

Abstract
Mitochondrial myopathy with lactic acidosis and sideroblastic anemia (MLASA) is an oxidative phosphorylation disorder, with primary clinical manifestations of myopathic exercise intolerance and a macrocytic sideroblastic anemia. One cause of MLASA is recessive mutations in PUS1, which encodes pseudouridine (Ψ) synthase 1 (Pus1p). Here we describe a mouse model of MLASA due to mutations in PUS1. As expected, certain Ψ modifications were missing in cytoplasmic and mitochondrial tRNAs from Pus1−/− animals. Pus1−/− mice were born at the expected Mendelian frequency and were non-dysmorphic. At 14 weeks the mutants displayed reduced exercise capacity. Examination of tibialis anterior (TA) muscle morphology and histochemistry demonstrated an increase in the cross sectional area and proportion of myosin heavy chain (MHC) IIB and low succinate dehydrogenase (SDH) expressing myofibers, without a change in the size of MHC IIA positive or high SDH myofibers. Cytochrome c oxidase activity was significantly reduced in extracts from red gastrocnemius muscle from Pus1−/− mice. Transmission electron microscopy on red gastrocnemius muscle demonstrated that Pus1−/− mice also had lower intermyofibrillar mitochondrial density and smaller mitochondria. Collectively, these results suggest that alterations in muscle metabolism related to mitochondrial content and oxidative capacity may account for the reduced exercise capacity in Pus1−/− mice.
Pseudouridine (Ψ), an abundant base modification1 found in snRNAs2, tRNAs3, and rRNAs4, strengthens base-pairing5,6,7,8 and stabilizes base stacking9. Ψ in snRNAs is required for pre-mRNA splicing10,11,12,13 and Ψ in tRNAs is involved in stop codon suppression14, recoding efficiency15, and translational specificity16. Furthermore, in the large ribosomal subunit rRNA, many of the Ψs are clustered in the peptidyl-transferase center and other functional regions17.
Pseudouridine synthases are responsible for the formation of Ψ in RNA in either a site-specific or snoRNA guide RNA-dependent manner. Pseudouridine synthase 1 (Pus1p) is a member of the TruA family18,19, and members of this family, and all other pseudouridine synthases, have a conserved aspartate in the active site20,21,22,23,24,25,26. Of the site-specific synthases, Pus1p is one of the best studied and, at a minimum, is responsible for the pseudouridylation of tRNAs18,27,28,29, U2 snRNA28,30, and steroid receptor RNA activator RNA (SRA)26,31,32,33. Pus1p is a co-activator of nuclear receptor-dependent transactivation, modifying SRA as a part of a promoter-bound complex26. The capacity of Pus1p to modify several different types of RNA is unusual, compared with other pseudouridine synthases, and may be the result of its lack of strict sequence recognition requirements34. Instead it relies on local secondary structure and the appropriate presentation of the uridine to be modified in the active site33,34,35.
Mitochondrial myopathy with lactic acidosis and sideroblastic anemia (MLASA; OMIM #600462 and 613561) is an autosomal recessive oxidative phosphorylation disorder that affects muscle and bone marrow, resulting primarily in exercise intolerance and anemia36,37,38,39. MLASA may also be associated with mental retardation and/or craniofacial abnormalities37,39. MLASA is the result of mutations in PUS140,41 or YARS2, which encodes mitochondrial tyrosyl-tRNA synthetase42,43,44,45. Cell lines derived from patients with MLASA who have mutations in PUS1, lack Pus1p activity as well as Pus1p-dependent pseudouridinylation46 and have alterations in gene expression47.
Although Ψ is important in the structure and function of RNA, it is unclear why the loss of Pus1p activity would result in the set of symptoms that are hallmarks of MLASA. Deletion of PUS1 in yeast results in no obvious phenotype27. However, deletion of PUS1 in combination with loss of PUS4, which modifies position 55 in nearly all tRNAs in yeast and is itself non-essential, is lethal, possibly due to a defect in nuclear export of certain tRNAs48. The loss of Pus1p in C. elegans results in a slight delay in maturation that does not result in a noticeable change in any other aspect of nematode morphology, locomotion, or metabolism49. In this report, we describe the phenotype of mice with a germline deletion of PUS1.
Materials and Methods
Assurances
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol (1775-100563-013012) was approved by the Institutional Animal Care and Use Committee (IACUC) of the University of South Carolina (Assurance number A3049-01) and that the methods were carried out in accordance with the approved protocol. At Children’s Hospital the protocol number 13-08-2476R was approved by the IACUC at Children’s Hospital Boston (Assurance number is A3303-01) and that the methods were carried out in accordance with the approved protocol. Human subjects were not used in these studies, at either the University of South Carolina or Children’s Hospital Boston.
PUS1 gene targeting
The targeting vector (Fig. 1) was constructed using standard recombinant DNA protocols. Briefly, the 5′ arm (2486 bp) was generated by PCR50 using primers: 5′TTTGTCGACATTTAAATAGAACCCAAGACTGTCCTG and 5′CTAGTCGACGACCCCAGATTCCTCTAAAG. The cloned fragment was inserted into the SalI site of pNTRLacZPGKNeoLoxp. The 3′ arm (2379 bp) was prepared similarly using the primers 5′TTTCGGCCGACAGCTGAAAGGTATGTGTCCTACTTG and 5′TTTGCGGCCGCAGTTTAAACCTGACACTGTGGTTCTGGTG and an EagI fragment was cloned into the NotI site in the vector. A thymidine kinase (TK) cassette was incorporated into the vector and the resulting targeting construct was linearized with Swa I and electroporated into mouse AB2.2 (129 SV/EV) ES cells (Darwin Transgenic Core, Baylor College of Medicine https://www.bcm.edu/research/advanced-technology-core-labs/lab-listing/mouse-embryonic-stem-cell-core/). Resulting G418 resistant/TK minus cell lines were screened for recombination51. Correctly targeted ES cell lines were identified, expanded, and two were injected into C57BL/6J blastocysts to generate chimeric mice and germline transmission obtained by breeding to C57BL/6 mice. The neomycin resistance cassette was removed by breeding to CMV-CRE mice (Jackson Laboratory- B6.C-Tg(CMV-cre)1Cgn/J, stock# 006054) and confirmed using the following primers: Pus1ex3F 5′TTGCAGAAGTGATGGTCAGC, Pus1ex3 R 5′AGTAGAAGCCGCAGGCAAGT, and Pus1-Neo KO F2 5′CCCCCTGAACCTGAAACATA. The wild-type allele produces a 249 bp fragment and mutant allele without the Neo cassette produces a 622 bp fragment. Genotypes were determined using the following primers: mPus1PCRscreenFor1, 5′TTTTGGGGAGTGGTTCTGAC; mPus1PCRscreenRev1, 5′GAAAGCAAAGGCCAGTGAAG; LacZFor, 5′TTCACTGGCCGTCGTTTTACAACGTCGTGA; and LacZRev, 5′ATGTGAGCGAGTAACAACCCGTCGGATTCT. The amplicons from the wild-type PUS1 locus and the LacZ in the mutant locus result in 940 bp and 364 bp fragments, respectively. The allele was subsequently backcrossed onto C57BL/6J by heterozygous x wild-type matings for greater than 5 generations prior to experimentation.
Figure 1. Mouse Pus1 gene targeting strategy and assay of genotype.

In panel (A) the targeting strategy is presented with a diagram of the wild-type gene on the top, indicating the positions of the initiator codon in exon II and the stop codon in exon VII with arrows. Exon III is indicated with an arrowhead because this exon contains the codon for the Asp that is essential for activity (see Results). This exon was targeted for disruption and the resulting mutant gene is shown in the lower diagram. In panel (B) an example of an ethidium bromide-stained gel employed to determine the genotypes of the mice is shown. The lane labeled C denotes a control where no DNA was added to the PCR reaction, +/+ is the result when DNA from wild-type mice is used, +/− is the result when DNA from heterozygous mice is used, and −/− is the result when DNA from homozygous mutant mice is used. M denotes the 1Kb+ marker (Promega, Madison, Wisconsin) with sizes in bp on the right. The sizes the bands resulting from the wild-type (940 bp) and mutant (364 bp) genes are indicated on the left of the panel. In panel (C) a 1.5% agarose gel stained with ethidium bromide showing the results of real time-PCR reactions of mRNA from three Pus1+/+ and three Pus1−/− mice with primers specific for Pus1p mRNA in the top portion of the gel and primers specific for PPIA in the bottom portion. The marker in the far-left lanes is the GeneRuler 50 bp DNA ladder (Life Technologies) with sizes represented on the left in the top portion. The far right lanes in both portions of the gel are the results of using no cDNA in the PCR reaction.
