

Abstract
Background
Preeclampsia is associated with the placental release of soluble fms-like tyrosine kinase 1 (sFlt-1) and soluble endoglin (sENG). These anti-angiogenic factors cause hypertension and multi-organ injury. Pravastatin decreases placental secretion of sFlt-1 in vitro and is currently being examined in clinical trials as a potential treatment for preeclampsia. However, it is possible that different classes of statins may be more potent at decreasing sFlt-1 secretion. We compared the relative potency of three different generations of statins on sFlt-1 and sENG secretion from human endothelial cells, trophoblast cells, and placenta explants.
Methods
We performed functional experiments using primary human umbilical vein endothelial cells, trophoblast cells and preterm preeclamptic placental explants to assess the affect of simvastatin, rosuvastatin and pravastatin on sFlt-1 and sENG secretion and compared the relative potency of each statin at reducing these factors (Inhibitory Concentration 50). Furthermore we assessed the effect of each statin on the antioxidant and cytoprotective enzyme, heme-oxygenase 1.
Results
All statins reduced sFlt-1 secretion from endothelial cells, trophoblasts and preterm preeclamptic placental explants. Simvastatin was the most potent inhibitor of sFlt-1 secretion from endothelial cells (IC 50 3.2 μM), trophoblast cells (IC 50 61.4 μM) and placental explants. Simvastatin was 28 times and 3 times more potent at reducing sFlt-1 secretion from endothelial cells and 85 times and 33 times more potent at reducing sFlt-1 secretion from trophoblast cells than pravastatin or rosuvastatin respectively.
All statins increased sENG secretion from endothelial cells however did not change secretion from placental explants.
While all statins up-regulated heme-oxygenase 1 in endothelial cells, only simvastatin up-regulated its expression in placenta from patients with preterm preeclampsia.
Conclusion
Simvastatin may be a more potent inhibitor of sFlt-1 secretion from endothelial cells, trophoblast cells and placenta from women with preterm preeclampsia than either pravastatin or rosuvastatin.
Keywords: Preeclampsia, Pregnancy, Statins, sFlt-1, Soluble endoglin
Background
Preeclampsia is associated with the placental release of soluble fms-like tyrosine kinase 1 (sFlt-1) [1] and soluble endoglin (sENG) [2] into the maternal circulation leading to hypertension, proteinuria and multi-system organ injury [3–7]. Currently there is no medical treatment that stabilises the disease pathophysiology and delivery, often at preterm gestations, is required to stop maternal disease progression [3, 4]. A therapeutic that stabilises the maternal disease process allowing preterm pregnancy continuation, or reducing progression to severe disease at term, would be beneficial [8].
Pravastatin has been proposed as a therapeutic candidate for preeclampsia [9–14]. It has been shown to reduce the preeclamptic phenotype in 4 different animal models of the disease [11–14]. We recently published a case series showing administration of pravastatin may stabilise biochemical features of the disease in 3 out of 4 cases of preterm preeclampsia [10]. We, and others have also shown pravastatin [10] and simvastatin [9] reduce sFlt-1 secretion from human placental tissue. This is a desirable feature of a preeclampsia therapeutic as sFlt-1 is a likely pathogenic protein of the disease [1]. Firstly, sFlt-1 is elevated in patients presenting with preeclampsia [15] compared to levels in normal pregnancy and secondly, when injected or overexpressed in mice and rats, hypertension and proteinuria occur [1, 2]. Importantly, when sFlt-1 levels were reduced in preeclamptic patients by plasma apheresis, there was a possible stabilisation of disease [16].
Pravastatin [10] and simvastatin [9] have both been shown to exert an effect on sFlt-1 secretion by inhibiting HMG CoA reductase, a key enzyme in the cholesterol synthesis pathway. There are many statins and they are divided into three different generations based on their relatively potencies in reducing low density lipoproteins, a surrogate marker for their inhibition of HMG CoA reductase [17]. It is therefore conceivable that more potent inhibitors of HMG CoA reductase, might produce greater reductions in sFlt-1 secretion.
Pravastatin is a first generation statin and in fact one of the least potent inhibitors of HMG CoA reductase [17]. Simvastatin is a second generation statin and a more potent inhibitor, whilst rosuvastatin is the most potent inhibitor of HMG CoA reductase and sole third generation statin [17]. Whilst in vivo studies [11–14] and clinical trials [18, 19] have focused on pravastatin as a preeclampsia therapeutic, it is conceivable that simvastatin or rosuvastatin might have a more pronounced effect on decreasing sFlt-1 secretion.
We compared the potency of simvastatin, rosuvastatin and pravastatin on sFlt-1 and sENG secretion in vitro using human endothelial cells, trophoblast cells and placental explants obtained from women with preterm preeclampsia. We assessed whether statins induced the cytoprotective enzyme, heme-oxygenase 1.
Methods
Ethics approval was obtained from the Mercy Health Human Research Ethics Committee and all women gave written informed consent prior to the collection and use of placenta and umbilical cord.
