

Abstract
Background
The classic AOX1 replacement approach is still one of the most often used techniques for expression of recombinant proteins in the methylotrophic yeast Pichia pastoris. Although this approach is largely successful, it frequently delivers clones with unpredicted production characteristics and a work-intense screening process is required to find the strain with desired productivity.
Results
In this project 845 P. pastoris clones, transformed with a GFP expression cassette, were analyzed for their methanol-utilization (Mut)-phenotypes, GFP gene expression levels and gene copy numbers. Several groups of strains with irregular features were identified. Such features include GFP expression that is markedly higher or lower than expected based on gene copy number as well as strains that grew under selective conditions but where the GFP gene cassette and its expression could not be detected. From these classes of strains 31 characteristic clones were selected and their genomes sequenced. By correlating the assembled genome data with the experimental phenotypes novel insights were obtained. These comprise a clear connection between productivity and cassette-to-cassette orientation in the genome, the occurrence of false-positive clones due to a secondary recombination event, and lower total productivity due to the presence of untransformed cells within the isolates were discovered. To cope with some of these problems, the original vector was optimized by replacing the AOX1 terminator, preventing the occurrence of false-positive clones due to the secondary recombination event.
Conclusions
Standard methods for transformation of P. pastoris led to a multitude of unintended and sometimes detrimental integration events, lowering total productivity. By documenting the connections between productivity and integration event we obtained a deeper understanding of the genetics of mutation in P. pastoris. These findings and the derived improved mutagenesis and transformation procedures and tools will help other scientists working on recombinant protein production in P. pastoris and similar non-conventional yeasts.
Electronic supplementary material
The online version of this article (doi:10.1186/s12934-016-0486-7) contains supplementary material, which is available to authorized users.
Keywords: Pichia pastoris, Recombinant protein production, AOX1 promoter, Genome sequencing, Insertion locus, Non-conventional yeast, Expression cassette orientation, False-positive
Background
Pichia (Komagatella) pastoris is a non-conventional methylotrophic yeast that is widely used as a host for recombinant protein production [1, 2]. Its capability to perform post-translational modifications such as disulphide isomerization or glycosylation, an efficient secretion apparatus and the relative ease of reaching high dry cell weights >100 g/L during bioreactor fermentation make this eukaryote a popular choice for protein expression in industry as well as in research [3–5]. Over 500 proteins, from industrial enzymes to biopharmaceuticals, have been expressed in P. pastoris [6]. A growing number of commercial products have reached the market in recent years [7]. Among them are the FDA-approved drugs Kalbitor® and Jetrea®, a kallikrein inhibitor and an aglycosylated protease, respectively [8].
The most common approach for heterologous protein expression in P. pastoris is the insertion of the target gene into the genome under the control of the AOX1 (alcohol oxidase 1) promoter (pAOX1). This approach offers tight regulation and a very strong, methanol-inducible expression [6]. Two different modes of homologous recombination-mediated insertion are typically used (i) ends–in insertion leads to additive insertion of the target gene and (ii) ends–out insertion facilitates the replacement of a genomic region, most commonly the native AOX1 gene [9]. Knock-out of AOX1 leads to the MutS-phenotype (methanol utilization slow), since only the lesser transcribed AOX2 gene remains. Clones with an additive insertion retain the native phenotype Mut+ (methanol utilization plus, full growth on methanol). The optimal phenotype for a given application can differ, with MutS-strains exhibiting higher productivity than Mut+-clones in some cases [10, 11]. Recently much progress has been made in understanding the regulation of pAOX1 as well as creating novel synthetic variants with improved characteristics, underlining that the importance of the promoter still holds [12–17].
A frequently encountered problem during generation of P. pastoris clones via homology-mediated integration of the expression cassette is the low targeting efficiency, being as low as <1 % in certain cases like the mannosyltransferase OCH1 [18]. In addition, an off-target insertion can lead to the disruption of a gene and potentially affect production characteristics. Different techniques have been proposed to improve the targeting efficiency, e.g. preventing random insertion due to non-homologous end-joining (NHEJ) via deletion of a KU70 homologue or increasing the genetic redundancy [18, 19]. While these methods help to reduce the number of untargeted insertions, they are better suited for genetic engineering studies rather than the generation of a production strain. Scientists working with P. pastoris are faced with the task of identifying the optimal producer from a diverse group of clones with varying production characteristics. Similar problems have been reported for other non-conventional yeasts that are often used for recombinant protein expression like Hansenula polymorpha, Yarrowia lipolytica and Kluyveromyces lactis [20–22].
The publicly available genome sequences for the most commonly used P. pastoris strains CBS 7435 [23] and GS115 [24] gave rise to multiple genome-scale experiments, most of which focused on better understanding metabolic pathways in order to improve yields in recombinant protein production [25–28]. However, to present no study has been published that investigates the effects of random insertions on the productivity in P. pastoris as well as the integration events on the genome scale. In essence a sort of “black box” is present during transformation of P. pastoris and it is uncertain if the clone with the desired characteristics will be generated. Unknown events during integration of the expression cassette can lead to drastically different production characteristics of clones from one transformation experiment.
For researchers working with P. pastoris, or similar non-conventional yeasts, it would be of great value to gain insights into what might cause unexpected expression levels. By correlating an insertion event seen on the genome with the production characteristics their interaction can be determined. Once these events are known steps can be taken to e.g. optimize vectors to prevent particular integration events.
Using methods previously described and established specifically for P. pastoris, a library of 845 clones was characterized for their expression levels and gene copy numbers (GCN) of GFPuv (cycle-3-GFP) [29] as well as their Mut-phenotypes [30–33]. Based on these characteristics the clones were grouped and the 31 most outstanding ones selected for genome sequencing. By correlating experimental and genome data novel insights into the integration event and its effect on productivity were discovered and the original vector optimized.
Results and discussion
Characterization and grouping of pAHBgl-GFP P. pastoris clones
In total, 845 P. pastoris clones transformed with the GFP expression cassette were characterized for their Mut-phenotypes, GFP gene expressions and GCN. The intent of the transformation strategy used in our study was to replace the native AOX1 gene with a single copy of the GFP expression cassette. Therefore a “regular” clone should have the MutS phenotype as well as GCN and GFP expression level of around 1. Overall, 347 out of all 845 clones fall into this category, accounting for approximately 41 % of all clones. This targeting efficiency for AOX1 is above previously reported values of around 25 % [34, 35]. It has to be considered that in the present study a different histidine auxotroph strain, CBS7435 (ΔHIS4) with a fully deleted HIS4 gene was used [19]. Thereby the background of spontaneous histidine prototrophy conversion clones found in GS115, in which histidine-auxotrophy is mediated by a single nucleotide polymorphism (c.1669C > T resulting in p.557Arg > Cys), is eliminated and the proportion of MutS strains increased. Strains with the Mut+ phenotype can exhibit negative traits due to illegitimate recombination of the expression cassette into the genome and require more methanol for continuous induction. Nevertheless, they might present suitable hosts for protein expression if adjusting process parameters accordingly [11]. Hence, Mut+ strains that otherwise displayed the same features as regular clones can also be considered suitable for most applications. To this end, they were added to the “regular” clones in this study.
All strains not falling into either of these categories were designated as “irregular”. They displayed certain properties that were not expected based on the transformation modus. Table 1 shows the distribution of strains based on these criteria and their Mut-phenotypes. While about a quarter of all clones exhibited irregular features only five of these were MutS strains, underlining the higher genetic variance of Mut+ clones. A total of 45 multi-copy clones (GCN ≥1.5) were found, accounting for ca. 5 % of all clones. Among them seven “jackpot” strains with a GCN >10 are present. All our subsequent analysis concentrated on the irregular clones, in order to elucidate the genetic cause of their aberrant properties.
Table 1.
Distribution of all 845 P. pastoris clones, transformed with the GFP expression cassette, based on “regular” and “irregular” properties as well as the Mut-phenotype
| Group | No. of clones/ % |
|---|---|
| Regular clones | |
| MutS | 347/41 |
| Mut+ | 308/36 |
| Combined | 655/77 |
| Irregular clones | |
| MutS | 5/1 |
| Mut+ | 185/22 |
| Combined | 190/23 |
| Total | 845/100 |
For a better insight into the diversity of the irregular clones, the relation between GFP expression level and GCN has to be looked at. P. pastoris is an industrially important host for recombinant protein expression, therefore these characteristics are at the forefront when it comes to determine whether a clone can meet the requirements of a production process. Interestingly, no clear correlation between GCN and GFP expression level could be seen evaluating all cones (Fig. 1). A wide distribution of clones is visible with no clear pattern. This is in contrast to previously reported results for the relation between GCN and expression level in P. pastoris for intracellular protein expression [36]. In other studies, a good linear correlation for intracellular expression was found [37, 38], while secretory expression showcased more complex correlations due to mechanisms like the UPR pathway (unfolded protein response) [34, 39, 40].