Assay of tRNA pseudouridylation
The CMCT/primer extension method52 was employed to assay for the presence of Ψ in a specific tRNA in total RNA from mouse kidney. The following primers we used: muscytoIleAAURev2Fluor 5′IRDye®700GATCGAACCCGCGACCTTGG and musmtHisGUGRevFluor 5′IRDye®800GGTTTATTTCCTGTTGTCAG. Amplicons were sequenced with the fmole® sequencing kit (Promega) according to the manufacturer’s recommendations. The template for the tRNAIle(AAU) sequencing reactions was a plasmid containing the entire coding sequence of the mouse cytoplasmic tRNAIle(AAU) gene in pGEMT. The template for the mouse mitochondrial tRNAHis(GUG) sequencing reactions was a gel purified PCR fragment, 65 bp in length, amplified from mouse genomic DNA with the following primers: musmtHisGUGFor, 5′GTGAATATAGTTTACAAAAAAC and musmtHisGUGRev1, 5′GAATAAGGAGGTTTATTTCC, using standard PCR protocols50. The samples were electrophoresed on 15% acrylamide:bis acrylamide (19:1) denaturing (8.3 M urea) gels and the fluorescence detected on a Li-Cor Odyssey CLX infrared imager (Instrumentation Resource Facility at the University of South Carolina School of Medicine (http://irf.med.sc.edu)) with no further treatment of the gel53. 1-cyclohexyl-3(2-morpholinoethyl) carbodiimide metho-p-toluenesulphonate (CMCT; was purchased from Sigma-Aldrich. The fluorescently-labeled primers were purchased from Integrated DNA Technologies (IDT; Coralville, Iowa) with IRDye®700 or IRDye®800 at the 5′ end, and these primers were HPLC purified by IDT.
Real time PCR
Total RNA was isolated from muscle tissue and cDNA was generated using iScript mix and Reverse transcriptase (BioRad, Inc.) as described in package inserts using 1 μg of total RNA per reaction, 1X iScript reaction mix and a 1:20 dilution of the iScript reverse transcriptase. The reactions were incubated at 25 °C for 5 minutes, at 42 °C for 30 minutes, and at 85 °C for 5 minutes. The cDNA was diluted 1:100 and 6 μl were used per real-time PCR reaction with iTaq Universal SYBR Green Supermix (BioRad) diluted 1:1 and 500 nM of each primer54. The two primers used to amplify from PUS1 mRNA are: forward, 5′AGCATCCTGCAAAGAGGGTC and Reverse, 5′GCCTGGACTAAGGCCGATAC. For peptidylprolyl isomerase A (PPIA) mRNA the two primers used were: forward, 5′GCAAATGCTGGACCAAACAC and reverse, 5′AGAGCTGTCCACAGTCGGAA. The parameters for the PUS1 primers were 95 °C for 3 mins, then 95 °C for 30 sec, 60.5 °C for 30 secs, 40×. The parameters for the PPIA reactions were the same except the annealing/extension temperature was increased to 63.9 °C.
DNA microarrays and gene expression analyses
Total RNA for gene expression analysis was isolated from frozen tissues (brain (cross section through whole brain), heart (cross section through atrium and ventricle), kidney (cross section), liver, and skeletal muscle, gastrocnemius, red (slow) and white (fast), using the mirVanaTM PARISTM kit. RNA quality was assessed using an Agilent 2100 Bioanalyzer and RNA Integrity Numbers (RIN) ranged from 7.1 to 9.0.
Microarray experiments were performed using Agilent’s platform. Total RNA samples were amplified and labeled using Agilent’s Low Input Quick Amp Labeling Kit according to the manufacturer recommendations. Briefly, samples contained 200 ng (brain, kidney, liver and white (fast) skeletal muscle) or 100 ng (heart and red (slow) skeletal muscle) of total RNA were converted into cDNA using a poly-dT primer that also contains the T7 RNA polymerase promoter sequence. Subsequently, T7 RNA polymerase was added to cDNA samples to amplify the original mRNA molecules and to simultaneously incorporate cyanine 3- or cyanine 5-labeled CTP (cRNA) into the amplification products. In addition, Agilent RNA spike-in controls (Cat. # 5188-5279) were added to samples prior to cDNA synthesis and used as experimental quality controls. In the next step, labeled RNA molecules were purified using Qiagen’s RNeasy Mini Kit. After spectrophotometric assessment of dye incorporation and cRNA yield, samples were stored at −80 °C until hybridization. Labeled cRNA samples were hybridized to SurePrint G3 Mouse GE 8 × 60 K Microarrays (Cat. # G4852A-028005) at 65 °C for 17 hours using Agilent’s Gene Expression Hybridization Kit according to the manufacturer’s recommendations. Wild-type and Pus1−/− (4 of each per tissue) samples were hybridized in a dye swap design. After washes, arrays were scanned using an Agilent DNA Microarray Scanner System.
Data was extracted from images with Feature Extractor Software version 10.7.3.1 (Agilent). In this process, background correction using additive and multiplicative detrending algorithms was performed. In addition, linear and LOWESS methods were used for dye normalization. Subsequently, data was uploaded into GeneSpring GX (Agilent), was log2 transformed, quantile normalized, and base line transformed using the median of all samples. Then, data was filtered by flags in a way that 75% of the samples in at least one of the two treatment groups have a “detected” flag and gene set enrichment/leading edge analysis was performed with the GSEA software55,56. The microarray data has been entered in the GEO database with the Superseries record number GSE77823.
Western blots
Mouse tissues (brain, heart, liver, red and white gastrocnemius skeletal muscle) in RIPA buffer (50 mM Tris pH 8, 150 mM NaCl, 0.1% SDS, 1.0% NP-40, 0.5% sodium deoxycholate, and 1:200 dilution of protease inhibitor (Sigma P8340)) were homogenized in Dounce homogenizers and extracted at 4 °C for 90 minutes on an orbital shaker. After a 12,000 rpm spin for 20 minutes at 4 °C (10,000xg)57, the protein concentration in the supernatant was determined and 40 μg of each sample was loaded on either a 12% polyacrylamide (29:1, acryl:bis) gel or an Any kD gel (BioRad, Inc). The gels were transferred to PVDF membrane, blocked, and reacted with primary and secondary antibodies as appropriate for the particular experiment.
Transmission electron microscopy
Liver and muscle samples were fixed at 4 °C in 2.5% glutaraldehyde in phosphate buffered saline and then processed using conventional techniques58,59. The samples were imaged on a JEOL 200CX transmission electron microscope in the Instrumentation Resource Facility (see above). Intermyofibrillar (IMF) density and size were determined by tracing the outline of IMF mitochondria at 10,000× magnification using Image J software (NIH, Bethesda, MD, USA). IMF mitochondria were defined as mitochondria greater than 1 μm from the plasma membrane and clearly integrated within the myofilaments60. IMF mitochondrial density is expressed as the percent of total myofiber area. An average of 105 mitochondria were traced per animal (N = 3/group) in a blinded fashion.
Run to Fatigue
The run to fatigue test was performed to examine endurance exercise capacity61. Mice were acclimated to the treadmill (both male and female experimenters were present for all tests) and the test began at 5 m/min at 5% grade. The speed was then increased after 5 minutes to 10 m/min, followed by 15 m/min after another 5 minutes. Thirty minutes into the test the speed was adjusted to a maximum of 25 m/min. The maximum time, in seconds, that the mice could run with gentle prods during the entire trial was recorded as the run to fatigue time.