Isolation and treatment of primary human umbilical vein endothelial cells (HUVECs)
Umbilical cords were collected from normal term placentas. The cord vein was infused with 10 ml (1 mg/ml) of collagenase (Worthington, Lakewood, New Jersey) and cells isolated as previously described [20]. Cells were cultured in M199 media (Life Technologies, Victoria, Australia) containing 10 % FCS (Sigma, St Louis, United States), 1 % antibiotic-antimycotic (Life Technologies) and 20 μg/ml endothelial cell growth factor (ECGS) (Sigma) and 100 μg/ml heparin (Sigma) and used between passages 2 to 4. Cells were plated at 24,000 /cm2 and treated at 80 % confluency with 0, 1, 2 or 5 μM simvastatin or rosuvastatin and 0, 2, 5 or 200, μM pravastatin for 24 h. Conditioned media was collected for assessment of sFlt-1 and sENG secretion and cell lysates collected for RNA extraction.
Isolation and treatment of primary human trophoblast cells
Term placentas were collected from women having elective caesarean sections. Human trophoblasts were isolated as previously described [20, 21]. Primary trophoblasts were cultured in DMEM high Glutamax (Life Technologies) containing 10 % FCS and 1 % antibiotic-antimycotic (Life Technologies) on fibronectin (10 μg/mL; BD Biosciences, New South Wales, Victoria) coated plates. Cells attached over 24 h before being washed in PBS [20]. Cells were treated with 0, 5, 50, 100 μM simvastatin, 0, 100, 200, 300 μM rosuvastatin or 0, 20, 200, 2000 μM pravastatin for 24 h under 8 % O2 and 5 % CO2 at 37 °C. Conditioned media was collected to assess sFlt-1 secretion.
Cell viability assay (MTS assay and calcein stain)
Cell viability assay were performed using CellTiter 96-Aquesous One solution (Promega, Madison WI) or calcein stain (Merk Millipore, Darmstadt, Germany) according to the manufacturer’s instructions. Fluostar omega fluorescent plate reader (BMG labtech, Victoria, Australia) was used to detect fluorescence (quantify adhesion).
Placental explant culture
Human placental tissue was collected from three women with severe early onset preeclampsia (delivered at ≤34 weeks gestation). Preeclampsia was defined using the 2013 American College of Obstetricians and Gynecologists (ACOG) guidelines: the presence of hypertension >140/90 on two occasions 4 h apart and any of the following: proteinuria >300 mg/day, renal insufficiency, impaired liver function, thrombocytopenia or visual disturbance [22].
Villous explants were prepared as previously described [10, 20] and cultured in DMEM high glutamax (Life Technologies) containing 1 % antibiotic-antimycotic (Life Technologies) and 10 % fetal calf serum (FCS) (Sigma). After 24 h placental explants were treated with 0, 100 μM simvastatin, 0, 100 μM rosuvastatin or 0, 2000 μM pravastatin (Sigma) for 72 h under 8 % O2 and 5 % CO2 at 37 °C. To assess sFlt-1 and sENG secretion, protein levels were normalized against wet placental explant weights. Tissue was collected for RNA extraction.
ELISA analysis
Concentrations of sFlt-1 and sENG were measured in conditioned cell/tissue culture media using the DuoSet VEGF R1/Flt-1 kit (R&D systems by Bioscience, Waterloo, Australia) and a DuoSet Human Endoglin CD/105 ELISA kit (R&D systems) according to manufacturer’s instructions.
RT-PCR
RNA was extracted from placental explants and HUVECs using an RNeasy mini kit (Qiagen, Valencia, CA) and quantified using the Nanodrop ND 1000 spectrophotometer (NanoDrop technologies Inc, Wilmington, DE). 0.2 μg of RNA was converted to cDNA using Superscript VILO cDNA synthesis kit (Life Technologies) as per manufacturer guidelines.
A taqman gene expression assay was performed for heme-oxygenase 1 (Life Technologies). Primer sequence for heme-oxygenase 1 was forward primer 5′-GGAGGAGGAGATTGAGCACAACA-3′ and reverse primer 5′-AGCGGTACAGCTGCTTGAAC-3′. RT-PCR was performed on the CFX 384 (Bio-Rad, Hercules, CA) using FAM-labeled Taqman universal PCR mastermix (Life Technologies) with the following run conditions: 50 °C for 2 min; 95 °C for 10 min, 95 °C for 15 s, 60 °C for 1 min (40 cycles). Sybr gene expression assay for sFlt-1 e15a and sFlt-1 i13 was used. Primers for sFlt-1 e15a (forward 5′-CTCCTGCGAAACCTCAGTG-3′ and reverse 5′-GACGATGGTGACGTTGATGT-3′) and sFlt-1 i13 (forward 5′-ACAATCAGAGGTGAGCACTGCAA-3′ and reverse 5′TCCGAGCCTGAAAGTTAGCAA-3′) were designed as previously described (Geneworks, South Australia, Australia) [23]. RT-PCR was performed using the following run conditions: 95 °C for 20 min; 95 °C for 0.01 min, 60 °C for 20 min, 95 °C for 1 min (39 cycles), melt curve 65 °C to 95 °C at 0.05 °C increments at 0.05 s.