Fig. 1.
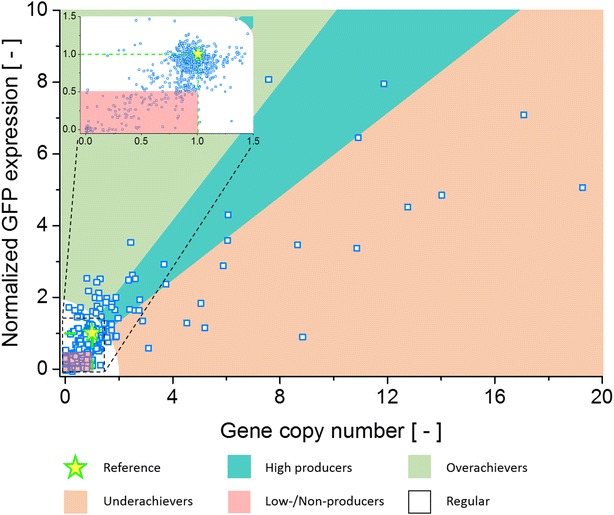
GCN and normalized GFP expression of all 845 characterized P. pastoris clones. The reference clone JPS066 with GCN and GFP expression = 1 is shown with a yellow star. Areas for clones categorized as high producers, overachievers, underachievers or low-/non-producers (shown in the cut-out) are highlighted. Regular clones are found in the white area
To facilitate a clearer understanding of the different kinds of irregular features, the clones were grouped. As shown in Fig. 1 irregular clones could be identified and grouped based on their GCN and GFP expression values. Strains that displayed high GFP expression levels that is paired with a high GCN, two very desirable features for a production strain, were categorized as “high producers”. On the other hand, clones that showed a distinct discrepancy between their GCN and GFP expression were designated “over-“or “underachievers”. Characteristic for these strains is an expression level markedly exceeding (overachiever) or falling below (underachiever) the expression level expected based on the GCN. Additionally, strains with an expression level or GCN notably below 1 or even at 0 were considered “low-/non-producers” making up the group least desirable as a production strain. Table 2 lists all groups the 190 irregular clones were divided into and the basic characteristics of each group. In Additional file 1: Table S1 the concrete criteria used for dividing clones into these groups can be found. By dividing the strains into groups with similar features, a strategy to identify clones of interest for genome sequencing could be conceived.
Table 2.
Grouping of all 190 irregular clones based on shared properties
| Group | No. of clones | Characteristics |
|---|---|---|
| High producers | 29 | High GCN and high expression |
| Overachievers | 21 | Low GCN and high expression |
| Underachievers | 40 | High GCN and low expression |
| Low-/non-producers | 100 | Low/no GCN and expression |
Selection of clones for sequencing and general sequencing results
Using a scoring system (Additional file 1: Table S1), the clones with the most outstanding characteristics from the various groups of irregular clones were selected for genome sequencing. In summary, the scoring system emphasized high deviations from the expected GCN or expression level of 1 and an expression level that did not correlate with the GCN. Based on the scoring system, 31 clones were submitted to genome sequencing. Of these 29 had the Mut+ and 2 the MutS phenotype.
The sequencing runs (2 × 300 bp) on the Illumina MiSeq platform resulted in 80,608,298 reads comprising 24.2 Gb of sequence information. De novo assemblies for each sample generated an average of 37 scaffolds, 76 scaffolded contigs and a size of 9.35 Mb. This represents an average sequence coverage of 83-fold. The average GC content (41.1 %) is in accordance with the findings of Küberl et al. [23], who reported the first genome of P. pastoris CBS 7435. Detailed sequencing statistics for each strain can be found in Additional file 2: Table S2.
After the sequencing and assembly phase, a “contig-length vs. read-count” plot analysis was performed gaining deeper insights into the composition of the 31 samples. Figure 2 shows an example for one of the Low-producer genomes. In general, assembled contigs can be classified in three different groups. Group I contigs (lower 0.5×) represent low amounts of additional P. pastoris isolates in the sample, with small changes in comparison to the main isolate identified by a BLAST approach. The subpopulations of group I, essentially represent mixed-cultures. These sometimes contained low amounts of untransformed P. pastoris CBS 7435 (ΔHIS4) cells that presumably were supplied with l-histidine from the transformed cells. Contigs of group II (0.5 × to 2×) represent the almost complete chromosomal genome. The contigs of groups III (above 2×) were mostly allocated to the more abundant mitochondrial DNA and the most abundant DNA encoding ribosomal RNAs (rRNA) or other repetitive elements.
Fig. 2.
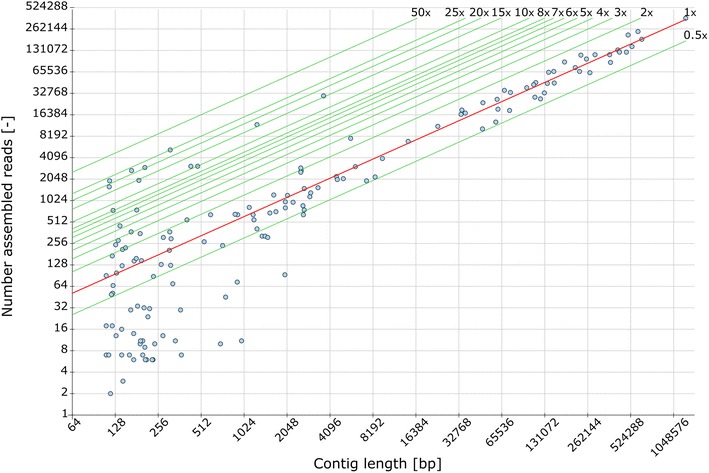
“Contig length vs. read-count” plot for the Low-producer strain JPS379 (EMBL FBTF01000000) contigs in log2 scale. Dots represent the length of a contig plotted against the number of reads assembled into that contig. The lines denote the different predicted coverage ranges within the genome. Underrepresented contigs (group I) are found at a coverage of 0.5 or lower and indicate a secondary isolate in the sequenced sample. Unique contigs are represented by the red line, indicating a onefold coverage. Together with the twofold coverage line, they account for group II contigs which make up the chromosome of the main isolate. Above the twofold coverage line contigs belonging to group III can be found. They represent more abundant DNA elements, e.g. mitochondrial DNA
While the majority of sequenced clones contained no subpopulation, the occurrence of mixed-cultures with e.g. untransformed cells could have been reduced or completely eliminated by performing dilution plating experiments with transformed strains. If the mixed-cultures were the result of two or more cells adhering to each other after transformation they could form a single colony on the plate, containing both cell types. Dilution plating on suitable plates should separate such mixtures, whereby untransformed cells would be removed. The sequencing results, and especially the “contig-length vs. read-count” analysis, emphasize the necessity of this procedure for P. pastoris experiments. Using the high accuracy and sequencing depth of next generation sequencing (NGS) even small contaminations (<5 %) can be identified and culture heterogeneity better understood.
Based on a BLAST approach, the expression cassette was identified in 26 strains, but five strains lacked the gfp gene. The amount of contigs for each GFP cassette varies between five and seven contigs, whereas JPS495 stands out with 10 contigs for the cassette. Via in silico finishing, all gaps between cassette contigs were closed. To this end, reads protruding contig ends were used to identify contigs that flank a certain source contig and were used to close the gaps between these contigs by applying CONSED [41]. Based on these results, further analysis was performed. By applying the “contig-length vs. read-count” plot analysis, the different amounts of inserted vector cassettes were calculated. Proportions between one inserted copy and about 20 copies were determined, as described in the methods section. Applying in silico finishing, four linear plasmids were closed and analyzed. Linear plasmids were identified by their left and right end. No additional reads were found that overlap at that position, whereas the coverage of the end contigs were often overrepresented in comparison to the chromosome. No connection to the chromosome and no chromosomal insertion side could be identified, therefore this sequences should represent linear plasmids. Possibly the linear plasmids are the result of expression cassettes being “looped out” from tandem arrays, resulting in instable GCN values for high copy P. pastoris clones [42], as illustrated in Additional file 3: Figure S2. Mainly such plasmids contain the vector cassette flanked by parts originating from the E. coli backbone of pAHBgl-GFP. The identified plasmids have a size of about 7–9 kb and include only 1–2 more genes on the non-vector cassette parts. On the other hand, it remains unclear how these linear plasmids would withstand degradation or segregational loss. Therefore more experiments are necessary to confirm their existence. In the following sections the various discovered integration events in sequenced P. pastoris strains are further discussed and the influence they might have on productivity analyzed.
