nuclear localization of p21 in Spry1−/−; Spry2−/− MEFs when compared to Spry1f/f; Spry2f/f MEFs (Figure 8C).
Figure 8.
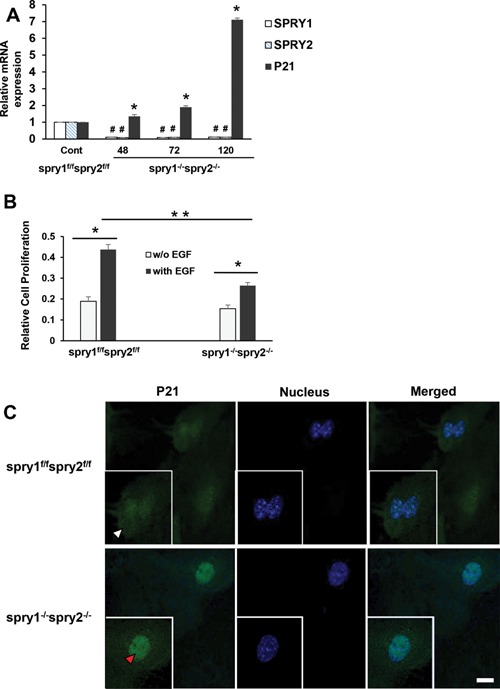
Spry1 and Spry2 deletion in mouse embryonic fibroblasts upregulates nuclear p21 expression and inhibits EGF‐dependent proliferation (A) Tamoxifen dependent recombination and a complete deletion of Spry1 and Spry2 in MEFs increases p21 expression. Spry1f/f; Spry2f/f and Spry1−/−; Spry2−/− MEFs were cultured for 48–120 h. Total RNA was extracted and mRNA transcripts of SPRY1, SPRY2, and p21 were assessed by RTPCR. Data represents means ± SD of three independent experiments, #,*P < 0.05, compared to control Spry1f/f; Spry2f/f MEFs (B) Deletion of Spry1 and Spry2 in MEFs reduces EGF‐dependent cell proliferation. Spry1f/f; Spry2f/f and Spry1−/−; Spry2−/− MEFs were plated and treated with vehicle or EGF (10 ng/ml) for 48 h. Cell proliferation was assessed by MTT assay. Groups compared; vehicle and EGF treated Spry1f/f; Spry2f/f cells, vehicle and EGF treated Spry1−/−; Spry2−/− cells and EGF treated Spry1−/−; Spry2−/− cells compared to EGF treated Spry1f/f; Spry2f/f cells. Data represents means ± SD of three independent experiments, *P < 0.05 (one way ANOVA). (C) Representative confocal images showing the expression of p21 in Spry1f/f; Spry2f/f and Spry1−/−; Spry2−/− MEFs. Cells were fixed with cold methanol, permeabilized, and blocked with normal goat serum. Sequentially, the cells were incubated with mouse anti‐p21 and Alexa Fluor 488‐conjugated goat anti‐mouse immunoglobulin. The cells were then stained with DAPI to label the nuclei. Images were obtained using a Leica TCS SP8 confocal laser scanning microscope. Cytoplasmic (white arrow head) and nuclear (red arrow head) localization of p21 is shown. All images were collected using identical microscope settings and magnification. The panels are representative of at least three independent experiments in which similar patterns were obtained for the indicated cells.
