

Case Report
A 29-year-old woman with a history of advanced adenoid cystic carcinoma (ACC) that was resistant to standard of care treatments presented to our phase I clinic seeking treatment with experimental therapeutics. The patient was diagnosed with ACC 11 years before presentation and had been treated with surgery, radiation therapy, and several lines of conventional treatments including platinums, antracyclines, and imatinib mesylate. Eleven months before being seen in our clinic, the patient had developed a brain metastasis that had been surgically resected. A personalized tumorgraft was successfully established from this lesion by the implantation of fragments of tumor materials in immunecompromised mice as described by our group.1 At the time of presentation, the patient had pulmonary and liver metastasis and, compared with a computed tomography (CT) scan performed 6 months before, was progressing with the growth of a preexisting liver metastasis (Fig 1, before baseline CT scan, black arrow) and development of a new liver lesion, as depicted in Figure 1 (upper panel). Brain magnetic resonance imaging showed a stable 2-mm brain lesion (Fig 1, lower panel, black arrows). The patient was asymptomatic with Eastern Cooperative Oncology Group performance status of 0 and normal liver, bone marrow, and kidney functions.
Fig 1.

To determine which phase I clinical studies could be more appropriate for the patient, we characterized her tumor for KRAS mutations and HER2 amplification and found the tumor to be KRAS wild type and not HER2 amplified, respectively (Table 1). Because the patient had a personalized tumorgraft model developed from her brain metastases, we used the model to evaluate a battery of anticancer agents, both conventional and experimental. Briefly, a tumor specimen obtained at the time of removal of her brain tumor had been transplanted and propagated in nude mice. Once the tumor specimen was in an exponential growth phase, cohorts of mice with tumor sizes of 0.15 to 0.3 mL were randomized to several treatment groups. The results of these studies are listed in Figure 2A (FGFR1, fibroblast growth factor receptor 1; IGF1R, insulin-like growth factor receptor 1) and Table 2. One of the most effective agents was a monoclonal antibody against IGF1R. As shown in Figure 2B, intraperitoneal administration of this agent at a dose of 40 mg/kg every 3 weeks resulted in a tumor-growth inhibition of approximately 76% compared with an untreated control after 49 days of treatment. Although we did not observe tumor regression, treatment resulted in a significant cytostatic effect, and we considered the tumor sensitive to treatment (P < .001; t-test analysis).
Table 1.
Molecular Markers
| Gene | Status |
|---|---|
| HER2/Neu | Not amplified |
| KRAS | WT |
Abbreviation: WT, wild type.
Fig 2.
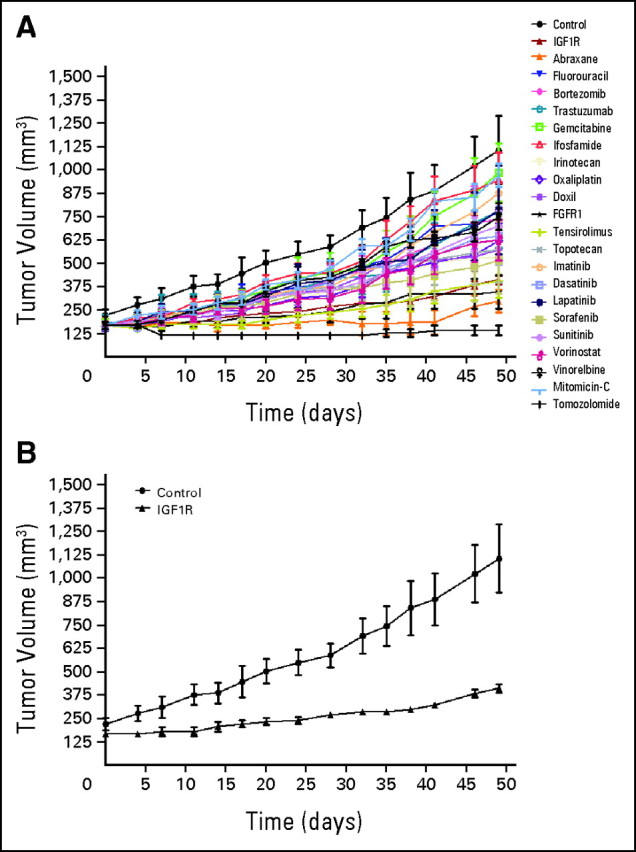
Table 2.
Drugs Tested on Tumorgraft
| Drug | Activity Rating (%) |
|---|---|
| IGF1R | 72 |
| Abraxane | 73 |
| Fluorouracil | 13 |
| Bortezomib | 19 |
| Trastuzumab | 12 |
| Gemcitabine | < 0 |
| Ifosfamide | < 0 |
| Irinotecan | 48 |
| Oxaliplatin | 34 |
| Doxil | 41 |
| FGFR1 | 74 |
| Temsirolimus | 67 |
| Topotecan | 41 |
| Tomozolomide | 102 |
| Imatinib | < 0 |
| Dasatinib | 27 |
| Lapatinib | 10 |
| Sorafenib | 50 |
| Sunitinib | 20 |
| Vorinostat | 31 |
| Vinorelbine | 14 |
| Mitomycin C | < 0 |
Abbreviations: FGFR1, fibroblast growth factor receptor 1; IGF1R, insulin-like growth factor receptor 1.