Rotorod Testing
Neuromuscular coordination was assessed with the Rotorod (Columbus Instruments, Columbus, OH) as previously described61. Mice were placed on the Rotorod apparatus and subjected to a ramping protocol test. This protocol increases speed from 0 to 25 rpm over a period of 90 seconds and continued at 25 rpm until the maximum possible time for each trial, 300 seconds, was reached. The average and maximum speed (rpm) and time (seconds) for three trials were recorded. Each mouse performed three trials with 2 minutes rest between trials.
Grip Strength
Total grip strength of the forelimbs was measured with the Grip Tester (Columbus Instruments, Columbus, OH) as previously described61,62. Mice were placed on a 45° angled grid connected to a force transducer. Mice were then pulled away from the grid by the tail until they could no longer hold the grid. Each mouse performed 2 sets of 5 trials with two minutes rest between sets. The highest and lowest values were removed, and the average values in Newtons (N) were calculated.
Tissue collection
Mice were euthanized by CO2 asphyxiation. Hindlimb skeletal muscles and organs were excised, rinsed in PBS, snap-frozen in liquid nitrogen, and stored at −80 °C until further analysis. The right tibialis anterior (TA) was placed in optimal cutting temperature (OCT) solution and frozen in isopentane cooled in liquid nitrogen for histological analysis.
Tibialis anterior morphology
Serial transverse muscle sections (10 μm) were cut from the mid-belly of the tibialis anterior on a cryostat at −20 °C and stored at −80 °C until further analysis. Hematoxylin and eosin (H&E) staining was performed on cross-sections to examine muscle morphology. Digital photographs were taken from each cross-section at 40x magnification with a Nikon spot camera. For cross-sectional area, approximately 125 fibers per animal were traced with imaging software (ImageJ-NIH) in a blinded fashion. Centralized nuclei, defined as nuclei found equidistant from a well-defined sarcolemma, were quantified from these images, and is expressed as the percent of centralized nuclei per total muscle fibers. The extracellular matrix area was quantified as previously described62,63. Images containing well-defined sarcolemma were traced and the extracellular matrix is expressed as the percentage of whole muscle.
Immunohistochemistry for myosin heavy chain IIA and IIB
Immunohistochemistry for myosin heavy chain IIA and IIB was performed as previously described64. Transverse muscle sections were air dried for 10 minutes, fixed in cold acetone for 1 minute, and washed in PBS for 5 minutes. Cross-sections were quenched in 0.3% H2O2-methanol solution for 20 minutes and rinsed in PBS for 5 minutes, three times. Sections were then blocked in 10% normal goat serum (Vectastain ABC kit, Vector Laboratories, Burlingame, CA) in PBS for 1 hour at room temperature and then incubated overnight at 4 °C with primary antibodies (IIA: SC-71 and IIB: BF-F3). Following overnight incubation, the sections were then washed three times in PBS. Secondary antibodies were applied to the sections and incubated at 37 °C for 1 hour. The sections were washed three times and the avidin-biotin complex system (ABC: Vector Laboratories) was used to detect the biotinylated secondary antibody (30 minutes at RT). Sections were washed three times in PBS and visualized by incubating in DAB solution for 6 minutes (Vectastain DAB kit). Slides were washed in dH2O, dried and mounted with Permount. Digital images were acquired as previously described (see Tibialis Anterior Morphology above).
Succinate Dehydrogenase Staining
Succinate dehydrogenase (SDH) enzyme activity was performed as previously described to determine myofiber oxidative capacity64. Frozen sections were air dried at room temperature for 10 min, followed by incubation in a solution containing 0.2 M phosphate buffer (pH 7.4), 0.1 M MgCl2, 0.2 M succinic acid, and 2.4 mM nitroblue tetrazolium at 37 °C for 45 min. Sections were then washed in dH2O for 3 min, dehydrated in 50% ethanol for 2 min, and mounted for viewing with mounting media. Digital images were acquired as previously described (see Tibialis Anterior Morphology above). The percentage of SDH positive fibers was determined at 20×. The SDH staining intensity was determined by subtracting the background from each slide to create an integrated optical density for each myofiber. The percentages of each stain (dark and light) were quantified and expressed as percent of total muscle fibers. High and low SDH activity myofibers were traced at a 40× magnification in a blinded fashion. Approximately 100 myofibers per animal were traced for both high and low SDH activity myofibers.
Cytochrome c oxidase (COX) activity
COX enzyme activity was used as a measure of mitochondrial content as previously described65. Whole gastrocnemius (red) muscle tissue (~10 mg) was homogenized on ice in extraction buffer (0.1 M KH2PO4 and 2 mM EDTA pH 7.2) and enzyme activity was determined by the maximal oxidation rate of completely reduced cytochrome c, evaluated as a change in absorbance at 550 nm using an Eppendorf Biophotometer.
Statistical Analysis
Data were analyzed using Graph Pad 6.0 software and values are reported as means ± standard error of the mean. Data were analyzed using Students t-test. Significance levels were set at p < 0.05.
Results
Gene targeting
Because the aspartate required for Pus1p activity is located in exon 366, we targeted that region for disruption (see Fig. 1A). Nearly the entire exon was replaced in mouse embryonic stem cells with an internal ribosome entry site (IRES)/LacZ sequence and a PGK Neo cassette flanked by loxP sites (see Materials and Methods). The PGK Neo cassette was excised by breeding to CMV-CRE transgenic animals (Materials and Methods). To simplify the characterization and analysis of the knockout, and because it was already known that heterozygous parents and siblings of MLASA patients are unaffected40,46, only wild-type and homozygous mutant (Pus1+/+ and Pus1−/−, respectively) mice derived from heterozygous (Pus1+/− x Pus1+/−) matings of animals backcrossed to C57BL/6J at least 5 generations, were used for experimental studies.
Confirmation of Pus1p enzyme deficiency
The results from a real time, reverse transcriptase PCR analysis of three Pus1+/+ and three Pus1−/− mice are shown in Fig. 1C. Samples from a real-time PCR protocol that employs one primer in the deleted exon, show the lack of a band for Pus1p mRNA in the Pus1−/− samples. However, all the samples with cDNA have a band for the loading control, PPIA, illustrating that the Pus1−/− mice lack mRNA that would code for an active enzyme. Pus1p modifies uridines at positions 27 and 28 in most cytoplasmic and mitochondrial tRNAs18,27,28,66 and position 30 in yeast and mouse pre-tRNAIle(UAU)28,29. To determine if Pus1p activity was indeed absent in the Pus1−/− mice, we assessed pseudouridinylation in cytoplasmic tRNAIle(AAU) and mitochondrial tRNAHis(GUG) (mt-tRNAHis(GUG); see Fig. 2A) using a reverse transcriptase (RT) primer-extension/CMCT assay in total RNA from kidney tissue of wild-type and knockout mice (Fig. 2). This method exploits the fact that Ψ induces a stop in reverse transcription when reacted with CMCT. Positions 27 and 30 in mouse tRNAIle are pseudouridylated, but only position 27 is modified by Pus1p28,29,67. As expected, in RNA samples from Pus1−/− mice we observed an absence of Ψ27 in tRNAIle(AAU) (Fig. 2B) as well as Ψ27 and 28 in mt-tRNAHis(GUG) (Fig. 2C). By contrast, also as expected, position 30 in tRNAIle(AAU) was modified in the Pus1−/− mice (Fig. 2B). Overall, these assays show that Pus1p activity is missing in the knockout mice and that there is no redundant activity that can modify these positions in the tRNAs.
Figure 2. Primer extension assays to test for Pus1p activity.

