

Abstract
Objective:
To estimate points along the spectrum of β-amyloid pathology at which rates of change of several measures of neuronal injury and cognitive decline begin to accelerate.
Methods:
In 460 patients with mild cognitive impairment (MCI), we estimated the points at which rates of florbetapir PET, fluorodeoxyglucose (FDG) PET, MRI, and cognitive and functional decline begin to accelerate with respect to baseline CSF Aβ42. Points of initial acceleration in rates of decline were estimated using mixed-effects regression.
Results:
Rates of neuronal injury and cognitive and even functional decline accelerate substantially before the conventional threshold for amyloid positivity, with rates of florbetapir PET and FDG PET accelerating early. Temporal lobe atrophy rates also accelerate prior to the threshold, but not before the acceleration of cognitive and functional decline.
Conclusions:
A considerable proportion of patients with MCI would not meet inclusion criteria for a trial using the current threshold for amyloid positivity, even though on average, they are experiencing cognitive/functional decline associated with prethreshold levels of CSF Aβ42. Future trials in early Alzheimer disease might consider revising the criteria regarding β-amyloid thresholds to include the range of amyloid associated with the first signs of accelerating rates of decline.
Understanding the sequence of pathophysiologic processes occurring during the progression of Alzheimer disease (AD) has been a primary goal of recent research. Evidence has accumulated pointing to β-amyloid deposition and hypometabolism occurring in early stages with subsequent gray matter atrophy and cognitive and functional decline.1–3 Hypothetical models have been proposed, though the details remain unclear.4
Understanding the timeframe of decline is required to facilitate targeting a specific process in order to slow progression.5 Possible reasons for recent failures of clinical trials of antiamyloid therapies include (1) targeting amyloid in patients who already exhibit substantial neurodegeneration and cognitive impairment and (2) failing to exclude patients without significant amyloid deposition.6–8 Given these concerns, several current trials target β-amyloid pathology in earlier stages of the disease and require significant β-amyloid pathology.9
Amyloid positivity thresholds are frequently defined to be the level of pathology that most accurately distinguishes cases of AD from cognitively normal controls.10–12 A recent report suggests that current Pittsburgh compound B thresholds are too high.13 With more exclusive thresholds, it is likely that certain pathophysiologic processes have advanced beyond what is optimal for clinical trials intending to recruit participants with little neurodegeneration or cognitive impairment. Several measures of neurodegeneration have been shown to occur years prior to established β-amyloid thresholds.14–16
A priori thresholds, commonly used to study the effect of amyloid on neurodegeneration or cognition, preclude the detection of the initial acceleration of decline with respect to the level of β-amyloid. The goal of this study was to estimate the points along the spectrum of β-amyloid pathology at which rates of neuronal injury and cognitive decline begin to accelerate.
METHODS
Standard protocol approvals, registrations, and patient consents.
This study was approved by the institutional review boards of all of the participating institutions. Informed written consent was obtained from all participants at each site.
Participants.
Data were obtained from the Alzheimer's Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu, www.adni-info.org). All sites had institutional review board approval to study human subjects. The population in this study included ADNI-2 participants enrolled into the mild cognitive impairment (MCI) cohort at screening, tested for CSF biomarkers, and followed longitudinally for at least one MRI, 18F-fluorodeoxyglucose (FDG) PET, 18F-florbetapir PET, or neuropsychological examination.
CSF biomarker concentrations.
CSF samples were collected at baseline by lumbar puncture. CSF methods have been described previously.10,17
MRI acquisition and processing.
Structural MRI brain scans were acquired using 3T MRI scanners with a standardized protocol.18 Quantification was performed in an automated pipeline using FreeSurfer software package version 5.1 (http://surfer.nmr.mgh.harvard.edu/fswiki).19 Detailed descriptions can be found at www.adni-info.org.
Regions of interest (ROIs) including the cingulate gyrus as well as the temporal, parietal, frontal, and occipital lobes were analyzed. Details about FreeSurfer parcellation can be found at https://surfer.nmr.mgh.harvard.edu/fswiki/CorticalParcellation. Left and right hemisphere volumes were averaged for each subregion.
FDG PET.
Methods to acquire and process FDG PET images were described previously. Full details of procedures and the standardized protocol are described at http://adni.loni.usc.eduqw/methods/pet-analysis/pre-processing/and at http://www.adni-info.org/Scientists/ADNIStudyProcedures.html. ROIs included in the analysis were the temporal, angular, and cingulate gyri.20,21
Florbetapir PET.
Similarly, methods to acquire and process ADNI florbetapir PET image data were described previously. Full details of acquisition and analysis can be found at http://adni.loni.usc.edu/methods/. ROIs included in the analysis were the temporal, parietal, and frontal lobes.11
Cognitive and functional outcomes.
Cognitive measures assessed included the Mini-Mental State Examination, Alzheimer's Disease Assessment Scale–cognitive subscale, both the 11- and 13-item versions (ADAS-11, ADAS-13), delayed memory recall from the Wechsler Memory Scale (Logical Memory II), delayed Rey Auditory Verbal Learning Test (dRAVLT), Trail Making Test parts A and B, Boston Naming Test, a cognitive composite (comprising the ADAS-11, Logical Memory II, and the Trail Making Test part B), Clinical Dementia Rating Sum of Boxes, and the Functional Assessment Questionnaire.22–29
Statistical analysis.