Correlation between sequencing and experimental results
Using the data obtained from the sequenced P. pastoris clones multiple integration events were discovered (Fig. 3). In addition to the expected replacement of AOX1 with the GFP cassette (Fig. 3a) the most common event were additional insertions of the expression cassette up- or downstream of the AOX1 locus (Fig. 3b1). Often not only the cassette, but the complete pAHBgl–GFP vector, was found integrated into the genome hinting at either incomplete digestion or in vivo re-annealing of previously separated restriction fragments (Fig. 3b2). The in vivo re-annealing of fragments after digestion and prior to integration into the chromosome is supported by the orientation of the E. coli backbone found in e.g. the MutS clone JPS233 (Fig. 3c); EMBL FBTV01000000). Here, the elements of the E. coli backbone are inverted in comparison to the original vector (Fig. 3a), while the adjacent expression cassettes have the same orientation as located on the plasmid. The observed organization cannot be the result of an incompletely digested vector, but rather was caused by P. pastoris ligating an inversed E. coli backbone to an expression cassette prior to integration. A gel purification step after plasmid digestion and prior to transformation would prevent the in vivo relegation between expression cassette and E. coli backbone.
Fig. 3.
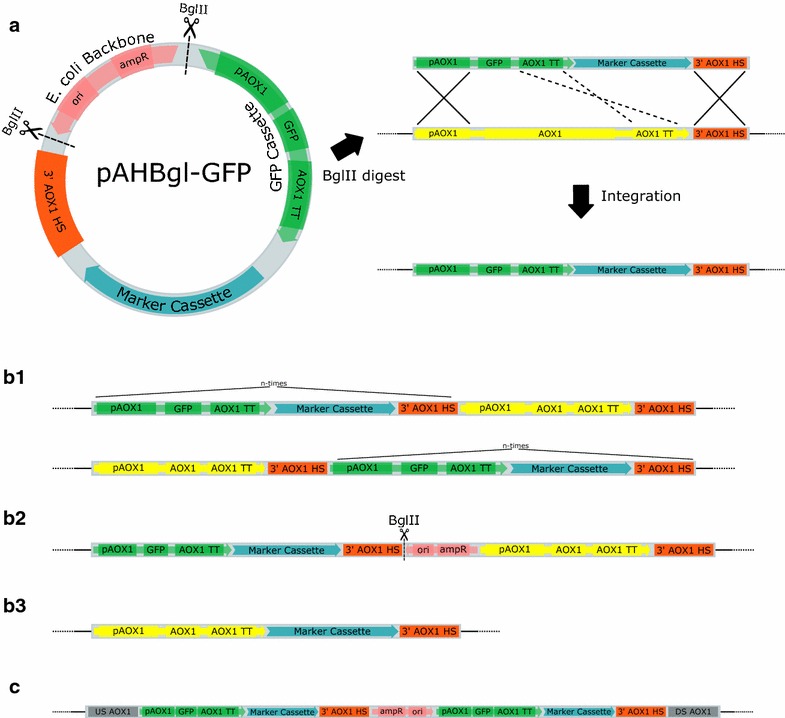
Overview of predicted and observed integration events in P. pastoris. a The plasmid pAHBgl-GFP is restricted with BglII, excising the expression cassette containing the gfp gene and a selection marker. The AOX1 promoter (pAOX1) and a homology sequence downstream of AOX1 (3′ AOX1 HS) facilitate a replacement of the chromosomal AOX1 locus with the expression cassette via double crossing over (solid crossed lines). After homologous recombination the expression cassette is stably integrated into the genome. b1 Via single cross over the expression cassette is inserted up- or downstream of the AOX1 locus. This event can occur multiple times, leading to tandem integrations. Other additive integrations can also happen multiple times up- or downstream of AOX1. b2 Additive integration of the whole pAHBgl-GFP plasmid at the AOX1 locus with highlighted BglII site. b3 Replacement of a region downstream of AOX1 with only the selection marker. The event is facilitated by a double crossing over event using 3′ AOX1 HS and AOX1 TT (dashed crossed lines in a). c Inversion of the E. coli backbone in comparison to the adjacent expression cassettes in the MutS clone JPS233
Multiple tandem-integrations of the cassette or the complete vector were found. This is the first genome-sequence based evidence for the prevalence of tandem-integrations as the genetic organization in multi-copy clones of P. pastoris, first postulated by Clare et al. [43]. No dispersed distribution of cassettes in multiple different loci on the chromosomes was discovered in the analyzed multi-copy strains. In the majority of sequenced clones with multiple tandem integrations, the head-to-tail order of cassettes was the only organization found. Tail-to-tail or head-to-head sequences were only present in five of the sequenced strains, and in all but one they occurred in equal quantities. The different cassette orders are shown in Fig. 4. Collectively, in 86 cases head-to-tail was encountered, tail-to-tail 14 times and head-to-head 15 times. The exclusivity of head-to-tail in comparison to the inclusivity of head-to-head with tail-to-tail hints at an “either/or” relationship between two main integration mechanisms in P. pastoris. Head-to-tail tandem insertions likely integrated via the mechanism described by Clare et al. [43]. A different path of integration was probably responsible for head-to-head and tail-to-tail insertions. Multiple adjacent head-to-head and tail-to-tail integrations would culminate in the observed equilibrium between both arrangements. They could be the result of consecutive integrations of singular expression cassettes, each one using the previous one has homology sequence for additive integration. Only in strain JPS300 (EMBL FBTO01000000), head-to-tail as well as head-to-head was found. How these paths differ and why they are seemingly exclusive to one another remains unclear. However, in the following sections the observed correlation between organization of cassettes and their GFP expression are discussed. Furthermore, other findings of the genome sequencing data are correlated with the experimental results for the different groups of irregular clones.
Fig. 4.
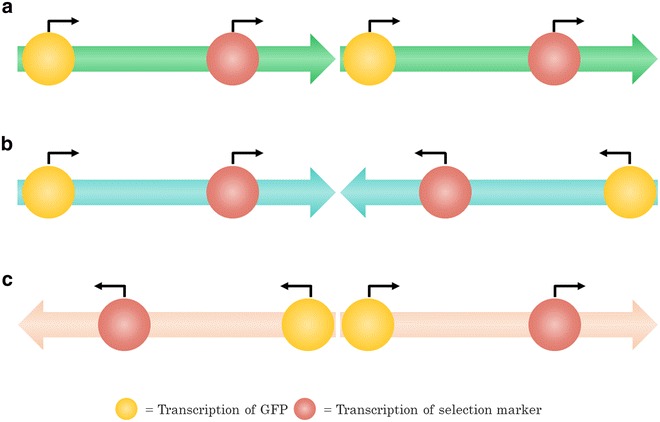
Illustration of the different cassette-to-cassette orientations of adjacent expression cassettes and the transcription start sites found on them. Cassettes are shown in the (a) head-to-tail, (b) tail-to-tail and (c) head-to-head organization
High producers
Strains in the high producer group displayed a markedly higher GCN and expression level than the reference clone. Multi-copy clones with up to 12 (±1.4) copies of the gfp gene and expression levels up to eightfold (±1.2) higher than the reference clone are present in this group. In addition, a linear correlation between GCN and GFP expression was apparent. In Fig. 5, a good agreement with R2 = 0.83 between experimental data is shown. The correlation found here is in accordance with previous reports for the relation between GCN and intracellular protein expression in P. pastoris [37, 38]. Notably, the slope of the regression line (0.73) is below 1. At a slope of 1 two copies would produce twice the amount of GFP. Especially clones with a high copy number are likely the cause of this lower incline. High copy clones put more stress on the protein synthesis apparatus of the cell, thereby diminishing the productivity per GFP cassette and lowering the slope of the linear regression curve.
Fig. 5.
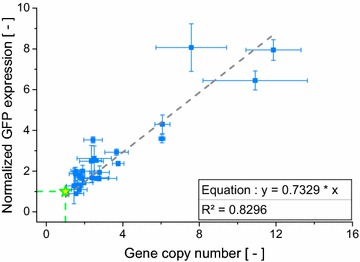
Experimental values for GCN and GFP expression as well as linear regression for clones belonging to the high producers. The reference clone JPS066 is shown as a green star. Error bars display the standard deviation, with n = 3 × 3
In total, six clones from this group were selected for genome sequencing. As was expected based on the qPCR experiments multiple copies of the cassette could be detected. Based on the read frequency, up to 15 copies of the gfp gene were present. In addition to the GFP cassette, in some cases the E. coli backbone of pAHBgl–GFP was additionally inserted in-between two cassettes or directly adjacent to another one. All integration events occurred at the AOX1 locus, even in Mut+ strains. While it can not be excluded that unsequenced high producer clones contain integrations at other loci, it appears no off-target integrations occurred in this group. Since high producers are most likely to be selected for production purposes, the lack of random gene disruptions increases their suitability for industrial applications.
