

Abstract
Prostate cancer is a common heterogeneous disease, and most patients diagnosed in the post prostate-specific antigen (PSA) era present with clinically localized disease, the majority of which do well regardless of treatment regimen undertaken. Overall, those with advanced prostate cancer at time of diagnosis do poorly after androgen withdrawal therapy. Understanding the biologic underpinning of prostate cancer is necessary to best determine the risk of disease progression and would be advantageous for the development of novel therapeutic approaches to impede or prevent disease. This review focuses on the recently identified common ETS and non-ETS gene rearrangements in prostate cancer. Although multiple molecular alterations have been detected in prostate cancer, a detailed understanding of gene fusion prostate cancer should help explain the clinical and biologic diversity, providing a rationale for a molecular subclassification of the disease.
INTRODUCTION
Prostate cancer is a major public health problem in the United States with more than 217,000 cases diagnosed and more than 32,000 deaths in 2010.1 Currently, a high percentage of men diagnosed through prostate-specific antigen (PSA) testing will die with prostate cancer and not from it. The aging population, with an expected increase to more than 500,000 diagnosed prostate cancers per year by 2015, presents a key clinical problem: the determination of risk factors in the development of aggressive prostate cancer and avoidance of unnecessary overtreatment. Although effective surgical and radiation treatments exist for clinically localized disease, metastatic prostate cancer remains essentially incurable, and most men diagnosed with metastatic disease will succumb over a period of months to years.
One of the challenges in understanding prostate cancer has been the clinical and molecular heterogeneity associated with this common disease. Hematologic malignancies, such as acute myeloid leukemia, are often subtyped on the basis of the recurrent cytogenetic or molecular aberration identified. Therefore, the recent and surprising discovery that at least 50% of prostate cancers harbor recurrent gene rearrangements resulting in the fusion of genes2 may enable molecular subtyping of prostate cancers, similar to what has been established for leukemias and lymphomas, thereby enabling the identification of patients with aggressive disease. Most often, these fusions juxtapose a hormone-specific promoter that acts as an “on” switch for the oncogene, conferring a distinct biology to this tumor. Although other molecular events play a role in prostate cancer development and progression, defining prostate cancer on the basis of the presence or absence of the different on switch that drives cancer development provides novel insight into disease heterogeneity. Despite the current lack of specific therapies to target the on switches created by the rearrangements, we contend that this hormonally controlled, clonal oncogenic event modulates tumor cells in a manner distinct from rearrangement-negative cases. The focus of this review is to determine the role of gene fusion in prostate cancer heterogeneity and provide a strong rationale for a molecular subclassification of this common tumor.
GENE FUSION PROSTATE CANCER: A PARADIGM SHIFT
Recurrent chromosomal aberrations were thought to be primarily characteristic of leukemias, lymphomas, and sarcomas. Epithelial tumors (ie, carcinomas), which are the most common human tumors contributing to a large percentage of morbidity and mortality associated with human cancer, comprised less than 1% of the known, disease-specific chromosomal rearrangements. Thus, the discovery of the ETS family transcription factor gene fusions by Tomlins et al2 in 2005 dramatically changed the field of solid tumor biology. The recurrent TMPRSS2-ERG fusion in prostate cancer is now the most common rearrangement described in any neoplasm, considering the large number of cases diagnosed in the world each year. The greatest surprise to the research community was that such a common rearrangement would be found in the most prevalent non–skin cancer to afflict men.
TMPRSS2-ETS Family Fusion Genes and Prostate Cancer
The key to the discovery of TMPRSS2-ETS gene fusions was the development of a simple, statistical approach termed “cancer outlier profile analysis” (COPA) to identify oncogene profiles in a subset of samples within publicly available cancer profiling data sets, characteristic of genes commonly associated with known genomic rearrangements (reviewed by Rubin and Chinnaiyan3 and Hanauer et al4). The application of COPA in prostate cancer microarray experiments revealed two consistently high-scoring and mutually exclusive candidates across 50% to 70% of prostate cancer samples that were members of the ETS family of transcription factors, ERG and ETV1. Further experiments revealing fusions of the 5′-untranslated region of TMPRSS2 (21q22.3) with the ETS transcription factor family members—ERG (21q22.2), ETV1 (7p21.2),2 or ETV45—were identified, suggesting a novel mechanism for overexpression of the ETS genes in prostate cancer. The discovery of a known family of oncogenic transcription factors driven by a hormonally regulated promoter offers critical therapeutic opportunities to target the on-off switches created by the rearrangement (Fig 1) and suggests that additional common epithelial cancers may harbor similar organ-specific rearrangements.
Fig 1.

Targeting prostate cancer by using the gene fusion “on/off” switches. Gene fusion prostate cancers present an opportunity to target specific promoters and ETS genes. Several approaches can be considered in targeting gene fusion prostate cancers. In this schematic, the TMPRSS2 5′ promoter acts as an on switch in the presence of androgens and, in some settings, estrogen. Therefore, targeting the androgen receptor (AR) site of TMPRSS2 with small molecules may effectively decrease the expression of the fusion transcript. Approaches to target the mRNA fusion transcript by using short interfering RNA (siRNA) might be an effective means of decreasing the specific oncogenic fusion transcript. Small molecules might also be generated against the specific 3′ ETS gene or putative collaborating lesions such as PTEN or MYC). DHT, dehydrotestosterone; Rx, drug therapy (eg, small molecules, siRNA).
ETS Gene Fusions in Prostate Cancer Progression
Prostate cancer, like other cancers, develops in the background of diverse genetic and environmental factors. Multiple, complex molecular events characterize prostate cancer initiation, unregulated growth, invasion, and metastasis (Fig 2). Distinct sets of genes and proteins dictate progression from precursor lesion to localized disease and finally to metastatic disease. Clinically localized prostate cancer can be effectively ablated by using surgical or radiation treatments. Metastatic disease, however, is invariably incurable and leads to death. Androgen ablation is the most common therapy for advanced prostate cancer, leading to massive apoptosis of androgen-dependent malignant cells and temporary tumor regression. In most cases, however, the tumor re-emerges and can proliferate independent of androgen signals. With the advent of global profiling strategies, a systematic analysis of genes involved in prostate cancer is now possible. There are multiple key signaling pathways associated with prostate cancer progression. The androgen receptor (AR) plays a central role in any model, but other key pathways include PTEN, NKX3.1, MYC, and GST-pi (Fig 2).
Fig 2.

Two models of prostate cancer progression. The standard view has been that prostate cancer progresses through a series of molecular lesions. In the linear model, molecular events, including mutations, deletions, and amplifications, occur in sequence corresponding to progression of disease from the morphologically appearing benign prostate tissue, moving to high-grade prostatic intraepithelial neoplasia (PIN), then progressing to invasive prostate cancer, and finally to local and distant metastatic spread. However, in this review, we support the view that prostate cancer progresses through a wide range of lesions that lead to several possible pathways, some of which may not progress at all. In the molecular diversity model, alterations occur that might be classified as gatekeeper lesions. Once these events occur, additional events may lead to PIN that does not have the capacity to progress or PIN that may progress. Accumulation of molecular alterations associated with aggressive disease such as the overexpression of EZH2 or PTEN mutations may lead to invasive disease that progresses to metastatic disease, whereas other lesions such as 5q or 6q gain and overexpression of AZGP1 might be seen most often in indolent disease. Mutations and alterations associated with p53 and the androgen receptor (AR) are probably late events and may play a key role in the development of castration-resistant disease. SNP, single-nucleotide polymorphism.