DISCUSSION
Mutational and non‐mutational perturbations account for increased tumor growth, survival, and resistance to known therapeutics. Dysregulation of RTK signaling has been found in a wide range of cancers. Therefore, RTKs have become an attractive therapeutic target. RTK pathways are regulated by endogenous antagonists that fine‐tune the signaling. SPRY proteins have been characterized as endogenous repressors of RTK signaling and thereby prevent tumorigenesis in breast, prostate, liver, and ovarian cancers 34. In the current investigation, SPRY2 suppression in colon cancer cells resulted in EGF‐dependent enhanced and sustained activation of EGFR, HER2, and MET receptors. Activation of HER2 and MET receptors following EGF treatment signifies EGFR heterodimerization with HER2 and MET that has been reported earlier in colon cancer cells 35, 36. Similarly, enhanced activation of ERKs and Akt following SPRY2 suppression represents a generalized role of SPRY2 in suppression of MAPKs and Akt downstream signaling. In response to a number of growth factor agonists, SPRY2 inhibited cell proliferation in HeLa cells, human umbilical vein endothelial cells, NIH3T3 cells and rat intestinal epithelial cells 34. However, in the present investigation, for the first time, SPRY2 suppression resulted in a significant decrease in EGF dependent cell proliferation in colon cancer cells. SPRY2 can enhance EGFR signaling by sequestering c‐CBL, an E3 ubiquitin ligase that otherwise interacts with the activated EGFR, promoting receptor degradation and signal termination. Therefore, resulting decrease in cell proliferation following SPRY2 suppression could be due to a reduction in EGFR expression and signaling. However, on the contrary, our studies demonstrated that SPRY2 downregulation resulted in increased EGFR signaling but no significant effect on total EGFR expression. Therefore, results indicate that SPRY2 inhibits signaling downstream of the EGFR receptors in Caco‐2 cells. Induction of cell cycle inhibitor p21 in SPRY2 KD cells could be responsible for the observed inhibition of cell proliferation. In this regard, we have earlier demonstrated that upregulation of SPRY2 by stable transfections in HCT116 colon cancer cells increased Cyclin D expression, an important positive regulator of G1/S phase cell cycle transition and cell proliferation 19. Hence, increased Cyclin D expression by SPRY2 upregulation and increased p21 expression by SPRY2 downregulation represents a tight control on cell proliferation by SPRY2. Nonetheless, our studies indicate that SPRY2 positively regulates cell proliferation in colon cancer cells. Induction of p21 is not limited to Caco‐2 cells as SW480 and SW620 colon cancer cell lines also demonstrated increased p21 expression following SPRY2 repression. Colon cancer cell lines are often classified on the basis of aneuploidy, MSI vs MSS and differentiation status, poorly differentiated vs well differentiated. However, a complete analysis of SPRY2 expression and its relationship with p21 expression is required in colon cancer cell lines with different genetic backgrounds in future investigations.
Role of p21 as a tumor suppressor is suggested as mice lacking p21 protein are more sensitive to tumorigenesis 37. However, this conclusion was later revised and it was shown that p21‐deficient mice are not characterized by elevated susceptibility to tumor formation 38. Cyclins, cyclin dependent kinases (CDKs) and CDK inhibitors regulate cell proliferation through cell cycle control. The induction of CDK inhibitor p21 leads to cell cycle arrest in G1, G2, or S‐phase depending on the cellular context. Interaction of p21 with CDK2/CyclinE and CDK2/CyclinA at N‐terminus and C‐terminus and interaction with PCNA on the C‐terminus are the important events in cell cycle inhibition 39. In the present investigation role of SPRY2 in p21 dependent inhibition and delayed G1 release requires further investigation. Loss of function of p21 has been linked to colonic tumor progression 40, 41. Inactivation of p21 protein tumor suppressor function is rarely due to mutations or deletions of the p21 gene, but commonly requires changes in transcriptional and post‐transcriptional mechanisms. The transcriptional regulation of p21 by p53‐dependent and ‐independent mechanisms has been extensively studied 42, 43. In the current investigation, transcriptional activation of p21 following SPRY2 suppression represents a p53 independent event as Caco‐2 cells harbor truncated nonfunctional p53 protein. Previous reports have indicated that enforced activation of ERKs lead to p21 induction in a p53 independent manner 44, 45. In the current investigation, suppression of SPRY2 significantly increased basal activation status of ERKs in Caco‐2 cells. Induction of p21 by activated ERKs in SPRY2 downregulated cells needs further investigation. Further, p21 is also regulated by post‐transcriptional mechanisms that include mRNA stabilization and translation in CRC 46, 47, 48, 49. Additionally, p21 can be regulated at post‐translational levels. In this regard, proteasome degradation of p21 can occur in both ubiquitin‐dependent and ‐independent mechanisms 50, 51. Whether such mechanisms are required for p21 regulation by SPRY2 is not clearly known, although there is precedence to it.