Except clone JPS535 (EMBL FBTY01000000), only head-to-tail tandem integrations were found in high producer clones, suggesting that this organization might be beneficial for productivity. For gaining further insight, the differences seen in comparison to over- and underachiever clones in the next section have to be included.
Over- and underachievers
Individual clones assigned to one of these groups displayed either a markedly higher or lower GFP expression as expected based on the GCN. In total, eight over- and nine underachiever clones were selected for genome sequencing. The discrepancy between experimental and theoretical expression level (calculated using the GCN and the equation shown in Fig. 5) are clearly visible in Fig. 6.
Fig. 6.
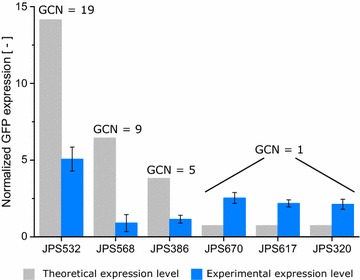
Comparison between theoretical and experimental GFP expression for selected over—(JPS532, JPS568 and JPS386) and underachiever (JPS670, JPS617, JPS320) clones. The theoretical values were calculated using the GCN and the equation shown in Fig. 5. Error bars represent the standard deviation, with n = 3 × 3
For certain overachiever clones the observed GFP expression was more than twofold increased in comparison to the theoretical value. On the other hand, some underachiever strains showcased expression levels far lower than half the expected GFP expression level. Notably, in three sequenced overachiever clones the abundances of the gfp gene calculated from sequence coverage indicate the presence of two copies. These clones would therefore have an expression level in line with their GCN and belong to the group of high producer strains. Multiple explanations are possible as to why the GCN of the affected clones was consistently lower in the qPCR experiments. Potentially, the PCR efficiency in these clones differed too much from the reference, thereby producing inaccurate qPCR predictions. Alternatively, the physical organization of the gfp locus in the isolated genomic DNA (gDNA) hindered the annealing of primers during qPCR. Nevertheless, in five sequenced overachiever strains only a single copy of gfp was found.
For an in-depth evaluation of the cause for deviant production characteristics the orientation of the cassettes to one another was analyzed. In strains of the overachiever group only head-to-tail tandem integrations were found. On the other hand underachiever clones showed the highest proportion of head-to-head and tail-to-tail integrations with 30 % of all tandem integrations. In some underachiever clones a GCN >10 was found. It is possible that in these clones the decreased productivity was due to the high gene dosage triggering cytosolic proteases. The distribution of different cassette orientations among the groups of high producers, as well as under- and overachievers is shown in Fig. 7.
Fig. 7.
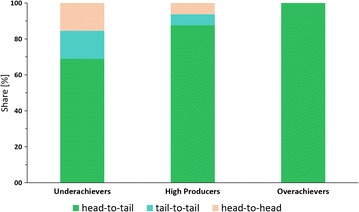
Distribution of different cassette orientations among the sequenced multi-copy clones of the under—and overachievers as well as high producers
In combination with the findings described for the high producer group a trend is visible. Good production characteristics correlate with a head-to-tail orientation of adjacent cassettes. Head-to-tail organization ensures that all cassettes are read in the same direction. The lower GFP expression of strains with head-to-head and tail-to-tail integrations potentially was due to physical obstructions between RNA polymerases on adjacent cassettes during transcription. Crampton et al. [44] demonstrated via atomic force microscopy the premature stop of RNA polymerases on DNA with convergent promoters due to collision events. For the tail-to-tail orientation a “Head-on collision” of RNA polymerases seems less relevant. In this constellation only the marker cassettes are directly convergent with two 3′ AOX1 HS (homology sequence downstream of AOX1, each 0.7 kb) separating them (Fig. 4b). However, in a head-to-head orientation the pAOX1 of neighboring cassettes are directly adjacent to one another (Fig. 4c). This could culminate in mutual obstruction of RNA polymerases binding during the initiation of transcription, resulting in lower expression of the target protein.
In consequence, the construction of vectors that contain multiple expression cassettes in the head-to-tail orientation should lead to clones with an on average higher productivity than alternative approaches for the generation of multi-copy clones. Vassileva et al. [38] demonstrated the successful application of this strategy for the production of hepatitis B surface antigen, displaying a good correlation between GCN and product titer. If the transformation technique allows for other tandem integrations of cassettes, especially the head-to-head orientation, clones with a lower productivity are to be expected. This could be an explanation for some of the discrepancies found in other studies between GCN and expression level, if the orientation of expression cassettes was not determined [36].
Low-/non-producers
Figure 1 displays a large number of clones producing only low amounts or no detectable GFP, the so called low- or non-producers. Six clones belonging to these groups were analyzed via genome sequencing. Many of these strains gave GCN values below 0.5 in qPCR experiments, indicating that a copy of the GFP cassette was present but the copy frequency was lower than that of the calibrator gene. Either the PCR efficiency for the gfp target was affected by e.g. the physical configuration of the GFP cassette or the mixed-culture phenomena caused a greater abundance of ARG4 genes compared to gfp genes. With the mixed-culture phenomena the lower production, illustrated with the Low-producer strain JPS379 (EMBL FBTF01000000) in Fig. 2, can also be explained. A higher ratio of untransformed cells would imply that more cells in the culture media consume methanol without producing GFP. Thus, while GCN values <1 during qPCR experiments still indicate the presence of a full length expression cassette, they might also be used to identify clones with a lower productivity than is to be expected with GCN = 1. The findings of the sequencing experiments of four low-producer clones support this theory. Cultures of these strains always contained likely untransformed cells, as discovered by “contig-length vs. read-count”-plots discussed earlier.
In contrast to the low-producer clones, non-producer clones exhibited no detectable GFP expression and also no detectable GCN. Since they also grew normally in minimal medium without histidine, they were not untransformed cells. Genome sequencing of the non-producer strains JPS056 and JPS060 (EMBL FBTH01000000 and FBVQ01000000) revealed that they shared the same genotype. Only the marker cassette region of pAHBgl-GFP had integrated in the AOX1 locus (Fig. 3b3). This insertion was most likely facilitated by a double crossover homologous recombination between the AOX1 terminator (AOX1 TT) and the 3′ AOX1 HS regions on the expression cassette and the AOX1 chromosomal locus. As shown in Fig. 3a with dashed lines, AOX1 TT presents a third homology sequence in addition to the two used for replacement of AOX1. In total, 68 clones exhibited the same phenotype (GCN and GFP expression level = 0) as JPS056 and JPS060. Therefore, this secondary homologous recombination event appeared to occur quite frequently. As a consequence of the insertion via AOX1 TT and 3′ AOX1 HS, an uncharacterized gene downstream of AOX1, the 810 bp long YDR514C, is disrupted. At the 5′ end of YDR514C, 165 bp are homologous to the AOX1 TT, while at the 3′ end 509 bp are homologous to 3′ AOX1 HS. Thus, base 510 to 645 of YDR514C are replaced with the marker cassette during this secondary homologous recombination event, which can be seen in the genome sequences of clone JPS056 and JPS060. Using this information, other clones with similar phenotype were analyzed via PCR for the integrity of YDR514C. A total of 64 strains with the same genotype as discovered in JPS056 and JPS060 could be identified. Thus, such false-positive clones accounted for approximately 8 % of all clones. They bypass the selection process and unnecessarily increase the workload e.g. in studies in which every clone is of interest. The irregular integration event seemed to depend on the close proximity of AOX1 TT and 3′ AOX1 HS on the expression cassette allowing insertion of the marker cassette without the target gene via homologous recombination. Resulting clones can grow in selective media and show the Mut+ phenotype, but cannot produce the target gene. For further analysis of this phenomenon a novel vector was constructed and transformed into P. pastoris.