Taken together, these molecular alterations represent events that may mutually add to the development and progression of prostate cancer. Although some investigators have favored a molecular model that includes linear accumulation of molecular lesions leading to prostate cancer progression,6 we favor a working model that includes multiple nodes for progression (Fig 3). This view is supported by the clinical and molecular heterogeneity identified in prostate cancer. Work by LaPointe et al7,8 and observations regarding gene fusion prostate cancer suggest that molecular alterations in prostate cancer do not accumulate in a linear manner but may, in fact, indicate differences in the ability to progress. As depicted in Figure 3, some molecular lesions may be seen in indolent tumors, whereas other tumors harboring a different set of alterations may progress to a metastatic state. Importantly, some molecular lesions may be associated with tumors that have little ability to progress beyond the in situ state. These theoretical considerations require the careful classification of tumors to aid in the determination of key factors in disease progression.
Fig 3.

Molecular events associated with prostate cancer development and progression. Recent work has identified several genes and pathways associated with prostate cancer progression. Critical pathways depicted in this schematic of a prostate cancer cell include disruption of the WNT signaling, PI3K/AKT/PTEN and RAS/RAF/MAPF kinase pathways. Other pathways may be involved in the inactivation of GST-pi through methylation and histone methylation by polycomb genes such as EZH2. The activation of alterations of the androgen receptor (AR) is also believed to play a central role in disease progression from the androgen-dependent state to the castration-resistant state observed in advanced disease. The ETS fusion cancers often harbor an upstream, hormonally regulated promoter (eg, TMPRSS2 or SLC45A3). These promoters are known androgen response elements (AREs) and act as amplifiers of the ETS gene expression in the setting of androgens. Interestingly, recent work has also demonstrated the presence of estrogen binding sites on the TMPRSS2 promoter site (not depicted), suggesting a mechanism for continued expression of the ETS fusion transcript in the castration state of low androgens. DHT, dehydrotestosterone.
UNDERSTANDING PROSTATE CANCER HETEROGENEITY THROUGH GENE FUSIONS
The clinical and molecular heterogeneity of prostate cancer represents a major challenge in developing adequate diagnostic and prognostic tools and creates a major hurdle in drug development. We propose that recognition of the complexity of gene fusion prostate cancers will lead to a better classification of a disease that, until now, has been treated as a single entity.
Multiple Types of Gene Fusions in Prostate Cancer
Since the initial discovery of ETS fusions in prostate cancer, several recent studies have identified fusion events involving additional ETS family members (ie, ELK4,9,10) novel 5′ (ie, upstream) partners, and a class of non–ETS-based fusions. On the basis of these discoveries, we have developed a classification system (Fig 4) comprising three categories: (1) fusions involving ETS gene family members (ERG, ETV1, ETV4, ETV5, and ELK4), (2) RAF kinase family fusions, and (3) SPINK1-positive cases.
Fig 4.
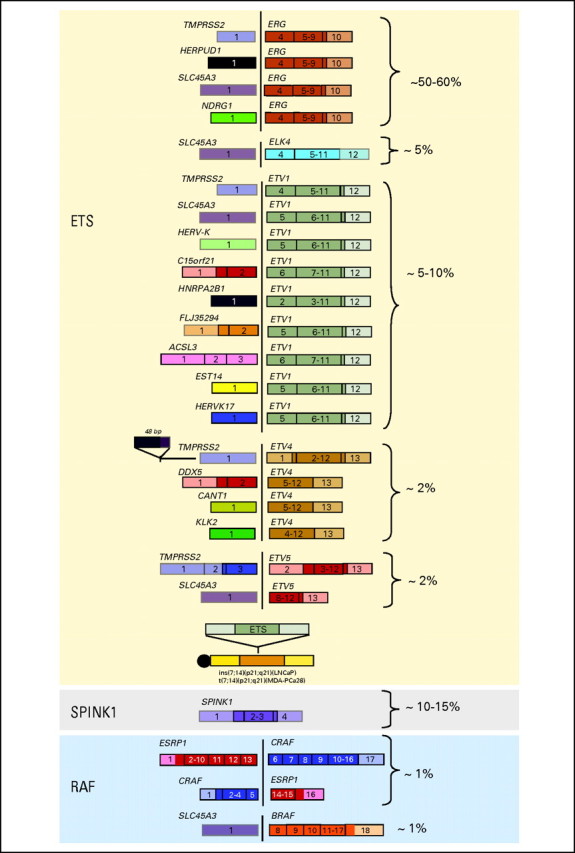
Prostate cancer gene fusion classification. The ongoing effort to screen prostate cancer patients for gene fusions, in combination with the recent technology advances, has resulted in a comprehensive gene fusion landscape. This schematic highlights all published gene fusions categorized into ETS rearrangements, RAF kinase gene fusions, and SPINK1-positive, ETS rearrangement–negative prostate cancers. The percentages highlight the estimated frequency of each gene fusion on the basis of published screens.
The largest category, ETS fusions, is composed of the highly recurrent TMPRSS2-ERG fusion, which contrasts with the remaining less common fusion events. Interestingly, the ETS family member fusions involve a diverse set of 5′ upstream partners, as exemplified by ETV1 having nine different fusion partners. In addition to TMPRSS2, three additional androgen responsive 5′ partners—SLC45A3,11,12 HERPUD1,13 and NDRG114—have been found to fuse with ERG. However, many of the 5′ partners appear to fuse to multiple ETS family members, such SLC45A3 (-ERG, -ELK4, -ETV1, and -ETV5) and TMPRSS2 (-ERG, -ETV1, -ETV4, and -ETV5), both of which are androgen responsive. Overall, the emerging trend is that most of these organ-specific promoters are driven initially by AR signaling. Thus, one hypothesis worthy of testing is that patients who harbor an androgen-induced gene fusion might be more responsive to hormonal treatment than those who harbor a constitutively active or androgen-repressed promoter.
Recent advances in next generation transcriptome sequencing facilitated the discovery of the second category—RAF kinase gene fusions SLC45A3-BRAF, ESRP1-RAF1, and RAF1-ESRP1 in advanced prostate cancers.15 Although rare, detected in approximately 1% to 2% of prostate cancers, RAF kinase fusions represent the first “driver” fusions in prostate cancers that do not involve an ETS family member. The third category, SPINK1-positive prostate cancers, is included in the classification since the outlier expression of SPINK1 occurs in ETS rearrangement–negative prostate cancers and therefore defines a specific subclass of prostate cancers.16 We presume that this is a first-generation classification and that future iterations will include other non-ETS gene fusions as well as driving molecular mutations as they are discovered.
A CALL FOR A MOLECULAR SUBCLASSIFICATION OF PROSTATE CANCER
Like hematologic and pediatric tumors, many neoplasms are defined by the genetic rearrangement they harbor as the defining oncogenic event; we believe the fusion of an androgen-driven promoter and an ETS family transcription factor should be a defining molecular event in prostate cancer. Here, we present supporting evidence based on the key role of ETS genes as oncogenic, phenotypic changes associated with the TMPRSS2-ERG fusion, in vitro and in vivo cell data, the early nature of this molecular event, the association with an aggressive natural history in the absence of treatment, and the presence of a defined molecular signature to justify the classification of TMPRSS2-ERG fusion cancers as a distinct subclass. We hope that future clinical and molecular studies will take into account the TMPRSS2-ERG fusion status and other subtypes as they become more clearly defined.