The relationship between longitudinal cognitive or imaging responses and baseline β-amyloid pathology was modeled in 2 steps. First, patient-specific rates for each response were estimated using all available data. Rates for responses with 3 or more observations per patient (cognition and MRI) were estimated using linear mixed effects regression with a random intercept and slope. Mixed effects models allow the number of responses to vary across individuals, adjusting for missingness. Longitudinal cognitive scores were regressed on time (years) since initial visit while adjusting for age, sex, and education. Volumes were regressed on time since initial visit while adjusting for age, sex, and intracranial volume. Rates for responses with only 2 observations per patient (FDG and florbetapir PET) were regressed on time between scans using linear regression for each patient, separately. Florbetapir PET values were adjusted for whole cerebellum uptake prior to the estimation of the rates. FDG and florbetapir PET rates were then adjusted for age and sex.
In the second step, patient-specific rates were regressed on baseline CSF Aβ42 using monotone penalized regression splines. Generalized cross-validation was used to tune the smoothing parameter and the Akaike information criterion was used to select the dimension of the basis used to represent the smooth term.30,31
Steps 1 and 2 were repeated in 500 bootstrap samples to estimate 95% confidence intervals for the association between CSF Aβ42 and each response using the 2.5th and 97.5th percentiles.
Permutation tests were performed to estimate the statistical significance of the association between CSF Aβ42 and each response. Responses were regressed on permuted values of CSF Aβ42 in each bootstrap sample to obtain a null distribution of F statistics. p Values were then calculated as the proportion of null F statistics that were equal to or greater than the observed F statistic estimated using the true CSF Aβ42 labels.
For responses with a significant association with CSF Aβ42, points of initial acceleration in rate of decline with respect to β-amyloid pathology were estimated. These points were taken to be the moment the curves dropped 1 SE below the mean response at the highest levels of CSF Aβ42. However, since the SE is sample size–dependent and the sample sizes varied with each model, we reweighted each SE to correspond to the average sample size. Standard deviations and 95% confidence intervals were estimated from the 500 bootstrap samples.
Baseline associations between demographics and CSF Aβ42 were assessed using Spearman correlation for age and education and the Wilcoxon rank-sum test for sex.
The association between baseline CSF Aβ42 and missing data were modeled using generalized mixed-effects regression for MRI and cognitive outcomes, with a binomial missing indicator for a missing visit. Similarly, logistic regression was used to assess missingness for FDG and florbetapir PET. p Values from permutation tests were adjusted for multiplicity using a false discovery rate correction.32 All analyses were done in R v3.1.1 (www.r-project.org).
RESULTS
Cohort characteristics.
A total of 460 patients with MCI were followed longitudinally up to 8 years. A total of 420 MCI participants were included in the MRI analysis, 460 in the analysis of cognition, 193 in the FDG PET analysis, and 258 in the florbetapir PET analysis. Participants underwent MRI scans an average of 4.1 times over an average of 1.5 years (maximum 3 years). Cognitive tests were administered an average of 7.3 times over an average of 2.6 years (maximum 8 years). Participants had 2 FDG PET and florbetapir PET scans each—one at baseline and another at their 2-year visit.
Overall, baseline CSF Aβ42 was highly associated with age (ρ = −0.20, p < 0.001) and marginally associated with sex (p = 0.09), with men having on average 8 ng/L less CSF Aβ42 compared with women. Years of education was not associated with baseline CSF Aβ42 (ρ = 0.02, p = 0.63). The 4 analysis groups (cognition, MRI, FDG, and florbetapir PET) were similar, with mean baseline CSF Aβ42 ranging from 174 to 181 ng/L, mean age ranging from 71 to 72 years, percent female ranging from 44% to 48%, mean years of education 16 for all groups, percent APOE ε4 carriers ranging from 43% to 49%, and mean ADAS-13 ranging from 14 to 15. All analyses were adjusted for age and sex and the cognitive analyses were also adjusted for education.
A 1 SD decrease in baseline CSF Aβ42 was associated with increased odds of missing FDG PET (log odds ratio [OR] = −0.20, SE = 0.10, p = 0.043), florbetapir PET (log OR = −0.25, SE = 0.10, p = 0.011), and marginally associated with increased odds of missing cognitive data (log OR = −0.09, SE = 0.05, p = 0.07) and MRI data (log OR = −0.11, SE = 0.08, p = 0.154).
MRI.
There was significant acceleration in atrophy rates with respect to CSF Aβ42 in all MRI ROIs, except the cingulate gyrus. The increase in atrophy rate was estimated to start at CSF Aβ42 = 213 ng/L in the temporal lobe and considerably later in the rest of the lobes (with the parietal and frontal lobes accelerating at intermediate CSF Aβ42 levels and the occipital lobe accelerating first at very low CSF Aβ42 levels). Results of the overall tests of association between CSF Aβ42 and MRI ROIs and also estimates of initial rate acceleration points and confidence intervals are summarized in the table. Rates of all ROIs are plotted against baseline CSF Aβ42, as well as estimates of points of the initial rate acceleration, in figure 1.
Table.