All data were normalized to GAPDH as an internal control and calibrated against the average Ct of the control samples. GAPDH mRNA expression was stable in control compared to statin treatment groups. Results are expressed as fold change from control.
Statistical analysis
Technical replicates were performed in triplicate for each experiment, with a minimum of three separate experiments repeated for each in vitro study. Data was tested for normal distribution and statistically analysed as appropriate. When three or more groups were compared a 1-way ANOVA (for parametric data) or Kruskal-Wallis test (for non-parametric data) was used. Post-hoc analysis was carried out using either the Tukey (parametric) or Dunn’s test (non-parametric). When two groups were analysed, either an unpaired t-test (parametric) or a Mann-Whitney test (non-parametric) was used. All data is expressed as mean ± SEM.
The IC50 values were determined by plotting the effect of the three statins on sFlt-1 secretion on the same graph. Doses of the statins were transformed and the log of the value recorded. The maximum, minimum and slope of the curve were shared between data sets and non-linear regression was performed. P-values <0.05 were considered significant. Statistical analysis was performed using GraphPad Prism 6 software (GraphPad Software, La Jolla, CA).
Results
Statin treatment of endothelial cells, primary trophoblasts and placental explants obtained from patients with preterm preeclampsia
We have previously reported the effect of pravastatin on endothelial cells, primary trophoblasts and preeclamptic placental explants [10]. Here we dose matched pravastatin to simvastatin and rosuvastatin and also included a dose that has an effect on sFlt-1, in order to perform a comparison of potency. The doses were chosen based on an ability to exert an effect on sFlt-1 whilst maintaining cell viability. Cell viability was maintained for all statins at the doses used in both placental and endothelial cells, except for 5 μM simvastatin treatment of endothelial cells, which reduced metabolic activity (a measure of cell viability) by 17 % (data not shown). Experiments using the three different statins were performed on the same tissues at the same time so comparisons could be made.
Simvastatin is the most potent inhibitor of sFlt-1 secretion from primary endothelial cells
We first examined the effect of simvastatin, rosuvastatin and pravastatin on sFlt-1 secretion from primary human umbilical vein endothelial cells (HUVECs). All statins induced a significant dose dependent reduction in sFlt-1 secretion from primary HUVECs (Fig. 1A). Simvastatin was the most potent inhibitor of sFlt-1 secretion with an IC50 (concentration of the drug required to reduce secretion by 50 %) of 3.1 μM. This was 28 fold more potent than pravastatin (IC50 88.5 μM) and 3 fold more potent than rosuvastatin (IC50 8.9 μM) (Fig. 1B).
Fig. 1.
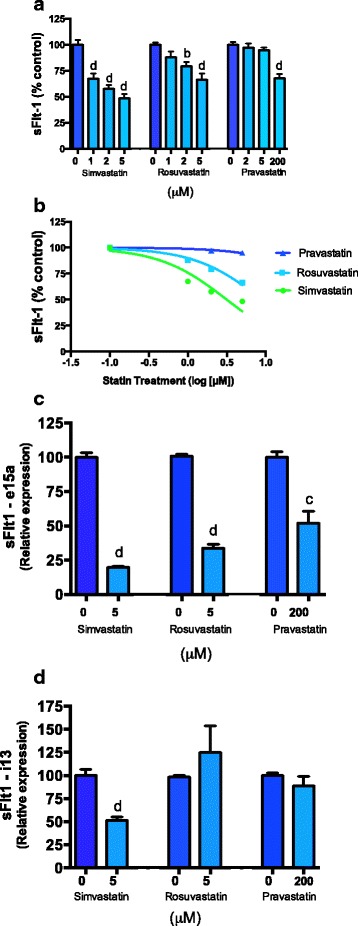
Effects of statins on sFlt-1 secretion and mRNA expression of isoforms e15a and i13 expression in primary HUVECs. a Simvastatin (0, 1, 2, 5 μM) rosuvastatin (0, 1, 2, 5 μM) and pravastatin (0, 2, 5, 200 μM) reduce sFlt-1 secretion from primary HUVECs. b Inhibitory concentration of sFlt-1 at 50 % (IC50) for simvastatin, rosuvastatin and pravastatin was determined. c Pravastatin, simvastatin and rosuvastatin reduce sFlt-1 e15a mRNA expression from primary HUVECs. d Simvastatin reduces sFlt-1 i13 mRNA expression from HUVECs whilst pravastatin and rosuvastatin have no effect. Data represents n = 3 separate experiments and is expressed as mean ± SEM. Dark blue bars = control, light blue bars = statin treatments. b = p < 0.01, d = p < 0.0001
Next we explored the effect of the different statins on the expression of two isoforms of sFlt-1. sFlt-1 is comprised of a number of different splice variants that differ in sequence at the c-terminal region. The most abundant variant in preeclampsia is sFlt-1 e15a which is primate specific and accounts for >80 % of sFlt-1 secreted from the placenta [24]. sFlt-1 i13 is the dominant sFlt-1 variant secreted by the endothelium [24]. We investigated whether the different generations of statins altered sFlt-1 e15a or i13 mRNA expression in primary HUVECs. All statins significantly reduced sFlt-1 e15a expression (Fig. 1C). Simvastatin also significantly reduced sFlt-1 i13 mRNA expression whilst pravastatin and rosuvastatin did not show any effect (Fig. 1D).