TMPRSS2-ETS fusions occur early and are present in the precursor lesion high-grade prostatic intraepithelial neoplasia. Microscopic examination of prostate cancers by using a fluorescent in situ hybridization (FISH) assay reveal that gene fusion occurs in neoplastic cells but not in adjacent benign nuclei or stromal cells.2,17,18 A larger study drawn from a wide spectrum of benign prostatic lesions and precursors of prostate cancer19 failed to detect TMPRSS2-ERG fusion in benign prostate tissue, benign prostatic hyperplasia, or proliferative inflammatory atrophy (also commonly referred to as focal prostate atrophy or prostate atrophy; reviewed in De Marzo et al20). The TMPRSS2-ERG fusion was observed in approximately 20% of high-grade prostatic intraepithelial neoplasia (PIN) lesions intermingled with prostate cancer that carried the same fusion pattern. This was the same frequency previously detected by Cerveira et al21 by using a reverse transcriptase polymerase chain reaction (RT-PCR) –based assay. We did not observe the TMPRSS2-ERG fusion in high-grade PIN lesions geographically distant to prostate cancer, even if the prostate cancer from the same individual demonstrated the TMPRSS2-ERG fusion. More recently, immunohistochemistry has been used to evaluate the gene fusions in situ.22 By using an antibody highly specific for ERG rearrangements, one can clearly see the earliest overexpression of the ERG oncogene in the morphologic area of high-grade PIN but not in directly adjacent benign prostate tissue (Fig 5). Hence, we believe these high-grade PIN lesions are a subset of true precursors for TMPRSS2-ERG–positive prostate cancer. A significant clinical implication for this finding is the potential utility of assessing the TMPRSS2-ERG fusion status in problematic prostate needle core biopsies with high-grade PIN and adjacent small atypical glands.
Fig 5.

ERG rearranged prostate cancer and high-grade prostatic intraepithelial neoplasia (HGPIN) express high levels of truncated ERG protein. Representative examples of prostate cancers and HGPIN (A and C) showing similar ERG protein expression by immunohistochemistry (a rabbit anti-ERG monoclonal antibody, clone EPR 3864, Epitomics, Burlingame, CA). Hematoxylin and eosin stain demonstrates prostatic cancerous glands (A) and another case with HGPIN (C). ERG protein expression by immunohistochemistry demonstrates strong expression in both cancer (B) and HGPIN (D). In (C), the PIN label indicates discrete demarcation between HGPIN and histologically benign luminal epithelial cells labeled B (×40).
Prevalence of Gene Fusions in Prostate Cancer
Several independent studies8,21,23–35 have corroborated the initial observation that TMPRSS2-ETS fusions are common in prostate cancer. Although most studies have focused on the dominant rearrangement TMPRSS2-ERG fusion, a variety of other fusions involving TMPRSS2 and other 5′ partners have been described (Fig 4) but appear to be less common.5,33,36–38 The prevalence of TMPRSS2-ERG prostate cancer has been reported to range from 40% to 70%, depending on the clinical cohorts investigated. The first large clinical study on a German prostatectomy cohort17 reported that approximately 50% of cases had a TMPRSS2-ERG fusion. Several retrospective studies24,30,32,36,39,40 from PSA-screened prostatectomy cohorts have reported frequencies of the TMPRSS2-ERG fusion between 35% and 50% when FISH assays were used to detect the rearrangement. Other smaller studies8,21,23,28,29 that used PCR-based methodology have reported higher frequencies. Only one study to date36 has comprehensively explored for the presence of other fusion partners and determined that an additional 5% to 10% of cases may harbor other gene fusions, including TMPRSS2-ETV1 and TMPRSS2-ETV4.
In two population-based studies from Sweden and the United Kingdom,30,41,42 15% to 20% of men diagnosed with incidental prostate cancer had tumors that harbored TMPRSS2-ERG. It is worth highlighting that the 354 incidental cancers from the Swedish cohorts were detected in five population-based cohorts before PSA screening.41 All of the tumors were detected on transurethral resection of the prostate (TURP) samples, which differs from the prostatectomy series. Although some have suggested that there may be a genetic component to this lower frequency in the Swedish population, we have determined that the frequency in a PSA-screened biopsy cohort from Örebro is approximately 45%, which is similar to that in all other PSA-screened hospital-based cohorts (Svensson and Rubin, manuscript in preparation). We have observed similar frequencies in gene fusion when examining hormone-naive primary and castration-resistant metastatic prostate cancers.43
As part of an Early Detection Research Network (EDRN) study sponsored by the National Cancer Institute, we prospectively determined that 46% of men with prostate cancer detected on 12 core needle biopsies by PSA screening harbor TMPRSS2-ERG fusion.44 This result is consistent with results in the surgical cohorts. Taken together, observations made over the past 3 years from several studies since the original description of TMPRRSS2-ETS prostate cancer suggest that the majority of prostate cancers currently detected by PSA screening harbor either the common TMPRSS2-ERG fusion (46%) or one of the less common fusions involving TMPRSS2 or other 5′ partners (5% to 10%). This has important clinical implications, because the TMPRSS2-ERG transcript can be detected in urine and represents a highly specific prostate cancer biomarker.
TMPRSS2-ERG and Association With a More Aggressive Clinical Outcome
The data generated in the search for associations with clinical outcome emerge from two types of studies: population-based watchful waiting cohorts and retrospective prostatectomy series. A review of the literature suggests that, in some instances, the TMPRSS2-ERG fusion is associated with a more aggressive clinical course but, conversely, others report the opposite result. We hope to clarify this confusion but concede that large population-based studies will be required to clarify this issue in the future.
Our group initially observed an enrichment in the TMPRSS2-ERG fusion in higher-stage prostate cancer.17 We then searched for associations between TMPRSS2-ERG fusion and clinical outcome in a population-based study.42 The Örebro watchful waiting cohort represents a treatment-naive population drawn from a strictly defined catchment area for 190,000 inhabitants living in Örebro. The TMPRSS2-ERG gene fusion was identified in 15% (17 of 111) of the patients' initial TURP biopsy samples and was significantly associated with prostate cancer–specific death (cumulative incidence ratio, 2.7; 95% CI, 1.3 to 5.8; P < .01). This is a well-defined population that dramatically differs from that in the retrospective prostatectomy series. First, this is a population-based cohort. All men with early prostate cancer (T1a-b, Nx, M0) diagnosed by TURP for symptomatic benign prostatic hyperplasia (ie, lower urinary tract symptoms) were included. There was no PSA screening in Sweden during the collection phase of this study. Second, the patients were followed expectantly (without curative treatment) and received clinical examinations, laboratory tests, and bone scans every 6 months during the first 2 years following diagnosis and subsequently at 12-month intervals. Third, the end point of the study was lethal prostate cancer, defined as development of distant metastases or prostate cancer as the underlying cause of death (median follow-up time, 9.1 years; maximum, 28 years). Therefore, this unique study design allows one to assess the biologic impact of TMPRSS2-ERG prostate cancer in the absence of early intervention.