An optimized vector to prevent false-positive clones
Based on the findings for non-producer clones a variant of pAHBgl-GFP was constructed, aiming to prevent the creation of false-positive clones due to the integration of only the marker cassette. Since the erroneous integration event was assumed to be facilitated by a homologous recombination between the AOX1 TT and 3′ AOX1 HS regions, they were the key optimization targets. By replacing the terminator and leaving the 3′ AOX1 HS unchanged, the core functionality of the vector would remain unaltered, thus this strategy was chosen. The CYC1 (cytochrome c iso-1) terminator (CYC1 TT) from S. cerevisiae was selected as replacement for multiple reasons: It shows no sequence similarities to the genome of P. pastoris CBS 7435 and is a well-studied, widely used terminator in S. cerevisiae [45, 46]. Additionally, CYC1 TT has been used before as part of P. pastoris vectors, albeit not in combination with pAOX1 [47, 48]. The resulting vector was named pAHBgl–GFP-CYC.
After transformation with pAHBgl-GFP-CYC, 120 clones were picked and characterized for their GFP expression using the same methods as described above. Their expression level was normalized to the same reference clone JPS066. In all 120 clones GFP expression was detectable. Using the Pearson’s Chi squared test, it was determined that the lack of false-positive clones was significant (Table 3).
Table 3.
Occurrence of false-positive clones when using pAHBgl-GFP compared to pAHBgl-GFP-CYC
| Vector | Total clones | False-positive clones (no./ %) | Χ2 value | p value |
|---|---|---|---|---|
| pAHBgl-GFP | 845 | 64/8 | – | – |
| pAHBgl-GFP-CYC | 120 | 0/0 | 10.91 | 0.0001–0.001 |
The absence of false-positive clones strongly suggests that the integration of the marker cassette found in JPS056 and JPS060 was indeed mediated by a double crossing over event using the AOX1 TT and 3′ AOX1 HS regions. Multiple commercial and non-commercial vectors for P. pastoris are targeted for AOX1 replacement, while putting the gene of interest under the control of pAOX1 and AOX1 TT [49, 50]. Integration of only the marker cassette by the mechanism described here can potentially also occur using these plasmids. Therefore it would seem advisable to switch to a different terminator for the gene of interest in order to prevent an increased workload for finding the producer strains, due to false-positive clones.
It has to be noted, however, that pAHBgl-GFP-CYC strains produced markedly lower amounts of GFP than pAHBgl-GFP clones on average (Additional file 3: Figure S3). Likely CYC1 TT is not strong enough as a terminator for the exceptionally strong pAOX1. Thereby, faulty transcription termination and inactive gene products occur. For high-level production, a different terminator ought to be used. In a recent study multiple terminators (and other regulatory elements) from P. pastoris were characterized, providing a good starting point for finding a more effective replacement terminator [51].
Conclusion
Multiple unexpected integration events were discovered during genome sequencing and correlated with the production characteristics of the clones. By analyzing the connection between genome sequence and classic characterization experiments, many novel insights were obtained. The findings demonstrate that the combination of both methods enables deeper understanding than using them separately. Previously postulated theories regarding the generation of multi-copy tandem head-to-tail integrations and in vivo ligation events prior to integration could be verified [43]. It was found that the head-to-tail modus is the dominant insertion pathway, markedly outweighing head-to-head and tail-to-tail integrations. Both pathways seem to be exclusive to one another. The data also suggests that head-to-head and tail-to-tail integrations have a negative impact on productivity. A likely cause is the close proximity of pAOX1 of neighboring head-to-head cassettes. As a result RNA polymerases obstruct each other during transcription. Therefore it seems advisable to use methods specifically generating head-to-tail multi-copy clones, if aiming to increase the product titer via the gene dosage.
In some sequenced strains the presence of multiple genotypes in the form of a mixed-culture was observed, sometimes containing untransformed cells likely provided with l-histidine by transformed cells. Using dilution plating procedures after transformation should eliminate most, if not all, of the mixed-cultures containing untransformed cells. Employing antibiotics like Zeocin for selection ought to reduce the risk of such contaminations as well. The discovery of these subpopulations via genome sequencing supports the validity of the dilution plating procedure, often used in experiments involving yeast.
A secondary double crossing over event using AOX1 TT and the 3′ AOX1 HS led to the integration of only the marker cassette and the creation of false positive clones in about 8 % of all clones. Such clones result in an increased workload when assaying transformed cells for their productivity. By replacing AOX1 TT with the non-homologous CYC1 TT, we could show that no more false-positive clones occurred after transformation. Thereby underlining the validity of theories derived from correlating experimental and genome sequencing results. However, productivity was markedly lower, likely caused by inefficient transcription termination, suggesting that a more suitable terminator needs to be implemented.
Notably, the expression cassette was always found at the AOX1 locus in the analyzed clones, for both MutS and Mut+ strains. Especially for high producers, the best suited strains for industrial applications, the apparent absence of integrations at other loci is desirable. Random integrations as a result of NHEJ have been described for P. pastoris. In the present study a vector system was used, designed to prevent off-target integration [5]. Additionally, the selection process for sequencing emphasized productivity characteristics. It is therefore possible that random integrations at other loci were overlooked as they had no or only a small impact on the expression of GFP. The majority of clones had a GCN and expression level of ca. 1. Potentially, many of these clones harbor an expression cassette integrated at a locus other than AOX1, which did not affect productivity.
Methods
Microorganisms and cultivation conditions
Escherichia coli KRX (Promega, USA) was used for plasmid construction and propagation work. KRX was cultivated in LB (Lysogeny Broth) medium supplemented with 100 µg/mL ampicillin. For experiments involving P. pastoris CBS 7435 (ΔHIS4), obtained from ACIB (Austrian Center of Industrial Biotechnology, Austria) as well as the wild type CBS 7435 (CECT 11047 at Spanish Type Culture Collection, Spain), were used. Saccharomyces cerevisiae wild type strain LBG H620 was provided by the Institute for Agricultural Bacteriology and Fermentation Biology, ETH Zurich, Switzerland. Yeast shake flask cultivations were carried out in BMD (Buffered Minimal Dextrose) [49] or YPD (Yeast Peptone Dextrose) medium, supplemented with 4 mg/L l-histidine when necessary. Experiments in 96-deep-well plates with 2.4 mL total volume (Eppendorf, Germany) used BMD, BMM2 (Buffered Minimal Methanol) and BMM10 as previously described by Weis et al. [30] and Hartner et al. [31]. In brief, BMD is used for the growth phase while BMM2 and BMM10 induce expression of the target gene by maintaining a 0.5 % (v/v) methanol content in the culture medium. The 96-deep-well plates contained up to 500 µL of culture media, were sealed with sterile Breathseal film (Greiner, Germany) and were shaken at 340 rpm at 28 °C.
Plasmid construction and transformation
Primers were designed using SnapGene (GSL Biotech, USA). The sequences of all primers used in this study can be found in Additional file 4: Table S3. In order to construct a vector for intracellular expression of GFPuv in P. pastoris, the plasmid pAHBgl from ACIB, Austria was used as the basis vector. pAHBgl allows for selection based on complementation of the histidine auxotrophy, lacks a secretion signal and can be used for ends-out insertion via linearization with BglII prior to transformation [5]. Using the Gibson assembly technique [52], the gfpuv gene was amplified via PCR from the plasmid pBAD-GFPuv [29] and inserted into linearized pAHBgl, resulting in pAHBgl–GFP. Similarly the AOX1 TT of pAHBgl–GFP was replaced by means of Gibson assembly with the CYC1 TT amplified from gDNA of S. cerevisiae LBG H620, creating pAHBgl-GFP-CYC.
pAHBgl-GFP and pAHBgl-GFP-CYC were amplified in E. coli KRX. For transformation into P. pastoris CBS 7435 (ΔHIS4) the plasmids were extracted with the Wizard® Plus SV Minipreps DNA Purification System (Promega, USA). Purified plasmids were digested with BglII to facilitate ends-out insertion of the GFP expression cassette targeted for replacement of the native AOX1. P. pastoris CBS 7435 (ΔHIS4) was transformed according to Wu and Letchworth [53], with 2–3 µg of digested plasmid DNA per transformation. Cells were spread immediately after transformation onto MD (Minimal Dextrose) plates [49] and incubated at 28 °C for 3–4 days before picking clones. The transformants were both used for the following characterization experiments and stored at −80 °C in 12.5 % (w/v) glycerol to serve as a master strain bank for later analysis.
Characterization of P. pastoris clones
For determination of the Mut-phenotype the plating test as described in the EasySelect™ Pichia Expression Kit Manual (Invitrogen, USA) was used, employing MD and MM (minimal methanol) plates [49].