At that time, we were conducting a phase I trial that evaluated the combination of figitumumab, which is an IGF1R monoclonal antibody, and PF00299804, which is a pan–human epidermal growth factor receptor (EGFR) inhibitor.2 On the basis of the mouse data obtained with the IGF1R inhibitor, and because the tumor was KRAS wild type, the patient was enrolled onto this trial. Figitumumab was administered intravenously every 3 weeks, and PF00299804 was administered on a daily orally administered schedule. After four cycles of treatment, the patient developed severe diarrhea, which was mostly related to the pan–human EGFR toxicity. Treatment with PF00299804 was discontinued, and the patient continued treatment with figitumumab alone. Treatment with the IGF1R inhibitor showed a good safety profile and resulted in a minor response in the rapidly growing liver lesion that lasted for 6 months. At that time, the patient progressed with a new brain lesion (Fig 1, lower panel, end of study magnetic resonance image, white arrow) and was taken off study. The tumor remained controlled outside the brain.
Discussion
ACCs are very rare variants of adenocarcinoma that most often arise from salivary glands. Approximately 500 new cases of ACCs are diagnosed in the United States each year.3 The natural history of the disease can be characterized either by an indolent growth in some patients or by an aggressive and rapidly progressive disease. The overall 10-year survival for patients is approximately 50%, but when metastases occur, the median duration of survival is approximately 3 years.4–6 Because of the rarity of this disease, there are few clinical trials that have investigated systemic therapy. Data from a recently published meta-analysis reported activity for a platinum- and/or doxorubicin-based regimen.7 Imatinib and EGFR inhibitors have been recently evaluated in this disease with limited success.8–11 There is no standard approach for patients who progress to conventional treatments., and thus, enrollment onto clinical trials with novel agents is an accepted approach. However, because of the paucity of molecular data on this cancer, the selection of an appropriate trial is generally done empirically. This situation is not limited to ACCs and is the norm for most solid tumors; patients are enrolled onto early clinical trials mostly on the basis of clinical and not molecular eligibility criteria. However, the failure rate in phase I clinical trials remains extremely high with most patients deriving no benefit. High response rates have only been shown in studies in which there was a clear connection between a genetic alteration in a target and an inhibitor of the target.12–14
During previous years, our group has been involved in the development of personalized mouse models from patients with cancer. These models are useful for drug screening and biomarker development and, thus, can be used to prioritize and rank effective agents against an individual cancer. As illustrated in this case, we were able to screen a large set of agents, both experimental and conventional, against the personalized tumorgraft model. Data from these studies suggested that this cancer may be sensitive to nanoparticle albumin-bound paclitaxel, temozolomide, and inhibitors of IGF1R, mammalian target of rapamycin, and FGFR, which are inhibitors that provide several therapeutic opportunities. Because the IGF1R-inhibitor trial was available, we elected to enroll the patient onto that study and to save the other conventional opportunities for the future. Overall, the data supports the use of a personalized tumorgraft as a model to test experimental agents before administration to patients. Furthermore, the personalized tumorgraft model, with a clinically validated susceptibility to the IGF1R blockade, is an interesting preclinical tool to comparatively explore other IGF1R inhibitors, design combinations, and investigate biomarkers. For example, this approach was illustrated in our recent report that showed that PALB2 mutations are strong candidate biomarkers of response to DNA damaging agents.15
However, there are some limitations that will need to be addressed before this strategy can be broadly implemented in phase I clinical studies. Patients need to have a personalized tumorgraft model established. For this patient, the model was accomplished by implanting excess tissue from a clinically indicated surgical resection. However, most phase I candidates do not need surgery, and thus, the collection of fresh tumor tissue for the generation of a personalized tumorgraft model can be a challenge. In addition, the development and propagation of the personalized tumorgraft model and drug testing takes 6 to 8 months, which is an amount of time that is often not available for most phase I candidates. Moreover, the failure rate in tumorgraft establishment and the possibility of molecular signature discordance between two lesions from different organs need to be considered. Thus, early selection of potential candidates is critical. Finally, the selection of agents to be tested needs to be considered. The number of drugs in development is too large, and there has to be a strategy to prioritize which drugs should be tested in the models. In this sense, the integration of molecular testing to rank-order candidates is critical.
The next prevailing concern is to test this strategy in a clinical trial to show that it is feasible and beneficial. A plausible design, for example, would include a patient with advanced colorectal cancer with accessible live metastasis from whom a personalized tumorgraft model can be generated and available at the time of second-line treatment. This tumor could be profiled with a battery of biomarkers to select a set of five to 10 agents to be tested in the model to select the ideal phase I study for the patient. Obviously, there are significant logistic and feasibility issues with such an approach, but the reality is that the field is stagnant and with little progress, and thus, new, albeit risky, approaches are needed.