The results of this study were supported by a report from the United Kingdom30 that identified associations between TMPRSS2-ERG fusion and survival of 445 men conservatively treated for prostate cancer. Overall, cancers lacking TMPRSS2-ERG fusion alterations demonstrated 90% survival at 8 years of clinical follow-up. The report also identified a novel association seen in TMPRSS2-ERG fusion prostate cancer in which the fusion of TMPRSS2 to ERG, along with interstitial deletion of sequences 5′ to ERG,17 was associated with a significantly worse cause-specific survival that took into account age, Gleason score, and pretreatment PSA. Supporting the hypothesis that overexpression of ERG is acting as an oncogene, the overall lowest cause-specific survival was associated with a duplication of the TMPRSS2-ERG fusion with an accompanying interstitial deletion (hazard ratio, 6.10; 95% CI, 3.33 to 11.15; P < .001; 25% survival at 8 years). On multivariate analysis, the duplication of the TMPRSS2-ERG fusion with associated deletion (referred to as “2+Edel”) was an independent predictor of clinical outcome that provided information in addition to Gleason score and pretreatment PSA level.
This study reported on 110 clinical T1 prostate cancer cases that had 20% TMPRSS2-ERG fusion similar to that in the Swedish watchful waiting cohort. This study supports the aggressive biologic significance of the TMPRSS2-ERG fusion. Two key observations from this study were that gain of ERG and the associated interstitial deletion of the 3-Mb region between TMPRSS2 and ERG on chromosome 21 are associated with more aggressive prostate cancer. Overexpression of ERG has been associated with poor clinical outcome in acute myeloid leukemia,45 and some of the genes located in the 3-Mb area of deletion (eg, HMGN1, ETS-2) may be acting as tumor suppressor genes.17
Several retrospective studies29,31,35 that sought an association between TMPRSS2-ERG and outcome following radical prostatectomy gave mixed results. It is difficult to compare results from a surgical study that used PSA biochemical failure as an end point with one that used observational studies with cancer-specific death as an outcome. One of the limitations of using an increase in PSA following prostatectomy as a surrogate end point comes from a single-institution study of men diagnosed with clinically localized prostate cancer in the pre-PSA-screening era. Porter et al46 observed 45.5% PSA biochemical failure in a radical prostatectomy series, but prostate cancer–specific death occurred in only 18.5% of the population with a follow-up time of up to 25 years. Carver et al47 reported that, in a population of high-risk men with T3 prostate cancer who underwent radical prostatectomy, 36% with PSA biochemical failure subsequently developed clinically relevant disease progression. Ward et al48 found that in a population of 3,897 radical prostatectomy patients, only 8.3% of the men with PSA biochemical failure died of prostate cancer with a median follow-up time of 10 years. An increase in PSA following surgery is associated with prostate cancer–specific death, but the majority of men with PSA biochemical failure will die of other causes. Therefore, we would argue for caution in overinterpreting the results of each of these types of clinical studies.
On the basis of the two large observational clinical studies with long-term follow-up, we would argue that left untreated, TMPRSS2-ERG prostate cancer will run a more aggressive clinical course than fusion-negative cancer. In the setting of surgical or other interventions immediately following diagnosis, there is insufficient data to make any reasonable conclusions.
Gene fusion is a key molecular event in prostate cancer development. Initial work exploring the role of the TMPRSS2-ERG fusions in cell lines demonstrates fairly consistent findings for overexpression of the ETS gene in benign epithelial cells. Studies that have overexpressed ETV1, ETV5, and ERG have demonstrated an increase in cell invasion capability, not an increase in proliferation or the ability to transform these cells into tumor cells.37,38,49 This was recently confirmed by Klezovitch et al,50 who demonstrated that the overexpression of ERG is associated with tumor cell migration through a proteolytic molecular program. These results suggest that ETS genes alone are insufficient to cause a transformation to cancer but may play a key role in the development of the invasive phenotype in the context of other underlying molecular alterations. It is also possible that these models do not capture the complexity of deregulation due to the fusion events. For example, could the decreased expression of ETS-2, located in the minimally deleted region of a translocated allele, in conjunction with ERG overexpression play a different role in vivo?
There are several published and unpublished mouse models that have been generated to recapitulate the overexpression of ERG49,50 and ETV1.37 All of these models demonstrate the ability of the trans gene to develop early molecular changes referred to as mouse PIN.51 These subtle changes have not reached the level of invasive cancer.52 This is similar to models of NKX3.1 and PTEN. Therefore, more recent efforts have focused on the identification of cooperating events in ETS-induced prostate carcinoma to rationalize combined therapies. For instance, Zong et al52 demonstrated that ERG overexpression cooperates with PI3K signaling to progress to invasive prostate adenocarcinoma. In addition, the combination of overexpressing both AR and ERG promoted the development of poorly differentiated invasive adenocarcinomas. These promising results support ongoing work to further elucidate the combination of other known prostate cancer oncogenes and to explore a cumulative effect. Therefore, the in vitro and in vivo models demonstrate that ETS genes have an effect on tumor progression but alone do not appear to be sufficient for transformation into cancer.
Gene fusion is a clonal event that aids understanding of prostate cancer heterogeneity. It is recognized that prostate cancer is multifocal. Both morphologic and molecular analysis have shown that by the time prostate cancer is diagnosed, more than 80% of prostates harbor multiple separate cancer foci.53–57 These discrete lesions have both biologic and clinical implications. The TMPRSS2-ERG fusion represents an excellent early clonal marker to provide insight into molecular heterogeneity.
TMPRSS2-ERG fusions, when present, are distributed evenly among all tumor nuclei within a discrete tumor lesion. We reported that 243 of 246 prostate cancer cases demonstrated homogeneity within a discrete tumor nodule.17 This observation was extended when multiple microdissected foci of cancer from individual patients were examined by RT-PCR for gene fusions and demonstrated that either all or no foci overexpressed ERG and its family members ETV1 and ETV4.58 Thus, within a discrete nodule, the fusion rearrangement must occur early because all of the tumor nuclei harbor the fusion when present. However, when we undertook studies to evaluate rearrangement among the multiple nodules within a single prostate gland from one individual, we found that discrete lesions may occur independently from one another. This has been observed in three independently conducted studies.59–61 For example, in the study by Barry et al,60 32 prostatectomy samples with clear-cut discrete tumors demonstrated fusion by balanced translocation and fusion by interstitial deletion occurring as distinct events, suggesting that these are clonal mechanisms for achieving TMPRSS2-ERG fusion. Interestingly, that study found a high rate of interfocal heterogeneity for fusion status (41%). These observations have both biologic and clinical implications. Biologically, the presence of multiple clonally distinct lesions suggests that, within a single gland, complex molecular events such as gene rearrangement can occur in some but not all lesions. This makes classifying prostate cancers more challenging. From a clinical perspective, how does one determine the most aggressive nodule to target? It has long been assumed that the dominant nodule harbors the most aggressive tumor and therefore dictates the clinical course. Therefore, if TMPRSS2-ERG prostate cancers are more biologically aggressive, strategies will be needed to detect them regardless of their size because these may be the tumors with the highest propensity for metastatic dissemination.44
DIAGNOSTIC AND CLINICAL THERAPY IMPLICATIONS
PSA has a diminished role in detecting prostate cancer, thus the requirement for a new molecular detection test. Several studies62–64 to date have demonstrated the detection of the TMPRSS2-ERG fusion transcripts in urine. These studies and other unpublished reports demonstrate a high specificity. Unlike PSA, which can be increased in benign conditions as well as in cancer, the presence of TMPRSS2-ERG transcripts has been reported only in neoplastic cells. In addition to the sensitive and specific detection of TMPRSS2-ERG in urine sediment,64 recent work has demonstrated improved detection of prostate cancer by using multiple biomarkers. Multiplexed detection of GOLM1, SPINK1, PCA3, and TMPRSS2-ERG was a more significant predictor of prostate cancer than serum PSA or PCA3 alone.64 These results are promising and, with some refinement, could be adopted as a clinical supplement to serum PSA for prostate cancer detection.