GFP expression was assayed in a 96-deep-well plate format using established protocols for P. pastoris [30, 31]. Clones were always cultivated in triplicate per plate, with clones belonging to high interest groups being cultivated on two additional deep-well plates. In order to normalize the GFP expression values of each clone, independent of the plate and experiment batch a reference clone had to be selected. The reference clone (JPS066) is a MutS, single-copy clone chosen from among the first 100 clones in a preliminary experiment. JPS066 exhibited a GFP fluorescence level closest to the mean of all single-copy clones of that test. Therefore the GFP fluorescence per OD600 of the reference clone was set as 1 and all other clones normalized to it. In the following experiments the reference clone as well as the untransformed CBS 7435 (ΔHIS4) were always cultivated on each deep-well plate for normalization. Eq. (1) was used for calculating the GFP expression level of each strain. Both GFP fluorescence (excitation 390 nm, emission 510 nm) and OD600 were measured using a SPECTRAFluor Plus microplate reader (Tecan, Switzerland). The value for the GFP expression level in the results and discussion section always represents the normalized expression level 60 h after the start of the methanol induction.
| 1 |
where GFPX is the normalized GFP expression level of clone X, RFUX/R/B the relative fluorescence units of GFP for clone X, the reference clone R or the blank [CBS 7435 (ΔHIS4)] and ODX/R/B is the OD600 value of clone X, the reference clone R or the blank (medium), respectively.
gDNA was extracted from P. pastoris using the MasterPure™ Yeast DNA Purification Kit (Epicentre, USA). The GCN was determined in qPCR experiments in technical duplicates according to previously reported methods [32] using the Rotor-Gene SYBR® Green PCR Kit (Qiagen, Germany) and a LightCycler® 96 system (Roche, Switzerland). Irregular clones were assayed in two additional biological replicates, each with technical duplicates. In brief, the GCN of the gfp gene was assayed using relative quantification based on the 2−ΔΔCt method [54]. The single copy gene ARG4 was chosen as calibrator gene. The primers designed for qPCR exhibited similar TM-values (59 ± 1 °C) and amplicon sizes (100 ± 1 bp). Their nucleotide sequences can be found in Additional file 4: Table S3. Using gDNA from the reference clone JPS066 in quadruple determination over 3.5 logs of copy quantity, a calibration curve for the ARG4 and gfp target was created to determine the PCR-efficiencies for each target (Additional file 3: Figure S1). These PCR-Efficiencies were used for the following assays. Approximately 1 ng gDNA was used per qPCR reaction.
Sequencing of 31 selected P. pastoris strains and genome assembly
gDNA of 31 selected P. pastoris strains was isolated for high throughput sequencing as described above. The quality of the DNA was assessed by gel-electrophoresis and the quantity was estimated by a fluorescence-based method using the Quant-iT PicoGreen dsDNA kit (Invitrogen, USA) and the Tecan Infinite 200 Microplate Reader (Tecan, Switzerland).
For sequencing of the P. pastoris strains, paired-end sequencing libraries (TruSeq sample preparation kit; Illumina, USA) were constructed according to the manufacturer’s protocol. The genome sequences of the P. pastoris strains were established on the Illumina MiSeq system by two paired-end sequencing runs (2 × 300 bp) with a distance range of about 500 bp. Upon sequencing and processing of the raw data, de novo assemblies were performed using the GS De Novo Assembler, software release version 2.8. (Roche, Switzerland) with default settings. The assembled draft sequences for the P. pastoris genomes were deposited in the EMBL-EBI database under the study id PRJEB12220.
Bioinformatic analyses of the 31 Pichia pastoris strains
First insights into the quality of the assemblies were provided by a “Contig-length vs. read-count plot” analysis [55–57]. In the plot the length of each contig (x-axis) is assessed against the number of assembled reads from the corresponding contig (y-axis). The values of lines representing the contig coverage were calculated from the contig length and the number of assembled reads of contigs longer than 10,000 bases, because the probability for these is higher than that for shorter contigs. The contig length distribution covers several orders of magnitude, so a double logarithmic scale was chosen for the axes. Both axes are logarithmic to the base two. This way even very long contigs can be presented clearly. For the calculation of the coverage lines also a double-logarithmic form was used.
Using the readcount and length values the relative abundance of contigs (e.g. the expression cassette) was calculated. With Eq. (2) and the median raw coverage (C) of all contigs longer than 10,000 bp the normalized coverage of a contig [cov(ci)] can be determined. Contigs longer than 10,000 bp are expected to have a single fold coverage.
| 2 |
cov(ci) values between 0.5 and 1.5 indicate that the corresponding contigs are represented once in the genome; ratios lower than 0.5 indicate underrepresented contigs and ratios higher 1.5 indicate overrepresented contigs (e.g. a multi-copy clone).
For more insight, genomic contigs resulting after the assembly were analyzed for large local similarities applying the BLASTn algorithm [58]. Each contig was compared to a local database including the pAHBgl-GFP vector sequence. Hits with an e-value >1 × 10−20 and a sequence identity of 100 % were analyzed in detail. In the first step, an in silico based finishing approach was used to close the gaps of vector sequence [23, 59, 60]. In the next step, the raw sequence coverage of the vector sequence was calculated and normalized based on “contig-length vs. read-count plot” analysis to detect the amount of inserted units. In addition, using the in silico based finishing approach the insertion site of each vector into the P. pastoris genome was identified. Annotation of relevant contigs from sequenced clones was done using SnapGene (GSL Biotech, USA).
Authors’ contributions
JPS, JK and KF designed, analyzed and interpreted wet lab experiments. JPS and TL performed wet lab experiments. AW performed genome sequencing work. DW analyzed and interpreted sequencing data. JPS and DW wrote the manuscript. JK and KF revised the manuscript. JPS, JK and KF conceived the study. JK and KF supervised the research. All authors read and approved the final manuscript.
Acknowledgements
Grants from the Federal State of North Rhine-Westphalia for the CLIB-Graduate Cluster Industrial Biotechnology are gratefully acknowledged.
The bioinformatics support of the BMBF-funded project “Bielefeld-Gießen Center for Microbial Bioinformatics—BiGi (Grant number 031A533)” within the German Network for Bioinformatics Infrastructure (de.NBI) is gratefully acknowledged.
We acknowledge support for the Article Processing Charge by the Deutsche Forschungsgemeinschaft and the Open Access Publication Fund of Bielefeld University.
Competing interests
The authors declare that they have no competing interests.
Abbreviations
- AOX1/2
alcohol oxidase 1/2
- pAOX1
alcohol oxidase 1 promoter
- AOX1 TT
alcohol oxidase 1 transcription terminator
- 3′ AOX1 HS
homology sequence downstream of AOX1
- BMD
buffered minimal dextrose
- BMM
buffered minimal methanol
- CYC1 TT
cytochrome c iso-1 transcription terminator
- GCN
gene copy number
- gDNA
genomic DNA
- LB
lysogeny broth
- MD
minimal dextrose
- MM
minimal methanol
- MutS/+
methanol utilization slow/plus
- NHEJ
non-homologous end-joining
- YPD
yeast peptone dextrose
Additional files
10.1186/s12934-016-0486-7Criteria used for sorting P. pastoris strains into different groups of “regular” and “irregular” clones, as well as scoring system for selection of clones for sequencing.
10.1186/s12934-016-0486-7Detailed genome sequencing statistics for all sequenced clones.
10.1186/s12934-016-0486-7Additional Figure 1 (qPCR calibration curve), Figure 2 (looping-out events) and Figure 3 (Comparison of GFP expression between pAHBgl-GFP and pAHBgl-GFP-CYC clones).
10.1186/s12934-016-0486-7Primer sequences.
Contributor Information
Jan-Philipp Schwarzhans, Email: jan-philipp.schwarzhans@uni-bielefeld.de.
Daniel Wibberg, Email: dwibberg@cebitec.uni-bielefeld.de.
Anika Winkler, Email: awinkler@cebitec.uni-bielefeld.de.
Tobias Luttermann, Email: tobias.luttermann@uni-bielefeld.de.
Jörn Kalinowski, Email: joern.kalinowski@cebitec.uni-bielefeld.de.
Karl Friehs, Email: karl.friehs@uni-bielefeld.de.