There is mounting evidence of the importance of post‐translational modifications in controlling p21 expression, localization and activity. The function of p21 protein depends on its localization in the cell as it plays different roles in nucleus and cytoplasm 52. Contrary to the contemporary view, oncogenic function of p21 in positive regulation of cell cycle is also reported if p21 is mainly expressed in the cytoplasm 53. Further, anti‐apoptotic activity of p21 protein is also manifested in the cytoplasm. Thus, both clinical studies and murine models suggest that tumor‐suppressing activity of p21 in the nucleus may reverse if localized in the cytoplasm 39. In the current study, increased nuclear localization of p21 with SPRY2 downregulation represents oncogenic function of SPRY2 in maintaining cytoplasmic p21. Further, rate of apoptosis of Caco‐2 cells following p21 induction was unaffected. Consequently, its paradoxical oncogenic activities are not materialized in this colon cancer model following SPRY2 downregulation. To support our notion, it is pertinent to mention here, that we have also demonstrated increased cell proliferation without affecting rate of apoptosis in SPRY2 stable transfectants of HCT116 cells 19. Nonetheless, our studies indicate that SPRY2 regulate cell proliferation without affecting basal rate of apoptosis in two diverse colon cancer cell culture models. In the current investigation, suppression of SPRY2 that causes a striking upregulation of p21 and inhibition of EGF‐induced cell proliferation specifies a unique but unreported mechanism of cell proliferation in colon cancer cells. Another adverse effect of increased expression of p21 in prostate, breast and cervical cancers is correlated with the augmented metastatic ability 54. Whether SPRY2 downregulation in these cancers would alter p21 dependent proliferative, apoptotic, and metastatic competency, is yet to be explored. Another deleterious effect of p21 is the induction of cellular senescence and escape from drug‐induced apoptosis with the use of chemotherapeutics in breast, prostate and colon cancers 55. Cancer cells were more susceptible to the treatment when p21 is inhibited. Studies, therefore, are needed to confirm the role of SPRY2 in cellular proliferation, senescence, and apoptosis with the use of chemotherapeutics in colon cancer cells.
Earlier we demonstrated increased SPRY2 expression in majority of adenocarcinomas 19 and others have shown that SPRY2 expression is indicative of poor prognosis in colon cancer patients 24. The biological and clinical implications of SPRY2 overexpression and its relationship with p21 expression in human colon cancers are poorly understood. A complete analysis of SPRY2 expression at various stages of tumor development with distinct mutational background is required to achieve a meaningful relationship with p21 expression. In this regard, p21 was detected in 75% adenomas and 31% carcinomas 56. In sporadic cases, a decrease in frequency of p21 expression accompanied adenoma development and progression to carcinomas. It is possible that SPRY suppression could be an effective strategy early on during colonic tumorigenesis to maintain a higher level of p21 expression. Furthermore, in most cases colon cancer tissues harbor mutant p53. Thus, p53 independent transcriptional upregulation of p21 following SPRY suppression further signifies an important mechanism of p21 regulation that could be exploited for inhibition of cancer progression from adenomas to carcinomas. To keep these results in perspective, molecular mechanisms of SPRY2 dependent but p53 independent induction of p21 in Caco‐2 cells may have implications in prevention/treatment of CRC, as inactivation of p53 is a frequent event in this malignancy. Several anti‐cancer agents such as histone deacetylase (HDAC) inhibitors and Statins exhibit profound anti‐proliferative capacity by inducing p21 57, 58. Further, p21 induction has been seen in colon cancer cell lines by chemopreventive 59 and chemotherapeutic anticancer agents 60 that interfere with RTK signaling. Thus, our observation that tweaking SPRY2 expression causes a striking upregulation of p21 and inhibition of proliferation in the presence of EGF may provide another potential mechanism for cancer prevention/treatment. Nonetheless, our study is an important first step in understanding how SPRY2 controls cell proliferation in CRC. However, the anti‐proliferative effect of SPRY2/p21 axis in CRC, though unique, does not preclude role for modulators other than p21. In conclusion, we demonstrated that SPRY2 suppression augments RTK dependent signaling but inhibits cell proliferation in CRC. This and our previous study lead us to put forth a supposition that unlike other human malignancies, in context dependent manner, SPRY2 may behave as a putative oncogene in CRC.