Given the heterogeneity demonstrated between tumor nodules, a positive TMPRSS2-ERG urine test and a biopsy negative for cancer would suggest that the cancer has been missed. If the cancer is detected but is fusion negative, the sampling would have missed the fusion cancer. The finding of interfocal heterogeneity for TMPRSS2-ERG fusion has direct relevance in the context of a urine test result that is positive for fusion and a subsequent prostate biopsy with cancer that is negative for fusion. Given the potential prognostic role of determining the mode of rearrangement (deletion through translocation v through interstitial deletion), a biopsy FISH test would allow for an accurate determination of the presence and type of gene fusions (Fig 6).
Fig 6.

The diagnostic predictive and prognostic implication of ETS fusion prostate cancer. The fusion of two genes to form a novel chimeric mRNA transcript represents a unique opportunity to develop a diagnostic test. Recent studies have demonstrated that the fusion transcript can be identified in the serum and urine from men with prostate cancer. The urine assay is being developed commercially with the goal of establishing a highly specific test. Prostate tissue derived from clinical biopsies, transurethral resection of the prostate samples, or radical prostatectomies can be used to detect the ETS rearrangement events by using fluorescent in situ hybridization (FISH) or reverse transcriptase polymerase chain reaction (PCR). The identification of ETSrearrangements may have prognostic implications in specific settings (eg, an active surveillance clinical trial) and may also be predictive of response to targeted therapies such as those targeting the androgen receptors. The significance of these clinical assays will largely depend on future studies that determine to what extent ETS rearrangement prostate cancers behave differently from nonrearranged prostate cancers. Bx, biopsy.
Recent trials in the setting of castration-resistant prostate cancer suggest that targeting androgen and estrogen might be an effective approach. Data suggest that low levels of intraprostatic testosterone or dehydrotestosterone are still present when men have undergone chemical castration with antiandrogens. Therefore, novel approaches have been developed to reduce these low levels of androgens and estrogens by blocking steroid synthesis. Abiraterone acetate is a selective small-molecule inhibitor of cytochrome P450 17 (CYP17), which effectively blocks the production of androgen and estrogen.65 It was recently tested in a phase I clinical trial, and it demonstrated a decrease in PSA following treatment in 50% of all men with castration-independent prostate cancer.66,67 In that study, 83% of men (5 of 6) with TMPRSS2-ERG fusion prostate cancer had a decrease in PSA following abiraterone treatment. Although that study was not designed to test the potential role of abiraterone with respect to TMPRSS2-ERG fusion status, future phase II and III studies will examine this hypothesis on the basis of these initial observations.
The RAF kinase fusions, although rare, are of immediate therapeutic significance given the numerous approved and investigational agents in the late stage of development. Palanisamy et al15 demonstrated that the RAF kinase fusions were sensitive to sorafenib, a US Food and Drug Administration (FDA) –approved RAF inhibitor that has also been demonstrated to target additional kinases.68 This suggests that screening patients for RAF fusions may identify a subset of the population that may benefit from existing targeted therapies similar to the current clinical application of ALK inhibitors to patients with EML-ALK4 non–small-cell lung carcinoma.69,70 We envision that other targetable gene fusions and driving mutations will be discovered in the coming years.
Ateeq et al71 recently demonstrated that SPINK1 prostate cancer can be targeted by using cetuximab, an epidermal growth factor receptor (EGFR) inhibitor. SPINK1 harbors a high homology with EGF. Preclinical models that use recombinant SPINK1 support targeting the extracellular domain of SPINK1. This early work provides a rationale for both the development of humanized monoclonal antibodies to SPINK1 and evaluation of EGFR inhibition in SPINK1-positive/ETS-negative prostate cancers.
EMERGING UNDERSTANDING OF PROSTATE CANCER GENOMIC COMPLEXITY
The emerging picture of prostate cancer genomic complexity demonstrates numerous rearrangements including the well-described ETS rearrangements.72 Some of these complex genomic alterations might lead to deregulation of important signaling pathways such as the MAGI2 inversions described by Berger et al72 that putatively lead to AKT activation. Understanding the underlying cause of these rearrangements may play a role in chemoprevention or selection of chemotherapies.
Genomic rearrangements appear to be nonrandom and locus-specific, and they depend, in part, on the proximity of chromosomal regions in the nucleus.73 Moreover, there is mounting evidence suggesting that transcription factors are associated with DNA double-strand breaks, thus predisposing transcribed regions to genomic rearrangements. For example, both androgen and estrogen signaling recruit the enzyme topoisomerase-2 beta (TOP-2b) to target gene promoters, which creates DNA double-strand breaks and facilitates transcription.74,75 AR and TOP-2b are coexpressed in human prostate cancer precursor lesions in which TMPRSS2-ERG rearrangements are known to occur, suggesting a critical role of TOP-2b in the recurrent ETS rearrangements. Three recent studies76–78 have also shown that androgen signaling promotes TMPRSS2-ERG fusion formation, in part, by recruiting DNA break-inducing enzymes such as activation of induced cytidine deaminase to translocation breakpoint sites.77 More recently, we demonstrated that rearrangement breakpoints were enriched near open chromatin, AR, and tERG DNA binding sites in the setting of the ETS gene fusion TMPRSS2-ERG but were inversely correlated with these regions in tumors lacking ETS fusions.72 Hence, transcription factors can contribute to the formation of genomic rearrangements by facilitating the juxtaposition of chromosomal loci and recruiting enzymatic machinery involved in DNA breaks to these target loci. This work also suggests that inhibitors of repair enzymes such as PARP1 and DNA-PK decrease the susceptibility to gene fusions. It also raises concerns that TOP-2b inhibitors such as etoposide or doxorubicin might facilitate gene fusions and rearrangements by enhancing double-stranded DNA breaks. Ongoing research is exploring the clinical implications of these observations.
In conclusion, gene fusion prostate cancer is among the most common genetic alterations identified in cancer. Although several ETS and non-ETS family members have been observed to be fused with TMPRSS2 or other 5′ partners, the vast majority of fusions involve TMPRSS2-ERG. This fusion can easily be studied, because it was identified in approximately 50% of all prostate cancers screened for PSA. Associations with disease-specific death have been made in clinical observation studies. The amplification of the TMPRSS2-ERG fusion and the interstitial deletion associated with the translocation add additional statistical power to predicting lethal prostate cancer. Morphologic features, functional in vitro and in vivo studies, and a specific gene signature support the view that the TMPRSS2-ERG fusion cancers represent a distinct molecular subclass. The more recent discovery of the RAF fusions also demonstrates that some of the gene fusions will be targets for clinical intervention.
Acknowledgment
We thank Jill Granger and Brigitte M. Terrier for assistance in editing this article; and Scott Tomlins, Sven Perner, and John Prensner for contributions and review of components of this review.