Estimates of initial acceleration points, 95% confidence intervals, and significance of permutation tests
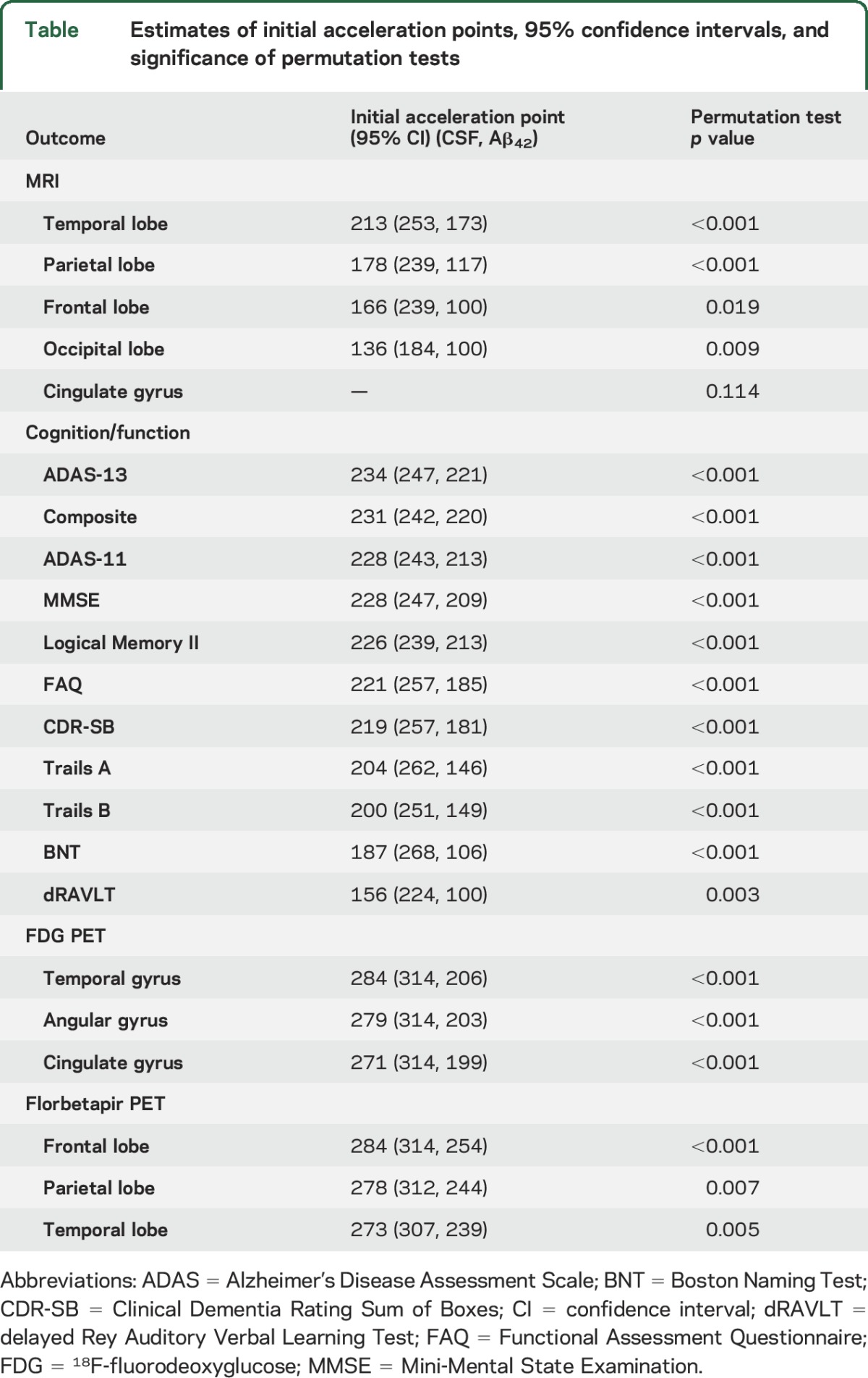
Figure 1. MRI rates of change.

Annual rates of change of MRI regions are plotted against baseline CSF Aβ42. Atrophy rates increase from top to bottom and β-amyloid pathology increases from left to right. Estimated curves are in red with 95% confidence intervals shaded in gray. The dashed blue line in the plot of the temporal lobe is the estimated curve without the monotonicity restriction, for comparison. The vertical dashed line in black is the estimate of the initial acceleration point, if it exists.
Cognitive and functional outcomes.
There was a significant acceleration in rate of decline in all 11 cognitive and functional measures across the span of baseline CSF Aβ42. Global measures of cognitive decline and delayed logical memory recall were estimated to begin accelerating near CSF Aβ42 = 230 ng/L, with functional measures immediately following. Hypothesis tests and estimates are summarized in the table. Plots of cognitive and functional outcomes are shown in figure 2.
Figure 2. Cognitive and functional rates of change.

Annual rates of change for cognitive and functional measures are plotted against baseline CSF Aβ42. Rates of cognition or function worsen from top to bottom and β-amyloid pathology increases from left to right. Estimated curves are in red with 95% confidence intervals shaded in gray. The vertical dashed line in black is the estimate of the initial acceleration point. ADAS = Alzheimer's Disease Assessment Scale; AVLT = Auditory Verbal Learning Test; CDR-SB = Clinical Dementia Rating Sum of Boxes; FAQ = Functional Assessment Questionnaire; MMSE = Mini-Mental State Examination.