Panel (A) shows the sequence and predicted secondary structure67 of mouse cytoplasmic tRNAIle(AAU) and mouse mitochondrial tRNAHis(GUG). The known Ψs at positions 27 and 30 in tRNAIle67 and predicted Ψs at positions 27 and 28 in tRNAHis are indicated by boxes. The additional modified nucleotides shown on tRNAIle are: D, dihydrouridine; I, inosine; m1A, 1-methyladenine; m5C, 5-methylcytidine; m5U, 5-methyluridine; m7G, 7-methylguanidine; m2,2G, N2,N2-dimethylguanidine; m2G, N2-methylguanidine; t6A, N6-threonylcarbamoyladenine71. In panels (B,C) a fluorescently-labeled primer specific for mouse tRNAIle (B) and a primer specific for mouse tRNAHis (C) were was used in primer extension reactions to determine the location of Ψ in samples of mouse kidney total RNA as described in Materials and Methods. In panels (B,C) lanes 5–7 contain wild-type control RNA (0) or RNA samples reacted with 0.042 or 0.167M CMCT (solid triangle), whereas lanes 8–10 are the result when RNA from Pus1−/− mice is used in the reactions. In panel (B) the sequence of tRNAIle in this region is shown in Lanes 1–4. In panel (C) the sequence of tRNAHis is shown in Lanes 1–4. For panels (B,C) the stops to RT that indicate Ψ in the primer extension reactions are shown on the right and positions of the modified uridines are indicated on the left, in the sequences. The band seen even in lanes not treated with CMCT, just below G26 in the sequence (lanes 5–10, panel B), is most likely due to the presence of N2,N2-dimethylguanidine (m2,2G, Fig. 2A) at position 26 in mouse cytoplasmic tRNAIle(AAU)71. This nucleotide modification has been observed by others to be a stop to RT even in untreated RNA72.
Pus1p deficiency phenotype
At seven weeks of age, female Pus1−/− mice were significantly smaller than their wild-type littermates. The gastrocnemius to body weight ratio was also significantly smaller, indicating not only growth retardation, but also disproportionally decreased muscle mass (Table 1). Complete blood count (CBC) analysis demonstrated no differences in white blood cells (WBC), hemoglobin (HGB) or hematocrit (HCT), or in red blood cell (RBC) parameters, including RBC number (RBC), mean corpuscular volume (MCV), mean corpuscular hemoglobin (MCH), or red blood cell distribution width (RDW) (Table 2). Siderocytes and ringed sideroblasts were not present on iron-stained smears of peripheral blood and bone marrow, respectively (not shown). In sum, in contrast to MLASA patients, there appears to be no overt hematological phenotype in Pus1p deficient mice.
Table 1. Body weight and relative gastrocnemius muscle mass in female wild-type and Pus1 −/− mice at 7 weeks of age.
| n | Body weight (BW;g)1 | Gastroc/BW (×100) | |
|---|---|---|---|
| WT | 7 | 19.4 ± 0.3 | 0.54 ± 0.01 |
| Pus1−/− | 5 | 15.8 ± 0.6† | 0.48 ± 0.02* |
Abbreviations: n, sample size. BW, body weight. g, grams. Gastroc, gastrocnemius.
1Entries are given as the mean plus/minus the standard error of the mean.
†Significant difference in the means of the two genotypes, p < 0.01.
*Significant difference in the means of the two genotypes, p < 0.05.
Table 2. Blood parameters in female wild-type and Pus1 −/− mice at 7 weeks of age.
| n | WBC ×103/μl1 | RBC ×10−3/μl | HGB g/dL | HCT% | MCV fL | MCH pg | RDW% | |
|---|---|---|---|---|---|---|---|---|
| WT | 6 | 6.05 ± 0.31 | 8.27 ± 0.23 | 13.0 ± 0.2 | 49.3 ± 1.5 | 59.6 ± 0.3 | 15.8 ± 0.3 | 12.6 ± 0.2 |
| Pus1−/− | 4 | 6.12 ± 1.67 | 8.13 ± 0.25 | 12.9 ± 0.3 | 47.0 ± 1.7 | 57.9 ± 0.9 | 16.0 ± 0.2 | 12.8 ± 0.3 |
Abbreviations: WT, wild-type. WBC, white blood cell. RBC, red blood cell. HGB, hemoglobin. HCT, hematocrit. MCV, mean corpuscular volume. MCH, mean corpuscular hemoglobin. RDW, Red blood cell distribution width. μl, microliter, g/dL. gram per deciliter. %, percent. fL, femtoliters (×10−15 L). pg, picogram.
1Entries are given as the mean plus/minus the standard error of the mean.
At 14 weeks of age, we re-examined body weight and muscle parameters in male and female Pus1−/− mice. At this age, overall there was no significant difference in body weight for Pus1−/− mice (Table 3), nor were there differences in the tibia length, or ratios for the tibialis anterior mass or gastrocnemius mass to body weight. We did observe significant differences in body weight and TA muscle mass between male wild-type and Pus1−/− mice at this age. Wild-type male mouse body weight average was 29.4 ± 0.7 g and the male Pus1−/− mice were 27.1 ± 0.6 g (p < 0.05) and the TA mass was 57.9 ± 1.5 for the wild type male mice and 50.8 ± 1.9 (p < 0.05) for the male Pus1−/− mice but these differences in muscle mass disappeared when corrected for body weight. No significant differences were observed between female genotypes on body weight or muscle mass at this age. These data indicated that although body weight and muscle mass were delayed in the mutant compared to the wild type, these parameters equilibrated as the mice aged.
Table 3. Body weight, tibia length and muscle mass to body weight ratios in wild-type and Pus1 −/− mice at 14 weeks of age.
| n* | Body weight (BW; g)1 | Tibia Length (mm) | TA mass/BW (×100) | Gastroc Mass/BW (×100) | |
|---|---|---|---|---|---|
| WT | 13 | 25.8 ± 1.4 | 16.8 ± 0.1 | 0.19 ± 0.004 | 0.51 ± 0.01 |
| Pus1−/− | 9 | 23.5 ± 1.4 | 16.4 ± 0.2 | 0.18 ± 0.004 | 0.50 ± 0.01 |
Abbreviations: n, sample size. WT, wild-type. BW, body weight. g, grams. mm, millimeter. TA, tibialis anterior. Gastroc, gastrocnemius.
1Entries are given as the mean ± standard error of the mean.
*For WT there were 8 males and 5 females, for Pus1−/− there were 4 males and 5 females. A body weight for a female Pus1−/− mouse was not taken at sacrifice (n = 1); therefore, data for 8 mice are presented in Table 3. A total of 9 mice were used throughout all other measures.
To examine the effect of the loss of Pus1p on musculoskeletal function we assessed strength, neuromuscular coordination, and endurance capacity using standard functional tests. There were no significant differences in grip strength or in maximum rotorod speed and time (Table 4), suggesting that the loss of Pus1p does not have a direct effect on neuromuscular coordination. However, in an exercise treadmill test, Pus1−/− mice fatigued more quickly exhibiting a 46% decrease in the run-to-fatigue time (p < 0.05, Fig. 3), suggesting an effect of the loss of Pus1p on endurance capacity.
Table 4. Average grip strength and max rotorod speed and time in male and female wild-type and Pus1 −/− mice.
| n1 | Grip Strength (N) | Max Rotorod Speed (rpm) | Max Rotorod Time (sec) | |
|---|---|---|---|---|
| WT | 13 | 2.0 ± 0.1 | 19.8 ± 1.2 | 111.8 ± 24.3 |
| Pus1−/− | 9 | 1.8 ± 0.1 | 20.6 ± 1.4 | 107.5 ± 26.0 |
Abbreviations: n, sample size. WT, wild-type. N, Newton. rpm, revolutions per minute. sec, seconds.
1Entries are given as the mean plus/minus the standard error of the mean. For WT there were 8 males and 5 females and for Pus1−/− there were 4 males and 5 females.
Figure 3. Endurance exercise capacity in wild-type and Pus1−/− mice.