Footnotes
Authors' disclosures of potential conflicts of interest and author contributions are found at the end of this article.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Although all authors completed the disclosure declaration, the following author(s) indicated a financial or other interest that is relevant to the subject matter under consideration in this article. Certain relationships marked with a “U” are those for which no compensation was received; those relationships marked with a “C” were compensated. For a detailed description of the disclosure categories, or for more information about ASCO's conflict of interest policy, please refer to the Author Disclosure Declaration and the Disclosures of Potential Conflicts of Interest section in Information for Contributors.
Employment or Leadership Position: None Consultant or Advisory Role: Mark A. Rubin, Gen-Probe (C), Ventana/Roche (C); Arul M. Chinnaiyan, Compendia Bioscience (C), Gen-Probe (C) Stock Ownership: None Honoraria: None Research Funding: None Expert Testimony: None Other Remuneration: None
AUTHOR CONTRIBUTIONS
Manuscript writing: All authors
Final approval of manuscript: All authors
REFERENCES
- 1.Jemal A, Siegel R, Xu J, et al. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Tomlins SA, Rhodes DR, Perner S, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–648. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 3.Rubin MA, Chinnaiyan AM. Bioinformatics approach leads to the discovery of the TMPRSS2:ETS gene fusion in prostate cancer. Lab Invest. 2006;86:1099–1102. doi: 10.1038/labinvest.3700477. [DOI] [PubMed] [Google Scholar]
- 4.Hanauer DA, Rhodes DR, Sinha-Kumar C, et al. Bioinformatics approaches in the study of cancer. Curr Mol Med. 2007;7:133–141. doi: 10.2174/156652407779940431. [DOI] [PubMed] [Google Scholar]
- 5.Tomlins SA, Mehra R, Rhodes DR, et al. TMPRSS2:ETV4 gene fusions define a third molecular subtype of prostate cancer. Cancer Res. 2006;66:3396–3400. doi: 10.1158/0008-5472.CAN-06-0168. [DOI] [PubMed] [Google Scholar]
- 6.Nelson WG, De Marzo AM, Isaacs WB. Prostate cancer. N Engl J Med. 2003;349:366–381. doi: 10.1056/NEJMra021562. [DOI] [PubMed] [Google Scholar]
- 7.Lapointe J, Li C, Higgins JP, et al. Gene expression profiling identifies clinically relevant subtypes of prostate cancer. Proc Natl Acad Sci U S A. 2004;101:811–816. doi: 10.1073/pnas.0304146101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lapointe J, Kim YH, Miller MA, et al. A variant TMPRSS2 isoform and ERG fusion product in prostate cancer with implications for molecular diagnosis. Mod Pathol. 2007;20:467–473. doi: 10.1038/modpathol.3800759. [DOI] [PubMed] [Google Scholar]
- 9.Maher CA, Kumar-Sinha C, Cao X, et al. Transcriptome sequencing to detect gene fusions in cancer. Nature. 2009;458:97–101. doi: 10.1038/nature07638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rickman DS, Pflueger D, Moss B, et al. SLC45A3-ELK4 is a novel and frequent erythroblast transformation-specific fusion transcript in prostate cancer. Cancer Res. 2009;69:2734–2738. doi: 10.1158/0008-5472.CAN-08-4926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Esgueva R, Perner S, J LaFargue C, et al. Prevalence of TMPRSS2-ERG and SLC45A3-ERG gene fusions in a large prostatectomy cohort. Mod Pathol. 2010;23:539–546. doi: 10.1038/modpathol.2009.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Han B, Mehra R, Dhanasekaran SM, et al. A fluorescence in situ hybridization screen for E26 transformation-specific aberrations: Identification of DDX5-ETV4 fusion protein in prostate cancer. Cancer Res. 2008;68:7629–7637. doi: 10.1158/0008-5472.CAN-08-2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maher CA, Palanisamy N, Brenner JC, et al. Chimeric transcript discovery by paired-end transcriptome sequencing. Proc Natl Acad Sci U S A. 2009;106:12353–12358. doi: 10.1073/pnas.0904720106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pflueger D, Rickman DS, Sboner A, et al. N-myc downstream regulated gene 1 (NDRG1) is fused to ERG in prostate cancer. Neoplasia. 2009;11:804–811. doi: 10.1593/neo.09572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Palanisamy N, Ateeq B, Kalyana-Sundaram S, et al. Rearrangements of the RAF kinase pathway in prostate cancer, gastric cancer and melanoma. Nat Med. 2010;16:793–798. doi: 10.1038/nm.2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tomlins SA, Rhodes DR, Yu J, et al. The role of SPINK1 in ETS rearrangement-negative prostate cancers. Cancer Cell. 2008;13:519–528. doi: 10.1016/j.ccr.2008.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Perner S, Demichelis F, Beroukhim R, et al. TMPRSS2:ERG fusion-associated deletions provide insight into the heterogeneity of prostate cancer. Cancer Res. 2006;66:8337–8341. doi: 10.1158/0008-5472.CAN-06-1482. [DOI] [PubMed] [Google Scholar]
- 18.Mosquera JM, Perner S, Genega EM, et al. Characterization of TMPRSS2-ERG fusion high-grade prostatic intraepithelial neoplasia and potential clinical implications. Clin Cancer Res. 2008;14:3380–3385. doi: 10.1158/1078-0432.CCR-07-5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perner S, Mosquera JM, Demichelis F, et al. TMPRSS2-ERG fusion prostate cancer: An early molecular event associated with invasion. Am J Surg Pathol. 2007;31:882–888. doi: 10.1097/01.pas.0000213424.38503.aa. [DOI] [PubMed] [Google Scholar]
- 20.De Marzo AM, Platz EA, Epstein JI, et al. A working group classification of focal prostate atrophy lesions. Am J Surg Pathol. 2006;30:1281–1291. doi: 10.1097/01.pas.0000213289.50660.be. [DOI] [PubMed] [Google Scholar]
- 21.Cerveira N, Ribeiro FR, Peixoto A, et al. TMPRSS2-ERG gene fusion causing ERG overexpression precedes chromosome copy number changes in prostate carcinomas and paired HGPIN lesions. Neoplasia. 2006;8:826–832. doi: 10.1593/neo.06427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Park K, Tomlins SA, Mudaliar KM, et al. Antibody-based detection of ERG rearrangement-positive prostate cancer. Neoplasia. 2010;12:590–598. doi: 10.1593/neo.10726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ahlers CM, Figg WD. ETS-TMPRSS2 fusion gene products in prostate cancer. Cancer Biol Ther. 2006;5:254–255. doi: 10.4161/cbt.5.3.2603. [DOI] [PubMed] [Google Scholar]
- 24.Clark J, Merson S, Jhavar S, et al. Diversity of TMPRSS2-ERG fusion transcripts in the human prostate. Oncogene. 2007;26:2667–2673. doi: 10.1038/sj.onc.1210070. [DOI] [PubMed] [Google Scholar]