ACKNOWLEDGMENTS
This work was supported by VA Merit Award (Project1171590), RO3 Award (CA150081), University of Missouri Research Board Award to SK; RO1 Award (DC010387) to KS.
[The copyright line for this article was updated in February 2016 after original online publication.]
REFERENCES
- 1. Hacohen N, Kramer S, Sutherland D, et al. Sprouty encodes a novel antagonist of fgf signaling that patterns apical branching of the drosophila airways. Cell 1998; 92:253–263. [DOI] [PubMed] [Google Scholar]
- 2. Leeksma OC, Van Achterberg TA, Tsumura Y, et al. Human sprouty 4, a new Ras antagonist on 5q31, interacts with the dual specificity kinase tesk1. Eur J Biochem 2002; 269:2546–2556. [DOI] [PubMed] [Google Scholar]
- 3. Su AI, Cooke MP, Ching KA, et al. Large‐scale analysis of the human and mouse transcriptomes. Proc Natl Acad Sci USA 2002; 99:4465–4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wong ES, Fong CW, Lim J, et al. Sprouty2 attenuates epidermal growth factor receptor ubiquitylation and endocytosis, and consequently enhances Ras/ErK signalling. EMBO J 2002; 21:4796–4808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gross I, Bassit B, Benezra M, et al. Mammalian sprouty proteins inhibit cell growth and differentiation by preventing ras activation. J Biol Chem 2001; 276:46460–46468. [DOI] [PubMed] [Google Scholar]
- 6. Impagnatiello MA, Weitzer S, Gannon G, et al. Mammalian sprouty‐1 and ‐2 are membrane‐anchored phosphoprotein inhibitors of growth factor signaling in endothelial cells. J Cell Biol 2001; 152:1087–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gross I, Armant O, Benosman S, et al. Sprouty2 inhibits Bdnf‐induced signaling and modulates neuronal differentiation and survival. Cell Death Differ 2007; 14:1802–1812. [DOI] [PubMed] [Google Scholar]
- 8. Ishida M, Ichihara M, Mii S, et al. Sprouty2 regulates growth and differentiation of human neuroblastoma cells through Ret tyrosine kinase. Cancer Sci 2007; 98:815–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yigzaw Y, Cartin L, Pierre S, et al. The C terminus of sprouty is important for modulation of cellular migration and proliferation. J Biol Chem 2001; 276:22742–22747. [DOI] [PubMed] [Google Scholar]
- 10. Lim J, Yusoff P, Wong ES, et al. The cysteine‐rich sprouty translocation domain targets mitogen‐activated protein kinase inhibitory proteins to phosphatidylinositol 4,5‐bisphosphate in plasma membranes. Mol Cell Biol 2002; 22:7953–7966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rubin C, Litvak V, Medvedovsky H, et al. Sprouty fine‐tunes Egf signaling through interlinked positive and negative feedback loops. Curr Biol 2003; 13:297–307. [DOI] [PubMed] [Google Scholar]
- 12. Lo TL, Yusoff P, Fong CW, et al. The Ras/mitogen‐activated protein kinase pathway inhibitor and likely tumor suppressor proteins, sprouty 1 and sprouty 2 are deregulated in breast cancer. Cancer Res 2004; 64:6127–6136. [DOI] [PubMed] [Google Scholar]