FDG PET.
Significant acceleration of rates of FDG PET was observed in all 3 ROIs. A 1 SE rate increase was estimated to occur near CSF Aβ42 = 280 ng/L in all 3 ROIs. Estimates are summarized in the table and plots are shown in the top row of figure 3.
Figure 3. 18F-fluorodeoxyglucose (FDG) and florbetapir PET rates of change.

Annual rates of change for FDG PET (top row) and florbetapir (bottom row) are plotted against baseline CSF Aβ42. Rates worsen from top to bottom and β-amyloid pathology increases from left to right. Estimated curves are in red with 95% confidence intervals shaded in gray. The dashed blue line in the plot of the temporal lobe is the estimated curve without the monotonicity restriction, for comparison. The vertical dashed line in black is the estimate of the initial acceleration point. SUVR = standardized uptake value ratio.
Florbetapir PET.
Significant acceleration of rates of florbetapir PET was observed in all ROIs. Similar to FDG PET, a 1 SE rate increase was estimated to occur near CSF Aβ42 = 280 ng/L. Estimates are summarized in the table and plots are shown in the bottom row of figure 3. All imaging and cognitive points of initial acceleration and confidence intervals are shown in figure 4.
Figure 4. Acceleration point estimates and 95% confidence intervals.

Estimates of the initial acceleration points and 95% confidence intervals for all outcomes are plotted against baseline CSF Aβ42. β-Amyloid pathology increases from left to right. The dashed black line is the conventional threshold for amyloid positivity (192 ng/L). ADAS = Alzheimer's Disease Assessment Scale; BNT = Boston Naming Test; CDR-SB = Clinical Dementia Rating Sum of Boxes; dRAVLT = delayed Rey Auditory Verbal Learning Test; FAQ = Functional Assessment Questionnaire; FDG = 18F-fluorodeoxyglucose; MMSE = Mini-Mental State Examination.
DISCUSSION
The main findings of this analysis are (1) rates of neuronal injury, cognitive decline, and even functional decline accelerate substantially before a conventional threshold for amyloid positivity, (2) acceleration points for florbetapir PET and FDG PET were estimated to occur at approximately the same level of CSF Aβ42, and (3) temporal lobe atrophy rates accelerate prior to the conventional threshold for amyloid positivity, confirming our previous report,16 but they did not precede the increase in rates of cognitive and functional decline.
Rates of multiple modes of neurodegeneration, cognitive decline, and functional decline are observed to accelerate prior to the current threshold for amyloid positivity. A considerable proportion of patients with MCI would not meet inclusion criteria for a clinical trial based on the current threshold for amyloid positivity, though on average, they are experiencing cognitive and functional decline associated with prethreshold levels of CSF Aβ42. In fact, these patients would never meet criteria for any stage of preclinical AD during the course of their progression because they demonstrated clinically significant cognitive and functional impairment prior to becoming amyloid-positive.33 If the efficacy of an antiamyloid treatment relies on mitigating the effect of amyloid at the initial downslope of cognitive decline, it is likely that trial inclusion criteria should be revised to include patients with substantially less β-amyloid pathology, especially in light of a recent report finding that those with emerging amyloid pathology (CSF Aβ42<225 ng/L, but still amyloid-negative) are at increased risk of becoming amyloid-positive in the near term.34
The cognitive and atrophy rate curves are flat at high levels of CSF Aβ42 before a distinct rate acceleration occurs. In contrast, the shape of the curves for FDG and florbetapir PET is linear without any plateau, suggesting a very early rate increase even at the least pathologic levels of CSF Aβ42. It is no surprise, given the high correlation between measures of β-amyloid in CSF and PET, that changes in florbetapir rates and CSF Aβ42 would occur close together. Recent reports suggest that CSF amyloid may decrease prior to an increase in amyloid deposition.3,4,35 FDG PET and synaptic dysfunction have also been identified as early-stage processes,36 although it is surprising to see changes in FDG PET, a measure of neurodegeneration, occur so closely with amyloid deposition. It is possible that there are too few observations at the highest levels of CSF Aβ42 to estimate the plateau in the FDG PET analysis, biasing the estimates of acceleration to higher levels of CSF Aβ42. There is also a considerable delay between the acceleration of FDG PET rates and the acceleration of cognitive decline, as shown in figure 4. The confidence intervals for FDG PET are wide and while the time between a reduction in brain glucose metabolism and the onset of cognitive decline is unknown, it is again possible that there is an upward bias for the FDG PET estimates of acceleration.
The third result of our analysis is the unexpected timing of the acceleration of atrophy rates compared to cognition. However, given the association between FDG PET and cognition, it makes sense that increased rates of cognitive decline would immediately follow worsening synaptic dysfunction, prior to the actual physical degeneration and loss of gray matter. It is also possible that the age adjustment may affect the estimates for atrophy acceleration more than other outcomes, given the high degree of association between aging and atrophy. The estimate of the point of initial acceleration of rate of temporal lobe atrophy (CSF Aβ42 = 213 ng/L) is similar to estimates previously reported in a different cohort.16 However, a plateau in atrophy at low levels of CSF Aβ42 was not replicated here, possibly due to a less progressed cohort studied here.