Footnotes
Clinical trial information can be found for the following: A7471004.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Although all authors completed the disclosure declaration, the following author(s) indicated a financial or other interest that is relevant to the subject matter under consideration in this article. Certain relationships marked with a “U” are those for which no compensation was received; those relationships marked with a “C” were compensated. For a detailed description of the disclosure categories, or for more information about ASCO's conflict of interest policy, please refer to the Author Disclosure Declaration and the Disclosures of Potential Conflicts of Interest section in Information for Contributors.
Employment or Leadership Position: Elizabeth Bruckheimer, Champions Oncology (C); David Sidransky, Champions Oncology (C) Consultant or Advisory Role: David Sidransky, Champions Oncology (C); Manuel Hidalgo, Champions Oncology (C) Stock Ownership: Elizabeth Bruckheimer, Champions Oncology, Cell Therapeutics; David Sidransky, Champions Oncology; Manuel Hidalgo, Champions Oncology Honoraria: None Research Funding: Emiliano Calvo, Pfizer Expert Testimony: None Other Remuneration: None
REFERENCES
- 1.Rubio-Viqueira B, Jimeno A, Cusatis G, et al. An in vivo platform for translational drug development in pancreatic cancer. Clin Cancer Res. 2006;12:4652–4661. doi: 10.1158/1078-0432.CCR-06-0113. [DOI] [PubMed] [Google Scholar]
- 2.Engelman JA, Zejnullahu K, Gale CM, et al. PF00299804, an irreversible pan-ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib. Cancer Res. 2007;67:11924–11932. doi: 10.1158/0008-5472.CAN-07-1885. [DOI] [PubMed] [Google Scholar]
- 3.Renehan A, Gleave EN, Hancock BD, et al. Long-term follow-up of over 1000 patients with salivary gland tumours treated in a single centre. Br J Surg. 1996;83:1750–1754. doi: 10.1002/bjs.1800831228. [DOI] [PubMed] [Google Scholar]
- 4.Spiro RH. Distant metastasis in adenoid cystic carcinoma of salivary origin. Am J Surg. 1997;174:495–498. doi: 10.1016/s0002-9610(97)00153-0. [DOI] [PubMed] [Google Scholar]
- 5.Spiro RH. Management of malignant tumors of the salivary glands. Oncology (Williston Park) 1998;12:671–680. discussion 683. [PubMed] [Google Scholar]
- 6.Terhaard CH, Lubsen H, Van der Tweel I, et al. Salivary gland carcinoma: Independent prognostic factors for locoregional control, distant metastases, and overall survival: Results of the Dutch head and neck oncology cooperative group. Head Neck. 2004;26:681–692. doi: 10.1002/hed.10400. discussion 692-693. [DOI] [PubMed] [Google Scholar]
- 7.Laurie SA, Ho AL, Fury MG, et al. Systemic therapy in the management of metastatic or locally recurrent adenoid cystic carcinoma of the salivary glands: A systematic review. Lancet Oncol. 2011;12:815–824. doi: 10.1016/S1470-2045(10)70245-X. [DOI] [PubMed] [Google Scholar]
- 8.Hotte SJ, Winquist EW, Lamont E, et al. Imatinib mesylate in patients with adenoid cystic cancers of the salivary glands expressing c-kit: A Princess Margaret Hospital phase II consortium study. J Clin Oncol. 2005;23:585–590. doi: 10.1200/JCO.2005.06.125. [DOI] [PubMed] [Google Scholar]
- 9.Pfeffer MR, Talmi Y, Catane R, et al. A phase II study of Imatinib for advanced adenoid cystic carcinoma of head and neck salivary glands. Oral Oncol. 2007;43:33–36. doi: 10.1016/j.oraloncology.2005.12.026. [DOI] [PubMed] [Google Scholar]
- 10.Locati LD, Bossi P, Perrone F, et al. Cetuximab in recurrent and/or metastatic salivary gland carcinomas: A phase II study. Oral Oncol. 2009;45:574–578. doi: 10.1016/j.oraloncology.2008.07.010. [DOI] [PubMed] [Google Scholar]
- 11.Agulnik M, Cohen EW, Cohen RB, et al. Phase II study of lapatinib in recurrent or metastatic epidermal growth factor receptor and/or erbB2 expressing adenoid cystic carcinoma and non adenoid cystic carcinoma malignant tumors of the salivary glands. J Clin Oncol. 2007;25:3978–3984. doi: 10.1200/JCO.2007.11.8612. [DOI] [PubMed] [Google Scholar]
- 12.Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809–819. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363:1693–1703. doi: 10.1056/NEJMoa1006448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Von Hoff DD, LoRusso PM, Rudin CM, et al. Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N Engl J Med. 2009;361:1164–1172. doi: 10.1056/NEJMoa0905360. [DOI] [PubMed] [Google Scholar]
- 15.Villarroel MC, Rajeshkumar NV, Garrido-Laguna I, et al. Personalizing cancer treatment in the age of global genomic analyses: PALB2 gene mutations and the response to DNA damaging agents in pancreatic cancer. Mol Cancer Ther. 2011;10:3–8. doi: 10.1158/1535-7163.MCT-10-0893. [DOI] [PMC free article] [PubMed] [Google Scholar]