- 13. Faratian D, Sims AH, Mullen P, et al. Sprouty 2 is an independent prognostic factor in breast cancer and may be useful in stratifying patients for trastuzumab therapy. PLoS ONE 2011; 6:e23772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Patel R, Gao M, Ahmad I, et al. Sprouty2, Pten, and Pp2a interact to regulate prostate cancer progression. J Clin Invest 2013; 123:1157–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sutterluty H, Mayer CE, Setinek U, et al. Down‐regulation of sprouty2 in non‐small cell lung cancer contributes to tumor malignancy via extracellular signal‐regulated kinase pathway‐dependent and ‐independent mechanisms. Mol Cancer Res 2007; 5:509–520. [DOI] [PubMed] [Google Scholar]
- 16. Shaw AT, Meissner A, Dowdle JA, et al. Sprouty‐2 regulates oncogenic K‐Ras in lung development and tumorigenesis. Genes Dev 2007; 21:694–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Minowada G, Miller YE. Overexpression of sprouty 2 in mouse lung epithelium inhibits urethane‐induced tumorigenesis. Am J Respir Cell Mol Biol 2009; 40:31–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fong CW, Chua MS, McKie AB, et al. Sprouty 2, an inhibitor of mitogen‐activated protein kinase signaling, is down‐regulated in hepatocellular carcinoma. Cancer Res 2006; 66:2048–2058. [DOI] [PubMed] [Google Scholar]
- 19. Holgren C, Dougherty U, Edwin F, et al. Sprouty‐2 controls C‐Met expression and metastatic potential of colon cancer cells: Sprouty/C‐Met upregulation in human colonic adenocarcinomas. Oncogene 2010; 29:5241–5253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Feng YH, Wu CL, Tsao CJ, et al. Deregulated expression of sprouty2 and microrna‐21 in human colon cancer: Correlation with the clinical stage of the disease. Cancer Biol Ther 2011; 11:111–121. [DOI] [PubMed] [Google Scholar]
- 21. Rhodes DR, Yu J, Shanker K, et al. Oncomine: A cancer microarray database and integrated data‐mining Platform. Neoplasia 2004; 6:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Barbachano A, Ordonez‐Moran P, Garcia JM, et al. Sprouty‐2 and E‐cadherin regulate reciprocally and dictate colon cancer cell tumourigenicity. Oncogene 2010; 29:4800–4813. [DOI] [PubMed] [Google Scholar]
- 23. Watanabe T, Kobunai T, Yamamoto Y, et al. Differential gene expression signatures between colorectal cancers with and without kras mutations: Crosstalk between the kras pathway and other signalling pathways. Eur J Cancer 2011; 47:1946–1954. [DOI] [PubMed] [Google Scholar]
- 24. Ordonez‐Moran P, Irmisch A, Barbachano A, et al. Sprouty2 Is a beta‐catenin and Foxo3a target gene indicative of poor prognosis in colon cancer. Oncogene 2014; 33:1975–1985. [DOI] [PubMed] [Google Scholar]
- 25. Mason JM, Morrison DJ, Basson MA, et al. Sprouty proteins: Multifaceted negative‐feedback regulators of receptor tyrosine kinase signaling. Trends Cell Biol 2006; 16:45–54. [DOI] [PubMed] [Google Scholar]
- 26. Basson MA, Akbulut S, Watson‐Johnson J, et al. Sprouty1 Is a critical regulator of Gdnf/Ret‐mediated kidney induction. Dev Cell 2005; 8:229–239. [DOI] [PubMed] [Google Scholar]
- 27. Shim K, Minowada G, Coling DE, et al. Sprouty2, a mouse deafness gene, regulates cell fate decisions in the auditory sensory epithelium by antagonizing Fgf signaling. Dev Cell 2005; 8:553–564. [DOI] [PubMed] [Google Scholar]