Simvastatin is the most potent inhibitor of sFlt-1 secretion from primary trophoblasts and preeclamptic explants
We next compared the effect of the different statins on sFlt-1 secretion from primary trophoblasts. All statins significantly reduced sFlt-1 secretion from primary trophoblasts at top doses (Fig. 2A). Simvastatin was the most potent inhibitor of sFlt-1 secretion with an IC50 of 61.4 μM. It was 33 times more potent than rosuvastatin (IC50 2029 μM) and 85 times more potent than pravastatin (IC50 5224 μM) (Fig. 2B).
Fig. 2.
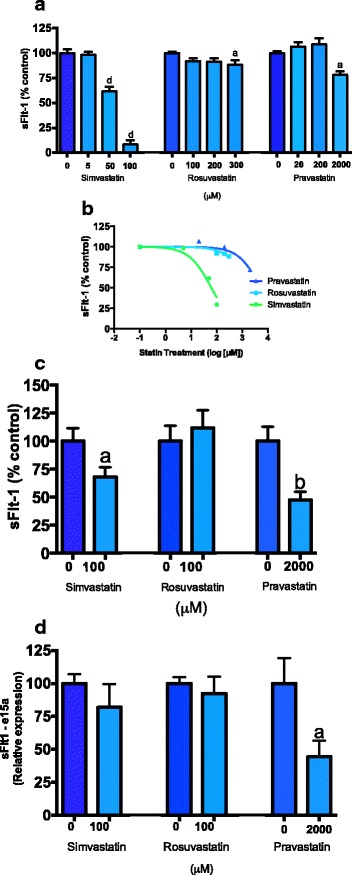
Effect of statins on sFlt-1 secretion and sFlt-1 e15a and i13 mRNA expression from placental tissues. a Simvastatin (0, 5, 50, 100 μM), rosuvastatin (0, 50, 100, 200, 300 μM) and pravastatin (0, 20, 200, 2000 μM) reduce sFlt-1 secretion from primary trophoblasts. b Inhibitory concentration of simvastatin, rosuvastatin and pravastatin was determined. c Simvastatin (100 μM) and pravastatin (2000 μM) significantly reduced sFlt-1 secretion from placental explants obtained from patients with preterm preeclampsia however rosuvastatin had no effect. d Pravastatin reduced sFlt-1 e15a mRNA expression from placental explants whilst simvastatin and rosuvastatin had no effect. Data represents n = 3-4 separate experiments and is expressed as mean ± SEM. Dark blue bars = control, light blue bars = statin treatments. a = p < 0.05, b = p < 0.01, d = p < 0.0001
We also investigated the effects of the different statins on sFlt-1 secretion from placental explants obtained from patients with preterm preeclampsia (Fig. 2C). Simvastatin was a more potent inhibitor of sFlt-1 secretion whilst at the same dose rosuvastatin did not show any effect and pravastatin only showed an effect at 20 times this dose (Fig. 2C).
In the same preterm preeclamptic explants, simvastatin and rosuvastatin did not change sFlt-1 e15a expression, however pravastatin significantly reduced its expression at a dose of 2000 μM (Fig. 2D).
All statins increase soluble endoglin (sENG) secretion from endothelial cells and do not affect secretion from placental explants
We next compared the effect of the statins on secretion of sENG from primary endothelial cells, and placental explants obtained from women with preterm preeclampsia. Previously we have shown that pravastatin significantly increases sENG secretion from endothelial cells [10], and here we show that simvastatin and rosuvastatin also increased its secretion. Simvastatin was the most potent inducer of sENG secretion followed by rosuvastatin and then pravastatin (Fig. 3A). In contrast, as previously demonstrated with pravastatin [10], there was no change in sENG secretion from placental explants from patients with preterm preeclampsia treated with the simvastatin or rosuvastatin (Fig. 3B).
Fig. 3.