References
- 1.Gasser B, Prielhofer R, Marx H, Maurer M, Nocon J, Steiger M, Puxbaum V, Sauer M, Mattanovich D. Pichia pastoris: protein production host and model organism for biomedical research. Future Microbiol. 2013;8:191–208. doi: 10.2217/fmb.12.133. [DOI] [PubMed] [Google Scholar]
- 2.Cregg J, Cereghino J, Shi J, Higgins D. Recombinant protein expression in Pichia pastoris. Mol Biotechnol. 2000;16:23–52. doi: 10.1385/MB:16:1:23. [DOI] [PubMed] [Google Scholar]
- 3.Jahic M, Rotticci-Mulder J, Martinelle M, Hult K, Enfors SO. Modeling of growth and energy metabolism of Pichia pastoris producing a fusion protein. Bioprocess Biosyst Eng. 2002;24:385–393. doi: 10.1007/s00449-001-0274-5. [DOI] [Google Scholar]
- 4.Bill RM. Playing catch-up with Escherichia coli: using yeast to increase success rates in recombinant protein production experiments. Front Microbiol. 2014;5(March):85. doi: 10.3389/fmicb.2014.00085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ahmad M, Hirz M, Pichler H, Schwab H. Protein expression in Pichia pastoris: recent achievements and perspectives for heterologous protein production. Appl Microbiol Biotechnol. 2014;98:5301–5317. doi: 10.1007/s00253-014-5732-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Macauley-Patrick S, Fazenda ML, McNeil B, Harvey LM. Heterologous protein production using the Pichia pastoris expression system. Yeast. 2005;22:249–270. doi: 10.1002/yea.1208. [DOI] [PubMed] [Google Scholar]
- 7.Bollok M, Resina D, Valero F, Ferrer P. Recent patents on the Pichia pastoris expression system: expanding the toolbox for recombinant protein production. Recent Pat Biotechnol. 2009;3:192–201. doi: 10.2174/187220809789389126. [DOI] [PubMed] [Google Scholar]
- 8.Meehl MA, Stadheim TA. Biopharmaceutical discovery and production in yeast. Curr Opin Biotechnol. 2014;30:120–127. doi: 10.1016/j.copbio.2014.06.007. [DOI] [PubMed] [Google Scholar]
- 9.Klinner U, Schäfer B. Genetic aspects of targeted insertion mutagenesis in yeasts. FEMS Microbiol Rev. 2004;28:201–223. doi: 10.1016/j.femsre.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 10.Krainer FW, Dietzsch C, Hajek T, Herwig C, Spadiut O, Glieder A. Recombinant protein expression in Pichia pastoris strains with an engineered methanol utilization pathway. Microb Cell Fact. 2012;11:22. doi: 10.1186/1475-2859-11-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pla IA, Damasceno LM, Vannelli T, Ritter G, Batt CA, Shuler ML. Evaluation of Mut+and MutSPichia pastoris phenotypes for high level extracellular scFv expression under feedback control of the methanol concentration. Biotechnol Prog. 2006;22:881–888. doi: 10.1021/bp060012+. [DOI] [PubMed] [Google Scholar]
- 12.Vogl T, Ruth C, Pitzer J, Kickenweiz T, Glieder A. Synthetic core promoters for Pichia pastoris. ACS Synth Biol. 2014;3:188–191. doi: 10.1021/sb400091p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vogl T, Glieder A. Regulation of Pichia pastoris promoters and its consequences for protein production. N Biotechnol. 2013;30:385–404. doi: 10.1016/j.nbt.2012.11.010. [DOI] [PubMed] [Google Scholar]
- 14.Lin-Cereghino GP, Godfrey L, de la Cruz BJ, Johnson S, Khuongsathiene S, Tolstorukov I, Yan M, Lin-Cereghino J, Veenhuis M, Subramani S, Cregg JM. Mxr1p, a key regulator of the methanol utilization pathway and peroxisomal genes in Pichia pastoris. Mol Cell Biol. 2006;26:883–897. doi: 10.1128/MCB.26.3.883-897.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parua PK, Ryan PM, Trang K, Young ET. Pichia pastoris 14-3-3 regulates transcriptional activity of the methanol inducible transcription factor Mxr1 by direct interaction. Mol Microbiol. 2012;85:282–298. doi: 10.1111/j.1365-2958.2012.08112.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kumar NV, Rangarajan PN. The zinc finger proteins Mxr1p and repressor of phosphoenolpyruvate carboxykinase (ROP) have the same DNA binding specificity but regulate methanol metabolism antagonistically in Pichia pastoris. J Biol Chem. 2012;287:34465–34473. doi: 10.1074/jbc.M112.365304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Capone S, Horvat J, Herwig C, Spadiut O. Development of a mixed feed strategy for a recombinant Pichia pastoris strain producing with a de-repression promoter. Microb Cell Fact. 2015;14:101. doi: 10.1186/s12934-015-0292-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen Z, Sun H, Li P, He N, Zhu T, Li Y. Enhancement of the gene targeting efficiency of non-conventional yeasts by increasing genetic redundancy. PLoS One. 2013;8:e57952. doi: 10.1371/journal.pone.0057952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Näätsaari L, Mistlberger B, Ruth C, Hajek T, Hartner FS, Glieder A. Deletion of the Pichia pastoris KU70 homologue facilitates platform strain generation for gene expression and synthetic biology. PLoS One. 2012;7:e39720. doi: 10.1371/journal.pone.0039720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saraya R, Krikken AM, Kiel JA, Baerends RJ, Veenhuis M, van der Klei IJ. Novel genetic tools for Hansenula polymorpha. FEMS Yeast Res. 2012;12:271–278. doi: 10.1111/j.1567-1364.2011.00772.x. [DOI] [PubMed] [Google Scholar]
- 21.Kretzschmar A, Otto C, Holz M, Werner S, Hübner L, Barth G. Increased homologous integration frequency in Yarrowia lipolytica strains defective in non-homologous end-joining. Curr Genet. 2013;59:63–72. doi: 10.1007/s00294-013-0389-7. [DOI] [PubMed] [Google Scholar]
- 22.Kegel A, Martinez P, Carter SD, Åström SU. Genome wide distribution of illegitimate recombination events in Kluyveromyces lactis. Nucleic Acids Res. 2006;34:1633–1645. doi: 10.1093/nar/gkl064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Küberl A, Schneider J, Thallinger GG, Anderl I, Wibberg D, Hajek T, Jaenicke S, Brinkrolf K, Goesmann A, Szczepanowski R, Pühler A, Schwab H, Glieder A, Pichler H. High-quality genome sequence of Pichia pastoris CBS7435. J Biotechnol. 2011;154:312–320. doi: 10.1016/j.jbiotec.2011.04.014. [DOI] [PubMed] [Google Scholar]
- 24.De Schutter K, Lin YC, Tiels P, Van Hecke A, Glinka S, Weber-Lehmann J, Rouzé P, Van de Peer Y, Callewaert N. Genome sequence of the recombinant protein production host Pichia pastoris. Nat Biotechnol. 2009;27:561–566. doi: 10.1038/nbt.1544. [DOI] [PubMed] [Google Scholar]
- 25.Stadlmayr G, Benakovitsch K, Gasser B, Mattanovich D, Sauer M. Genome-scale analysis of library sorting (GALibSo): isolation of secretion enhancing factors for recombinant protein production in Pichia pastoris. Biotechnol Bioeng. 2010;105:543–555. doi: 10.1002/bit.22573. [DOI] [PubMed] [Google Scholar]
- 26.Chung BKS, Selvarasu S, Camattari A, Ryu J, Lee H, Ahn J, Lee H, Lee D. Genome-scale metabolic reconstruction and in silico analysis of methylotrophic yeast Pichia pastoris for strain improvement. Microb Cell Fact. 2010;9:50. doi: 10.1186/1475-2859-9-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Caspeta L, Shoaie S, Agren R, Nookaew I, Nielsen J. Genome-scale metabolic reconstructions of Pichia stipitis and Pichia pastoris and in silico evaluation of their potentials. BMC Syst Biol. 2012;6:24. doi: 10.1186/1752-0509-6-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sohn SB, Graf AB, Kim TY, Gasser B, Maurer M, Ferrer P, Mattanovich D, Lee SY. Genome-scale metabolic model of methylotrophic yeast Pichia pastoris and its use for in silico analysis of heterologous protein production. Biotechnol J. 2010;5:705–715. doi: 10.1002/biot.201000078. [DOI] [PubMed] [Google Scholar]
- 29.Crameri A, Whitehorn E, Tate E, Stemmer WPC. Improved green fluorescent protein by molecular evolution using DNA Shuffling. Nat Biotechnol. 1996;14:315–319. doi: 10.1038/nbt0396-315. [DOI] [PubMed] [Google Scholar]