- 28. Soriano P. Generalized lacz expression with the Rosa26 Cre reporter strain. Nat Genet 1999; 21:70–71. [DOI] [PubMed] [Google Scholar]
- 29. Hayashi S, McMahon AP. Efficient recombination in diverse tissues by a tamoxifen‐inducible form of Cre: A tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol 2002; 244:305–318. [DOI] [PubMed] [Google Scholar]
- 30. Khare S, Holgren C, Samarel AM. Deoxycholic acid differentially regulates focal adhesion kinase phosphorylation: Role of tyrosine phosphatase Shp2. Am J Physiol Gastrointest Liver Physiol 2006; 291:G1100–G1112. [DOI] [PubMed] [Google Scholar]
- 31. Khare S, Mustafi R, Cerda S, et al. Ursodeoxycholic acid suppresses Cox‐2 expression in colon Ccancer: Roles of Ras, P38, and Ccaat/enhancer‐binding protein. Nutr Cancer 2008; 60:389–400. [DOI] [PubMed] [Google Scholar]
- 32. Gartel AL, Radhakrishnan SK. Lost in transcription: P21 repression, mechanisms, and consequences. Cancer Res 2005; 65:3980–3985. [DOI] [PubMed] [Google Scholar]
- 33. Piccolo MT, Crispi S. The dual role played by P21 may influence the apoptotic or anti‐apoptotic fate in cancer. J Cancer Res Updates 2012; 1:189–202. [Google Scholar]
- 34. Masoumi‐Moghaddam S, Amini A, Morris DL. The developing story of sprouty and cancer. Cancer Metastasis Rev 2014; 33:695–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Riese DJ, 2nd , Cullum Epiregulin: RL . Roles in normal physiology and cancer. Semin Cell Dev Biol 2014; 28:49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Troiani T, Martinelli E, Napolitano S, et al. Increased Tgf‐Alpha as a mechanism of acquired resistance to the anti‐Egfr inhibitor cetuximab through Egfr‐Met interaction and activation of Met signaling in colon cancer cells. Clin Cancer Res 2013; 19:6751–6765. [DOI] [PubMed] [Google Scholar]
- 37. Martin‐Caballero J, Flores JM, Garcia‐Palencia P, et al. Tumor susceptibility of P21(Waf1/Cip1)‐deficient mice. Cancer Res 2001; 61:6234–6238. [PubMed] [Google Scholar]
- 38. Deng C, Zhang P, Harper JW, et al. Mice lacking P21cip1/Waf1 undergo normal development, but are defective in G1 checkpoint control. Cell 1995; 82:675–684. [DOI] [PubMed] [Google Scholar]
- 39. Warfel NA, El‐Deiry WS. P21waf1 and tumourigenesis: 20 years after. Curr Opin Oncol 2013; 25:52–58. [DOI] [PubMed] [Google Scholar]
- 40. Xiong Y, Hannon GJ, Zhang H, et al. P21 Is a universal inhibitor of cyclin kinases. Nature 1993; 366:701–704. [DOI] [PubMed] [Google Scholar]
- 41. Ogino S, Nosho K, Shima K, et al. P21 expression in colon cancer and modifying effects of patient age and body mass index on prognosis. Cancer Epidemiol Biomarkers Prev 2009; 18:2513–2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gartel AL, Tyner AL. Transcriptional regulation of the P21((Waf1/Cip1)) Gene. Exp Cell Res 1999; 246:280–289. [DOI] [PubMed] [Google Scholar]
- 43. el‐Deiry WS, Tokino T, Velculescu VE, et al. Waf1, a Potential Mediator of P53 tumor suppression. Cell 1993; 75:817–825. [DOI] [PubMed] [Google Scholar]