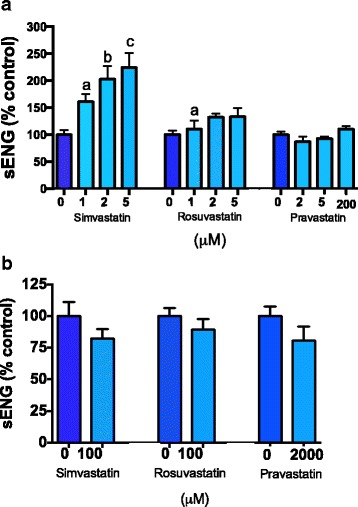
Effect of statins on soluble endoglin secretion from primary human tissues. a Simvastatin (0, 1, 2, 5 μM), rosuvastatin (0, 1, 2, 5, μM) and pravastatin (0, 2, 5, 200 μM) all caused an increase in soluble endoglin secretion from primary HUVECs. b Statin treatment did not alter sEng secretion from preterm preeclamptic explants. Data represents n = 3–4 separate experiments and is expressed as mean ± SEM. Dark blue bars = control, light blue bars = statin treatments. a = p < 0.05, b = p < 0.01, d = p < 0.0001
All statins upregulate heme-oxygenase 1 in primary endothelial cells
Preeclampsia is thought to be associated with increased oxidative stress [3]. Therefore it would be beneficial if statins could up-regulate endogenous antioxidant defenses. We have previously shown pravastatin up-regulates heme-oxygenase 1 in endothelial cells however has no effect on its expression in placental explants [10]. Here we assessed whether simvastatin and rosuvastatin regulate heme-oxygenase 1 expression in either endothelial cells or placental explants from women with preterm preeclampsia. Indeed, we found all statins increased heme-oxygenase 1 mRNA expression in endothelial cells (Fig. 4A). Simvastatin significantly increased heme-oxygenase 1 mRNA expression in placental explants taken from women with preterm preeclampsia (Fig. 4B) however rosuvastatin and pravastatin had no effect on its expression.
Fig. 4.
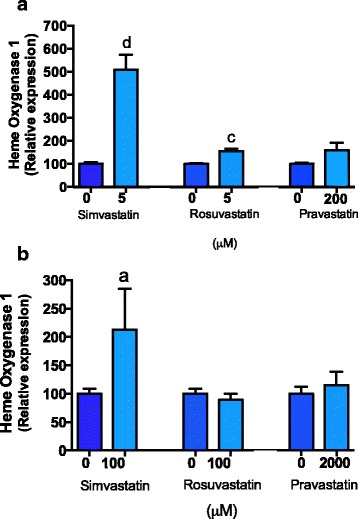
Effect of statins on heme-oxygenase 1 mRNA expression. a Simvastatin (0, 5 μM), rosuvastatin (0, 5 μM) and Pravastatin (0, 200 μM) up-regulated heme-oxygenase 1 expression by HUVECs. b Simvastatin 100 μM up-regulated heme-oxygenase 1 in preeclamptic placental explants, whist pravastatin 2000 μM and rosuvastatin 100 μM had no significant effect. Data represents n = 3–4 separate experiments and is expressed as mean ± SEM. Dark blue bars = control, light blue bars = statin treatments. a = p < 0.05, c = p < 0.001, d = p < 0.0001
Discussion
Statins have been proposed as possible therapeutics for preeclampsia. Our group [10] and Cudmore et al. [9], have shown pravastatin and simvastatin reduced sFlt-1 secretion from placental explants and endothelial cells respectively. The decrease in sFlt-1 caused by these statins was directly mediated through HMG CoA reductase [9, 10]. Pravastatin is the weakest inhibitor of HMG CoA reductase and it is conceivable that the more potent inhibitors of this enzyme (simvastatin or rosuvastatin) [17] may have a more pronounced effect on sFlt-1 secretion. Hence in this study, we examined the relative potency of three generations of statins on sFlt-1 secretion.
We found simvastatin was the most potent inhibitor of sFlt-1 secretion from all primary tissues examined. It was 28–85 times more potent than pravastatin and 3–33 times more potent than rosuvastatin at reducing sFlt-1 secretion from placental and endothelial cells respectively. Whilst simvastatin is known to be a more potent inhibitor of HMG CoA reductase than pravastatin, it is not as potent as rosuvastatin [17]. Perhaps the more pronounced effect of simvastatin on sFlt-1 secretion is a result of the hydrophobicity of the molecule. In contrast to pravastatin and rosuvastatin, simvastatin is hydrophobic and is readily able to cross the cellular lipid bilayer membrane [25, 26]. Therefore it is possible that this characteristic enhanced its capacity to enter the cells and reduce sFlt-1, relative to hydrophilic compounds, pravastatin and rosuvastatin. This effect was particularly apparent in endothelial cells, where simvastatin-induced a significant decrease in both sFlt-1 e15a and sFlt-1 i13 isoforms, culminating in reduced sFlt-1 secretion. This is in contrast to rosuvastatin, which only reduced sFlt-1 e15a, and pravastatin that was only effective at reducing these variants at much higher doses [9, 10].