The results of this analysis provide some evidence for the ordering of markers for neurodegeneration and cognitive decline with respect to CSF Aβ42, but not formal statistical tests. Figure 4 shows wide confidence intervals for all markers, with the exception of the global cognitive measures and delayed logical memory, which are surprisingly narrow. We opted to include all available data for each outcome in order to get the most precise estimates of rate changes; however, this resulted in varying sample sizes across outcomes. Additional studies will be required to make formal conclusions about the specifics of the order; however, it appears that the rates of nearly all the biomarkers in this study begin to increase prior to what is considered the threshold for amyloid positivity.
These analyses do not provide evidence that amyloid plays a causal role in the increase of neurodegeneration and cognitive decline. It is possible that neuronal injury and cognitive decline are due to increased amyloid deposition; however, it is also possible that amyloid deposition is a downstream effect of other unobserved factors. It is likely that the amyloid burden required to elicit increased neuronal injury varies greatly across patients in this cohort. Some may be susceptible to minimal levels of amyloid deposition, while others may only show signs of neurodegeneration after prolonged amyloid deposition. Or some patients may exhibit substantial neurodegeneration prior to even minimal amyloid deposition. Our models are a simplified version of the relationships among biomarkers and cognition. A more complex model, one that adjusted the association between CSF Aβ42 and cognition for cortical atrophy, for example, may yield different results with different estimates of points of initial rate acceleration. Additional risk factors not considered in this analysis are also likely at play, including cerebrovascular disease, and other proteinopathies including α-synuclein and TDP-43.37,38 Another consideration is the heterogeneity of the MCI population. While we focus on estimates averaged over the entire cohort, it is likely that some patients' rates begin to increase after the conventional threshold for amyloid positivity. Floor effects of some measures may also bias estimates of acceleration. For example, the estimate of acceleration observed in dRAVLT, a measure thought to decline early, is noticeably later than other measures of cognition, especially measures including delayed memory recall. The late estimate of acceleration is likely due to many MCI patients already scoring near the floor of the measure with no room to decline.
While this study only examined patients with MCI, understanding the sequence of biomarker change in a cognitively normal population will be important for the design of clinical trials in a presymptomatic population. A large population of amyloid-positive cognitively normal people resist the cognitive decline seen in their MCI-affected peers, and a large proportion has done it with a considerably larger amyloid burden, given the low levels of amyloid shown to be associated with decline in this study. The ability of some to tolerate large amounts of amyloid, while others demonstrate decline much earlier in the process of amyloid accumulation, remains a gray area in understanding the progression of AD and an obstacle for the design of clinical trials. By making amyloid thresholds more liberal, there may be some loss in specificity; however, this loss may be worth the gains made by treating a less progressed population.
Even with an optimal amyloid threshold, considerable variability will remain regarding the association between Aβ pathology and cognition in elders, given that a similar degree of Aβ pathology may be seen in people who are cognitively normal, slightly impaired, or with full dementia. This variability highlights the importance of assessing other screening characteristics besides Aβ status, including specific markers for neurodegeneration such as FDG PET, tau pathology, and gray matter atrophy, when selecting preclinical populations for trial enrichment. It is possible that a threshold for the risk of progression should not be based on Aβ or neurodegeneration alone, but rather inclusion into a trial requires some interdependent minimal level of both types of pathology.
Future clinical trials in early AD might consider revising the criteria regarding β-amyloid thresholds to include the range of amyloid associated with the first signs of accelerating rates of decline.
Supplementary Material
GLOSSARY
- AD
Alzheimer disease
- ADAS
Alzheimer's Disease Assessment Scale
- ADNI
Alzheimer's Disease Neuroimaging Initiative
- dRAVLT
delayed Rey Auditory Verbal Learning Test
- FDG
18F-fluorodeoxyglucose
- MCI
mild cognitive impairment
- OR
odds ratio
- ROI
region of interest
Footnotes
Supplemental data at Neurology.org
Contributor Information