- 25.Hermans KG, van Marion R, van Dekken H, et al. TMPRSS2:ERG fusion by translocation or interstitial deletion is highly relevant in androgen-dependent prostate cancer, but is bypassed in late-stage androgen receptor-negative prostate cancer. Cancer Res. 2006;66:10658–10663. doi: 10.1158/0008-5472.CAN-06-1871. [DOI] [PubMed] [Google Scholar]
- 26.Iljin K, Wolf M, Edgren H, et al. TMPRSS2 fusions with oncogenic ETS factors in prostate cancer involve unbalanced genomic rearrangements and are associated with HDAC1 and epigenetic reprogramming. Cancer Res. 2006;66:10242–10246. doi: 10.1158/0008-5472.CAN-06-1986. [DOI] [PubMed] [Google Scholar]
- 27.Liu W, Chang B, Sauvageot J, et al. Comprehensive assessment of DNA copy number alterations in human prostate cancers using Affymetrix 100K SNP mapping array. Genes Chromosomes Cancer. 2006;45:1018–1032. doi: 10.1002/gcc.20369. [DOI] [PubMed] [Google Scholar]
- 28.Soller MJ, Isaksson M, Elfving P, et al. Confirmation of the high frequency of the TMPRSS2/ERG fusion gene in prostate cancer. Genes Chromosomes Cancer. 2006;45:717–719. doi: 10.1002/gcc.20329. [DOI] [PubMed] [Google Scholar]
- 29.Wang J, Cai Y, Ren C, et al. Expression of variant TMPRSS2/ERG fusion messenger RNAs is associated with aggressive prostate cancer. Cancer Res. 2006;66:8347–8351. doi: 10.1158/0008-5472.CAN-06-1966. [DOI] [PubMed] [Google Scholar]
- 30.Attard G, Clark J, Ambroisine L, et al. Duplication of the fusion of TMPRSS2 to ERG sequences identifies fatal human prostate cancer. Oncogene. 2008;27:253–263. doi: 10.1038/sj.onc.1210640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nam RK, Sugar L, Wang Z, et al. Expression of TMPRSS2 ERG gene fusion in prostate cancer cells is an important prognostic factor for cancer progression. Cancer Biol Ther. 2007;6:40–45. doi: 10.4161/cbt.6.1.3489. [DOI] [PubMed] [Google Scholar]
- 32.Rajput AB, Miller MA, De Luca A, et al. Frequency of the TMPRSS2:ERG gene fusion is increased in moderate to poorly differentiated prostate cancers. J Clin Pathol. 2007;60:1238–1243. doi: 10.1136/jcp.2006.043810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Winnes M, Lissbrant E, Damber JE, et al. Molecular genetic analyses of the TMPRSS2-ERG and TMPRSS2-ETV1 gene fusions in 50 cases of prostate cancer. Oncol Rep. 2007;17:1033–1036. [PubMed] [Google Scholar]
- 34.Yoshimoto M, Ludkovski O, Bayani J, et al. Microdeletion and concurrent translocation associated with a complex TMPRSS2:ERG prostate cancer gene fusion. Genes Chromosomes Cancer. 2007;46:861–863. doi: 10.1002/gcc.20470. [DOI] [PubMed] [Google Scholar]
- 35.Saramäki OR, Harjula AE, Martikainen PM, et al. TMPRSS2:ERG fusion identifies a subgroup of prostate cancers with a favorable prognosis. Clin Cancer Res. 2008;14:3395–3400. doi: 10.1158/1078-0432.CCR-07-2051. [DOI] [PubMed] [Google Scholar]
- 36.Mehra R, Tomlins SA, Shen R, et al. Comprehensive assessment of TMPRSS2 and ETS family gene aberrations in clinically localized prostate cancer. Mod Pathol. 2007;20:538–544. doi: 10.1038/modpathol.3800769. [DOI] [PubMed] [Google Scholar]
- 37.Tomlins SA, Laxman B, Dhanasekaran SM, et al. Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature. 2007;448:595–599. doi: 10.1038/nature06024. [DOI] [PubMed] [Google Scholar]
- 38.Helgeson BE, Tomlins SA, Shah N, et al. Characterization of TMPRSS2:ETV5 and SLC45A3:ETV5 gene fusions in prostate cancer. Cancer Res. 2008;68:73–80. doi: 10.1158/0008-5472.CAN-07-5352. [DOI] [PubMed] [Google Scholar]
- 39.Yoshimoto M, Joshua AM, Chilton-Macneill S, et al. Three-color FISH analysis of TMPRSS2/ERG fusions in prostate cancer indicates that genomic microdeletion of chromosome 21 is associated with rearrangement. Neoplasia. 2006;8:465–469. doi: 10.1593/neo.06283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tu JJ, Rohan S, Kao J, et al. Gene fusions between TMPRSS2 and ETS family genes in prostate cancer: Frequency and transcript variant analysis by RT-PCR and FISH on paraffin-embedded tissues. Mod Pathol. 2007;20:921–928. doi: 10.1038/modpathol.3800903. [DOI] [PubMed] [Google Scholar]
- 41.Setlur SR, Mertz KD, Hoshida Y, et al. Estrogen-dependent signaling in a molecularly distinct subclass of aggressive prostate cancer. J Natl Cancer Inst. 2008;100:815–825. doi: 10.1093/jnci/djn150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Demichelis F, Fall K, Perner S, et al. TMPRSS2:ERG gene fusion associated with lethal prostate cancer in a watchful waiting cohort. Oncogene. 2007;26:4596–4599. doi: 10.1038/sj.onc.1210237. [DOI] [PubMed] [Google Scholar]
- 43.Mehra R, Tomlins SA, Yu J, et al. Characterization of TMPRSS2-ETS gene aberrations in androgen-independent metastatic prostate cancer. Cancer Res. 2008;68:3584–3590. doi: 10.1158/0008-5472.CAN-07-6154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mosquera JM, Mehra R, Regan MM, et al. Prevalence of TMPRSS2-ERG fusion prostate cancer among men undergoing prostate biopsy in the United States. Clin Cancer Res. 2009;15:4706–4711. doi: 10.1158/1078-0432.CCR-08-2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marcucci G, Baldus CD, Ruppert AS, et al. Overexpression of the ETS-related gene, ERG, predicts a worse outcome in acute myeloid leukemia with normal karyotype: A Cancer and Leukemia Group B study. J Clin Oncol. 2005;23:9234–9242. doi: 10.1200/JCO.2005.03.6137. [DOI] [PubMed] [Google Scholar]
- 46.Porter CR, Kodama K, Gibbons RP, et al. 25-year prostate cancer control and survival outcomes: A 40-year radical prostatectomy single institution series. J Urol. 2006;176:569–574. doi: 10.1016/j.juro.2006.03.094. [DOI] [PubMed] [Google Scholar]
- 47.Carver BS, Bianco FJ, Jr, Scardino PT, et al. Long-term outcome following radical prostatectomy in men with clinical stage T3 prostate cancer. J Urol. 2006;176:564–568. doi: 10.1016/j.juro.2006.03.093. [DOI] [PubMed] [Google Scholar]
- 48.Ward JF, Blute ML, Slezak J, et al. The long-term clinical impact of biochemical recurrence of prostate cancer 5 or more years after radical prostatectomy. J Urol. 2003;170:1872–1876. doi: 10.1097/01.ju.0000091876.13656.2e. [DOI] [PubMed] [Google Scholar]
- 49.Tomlins SA, Laxman B, Varambally S, et al. Role of the TMPRSS2-ERG gene fusion in prostate cancer. Neoplasia. 2008;10:177–188. doi: 10.1593/neo.07822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Klezovitch O, Risk M, Coleman I, et al. A causal role for ERG in neoplastic transformation of prostate epithelium. Proc Natl Acad Sci U S A. 2008;105:2105–2110. doi: 10.1073/pnas.0711711105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shappell SB, Thomas GV, Roberts RL, et al. Prostate pathology of genetically engineered mice: Definitions and classification—The consensus report from the Bar Harbor meeting of the Mouse Models of Human Cancer Consortium Prostate Pathology Committee. Cancer Res. 2004;64:2270–2305. doi: 10.1158/0008-5472.can-03-0946. [DOI] [PubMed] [Google Scholar]