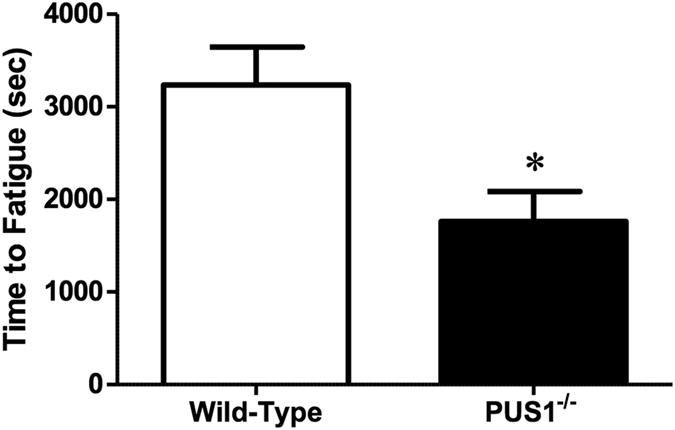
Total time during a run to fatigue exercise test is presented in seconds (sec) in wild-type and Pus1−/− mice. A total of 13 wild-type and 9 Pus1−/− mice were examined. Data were analyzed with t-Tests and significance was set at p < 0.05. An asterisk (*) indicates the results for Pus1−/− mice are significantly different from wild-type mice at p < 0.05.
To examine the effect of the loss of Pus1p on muscle morphology, fiber type, and oxidative capacity, we performed quantitative morphometry on serial cross sections taken from the TA muscle. H&E stained muscle sections were used to examine general muscle morphology. There was a 24% increase in mean cross-sectional myofiber area in the tibialis anterior muscle in the Pus1−/− mice compared to wild-type mice (Fig. 4A). However, there were no changes in the percentage of extracellular matrix area or centralized nuclei (Fig. 4B,C). Overall, this suggests the loss of Pus1p has an effect on skeletal muscle myofiber area that is not related to the degeneration and regeneration of muscle fibers.
Figure 4. Tibialis anterior morphology in wild-type and Pus1−/− mice.

(A) Mean cross sectional area (CSA) of myofibers in the tibialis anterior in wild-type and Pus1−/− mice. Data are expressed as the percentage of wild-type to account for differences in size between male and female mice. (B) The percentage of extracellular matrix area in the tibialis anterior in wild-type and Pus1−/− mice. Data are expressed as the percentage of extracellular matrix area to the total myofiber area. (C) The percentage of centralized nuclei in the tibialis anterior in wild-type and Pus1−/− mice. Data are expressed as the percentage of total myofibers containing centralized nuclei. Muscles from 13 wild-type and 9 Pus1−/− were examined. Data were analyzed with t-Tests and significance was set at p < 0.05. An asterisk (*) indicates the results for Pus1−/− mice are significantly different from wild-type mice at p < 0.05.
Muscle sample cross sections from the two genotypes were also stained with Gomori trichrome and NADH reductase stains (see Supplemental Figure S1) and the muscle from Pus1−/− mice shows distinctly increased coarse red staining with Gomori Trichrome, and coarse reaction product with NADH reductase histochemistry. To further examine the myofiber changes, we examined the percentage and cross-sectional area of fibers stained positive for MHC type IIA and IIB. We found that the proportion of MHC IIA positive fibers decreased (37% versus 21%) and MHC IIB positive fibers reciprocally increased (58% versus 71%) in the Pus1−/− mice (Fig. 5A,B). Interestingly while there was no change in the mean cross-sectional area of MHC IIA myofibers, there was a significant increase (17%) in the mean cross-sectional area of IIB myofibers in Pus1−/− mice. This indicates that the histological changes in myofiber cross sectional area can be accounted for entirely by alterations in MHC IIB myofibers.
Figure 5. Tibialis anterior fiber type percentages and myofiber areas in wild-type and Pus1−/− mice.

(A) Representative images (10×) of tibialis anterior MHC IIA and IIB myofibers in wild-type and Pus1−/− mice. (B) Percentage of MHC IIA and IIB myofibers in the tibialis anterior of wild-type and Pus1−/− mice. Data are expressed as the percentage of total myofibers stained positive for MHC IIA and IIB. (C) Mean cross sectional area of MHC IIA and IIB myofibers in the tibialis anterior of wild-type and Pus1−/− mice. Data are expressed as the percentage of wild-type to account for differences in size between male and female mice. Muscles from 13 wild-type and 9 Pus1−/− were examined. Data were analyzed with t-Tests and significance was set at p < 0.05. An asterisk (*) indicates the results for Pus1−/− mice are significantly different from wild-type mice at p < 0.05.
To examine whether the absence of Pus1p has an effect on oxidative capacity, SDH staining was utilized to quantify the percentage and cross-sectional area of high SDH activity myofibers. There was a 38% decrease in the number of high SDH activity myofibers in the Pus1−/− mice (Fig. 6A,B). Similar to the observations in the fiber typing (Fig. 5), there was no difference in the mean cross-sectional area of high SDH activity myofibers, but there was an increase in the mean cross-sectional area of low SDH activity myofibers (Fig. 6C). Additional analysis demonstrated that fibers staining for low SDH activity were also type IIB positive. In toto, these data support the conclusion that while there is an overall maintenance of muscle mass, there is a reduction in oxidative capacity that is related to lower high SDH activity myofibers and increased percentage and size of type IIB, glycolytic fibers.
Figure 6. Tibialis anterior succinate dehydrogenase (SDH) activity in wild-type and Pus1−/− mice.

(A) Representative images (10×) of tibialis anterior SDH staining in wild-type and Pus1−/− mice. (B) The percentage of high SDH activity myofibers in the tibialis anterior of wild-type and Pus1−/− mice. (C) Mean cross sectional area of tibialis anterior high and low SDH activity myofibers in wild-type and Pus1−/− mice. Data are expressed as the percentage of wild-type to account for differences in size between male and female mice. Muscles from 13 wild-type and 9 Pus1−/− were examined. Data were analyzed with t-Tests and significance was set at p < 0.05. An asterisk (*) indicates the results for Pus1−/− mice are significantly different from wild-type mice at p < 0.05.
Western blots of protein samples extracted from tissues (red and white gastrocnemius muscle, heart, liver, and brain) were probed with the Mitoprofile (Abcam, Inc.) cocktail of antibodies. This cocktail of five mono-clonal antibodies recognizes the most labile component of each of the five oxidative phosphorylation complexes, which can be used to determine differences in the levels of each complex between tissues from wild-type and Pus1−/− mice. The only significant difference seen in all of the blots probed with Mitoprofile was in the heart, as Complex IV was elevated (41% higher; p < 0.05) in the Pus1−/− mice (see Supplemental Figure S2). Given the SDH activity results presented above, we anticipated that the level of the band at 30 KDa, which is the Succinate dehydrogenase [ubiquinone] iron-sulfur subunit, mitochondrial (SDHB) protein, might be different between the two genotypes, but it was not.
Transmission electron microscopy of red and white gastrocnemius muscle from the wild-type and Pus1−/− mice confirmed, as suggested by histochemical studies, that skeletal muscle mitochondria are affected by the loss of Pus1p. As shown in Fig. 7A, the mitochondria in the Pus1−/− animals appear smaller and less abundant than those in the muscles from wild-type mice. None of the mitochondria, even those in the subsarcolemmal regions, showed evidence of paracrystalline inclusion bodies similar to those seen muscle biopsies from MLASA patients36,39. Unlike skeletal muscle mitochondria, liver mitochondria in Pus1−/− mice are not ultrastructurally abnormal (Fig. 7A). These differences in muscle mitochondria are manifest as a 45% reduction in mitochondrial density (p < 0.05, Fig. 7B) and a 33% reduction in average mitochondrial size (p < 0.05, Fig. 7C). There is an increase in the number of very small mitochondria in the Pus1−/− mice (Fig. 7D). COX activity was also measured in crude homogenates of the red gastrocnemius muscle to provide an assessment of mitochondrial function. Pus1−/− mice demonstrated a 63% reduction in COX activity compared to wild-type mice (p < 0.0001; Fig. 7E).
Figure 7. Transmission electron micrographs of liver and muscle samples in wild-type and Pus1−/− mice.