- 44. Woods D, Parry D, Cherwinski H, et al. Raf‐Induced proliferation or cell cycle arrest is determined by the level of Raf activity with arrest mediated by P21cip1. Mol Cell Biol 1997; 17:5598–5611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sewing A, Wiseman B, Lloyd AC, et al. High‐intensity Raf signal causes cell cycle arrest mediated by P21cip1. Mol Cell Biol 1997; 17:5588–5597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hu S, Dong TS, Dalal SR, et al. The Microbe‐derived short chain fatty acid butyrate targets mirna‐dependent P21 gene expression in human colon cancer. PLoS ONE 2011; 6:e16221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jiang Y, Zhang M, Qian Y, et al. Rbm24, an Rna‐binding protein and a target of P53, regulates P21 expression via Mrna stability. J Biol Chem 2014; 289:3164–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bauer J, Sporn JC, Cabral J, et al. Effects of activin and Tgfbeta on P21 in colon cancer. PLoS ONE 2012; 7:‐e39381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ivanovska I, Ball AS, Diaz RL, et al. Micrornas in the Mir‐106b family regulate P21/Cdkn1a and promote cell cycle progression. Mol Cell Biol 2008; 28:2167–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bornstein G, Bloom J, Sitry‐Shevah D, et al. Role of the Scfskp2 ubiquitin ligase in the degradation of P21cip1 in S phase. J Biol Chem 2003; 278:25752–25757. [DOI] [PubMed] [Google Scholar]
- 51. Sheaff RJ, Singer JD, Swanger J, et al. Proteasomal turnover of P21cip1 does not require P21cip1 ubiquitination. Mol Cell 2000; 5:403–410. [DOI] [PubMed] [Google Scholar]
- 52. Cmielova J, Rezacova M. P21cip1/Waf1 protein and its function based on a subcellular localization [corrected]. J Cell Biochem 2011; 112:3502–3506. [DOI] [PubMed] [Google Scholar]
- 53. Romanov VS, Pospelov VA, Pospelova TV. Cyclin‐dependent kinase inhibitor P21(Waf1): Contemporary view on its role in senescence and oncogenesis. Biochemistry (Mosc) 2012; 77:575–584. [DOI] [PubMed] [Google Scholar]
- 54. Abbas T, Dutta A. P21 in Cancer: Intricate networks and multiple activities. Nat Rev Cancer 2009; 9:400–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gartel AL. P21(Waf1/Cip1) and cancer: A shifting paradigm?. Biofactors 2009; 35:161–164. [DOI] [PubMed] [Google Scholar]
- 56. Sinicrope FA, Roddey G, Lemoine M, et al. Loss of P21waf1/Cip1 protein expression accompanies progression of sporadic colorectal neoplasms but not hereditary nonpolyposis colorectal cancers. Clin Cancer Res 1998; 4:1251–1261. [PubMed] [Google Scholar]
- 57. Ukomadu C, Dutta A. P21‐Dependent inhibition of colon cancer cell growth by mevastatin is independent of inhibition of G1 cyclin‐dependent kinases. J Biol Chem 2003; 278:43586–43594. [DOI] [PubMed] [Google Scholar]
- 58. Ocker M, Schneider‐Stock R. Histone deacetylase inhibitors: Signalling towards P21cip1/Waf1. Int J Biochem Cell Biol 2007; 39:1367–1374. [DOI] [PubMed] [Google Scholar]
- 59. Roy HK, Koetsier JL, Tiwari AK, et al. Involvement of P21cip1/Waf1 in the anti‐proliferative effects of polyethylene glycol in colon carcinogenesis. Int J Oncol 2011; 38:529–536. [DOI] [PubMed] [Google Scholar]
- 60. Owa T, Yoshino H, Yoshimatsu K, et al. Cell cycle regulation in the G1 phase: A promising target for the development of new chemotherapeutic anticancer agents. Curr Med Chem 2001; 8:1487–1503. [DOI] [PubMed] [Google Scholar]