The effect of statins on the secretion of soluble endoglin is concerning. We previously demonstrated pravastatin up-regulates soluble endoglin secretion from endothelial cells [10]. Here we demonstrate that both simvastatin and rosuvastatin also upregulate soluble endoglin secretion from endothelial cells. Intriguingly, the effect on soluble endoglin appears also to relate to statin potency, with simvastatin inducing the most potent increase. Atorvastatin has previously been shown to upregulate membrane bound endoglin in endothelial cells, thus it is possible the increased soluble endoglin secretion we observed is a result of the same mechanism [27]. Reassuringly, we did not observe an increase in soluble endoglin secretion from preeclamptic explants treated with statins. Therefore, we conclude that the effect statins have on soluble endoglin secretion in vivo warrants further exploration and should be measured in future studies.
Of interest, the three statins studied herein had tissue specific effects on heme-oxygenase 1. Whilst robust up-regulation was observed in primary endothelial cells treated with all three classes of statins, only simvastatin increased heme-oxygenase 1 expression in placental explants obtained from women with preterm preeclampsia. This supports the premise that heme-oxygenase 1 may not regulate sFlt-1 secretion [28]; as pravastatin reduced sFlt-1 secretion with no change to heme-oxygeanse 1 expression in preterm preeclamptic placental explants [28]. Up-regulating an antioxidant molecule may be beneficial however and this further supports the beneficial effects of simvastatin as a possible preeclampsia therapeutic.
Although our data suggest simvastatin is the most potent statin at reducing sFlt-1 secretion, it is important to note that there have been concerns regarding its safety profile as a therapeutic for preeclampsia. This stems from the retrospective observational study performed by Edison [29] reporting an increase in limb defects in fetuses exposed to simvastatin in the first trimester. This finding has been challenged by other large retrospective observational studies [30, 31] showing no increased risk of fetal malformation in simvastatin exposed pregnancies compared to baseline. Furthermore, Gibb [32] questioned the validity of observations by Edison et al. [29] as there was no distinctive pattern to the limb defects described and the population baseline risk was not taken into account. As clinical onset of preeclampsia occurs in the second and third trimester after the majority of organogenesis has occurred, and the current treatment of preeclampsia consists solely of delivery, inflicting high rates of death and disability due to prematurity, we propose that the possible benefits of simvastatin administration may outweigh the theoretical risk.
Conclusion
In conclusion, we have performed a direct comparison of three different generations of statins on key biochemical features of preeclampsia using primary human cells and tissues. Simvastatin was the most potent inhibitor of sFlt-1 secretion and the most potent agent in up-regulating heme-oxygenase 1 expression. Therefore, it is possible that it may be more efficacious as a treatment for preeclampsia compared to other statins.
Acknowledgments
We would like to thank the research midwives, Gabrielle Pell, Genevieve Christophers, Rachel Murdoch and Debra Jinks, and patients at Mercy Hospital for Women for participating in this research.
Sources of funding
This work was funded by The National Health and Medical Research Council of Australia (NHMRC; #1048707, #1046484) and an Arthur Wilson RANZCOG scholarship. FB was supported by an Australian Postgraduate Award and an AVANT scholarship. The NHMRC provided salary support (#1050765 to ST #1062418 to TKL). The University of Melbourne provided salary support to NJH (CR Roper Fellowship). The funders had no role in study design, data collection, analysis, decision to publish or preparation of the manuscript.
Authors’ contributions
FCB devised the study, conducted the experiments, performed statistical calculations, created the figures and wrote the initial draft of the paper. ST and TK-L assisted with devising experiments, statistical calculations and drafting the paper. NH, RH and PC assisted with performing the experiments. All authors read and approved the final manuscript.
Authors’ information
FB has a MBBS, ST a MBBS and PhD, NH a B Sci and PhD, RH a BBiomed Sc, PC a B Sci, TKL a B Sci and PhD.
Competing interests
The authors declare that they have no competing interests.
Details of ethics approval
Ethics approval was obtained from the Mercy Health Human Research Ethics Committee on the 22nd December 2014 (Document number R11/34) and all women gave written informed consent prior to the collection and use of placenta and umbilical cord.