Collaborators: Alzheimer's Disease Neuroimaging Initiative, Michael Weiner, Paul Aisen, Michael Weiner, Paul Aisen, Ronald Petersen, Clifford R. Jack, Jr, William Jagust, John Q. Trojanowki, Arthur W. Toga, Laurel Beckett, Robert C. Green, Andrew J. Saykin, John Morris, Enchi Liu, Robert C. Green, Tom Montine, Ronald Petersen, Paul Aisen, Anthony Gamst, Ronald G. Thomas, Michael Donohue, Sarah Walter, Devon Gessert, Tamie Sather, Laurel Beckett, Danielle Harvey, Anthony Gamst, Michael Donohue, John Kornak, Clifford R. Jack, Jr, Anders Dale, Matthew Bernstein, Joel Felmlee, Nick Fox, Paul Thompson, Norbert Schuff, Gene Alexander, Charles DeCarli, William Jagust, Dan Bandy, Robert A. Koeppe, Norm Foster, Eric M. Reiman, Kewei Chen, Chet Mathis, John Morris, Nigel J. Cairns, Lisa Taylor-Reinwald, J.Q. Trojanowki, Les Shaw, Virginia M.Y. Lee, Magdalena Korecka, Arthur W. Toga, Karen Crawford, Scott Neu, Andrew J. Saykin, Tatiana M. Foroud, Steven Potkin, Li Shen, Zaven Kachaturian, Richard Frank, Peter J. Snyder, Susan Molchan, Jeffrey Kaye, Joseph Quinn, Betty Lind, Sara Dolen, Lon S. Schneider, Sonia Pawluczyk, Bryan M. Spann, James Brewer, Helen Vanderswag, Judith L. Heidebrink, Joanne L. Lord, Ronald Petersen, Kris Johnson, Rachelle S. Doody, Javier Villanueva-Meyer, Munir Chowdhury, Yaakov Stern, Lawrence S. Honig, Karen L. Bell, John C. Morris, Beau Ances, Maria Carroll, Sue Leon, Mark A. Mintun, Stacy Schneider, Daniel Marson, Randall Griffith, David Clark, Hillel Grossman, Effie Mitsis, Aliza Romirowsky, Leyla deToledo-Morrell, Raj C. Shah, Ranjan Duara, Daniel Varon, Peggy Roberts, CNA, Marilyn Albert, Chiadi Onyike, Stephanie Kielb, Henry Rusinek, Mony J de Leon, Lidia Glodzik, Susan De Santi, P. Murali Doraiswamy, Jeffrey R. Petrella, R. Edward Coleman, Steven E. Arnold, Jason H. Karlawish, David Wolk, Charles D. Smith, Greg Jicha, Peter Hardy, Oscar L. Lopez, MaryAnn Oakley, Donna M. Simpson, Anton P. Porsteinsson, Bonnie S. Goldstein, Kim Martin, Kelly M. Makino, M. Saleem Ismail, Connie Brand, Ruth A. Mulnard, Gaby Thai, Catherine Mc-Adams-Ortiz, Kyle Womack, Dana Mathews, Mary Quiceno, Ramon Diaz-Arrastia, Richard King, Myron Weiner, Kristen Martin-Cook, Michael DeVous, Allan I. Levey, James J. Lah, Janet S. Cellar, Jeffrey M. Burns, Heather S. Anderson, Russell H. Swerdlow, Liana Apostolova, Po H. Lu, George Bartzokis, Daniel H.S. Silverman, Neill R Graff-Radford, MBBCH, Francine Parfitt, Heather Johnson, Martin R. Farlow, Ann Marie Hake, Brandy R. Matthews, Scott Herring, Christopher H. van Dyck, Richard E. Carson, Martha G. MacAvoy, Howard Chertkow, Howard Bergman, Chris Hosein, Sandra Black, Bojana Stefanovic, Curtis Caldwell, Ging-Yuek Robin Hsiung, Howard Feldman, Benita Mudge, Michele Assaly, Andrew Kertesz, John Rogers, Dick Trost, Charles Bernick, Donna Munic, Diana Kerwin, Marek-Marsel Mesulam, Kristina Lipowski, Chuang-Kuo Wu, Nancy Johnson, Carl Sadowsky, Walter Martinez, Teresa Villena, Raymond Scott Turner, Kathleen Johnson, Brigid Reynolds, Reisa A. Sperling, Keith A. Johnson, Gad Marshall, Meghan Frey, Jerome Yesavage, Joy L. Taylor, Barton Lane, Allyson Rosen, Jared Tinklenberg, Marwan Sabbagh, Christine Belden, Sandra Jacobson, Neil Kowall, Ronald Killiany, Andrew E. Budson, Alexander Norbash, Patricia Lynn Johnson, Thomas O. Obisesan, Saba Wolday, Salome K. Bwayo, Alan Lerner, Leon Hudson, Paula Ogrocki, Evan Fletcher, Owen Carmichael, John Olichney, Charles DeCarli, Smita Kittur, Michael Borrie, T-Y Lee, Rob Bartha, Sterling Johnson, Sanjay Asthana, Cynthia M. Carlsson, Steven G. Potkin, Adrian Preda, Dana Nguyen, Pierre Tariot, Adam Fleisher, Stephanie Reeder, Vernice Bates, Horacio Capote, Michelle Rainka, Douglas W. Scharre, Maria Kataki, Earl A. Zimmerman, Dzintra Celmins, Alice D. Brown, Godfrey D. Pearlson, Karen Blank, Karen Anderson, Andrew J. Saykin, Robert B. Santulli, Eben S. Schwartz, Kaycee M. Sink, Jeff D. Williamson, Pradeep Garg, Franklin Watkins, Brian R. Ott, Henry Querfurth, Geoffrey Tremont, Stephen Salloway, Paul Malloy, Stephen Correia, Howard J. Rosen, Bruce L. Miller, Jacobo Mintzer, Crystal Flynn Longmire, Kenneth Spicer, Elizabether Finger, Irina Rachinsky, John Rogers, Andrew Kertesz, Dick Drost, Nunzio Pomara, Raymundo Hernando, Antero Sarrael, Susan K. Schultz, Laura L. Boles Ponto, Hyungsub Shim, Karen Elizabeth Smith, Norman Relkin, Gloria Chaing, Lisa Raudin, Amanda Smith, Kristin Fargher, and Balebail Ashok Raj
AUTHOR CONTRIBUTIONS
P.S. Insel drafted and revised the manuscript for content, contributed to the study design, and analyzed and interpreted the data. Dr. Mattsson revised the manuscript for content, contributed to the study design, and interpreted the data. Dr. Mackin revised the manuscript for content and interpreted the data. Dr. Schöll revised the manuscript for content. Dr. Nosheny revised the manuscript for content. Dr. Tosun revised the manuscript for content. Dr. Donohue revised the manuscript for content, contributed to the study design, and interpreted the data. Dr. Aisen revised the manuscript for content. Dr. Jagust revised the manuscript for content. Dr. Weiner revised the manuscript for content.