- 30.Weis R, Luiten R, Skranc W, Schwab H, Wubbolts M, Glieder A. Reliable high-throughput screening with Pichia pastoris by limiting yeast cell death phenomena. FEMS Yeast Res. 2004;5:179–189. doi: 10.1016/j.femsyr.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 31.Hartner FS, Ruth C, Langenegger D, Johnson SN, Hyka P, Lin-Cereghino GP, Lin-Cereghino J, Kovar K, Cregg JM, Glieder A. Promoter library designed for fine-tuned gene expression in Pichia pastoris. Nucleic Acids Res. 2008;36:e76. doi: 10.1093/nar/gkn369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abad S, Kitz K, Hörmann A, Schreiner U, Hartner FS, Glieder A. Real-time PCR-based determination of gene copy numbers in Pichia pastoris. Biotechnol J. 2010;5:413–420. doi: 10.1002/biot.200900233. [DOI] [PubMed] [Google Scholar]
- 33.Cregg JM, Madden KR, Barringer KJ, Thill GP, Stillman CA. Functional characterization of the two alcohol oxidase genes from the yeast Pichia pastoris. Mol Cell Biol. 1989;9:1316–1323. doi: 10.1128/MCB.9.3.1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Clare JJ, Romanos MA, Rayment FB, Rowedder JE, Smith MA, Payne MM, Sreekrishna K, Henwood CA. Production of mouse epidermal growth factor in yeast: high-level secretion using Pichia pastoris strains containing multiple gene copies. Gene. 1991;105:205–212. doi: 10.1016/0378-1119(91)90152-2. [DOI] [PubMed] [Google Scholar]
- 35.Daly R, Hearn MTW. Expression of heterologous proteins in Pichia pastoris: a useful experimental tool in protein engineenring and production. J Mol Recognit. 2005;18:119–138. doi: 10.1002/jmr.687. [DOI] [PubMed] [Google Scholar]
- 36.Aw R, Polizzi KM. Can too many copies spoil the broth? Microb Cell Fact. 2013;12:128. doi: 10.1186/1475-2859-12-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marx H, Mecklenbräuker A, Gasser B, Sauer M, Mattanovich D. Directed gene copy number amplification in Pichia pastoris by vector integration into the ribosomal DNA locus. FEMS Yeast Res. 2009;9:1260–1270. doi: 10.1111/j.1567-1364.2009.00561.x. [DOI] [PubMed] [Google Scholar]
- 38.Vassileva A, Chugh DA, Swaminathan S, Khanna N. Effect of copy number on the expression levels of hepatitis B surface antigen in the methylotrophic yeast Pichia pastoris. Protein Expr Purif. 2001;21:71–80. doi: 10.1006/prep.2000.1335. [DOI] [PubMed] [Google Scholar]
- 39.Inan M, Aryasomayajula D, Sinha J, Meagher MM. Enhancement of protein secretion in Pichia pastoris by overexpression of protein disulfide isomerase. Biotechnol Bioeng. 2006;93:771–778. doi: 10.1002/bit.20762. [DOI] [PubMed] [Google Scholar]
- 40.Zhu T, Guo M, Tang Z, Zhang M, Zhuang Y, Chu J, Zhang S. Efficient generation of multi-copy strains for optimizing secretory expression of porcine insulin precursor in yeast Pichia pastoris. J Appl Microbiol. 2009;107:954–963. doi: 10.1111/j.1365-2672.2009.04279.x. [DOI] [PubMed] [Google Scholar]
- 41.Gordon D, Abajian C, Green P. Consed: a graphical tool for sequence finishing. Genome Res. 1998;8:195–202. doi: 10.1101/gr.8.3.195. [DOI] [PubMed] [Google Scholar]
- 42.Zhu T, Guo M, Sun C, Qian J, Zhuang Y, Chu J, Zhang S. A systematical investigation on the genetic stability of multi-copy Pichia pastoris strains. Biotechnol Lett. 2009;31:679–684. doi: 10.1007/s10529-009-9917-4. [DOI] [PubMed] [Google Scholar]
- 43.Clare J, Rayment F, Sreekrishna K, Romanos MA. High-level expression of tetanus toxin fragment C in Pichia pastoris strains containing multiple tandem integrations of the gene. Nat Biotechnol. 1991;9:455–460. doi: 10.1038/nbt0591-455. [DOI] [PubMed] [Google Scholar]
- 44.Crampton N, Bonass WA, Kirkham J, Rivetti C, Thomson NH. Collision events between RNA polymerases in convergent transcription studied by atomic force microscopy. Nucleic Acids Res. 2006;34:5416–5425. doi: 10.1093/nar/gkl668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mumberg D, Müller R, Funk M. Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Genetics. 1995;156:119–122. doi: 10.1016/0378-1119(95)00037-7. [DOI] [PubMed] [Google Scholar]
- 46.Zaret K, Sherman F. Mutationally altered 3′ ends of yeast CYC1 mRNA affect transcript stability and translational efficiency. J Mol Biol. 1984;176:107–135. doi: 10.1016/0022-2836(84)90060-3. [DOI] [PubMed] [Google Scholar]
- 47.Stadlmayr G, Mecklenbräuker A, Rothmüller M, Maurer M, Sauer M, Mattanovich D, Gasser B. Identification and characterisation of novel Pichia pastoris promoters for heterologous protein production. J Biotechnol. 2010;150:519–529. doi: 10.1016/j.jbiotec.2010.09.957. [DOI] [PubMed] [Google Scholar]
- 48.Prielhofer R, Maurer M, Klein J, Wenger J, Kiziak C, Gasser B, Mattanovich D. Induction without methanol: novel regulated promoters enable high-level expression in Pichia pastoris. Microb Cell Fact. 2013;12:5. doi: 10.1186/1475-2859-12-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Invitrogen: EasySelect pichia expression kit. 2010:95.
- 50.Vogl T, Ahmad M, Krainer FW, Schwab H, Glieder A. Restriction site free cloning (RSFC) plasmid family for seamless, sequence independent cloning in Pichia pastoris. Microb Cell Fact. 2015;14:103. doi: 10.1186/s12934-015-0293-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vogl T, Sturmberger L, Kickenweiz T, Wasmayer R, Schmid C, Hatzl AM, Gerstmann MA, Pitzer J, Wagner M, Thallinger GG, Geier M, Glieder A. A toolbox of diverse promoters related to methanol utilization—functionally verified parts for heterologous pathway expression in Pichia pastoris. ACS Synth Biol. 2016;5:172–186. doi: 10.1021/acssynbio.5b00199. [DOI] [PubMed] [Google Scholar]
- 52.Gibson DG, Young L, Chuang R, Venter JC, Iii CAH, Smith HO. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 2009;6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 53.Wu S, Letchworth GJ. High efficiency transformation by electroporation of Pichia pastoris pretreated with lithium acetate and dithiothreitol. Biotechniques. 2004;36:152–154. doi: 10.2144/04361DD02. [DOI] [PubMed] [Google Scholar]
- 54.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 55.Heinl S, Wibberg D, Eikmeyer F, Szczepanowski R, Blom J, Linke B, Goesmann A, Grabherr R, Schwab H, Pühler A, Schlüter A. Insights into the completely annotated genome of Lactobacillus buchneri CD034, a strain isolated from stable grass silage. J Biotechnol. 2012;161:153–166. doi: 10.1016/j.jbiotec.2012.03.007. [DOI] [PubMed] [Google Scholar]
- 56.Schwientek P, Szczepanowski R, Rückert C, Kalinowski J, Klein A, Selber K, Wehmeier UF, Stoye J, Pühler A. The complete genome sequence of the acarbose producer Actinoplanes sp. SE50/110. BMC Genomics. 2012;13:112. doi: 10.1186/1471-2164-13-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wibberg D, Jelonek L, Rupp O, Hennig M, Eikmeyer F, Goesmann A, Hartmann A, Borriss R, Grosch R, Pühler A, Schlüter A. Establishment and interpretation of the genome sequence of the phytopathogenic fungus Rhizoctonia solani AG1-IB isolate 7/3/14. J Biotechnol. 2013;167:142–155. doi: 10.1016/j.jbiotec.2012.12.010. [DOI] [PubMed] [Google Scholar]
- 58.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Maus I, Stantscheff R, Wibberg D, Stolze Y, Winkler A, Pühler A, König H, Schlüter A. Complete genome sequence of the methanogenic neotype strain Methanobacterium formicicum MFT. J Biotechnol. 2014;192:40–41. doi: 10.1016/j.jbiotec.2014.09.018. [DOI] [PubMed] [Google Scholar]
- 60.Wibberg D, Blom J, Jaenicke S, Kollin F, Rupp O, Scharf B, Schneiker-Bekel S, Sczcepanowski R, Goesmann A, Setubal JC, Schmitt R, Pühler A, Schlüter A. Complete genome sequencing of Agrobacterium sp. H13-3, the former Rhizobium lupini H13-3, reveals a tripartite genome consisting of a circular and a linear chromosome and an accessory plasmid but lacking a tumor-inducing Ti-plasmid. J Biotechnol. 2011;155:50–62. doi: 10.1016/j.jbiotec.2011.01.010. [DOI] [PubMed] [Google Scholar]