Abbreviations
- IC50
inhibitory concentration 50
- sENG
soluble endoglin
- sFlt-1
soluble fms-like tyrosine kinase 1
References
- 1.Maynard S, Min JY, Merchan J, Lim KH, Li J, Mondal S, Libermann TA, Morgan JP, Sellke FW, Stillman IE, Epstein FH, Sukhatme VP, Ananth Karumanchi S. Excess placental soluble fms-like tyrosine kinase 1 (sFlt-1) may contribute to endothelial dysfunction, hypertension, and proteinuria in pre-eclampsia. J Clin Invest. 2003;111(5):649–658. doi: 10.1172/JCI17189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Venkatesha S, Toporsian M, Lam C, Hanai J, Mammoto T, Kim YM, Bdolah Y, Lim KH, Yuan HT, Libermann TA, et al. Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat Med. 2006;12(6):642–649. doi: 10.1038/nm1429. [DOI] [PubMed] [Google Scholar]
- 3.Redman CW, Sargent IL. Latest advances in understanding preeclampsia. Science. 2005;308(5728):1592–1594. doi: 10.1126/science.1111726. [DOI] [PubMed] [Google Scholar]
- 4.Sibai B, Dekker G, Kupferminc M. Pre-eclampsia. Lancet. 2005;365(9461):785–799. doi: 10.1016/S0140-6736(05)71003-5. [DOI] [PubMed] [Google Scholar]
- 5.Powe CE, Levine RJ, Karumanchi SA. Preeclampsia, a disease of the maternal endothelium: the role of antiangiogenic factors and implications for later cardiovascular disease. Circulation. 2011;123(24):2856–2869. doi: 10.1161/CIRCULATIONAHA.109.853127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Young BC, Levine RJ, Karumanchi SA. Pathogenesis of preeclampsia. Annu Rev Pathol. 2010;5:173–192. doi: 10.1146/annurev-pathol-121808-102149. [DOI] [PubMed] [Google Scholar]
- 7.Chaiworapongsa T, Chaemsaithong P, Yeo L, Romero R. Pre-eclampsia part 1: current understanding of its pathophysiology. Nat Rev Nephrol. 2014;10(8):466–480. doi: 10.1038/nrneph.2014.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chaiworapongsa T, Chaemsaithong P, Korzeniewski SJ, Yeo L, Romero R. Pre-eclampsia part 2: prediction, prevention and management. Nat Rev Nephrol. 2014;10(9):531–540. doi: 10.1038/nrneph.2014.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cudmore M, Ahmad S, Al-Ani B, Fujisawa T, Coxall H, Chudasama K, Devey LR, Wigmore SJ, Abbas A, Hewett PW, et al. Negative regulation of soluble Flt-1 and soluble endoglin release by heme oxygenase-1. Circulation. 2007;115(13):1789–1797. doi: 10.1161/CIRCULATIONAHA.106.660134. [DOI] [PubMed] [Google Scholar]
- 10.Brownfoot FC, Tong S, Hannan NJ, Binder NK, Walker SP, Cannon P, Hastie R, Onda K, Kaitu’u-Lino TJ. Effects of pravastatin on human placenta, endothelium, and women with severe preeclampsia. Hypertens. 2015;66(3):687-97. [DOI] [PubMed]
- 11.Costantine MM, Tamayo E, Lu F, Bytautiene E, Longo M, Hankins GD, Saade GR. Using pravastatin to improve the vascular reactivity in a mouse model of soluble fms-like tyrosine kinase-1-induced preeclampsia. Obstet Gynecol. 2010;116(1):114–120. doi: 10.1097/AOG.0b013e3181e10ebd. [DOI] [PubMed] [Google Scholar]
- 12.Fox KA, Longo M, Tamayo E, Kechichian T, Bytautiene E, Hankins GD, Saade GR, Costantine MM. Effects of pravastatin on mediators of vascular function in a mouse model of soluble Fms-like tyrosine kinase-1-induced preeclampsia. Am J Obstet Gynecol. 2011;205(4):366. doi: 10.1016/j.ajog.2011.06.083. [DOI] [PubMed] [Google Scholar]
- 13.Kumasawa K, Ikawa M, Kidoya H, Hasuwa H, Saito-Fujita T, Morioka Y, Takakura N, Kimura T, Okabe M. Pravastatin induces placental growth factor (PGF) and ameliorates preeclampsia in a mouse model. Proc Natl Acad Sci U S A. 2011;108(4):1451–1455. doi: 10.1073/pnas.1011293108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bauer AJ, Banek CT, Needham K, Gillham H, Capoccia S, Regal JF, Gilbert JS. Pravastatin attenuates hypertension, oxidative stress, and angiogenic imbalance in rat model of placental ischemia-induced hypertension. Hypertension. 2013;61(5):1103–1110. doi: 10.1161/HYPERTENSIONAHA.111.00226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Levine RJ, Qian C, Maynard SE, Yu KF, Epstein FH, Karumanchi SA. Serum sFlt1 concentration during preeclampsia and mid trimester blood pressure in healthy nulliparous women. Am J Obstet Gynecol. 2006;194(4):1034–1041. doi: 10.1016/j.ajog.2005.10.192. [DOI] [PubMed] [Google Scholar]
- 16.Thadhani R, Kisner T, Hagmann H, Bossung V, Noack S, Schaarschmidt W, Jank A, Kribs A, Cornely OA, Kreyssig C, et al. Pilot study of extracorporeal removal of soluble fms-like tyrosine kinase 1 in preeclampsia. Circulation. 2011;124(8):940–950. doi: 10.1161/CIRCULATIONAHA.111.034793. [DOI] [PubMed] [Google Scholar]
- 17.Kapur NK, Musunuru K. Clinical efficacy and safety of statins in managing cardiovascular risk. Vasc Health Risk Manag. 2008;4(2):341–353. doi: 10.2147/vhrm.s1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.The StAmP Trial: A Proof of Principle, Double-Blind, Randomised Placebo-Controlled, Multi Centre Trial of pravaStatin to Ameliorate Early Onset Pre-eclampsia. 2012.