(A) Representative transmission electron micrographs (10,000X) of liver (left column), and the primarily red (middle column) and white portions (right column) of the gastrocnemius muscle from wild-type and Pus1−/− mice. The scale bar on the photograph represents 2 microns. (B) The intermyofibrillar (IMF) mitochondrial density of the red portion of the gastrocnemius muscle in wild-type and Pus1−/− mice. Data are presented as the percentage of mitochondrial area per total fiber area. (C) The IMF mitochondria size (μm2) of the red portion of the gastrocnemius muscle in wild-type and Pus1−/− mice. Data are presented as the total mitochondrial size divided by the total fiber area. (D) The IMF mitochondria size (μm2) distribution of red gastrocnemius muscle in wild-type and Pus1−/− mice. Muscles from 3 animals per group were examined (~100 IMF mitochondria per animal). An asterisk (*) indicates the results for Pus1−/− mice are significantly different from wild-type mice at p < 0.05. (E) Cytochrome c oxidase (COX) activity of the red portion of the gastrocnemius muscle in wild-type and Pus1−/− mice. An asterisk (*) indicates the results for Pus1−/− mice are significantly different from wild-type mice at p < 0.05.
The results from microarray analysis (see Methods for experimental specifics) from six tissues (brain, heart, kidney, liver, red gastrocnemius muscle, and white gastrocnemius muscle) of eight mice (four wild-type and four Pus1−/− mice, two males and two females of each genotype) show patterns that correlate with the above results. Leading Edge Analysis shows that several pathways, such as those involved in mitochondrial biogenesis, respiration, and oxidative phosphorylation, for example, are affected in all the tissues tested (see Table S1 in Supplemental Material). Many of the genes listed in the table are enriched in the tissues from Pus1−/− mice when compared with the tissues from wild-type mice (see enrichment scores in columns for brain, heart, kidney, liver, and to some extent, red muscle in Table S1). But in white skeletal muscle the levels of many of those genes are enriched in the wild-type mice relative to the Pus1−/− mice. In other words, the Pus1−/− mice appear to be compensating for the loss of Pus1p by increasing the expression of genes involved in mitochondrial biogenesis and respiration in brain, heart, kidney, liver, and to some extent in red skeletal muscle, but not in white skeletal muscle. This predicts that white skeletal muscle will be the most affected in these pathways, which supports the histological and biochemical analysis outlined above. The top 50 features from the Gene Set Enrichment Analysis that are enriched in the tissues from wild-type mice and enriched in the Pus1−/− mice, are given in heat maps in Supplemental Figure S3A–F. For each tissue these are the 50 most enriched genes in the two genotypes of mice. An interesting feature in the heat map for Brain samples (Figure S3A) for instance, is that there are five aminoacyl tRNA-synthetases that are enriched in the Pus1−/− mouse brain, significant because the other cause for MLASA is the mutation of an aminoacyl tRNA-synthetase (see Introduction).
Taken collectively, these data demonstrate that Pus1p deficiency alters mouse skeletal muscle metabolism related to mitochondrial content and oxidative capacity, and these alterations may account for impaired exercise capacity independent of changes in muscle mass.
Discussion
MLASA is caused by mutations of either the Pus1 or the YARS2 genes, encoding Pus1p40,41 or mitochondrial tyrosyl-tRNA synthetase42,43,44,45, respectively. It is relatively straight forward to understand how the reduction in charging efficiency of an amino acyl-tRNA synthetase (ARS) found in mitochondria, combined with a lower level of expression of ARS in skeletal muscle, would negatively impact mitochondrial protein synthesis and lead to a oxidative phosphorylation disorder such as MLASA42. It is more difficult to grasp why the loss of Pus1p, an enzyme that forms Ψ on most cytoplasmic as well as the majority of mitochondrial tRNAs46, would result in the MLASA phenotype, affecting primarily skeletal muscle and bone marrow38. The Pus1 knockout mouse model described will help us answer this question in ways that MLASA patient-derived cell lines cannot46,47. In addition, the mouse model will provide a way to test interventions that will benefit MLASA patients.
Lacking a full complement of modifications, the tRNAs found in Pus1−/− mice will probably be affected in a number of ways. Transport of tRNAs out of the nucleus may be compromised, as was seen with yeast Pus1/Pus4 double mutants48. In preliminary experiments, RNA samples isolated from tissues of wild-type and Pus1−/− mice have been hybridized to tRNA microarrays68, and the results indicate that the levels of certain tRNAs are modulated in the Pus1−/− mice (unpublished results). In situ hybridization analysis of affected tRNAs in tissue samples from the knockout mice will be instrumental in determining if these tRNAs are retained in the nucleus, as the yeast evidence would suggest.
With regard to the differences seen in the muscle fibers, the TA muscle of the Pus1−/− mice showed an increase in the percentage of fibers expressing myosin heavy chain IIB and a decrease in IIA positive fibers relative to wild-type muscle. The overall increase in the mean cross sectional area in the muscles from the Pus1−/− mice is highly influenced by the increased percentage of large Type IIB fibers. This is indicative of a shift to a faster, more glycolytic muscle phenotype and would be expected when one considers the characteristics of MLASA patients: exercise intolerance and elevated levels of lactic acid. The timing and causes for these shifts in fiber composition are not known, but given exercise intolerance was seen in the Pus1−/− mice, we will be able to begin to understand the processes involved with this model of MLASA.
The decrease in COX activity and the percentage of high SDH activity fibers in the TA muscle in the Pus1−/− mice when compared to wild-type mice is indicative of a reduction in oxidative capacity in the muscles of the knockout mice. SDH is part of complex II and MLASA case studies have not reported irregularities with complex II from muscle biopsies but they have noted deficiencies in complexes I and IV38,39,41 in samples from MLASA patients. With this new model, it will now be possible to isolate mitochondria from fast and slow muscle types as well as other tissues, such as heart, liver and brain, for comparison of inner membrane complexes and respiration capacity.
Why is there no sign of anemia in the Pus1−/− mice? In published case reports of MLASA patients, the age where anemia is diagnosed is variable. In one report of two brothers, both were in their late teens (18 and 19) before anemia was added to the myopathy diagnosis36. Inbal et al.37 reported that a sister and brother were diagnosed with anemia at 14 and 10 respectively. Additional patients with MLASA and with the same ethnic background38,39, were diagnosed with anemia at 8, 12, 17, and 19 years of age, with an additional patient requiring transfusions by 16 years old due to severe anemia (this patient was at least 11 years old before anemia diagnosis). In these published cases, exercise intolerance is reported as an initiating symptom and anemia is either present initially or added to the diagnosis subsequently. The severity of the sideroblastic anemia fluctuated in two of the described cases36. It is possible that the mice will eventually develop anemia as they age. However, it is equally probable that due to differences in mitochondrial iron metabolism in mice and humans, including the absence of ring sideroblasts in most mouse models of sideroblastic anemia, that mice are simply not susceptible to this disorder69,70.
Overall, these results demonstrate that there are alterations in muscle metabolism related to mitochondrial content and oxidative capacity in the Pus1−/− mice. The Pus1 knockout model will eventually be used to test interventions to alleviate the symptoms of MLASA. The efficacy of a number of pharmaceuticals that are in clinical trials could be tested in this model and perhaps yield a treatment for this devastating disease.
Additional Information
How to cite this article: Mangum, J. E. et al. Pseudouridine synthase 1 deficient mice, a model for Mitochondrial Myopathy with Sideroblastic Anemia, exhibit muscle morphology and physiology alterations. Sci. Rep. 6, 26202; doi: 10.1038/srep26202 (2016).
Supplementary Material
Acknowledgments
This project was supported by the Mouse Embryonic Stem Cell Core at Baylor College of Medicine, a Research Development Fund Grant from the University of South Carolina School of Medicine and a South Carolina INBRE Pilot Grant to JRP, a National Institutes of Health (NIH; DK074368-01) grant to NF-G, and a NIH/National Cancer Institute research grant R01 CA121249 to JAC. Boston Chidren’s Hospital Center for Molecular Developmental Hematopoiesis, supported by NIH P30 DK49216, performed blastocyst injections. This work was supported by NIH R01 DK087992 to MDF.
Footnotes
Author Contributions J.E.M., J.P.H., D.K.F., M.J.P., J.E., D.A., Y.B., D.R.C., A.K.S. and J.R.P. planned and performed experiments and analyzed data. P.J.S. analyzed all of the iron parameters (serum and tissue) and CBC for the animals and H.G.W.L. performed and interpreted histology of samples. The PUS1 KO mouse was derived by J.R.P., Y.B., N.F.-G., M.D.F. and D.R.C. S.C.B. performed necropsies and interpreted histological specimens of the PUS1 KO mice. J.R.P, J.E.M, J.P.H. and D.R.C wrote the main manuscript text with additional contributions from Y.B., N.F.-G., M.D.F. and J.A.C. All authors reviewed and edited the manuscript.