- 52.Zong Y, Xin L, Goldstein AS, et al. ETS family transcription factors collaborate with alternative signaling pathways to induce carcinoma from adult murine prostate cells. Proc Natl Acad Sci U S A. 2009;106:12465–12470. doi: 10.1073/pnas.0905931106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Arora R, Koch MO, Eble JN, et al. Heterogeneity of Gleason grade in multifocal adenocarcinoma of the prostate. Cancer. 2004;100:2362–2366. doi: 10.1002/cncr.20243. [DOI] [PubMed] [Google Scholar]
- 54.Cheng L, Song SY, Pretlow TG, et al. Evidence of independent origin of multiple tumors from patients with prostate cancer. J Natl Cancer Inst. 1998;90:233–237. doi: 10.1093/jnci/90.3.233. [DOI] [PubMed] [Google Scholar]
- 55.Greene DR, Wheeler TM, Egawa S, et al. A comparison of the morphological features of cancer arising in the transition zone and in the peripheral zone of the prostate. J Urol. 1991;146:1069–1076. doi: 10.1016/s0022-5347(17)38003-5. [DOI] [PubMed] [Google Scholar]
- 56.Qian J, Bostwick DG, Takahashi S, et al. Chromosomal anomalies in prostatic intraepithelial neoplasia and carcinoma detected by fluorescence in situ hybridization. Cancer Res. 1995;55:5408–5414. [PubMed] [Google Scholar]
- 57.Sakr WA, Macoska JA, Benson P, et al. Allelic loss in locally metastatic, multisampled prostate cancer. Cancer Res. 1994;54:3273–3277. [PubMed] [Google Scholar]
- 58.Tomlins SA, Mehra R, Rhodes DR, et al. Integrative molecular concept modeling of prostate cancer progression. Nat Genet. 2007;39:41–51. doi: 10.1038/ng1935. [DOI] [PubMed] [Google Scholar]
- 59.Clark J, Attard G, Jhavar S, et al. Complex patterns of ETS gene alteration arise during cancer development in the human prostate. Oncogene. 2008;27:1993–2003. doi: 10.1038/sj.onc.1210843. [DOI] [PubMed] [Google Scholar]
- 60.Barry M, Perner S, Demichelis F, et al. TMPRSS2-ERG fusion heterogeneity in multifocal prostate cancer: Clinical and biologic implications. Urology. 2007;70:630–633. doi: 10.1016/j.urology.2007.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mehra R, Han B, Tomlins SA, et al. Heterogeneity of TMPRSS2 gene rearrangements in multifocal prostate adenocarcinoma: Molecular evidence for an independent group of diseases. Cancer Res. 2007;67:7991–7995. doi: 10.1158/0008-5472.CAN-07-2043. [DOI] [PubMed] [Google Scholar]
- 62.Hessels D, Smit FP, Verhaegh GW, et al. Detection of TMPRSS2-ERG fusion transcripts and prostate cancer antigen 3 in urinary sediments may improve diagnosis of prostate cancer. Clin Cancer Res. 2007;13:5103–5108. doi: 10.1158/1078-0432.CCR-07-0700. [DOI] [PubMed] [Google Scholar]
- 63.Laxman B, Tomlins SA, Mehra R, et al. Noninvasive detection of TMPRSS2:ERG fusion transcripts in the urine of men with prostate cancer. Neoplasia. 2006;8:885–888. doi: 10.1593/neo.06625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Laxman B, Morris DS, Yu J, et al. A first-generation multiplex biomarker analysis of urine for the early detection of prostate cancer. Cancer Res. 2008;68:645–649. doi: 10.1158/0008-5472.CAN-07-3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Barrie SE, Potter GA, Goddard PM, et al. Pharmacology of novel steroidal inhibitors of cytochrome P450(17) alpha (17 alpha-hydroxylase/C17-20 lyase) J Steroid Biochem Mol Biol. 1994;50:267–273. doi: 10.1016/0960-0760(94)90131-7. [DOI] [PubMed] [Google Scholar]
- 66.Attard G, Reid AH, Yap TA, et al. Phase I clinical trial of a selective inhibitor of CYP17, abiraterone acetate, confirms that castration-resistant prostate cancer commonly remains hormone driven. J Clin Oncol. 2008;26:4563–4571. doi: 10.1200/JCO.2007.15.9749. [DOI] [PubMed] [Google Scholar]
- 67.Attard G, Swennenhuis JF, Olmos D, et al. Characterization of ERG, AR and PTEN gene status in circulating tumor cells from patients with castration-resistant prostate cancer. Cancer Res. 2009;69:2912–2918. doi: 10.1158/0008-5472.CAN-08-3667. [DOI] [PubMed] [Google Scholar]
- 68.Wilhelm SM, Adnane L, Newell P, et al. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol Cancer Ther. 2008;7:3129–3140. doi: 10.1158/1535-7163.MCT-08-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Koivunen JP, Mermel C, Zejnullahu K, et al. EML4-ALK fusion gene and efficacy of an ALK kinase inhibitor in lung cancer. Clin Cancer Res. 2008;14:4275–4283. doi: 10.1158/1078-0432.CCR-08-0168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561–566. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- 71.Ateeq B, Tomlins SA, Laxman B, et al. Therapeutic targeting of SPINK1-positive prostate cancer. Sci Transl Med. 2011;3:72ra17. doi: 10.1126/scitranslmed.3001498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Berger MF, Lawrence MS, Demichelis F, et al. The genomic complexity of primary human prostate cancer. Nature. 2011;470:214–220. doi: 10.1038/nature09744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mani RS, Chinnaiyan AM. Triggers for genomic rearrangements: Insights into genomic, cellular and environmental influences. Nat Rev Genet. 2010;11:819–829. doi: 10.1038/nrg2883. [DOI] [PubMed] [Google Scholar]
- 74.Ju BG, Lunyak VV, Perissi V, et al. A topoisomerase IIbeta-mediated dsDNA break required for regulated transcription. Science. 2006;312:1798–1802. doi: 10.1126/science.1127196. [DOI] [PubMed] [Google Scholar]
- 75.Haffner MC, Aryee MJ, Toubaji A, et al. Androgen-induced TOP2B-mediated double-strand breaks and prostate cancer gene rearrangements. Nat Genet. 2010;42:668–675. doi: 10.1038/ng.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bastus NC, Boyd LK, Mao X, et al. Androgen-induced TMPRSS2:ERG fusion in nonmalignant prostate epithelial cells. Cancer Res. 2010;70:9544–9548. doi: 10.1158/0008-5472.CAN-10-1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lin C, Yang L, Tanasa B, et al. Nuclear receptor-induced chromosomal proximity and DNA breaks underlie specific translocations in cancer. Cell. 2009;139:1069–1083. doi: 10.1016/j.cell.2009.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mani RS, Tomlins SA, Callahan K, et al. Induced chromosomal proximity and gene fusions in prostate cancer. Science. 2009;326:1230. doi: 10.1126/science.1178124. [DOI] [PMC free article] [PubMed] [Google Scholar]