- 19.Costantine MM, Cleary K. Pravastatin for the prevention of preeclampsia in high-risk pregnant women. Obstet Gynecol. 2013;121(2 Pt 1):349–353. doi: 10.1097/AOG.0b013e31827d8ad5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brownfoot FC, Hannan N, Onda K, Tong S, Kaitu’u-Lino T. Soluble endoglin production is upregulated by oxysterols but not quenched by pravastatin in primary placental and endothelial cells. Placenta. 2014;35(9):724–731. doi: 10.1016/j.placenta.2014.06.374. [DOI] [PubMed] [Google Scholar]
- 21.Kaitu’u-Lino TJ, Tong S, Beard S, Hastie R, Tuohey L, Brownfoot F, Onda K, Hannan NJ. Characterization of protocols for primary trophoblast purification, optimized for functional investigation of sFlt-1 and soluble endoglin. Pregnancy Hypertens. 2014;4(4):287–295. doi: 10.1016/j.preghy.2014.09.003. [DOI] [PubMed] [Google Scholar]
- 22.ACOG Report of the american college of obstetricians and gynecologists’ task force on hypertension in pregnancy. Obstet and gynecol. 2013;122:1122–1131. doi: 10.1097/01.AOG.0000437382.03963.88. [DOI] [PubMed] [Google Scholar]
- 23.Whitehead CL, Palmer KR, Nilsson U, Gao Y, Saglam B, Lappas M, Tong S. Placental expression of a novel primate-specific splice variant of sFlt-1 is upregulated in pregnancies complicated by severe early onset pre-eclampsia. BJOG. 2011;118(10):1268–1271. doi: 10.1111/j.1471-0528.2011.02962.x. [DOI] [PubMed] [Google Scholar]
- 24.Jebbink J, Keijser R, Veenboer G, van der Post J, Ris-Stalpers C, Afink G. Expression of placental FLT1 transcript variants relates to both gestational hypertensive disease and fetal growth. Hypertension. 2011;58(1):70–76. doi: 10.1161/HYPERTENSIONAHA.110.164079. [DOI] [PubMed] [Google Scholar]
- 25.Lecarpentier EMO, Fournier T, Elefant E, Chavatte-Palmer P, Tsatsaris V. Statins and pregnancy. Drugs. 2012;72(6):773–788. doi: 10.2165/11632010-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 26.Nanovskaya TN, Patrikeeva SL, Paul J, Costantine MM, Hankins GD, Ahmed MS. Transplacental transfer and distribution of pravastatin. Am J Obstet Gynecol. 2013;209(4):373. doi: 10.1016/j.ajog.2013.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nachtigal P, Pospisilova N, Vecerova L, Micuda S, Brcakova E, Pospechova K, Semecky V. Atorvastatin increases endoglin, SMAD2, phosphorylated SMAD2/3 and eNOS expression in ApoE/LDLR double knockout mice. J Atheroscler Thromb. 2009;16(3):265–274. doi: 10.5551/jat.E745. [DOI] [PubMed] [Google Scholar]
- 28.Tong S, Kaitu’u-Lino TJ, Onda K, Beard S, Hastie R, Binder NK, Cluver C, Tuohey L, Whitehead C, Brownfoot F, et al. Heme oxygenase-1 is not decreased in preeclamptic placenta and does not negatively regulate placental soluble fms-like tyrosine kinase-1 or soluble endoglin secretion. Hypertension. 2015;66(5):1073–1081. doi: 10.1161/HYPERTENSIONAHA.115.05847. [DOI] [PubMed] [Google Scholar]
- 29.Edison RJ, Muenke M. Mechanistic and epidemiologic considerations in the evaluation of adverse birth outcomes following gestational exposure to statins. Am J Med Genet A. 2004;131(3):287–298. doi: 10.1002/ajmg.a.30386. [DOI] [PubMed] [Google Scholar]
- 30.Petersen EE, Mitchell AA, Carey JC, Werler MM, Louik C, Rasmussen SA, National Birth Defects Prevention S Maternal exposure to statins and risk for birth defects: a case-series approach. Am J Med Genet A. 2008;146A(20):2701–2705. doi: 10.1002/ajmg.a.32493. [DOI] [PubMed] [Google Scholar]
- 31.Bateman BT, Hernandez-Diaz S, Fischer MA, Seely EW, Ecker JL, Franklin JM, Desai RJ, Allen-Coleman C, Mogun H, Avorn J, et al. Statins and congenital malformations: cohort study. BMJ. 2015;350:h1035. doi: 10.1136/bmj.h1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gibb H, Scialli AR. Statin drugs and congenital anomalies. Am J Med Genet A. 2005;135(2):230–231. doi: 10.1002/ajmg.a.30685. [DOI] [PubMed] [Google Scholar]