STUDY FUNDING
Data collection and sharing for this project was funded by the Alzheimer's Disease Neuroimaging Initiative (ADNI) (NIH grant U01 AG024904). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through contributions from the following: Alzheimer's Association; Alzheimer's Drug Discovery Foundation; BioClinica, Inc.; Biogen Idec Inc.; Bristol-Myers Squibb Company; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; F. Hoffmann-La Roche Ltd. and its affiliated company Genentech, Inc.; GE Healthcare; Innogenetics, N.V.; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC; Johnson & Johnson Pharmaceutical Research & Development LLC; Medpace, Inc.; Merck & Co., Inc.; Meso Scale Diagnostics, LLC; NeuroRx Research; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Synarc Inc.; and Takeda Pharmaceutical Company. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the NIH (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated by the Laboratory for Neuroimaging at the University of Southern California. This research was also supported by NIH grants P30 AG010129 and K01 AG030514.
DISCLOSURE
P. Insel and N. Mattsson report no disclosures relevant to the manuscript. R. Mackin has financial support from NIMH R01 0977669 and NIMH K08 MH081065. M. Schöll, R. Nosheny, and D. Tosun report no disclosures relevant to the manuscript. M. Donohue was a consultant for Bristol-Myers Squibb. P. Aisen serves on a scientific advisory board for NeuroPhage; has served as a consultant to Elan, Wyeth, Eisai, Schering-Plough, Bristol-Myers Squibb, Eli Lilly and Company, NeuroPhage, Merck, Roche, Amgen, Genentech, Abbott, Pfizer, Novartis, Bayer, Astellas, Dainippon, Biomarin, Solvay, Otsuka, Daiichi, AstraZeneca, Janssen, Medivation, Ichor, Toyama, Lundbeck, Biogen Idec, iPerian, Probiodrug, Somaxon, Biotie, Cardeus, Anavex, Kyowa Hakko Kirin Pharma, and Medtronic; and receives research support from Eli Lilly and Baxter and the NIH (NIA U01-AG10483 [PI], NIA U01-AG024904 [Coordinating Center Director], NIA R01-AG030048 [PI], and R01-AG16381 [Co-I]). W. Jagust reports no disclosures relevant to the manuscript. M. Weiner has been on scientific advisory boards for Pfizer and BOLT International; has been consultant for Pfizer Inc., Janssen, KLJ Associates, Easton Associates, Harvard University, inThought, INC Research, Inc., University of California, Los Angeles, Alzheimer's Drug Discovery Foundation, and Sanofi-Aventis Group; has received funding for travel from Pfizer, ADPD meeting, Paul Sabatier University, Novartis, Tohoku University, MCI Group, France, Travel eDreams, Inc., Neuroscience School of Advanced Studies (NSAS), Danone Trading, BV, and CTAD ANT Congres; serves as an associated editor of Alzheimer's & Dementia; has received honoraria from Pfizer, Tohoku University, and Danone Trading BV; has research support from Merck, Avid, DOD and VA; and has stock options in Synarc and Elan. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Donohue MC, Jacqmin-Gadda H, Le Goff M, et al. Estimating long-term multivariate progression from short-term data. Alzheimers Dement 2014;10:S400–S410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jack CR, Knopman DS, Jagust WJ, et al. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol 2010;9:119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bateman RJ, Xiong C, Benzinger TL, et al. Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N Engl J Med 2012;367:795–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jack CR, Knopman DS, Jagust WJ, et al. Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol 2013;12:207–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hyman BT. Amyloid-dependent and amyloid-independent stages of Alzheimer disease. Arch Neurol 2011;68:1062–1064. [DOI] [PubMed] [Google Scholar]
- 6.Doody RS, Thomas RG, Farlow M, et al. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer's disease. N Engl J Med 2014;370:311–321. [DOI] [PubMed] [Google Scholar]
- 7.Sperling RA, Jack CR, Aisen PS. Testing the right target and right drug at the right stage. Sci Translational Med 2011;3:111cm33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Karran E, Hardy J. Antiamyloid therapy for Alzheimer's disease: are we on the right road?. N Engl J Med 2014;370:377–378. [DOI] [PubMed] [Google Scholar]