References
- Davis F. F. & Allen F. W. Ribonucleic acids from yeast which contain a fifth nucleotide. J Biol Chem 227, 907–915 (1957). [PubMed] [Google Scholar]
- Massenet S. A., Mougin A. & Branlant C. In Modification and Editing of RNA (eds Grosjean H. & Benne R.) 201–227 (ASM Press, 1998). [Google Scholar]
- Auffinger P. & Westhof E. In Modification and Editing of RNA (eds Grosjean H. & Benne R.) 569–576 (ASM Press, 1998). [Google Scholar]
- Ofengand J. Ribosomal RNA pseudouridines and pseudouridne synthases. FEBS Lett. 514, 17–25 (2002). [DOI] [PubMed] [Google Scholar]
- Davis D. R., Veltri C. A. & Nielsen L. An RNA model system for investigation of pseudouridine stabilization of the codon-anticodon interaction in tRNALys, tRNAHis and tRNATyr. J Biomol Struct Dyn 15, 1121–1132 (1998). [DOI] [PubMed] [Google Scholar]
- Davis D. R. & Poulter C. D. 1H-15N NMR studies of Escherichia coli tRNA(Phe) from hisT mutants: a structural role for pseudouridine. Biochemistry 30, 4223–4231 (1991). [DOI] [PubMed] [Google Scholar]
- Hudson G. A., Bloomingdale R. J. & Znosko B. M. Thermodynamic contribution and nearest-neighbor parameters of pseudouridine-adenosine base pairs in oligoribonucleotides. RNA 19, 1474–1482 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilbille Y. et al. The structure of the human tRNALys3 anticodon bound to the HIV genome is stabilized by modified nucleosides and adjacent mismatch base pairs. Nucl Acids Res 37, 3342–3353 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis D. R. Stabilization of RNA stacking by pseudouridine. Nucleic Acids Res 23, 5020–5026 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segault V., Will C. L., Sproat B. S. & Luhrmann R. In vitro reconstitution of mammalian U2 and U5 snRNPs active in splicing: Sm proteins are functionally interchangeable and are essential for the formation of functional U2 and U5 snRNPs. Embo J 14, 4010–4021 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y. T., Shu M. D. & Steitz J. A. Modifications of U2 snRNA are required for snRNP assembly and pre-mRNA splicing. Embo J 17, 5783–5795 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X. & Yu Y.-T. Pseudouridines in and near the branch site recognition region of U2 snRNA are required for snRNP biogenesis and pre-mRNA splicing in Xenopus oocytes. RNA 10, 681–690 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donmez G., Hartmuth K. & Luhrmann R. Modified nucleotides at the 5′ end of human U2 snRNA are required for spliceosomal E-complex formation. RNA 10, 1925–1933 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerfass K. & Beier H. Pseudouridine in the anticodon G psi A of plant cytoplasmic tRNA(Tyr) is required for UAG and UAA suppression in the TMV-specific context. Nucleic Acids Res 20, 5911–5918 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecointe F. et al. Lack of pseudouridine 38/39 in the anticodon arm of yeast cytoplasmic tRNA decreases in vivo recoding efficiency. J Biol Chem 277, 30445–30453 (2002). [DOI] [PubMed] [Google Scholar]
- Perret V. et al. Relaxation of a transfer RNA specificity by removal of modified nucleotides. Nature 344, 787–789 (1990). [DOI] [PubMed] [Google Scholar]
- Ofengand J. & Fournier M. J. In Modification and Editing of RNA (eds Grosjean H. & Benne R.) 229–253 (ASM Press, 1998). [Google Scholar]
- Chen J. & Patton J. R. Cloning and characterization of a mammalian pseudouridine synthase. RNA 5, 409–419 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J. & Patton J. R. Pseudouridine Synthase 3 from Mouse Modifies the Anticodon Loop of tRNA. Biochemistry 39, 12723–12730 (2000). [DOI] [PubMed] [Google Scholar]
- Foster P. G., Huang L., Santi D. V. & Stroud R. M. The structural basis for tRNA recognition and pseudouridine formation by pseudouridine synthase I. Nat Struct Biol 7, 23–27 (2000). [DOI] [PubMed] [Google Scholar]
- Huang L., Pookanjanatavip M., Gu X. & Santi D. V. A conserved aspartate of tRNA pseudouridine synthase is essential for activity and a probable nucleophilic catalyst. Biochemistry 37, 344–351 (1998). [DOI] [PubMed] [Google Scholar]
- Ramamurthy V., Swann S. L., Paulson J. L., Spedaliere C. J. & Mueller E. G. Critical aspartic acid residues in pseudouridine synthases. J Biol Chem 274, 22225–22230 (1999). [DOI] [PubMed] [Google Scholar]
- Raychaudhuri S., Niu L., Conrad J., Lane B. G. & Ofengand J. Functional effect of deletion and mutation of the Escherichia coli ribosomal RNA and tRNA pseudouridine synthase RluA. J Biol Chem 274, 18880–18886 (1999). [DOI] [PubMed] [Google Scholar]
- Zebarjadian Y., King T., Fournier M. J., Clarke L. & Carbon J. Point mutations in yeast CBF5 can abolish in vivo pseudouridylation of rRNA. Mol Cell Biol 19, 7461–7472 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Campo M., Kaya Y. & Ofengand J. Identification and site of action of the remaining four putative pseudouridine synthases in Escherichia coli. RNA 7, 1603–1615. (2001). [PMC free article] [PubMed] [Google Scholar]
- Zhao X. et al. Regulation of nuclear receptor activity by a pseudouridine synthase through posttranscriptional modification of Steriod Receptor RNA Activator. Mol Cell 15, 549–558 (2004). [DOI] [PubMed] [Google Scholar]
- Simos G. et al. Nuclear pore proteins are involved in the biogenesis of functional tRNA. Embo J 15, 2270–2284 (1996). [PMC free article] [PubMed] [Google Scholar]
- Behm-Ansmant I. et al. A previously unidentified activity of yeast and mouse RNA:pseudouridine synthases 1 (Pus1p) on tRNAs. RNA 12, 1583–1593 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibert B. S., Fischel-Ghodsian N. & Patton J. R. Partial activity is seen with many substitutions of highly conserved active site residues in human Pseudouridine synthase 1. RNA 14, 1895–1906 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massenet S. et al. Pseudouridine mapping in the Saccharomyces cerevisiae spliceosomal U small nuclear RNAs (snRNAs) reveals that pseudouridine synthase pus1p exhibits a dual substrate specificity for U2 snRNA and tRNA. Mol Cell Biol 19, 2142–2154 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X. et al. Pus3p- and Pus1p-dependent pseudouridylation of steroid receptor RNA activator controls a functional switch that regulates nuclear receptor signaling. Mol Endocrinol 21, 686–699 (2007). [DOI] [PubMed] [Google Scholar]
- Ghosh S. K., Patton J. R. & Spanjaard R. A. A small RNA derived from RNA coactivator SRA blocks steroid receptor signaling via inhibition of Pus1p-mediated pseudouridylation of SRA: evidence of a novel RNA binding domain in the N-terminus of steroid receptors. Biochemistry 51, 8163–8172 (2012). [DOI] [PubMed] [Google Scholar]
- Huet T., Miannay F. A., Patton J. R. & Thore S. Steroid Receptor RNA Activator (SRA) Modification by the Human Pseudouridine Synthase 1 (hPus1p): RNA Binding, Activity, and Atomic Model. PloS One 9, e94610 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibert B. S. & Patton J. R. Pseudouridine synthase 1: a site-specific synthase without strict sequence recognition requirements. Nucleic Acids Res 40, 2107–2118 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czudnochowski N., Wang A. L., Finer-Moore J. & Stroud R. M. In human pseudouridine synthase 1 (hPus1), a C-terminal helical insert blocks tRNA from binding in the same orientation as in the Pus1 bacterial homologue TruA, consistent with their different target selectivities. J Mol Biol 425, 3875–3887 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawles J. M. & Weller R. O. Familial association of metabolic myopathy, lactic acidosis and sideroblastic anemia. Am J Med 56, 891–897 (1974). [DOI] [PubMed] [Google Scholar]