- 9.Sperling RA, Rentz DM, Johnson KA, et al. The A4 study: stopping AD before symptoms begin?. Sci Transl Med 2014;6:228fs13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shaw LM, Vanderstichele H, Knapik-Czajka M, et al. Cerebrospinal fluid biomarker signature in Alzheimer's disease neuroimaging initiative subjects. Ann Neurol 2009;65:403–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Landau SM, Mintun MA, Joshi AD, et al. Amyloid deposition, hypometabolism, and longitudinal cognitive decline. Ann Neurol 2012;72:578–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jack CR, Vemuri P, Wiste HJ, et al. Evidence for ordering of Alzheimer disease biomarkers. Arch Neurol 2011;68:1526–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Villenueve S, Rabinovici GD, Cohn-Sheehy BI, et al. Existing Pittsburgh compound B positron emission tomography thresholds are too high: statistical and pathological evaluation. Brain 2015;138:2020–2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reiman EM, Quiroz YT, Fleisher AS, et al. Brain imaging and fluid biomarker analysis in young adults at genetic risk for autosomal dominant Alzheimer's disease in the presenilin 1 E280A kindred: a case-control study. Lancet Neurol 2012;11:1048–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Filippini N, MacIntosh BJ, Hough MG, et al. Distinct patterns of brain activity in young carriers of the APOE-ε4 allele. Proc Natl Acad Sci USA 2009;106:7209–7214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Insel PS, Mattsson N, Donohue MC, et al. The transitional association between β-amyloid pathology and regional brain atrophy. Alzheimers Dement 2015;11:1171–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Olsson A, Vanderstichele H, Andreasen N, et al. Simultaneous measurement of beta-amyloid(1-42), total tau, and phosphorylated tau (Thr181) in cerebrospinal fluid by the xMAP technology. Clin Chem 2005;51:336–345. [DOI] [PubMed] [Google Scholar]
- 18.Jack CR, Bernstein MA, Fox NC, et al. The Alzheimer's Disease Neuroimaging Initiative (ADNI): MRI methods. J Magn Reson Imaging 2008;27:685–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fischl B, Salat DH, Busa E, et al. Whole brain segmentation: automated labeling of neuroanatomical structures in the human brain. Neuron 2002;33:341–355. [DOI] [PubMed] [Google Scholar]
- 20.Jagust WJ, Bandy D, Chen K, Foster NL, Landau SM, Mathis CA. The Alzheimer's Disease Neuroimaging Initiative positron emission tomography core. Alzheimers Dement 2010;6:221–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Landau SM, Harvey D, Madison CM, et al. Associations between cognitive, functional, and FDG-PET measures of decline in AD and MCI. Neurobiol Aging 2011;32:1207–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reitan R. Validity of the Trail-Making test as an indication of organic brain damage. Percept Mot Skills 1958;8:271–276. [Google Scholar]
- 23.Wechsler DA. Wechsler Adult Intelligence Scale–Revised. New York: Psychological Corporation; 1987. [Google Scholar]
- 24.Kaplan EF, Goodglass H, Weintraub S. The Boston Naming Test, 2nd ed Philadelphia: Lea & Febiger; 1982. [Google Scholar]
- 25.Rey A. l'Examen Clinique En Psychologie. Paris: Presses Universitaires de France; 1964. [Google Scholar]
- 26.Rosen WG, Mohs RC, Davis KL. A new rating scale for Alzheimer's disease. Am J Psychiatry 1984;141:1356–1364. [DOI] [PubMed] [Google Scholar]
- 27.Pfeffer RI, Kurosaki TT, Harrah CH, Chance JM, Filos S. Measurement of functional activities of older adults in the community. J Gerontol 1982;37:323–329. [DOI] [PubMed] [Google Scholar]
- 28.Morris JC. Clinical dementia rating. Neurology 1993;43:2412–2414. [DOI] [PubMed] [Google Scholar]
- 29.Insel PS, Mattsson N, Mackin RS, et al. Biomarkers and cognitive endpoints to optimize trials in Alzheimer's disease. Ann Clin Transl Neurol 2015;2:534–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wood SN. Generalized Additive Models: An Introduction with R. Boca Raton, FL: Chapman and Hall/CRC Press; 2006. [Google Scholar]
- 31.Wood SN. Monotonic smoothing splines fitted by cross validation. SIAM J Sci Comput 1994;15:1126–1133. [Google Scholar]
- 32.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B 1995;57:289–300. [Google Scholar]
- 33.Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011;7:280–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mattsson N, Insel PS, Donohue M, et al. Predicting reduction of cerebrospinal fluid β-amyloid 42 in cognitively healthy controls. JAMA Neurol 2015;72:554–560. [DOI] [PubMed] [Google Scholar]
- 35.Mattsson N, Insel PS, Donohue M, et al. Independent information from cerebrospinal fluid amyloid-β and florbetapir imaging in Alzheimer's disease. Brain 2015;138:772–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reiman EM, Caselli RJ, Chen K, Alexander GE, Bandy D, Frost J. Declining brain activity in cognitively normal apolipoprotein E ɛ4 heterozygotes: a foundation for using positron emission tomography to efficiently test treatments to prevent Alzheimer's disease. Proc Natl Acad Sci 2001;98:3334–3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mackin RS, Insel P, Zhang J, et al. Cerebrospinal fluid α-synuclein and Lewy body-like symptoms in normal controls, mild cognitive impairment, and Alzheimer's disease. J Alzheimers Dis 2015;43:1007–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Keage HA, Hunter S, Matthews FE, et al. TDP-43 pathology in the population: prevalence and associations with Dementia and age. J Alzheimers Dis 2014;42:641–650. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.