- Inbal A. et al. Myopathy, lactic acidosis, and sideroblastic anemia: a new syndrome. Am J Med Genet 55, 372–378 (1995). [DOI] [PubMed] [Google Scholar]
- Casas K. A. & Fischel-Ghodsian N. Mitochondrial myopathy and sideroblastic anemia. Am J Med Genet 125A, 201–204 (2004). [DOI] [PubMed] [Google Scholar]
- Zeharia A. et al. Mitochondrial myopathy, sideroblastic anemia, and lactic acidosis: an autosomal recessive syndrome in Persian Jews caused by a mutation in the PUS1 gene. J Child Neurol 20, 449–452 (2005). [DOI] [PubMed] [Google Scholar]
- Bykhovskaya Y., Casas K., Mengesha E., Inbal A. & Fischel-Ghodsian N. Missense mutation in pseudouridine synthase 1 (PUS1) causes mitochondrial myopathy and sideroblastic anemia (MLASA). Am J Hum Genet 74, 1303–1308 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Vizarra E., Berardinelli A., Valente L., Tiranti V. & Zeviani M. Nonsense mutation in pseudouridylate synthase 1 (PUS1) in two brothers affected by myopathy, lactic acidosis and sideroblastic anaemia (MLASA). J Med Genet 44, 173–180 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley L. G. et al. Mutation of the mitochondrial tyrosyl-tRNA synthetase gene, YARS2, causes myopathy, lactic acidosis, and sideroblastic anemia–MLASA syndrome. Am J Hum Genet 87, 52–59 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasarman F., Nishimura T., Thiffault I. & Shoubridge E. A. A novel mutation in YARS2 causes myopathy with lactic acidosis and sideroblastic anemia. Hum Mutat 33, 1201–1206 (2012). [DOI] [PubMed] [Google Scholar]
- Shahni R. et al. A distinct mitochondrial myopathy, lactic acidosis and sideroblastic anemia (MLASA) phenotype associates with YARS2 mutations. Am J Med Genet Part A 161, 2334–2338 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley L. G. et al. Phenotypic variability and identification of novel YARS2 mutations in YARS2 mitochondrial myopathy, lactic acidosis and sideroblastic anaemia. Orphanet J Rare Dis 8, 193 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton J. R., Bykhovskaya Y., Mengesha E., Bertolotto C. & Fischel-Ghodsian N. Mitochondrial myopathy and sideroblastic anemia (MLASA): missense mutation in the pseudouridine synthase 1 (PUS1) gene is associated with the loss of tRNA pseudouridylation. J Biol Chem 280, 19823–19828 (2005). [DOI] [PubMed] [Google Scholar]
- Bykhovskaya Y., Mengesha E. & Fischel-Ghodsian N. Pleiotropic effects and compensation mechanisms determine tissue specificity in mitochondrial myopathy and sideroblastic anemia (MLASA). Mol Genet Metabol 91, 148–156 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosshans H., Lecointe F., Grosjean H., Hurt E. & Simos G. Pus1p-dependent tRNA pseudouridinylation becomes essential when tRNA biogenesis is compromised in yeast. J Biol Chem 276, 46333–46339. (2001). [DOI] [PubMed] [Google Scholar]
- Patton J. R. & Padgett R. W. Caenorhabditis elegans pseudouridine synthase 1 activity in vivo: tRNA is a substrate but not U2 small nuclear RNA. Biochem J 372, 595–602 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullis K. B. & Faloona F. A. Specific synthesis of DNA in vitro via a polymerase-catalyzed chain reaction. Methods Enzymol 155, 335–350 (1987). [DOI] [PubMed] [Google Scholar]
- Southern E. M. Detection of specific sequences among DNA fragments separated by gel electrophoresis. J Mol Biol 98, 503–517 (1975). [DOI] [PubMed] [Google Scholar]
- Bakin A. V. & Ofengand J. Mapping of pseudouridine residues in RNA to nucleotide resolution. Methods Mol Biol 77, 297–309 (1998). [DOI] [PubMed] [Google Scholar]
- Ying B. W., Fourmy D. & Yoshizawa S. Substitution of the use of radioactivity by fluorescence for biochemical studies of RNA. RNA 13, 2042–2050 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl M. W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29, e45 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha V. K. et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nature Genet 34, 267–273 (2003). [DOI] [PubMed] [Google Scholar]
- Subramanian A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Nat Acad Sci USA 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller A. L., Desmond P. F., Hsia R. C. & Roche J. A. Improved immunoblotting methods provide critical insights into phenotypic differences between two murine dysferlinopathy models. Muscle Nerve 50, 286–289 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- White J. P. et al. IL-6 regulation on skeletal muscle mitochondrial remodeling during cancer cachexia in the ApcMin/+ mouse. Skelet Muscle 2, 14 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price R. L., Chintanowonges C., Shiraishi I., Borg T. K. & Terracio L. Local and regional variations in myofibrillar patterns in looping rat hearts. Anat Rec 245, 83–93 (1996). [DOI] [PubMed] [Google Scholar]
- Toth M. J., Miller M. S., Ward K. A. & Ades P. A. Skeletal muscle mitochondrial density, gene expression, and enzyme activities in human heart failure: minimal effects of the disease and resistance training. J Appl Physiol 112, 1864–1874 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puppa M. J. et al. The effect of exercise on IL-6-induced cachexia in the Apc (Min/+) mouse. J Cachexia, Sarcopenia Muscle 3, 117–137 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardee J. P. et al. The Effect of Radiation Dose on Mouse Skeletal Muscle Remodeling. Radiol Oncol 48, 247–256 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClung J. M., Davis J. M., Wilson M. A., Goldsmith E. C. & Carson J. A. Estrogen status and skeletal muscle recovery from disuse atrophy. J Appl Physiol 100, 2012–2023 (2006). [DOI] [PubMed] [Google Scholar]
- White J. P. et al. Muscle oxidative capacity during IL-6-dependent cancer cachexia. Am J Physiol Regul Integr Comp Physiol 300, R201–211 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vainshtein A., Kazak L. & Hood D. A. Effects of endurance training on apoptotic susceptibility in striated muscle. J Appl Physiol 110, 1638–1645 (2011). [DOI] [PubMed] [Google Scholar]
- Chen J. & Patton J. R. Mouse pseudouridine synthase 1: gene structure and alternative splicing of pre-mRNA. Biochem J 352, 465–473 (2000). [PMC free article] [PubMed] [Google Scholar]
- Juhling F. et al. tRNAdb 2009: compilation of tRNA sequences and tRNA genes. Nucleic Acids Res 37, D159–162 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittmar K. A., Mobley E. M., Radek A. J. & Pan T. Exploring the regulation of tRNA distribution on the genomic scale. J Mol Biol 337, 31–47 (2004). [DOI] [PubMed] [Google Scholar]
- Fleming M. D., Campagna D. R., Haslett J. N., Trenor C. C. 3rd & Andrews N. C. A mutation in a mitochondrial transmembrane protein is responsible for the pleiotropic hematological and skeletal phenotype of flexed-tail (f/f) mice. Genes Dev 15, 652–657 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harigae H. et al. Aberrant iron accumulation and oxidized status of erythroid-specific delta-aminolevulinate synthase (ALAS2)-deficient definitive erythroblasts. Blood 101, 1188–1193 (2003). [DOI] [PubMed] [Google Scholar]
- Shinriki N. et al. Mode of degradation of tRNAs with ozone. Nucleic Acids Symp Ser 211–214 (1981). [PubMed] [Google Scholar]
- Motorin Y., Muller S., Behm-Ansmant I. & Branlant C. Identification of modified residues in RNAs by reverse transcription-based methods. Methods Enzymol 425, 21–53 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.