

Abstract
Transthyretin (TTR) related cardiac amyloidosis is a progressive infiltrative cardiomyopathy that mimics hypertensive, hypertrophic heart disease and often goes undiagnosed. In the United States, the hereditary form disproportionately afflicts Black Americans, who when compared to Caucasians with wild type TTR amyloidosis, a phenotypically similar condition, present with more advanced disease despite having a non-invasive method for early identification (genetic testing). While reasons for this are unclear, this begs to consider the inadequate access to care, societal factors or a biological basis. In an effort to improve awareness and explore unique characteristics, we review the pathophysiology, epidemiology and therapeutic strategies for TTR amyloidosis, and highlight diagnostic pitfalls and clinical pearls for identifying patients with amyloid heart disease.
Keywords: heart failure, amyloidosis, black, restrictive cardiomyopathy, race and ethnicity
Black Americans are at higher risk of developing congestive heart failure and their clinical outcomes with heart failure are less favorable than Caucasians. While poor cardiovascular outcomes in Black Americans have been associated with societal factors such as decreased health care literacy, poor access to care, decreased trust in health care providers or health systems, there are biological differences noted.1–3 The onset of heart failure is earlier in Black patients compared to Caucasians and strongly associated with higher blood pressure and greater left ventricular hypertrophy.4,5 Blacks typically have a higher prevalence of obesity and diabetes.5 In clinical studies, Black patients exhibit biologically distinct responses to heart failure therapies from Whites with differential responses to oral vasodilators and diuretics for the treatment and prevention of heart failure.6,7
On this backdrop, the Black patient with heart failure has been ascribed a phenotype depicting a hypertensive, salt-sensitive, diabetic with left ventricular hypertrophy. Transthyretin (TTR) related cardiac amyloidosis is a progressive infiltrative cardiomyopathy that mimics hypertensive, hypertrophic heart disease and may go undiagnosed when a clinician attributes ventricular hypertrophy or heart failure to other prevalent pathologies (i.e. hypertension, diabetes, obesity). Studies evaluating senile, or wild type transthyretin (ATTRwt) almost exclusively consisted of White patients, however population based data from the United Kingdom reported an equal prevalence of ATTRwt among races.8 Under-recognition of disease may explain why Blacks are poorly represented in American studies of senile cardiac amyloidosis.9
Furthermore, in the United States, the hereditary form of TTR amyloidosis (mutated TTR) is predominantly a disease of older Black Americans. Intrinsic features of hereditary amyloidosis could contribute a biological explanation for the disparate heart failure outcomes for Blacks, with several reports associating a race specific TTR variant with congestive heart failure and high frequency of this mutation observed in Black Americans (~1:29).10–12
While we continue to manage the burden of comorbidities such as hypertensive heart disease, discrepant heart failure outcomes for Blacks may be, at least partly, explained by an inherited genetic mutation and/or under-recognition of TTR amyloidosis, a masquerader of hypertensive heart disease. The purpose of this manuscript is to increase awareness of transthyretin cardiac amyloidosis in Black Americans.
Amyloid Heart Disease
The term “amyloid” describes the accumulation of proteinaceous material that deposits in otherwise normal organs. Amyloid is derived from the misfolding of endogenous proteins into an insoluble beta-pleated sheet conformation which is resistant to proteolysis and reabsorption. The deposits disrupt normal organ structure and function (e.g. amyloidosis). Over thirty proteins have been identified as being amyloidogenic, of which five can cause cardiac amyloidosis: immunoglobulin light chain (AL or primary amyloidosis), immunoglobulin heavy chain (AH), transthyretin (ATTR), serum amyloid A (AA), and apolipoprotein A I (AApoA1).13
The causative protein determines the organ involvement and clinical presentation of the disease. Early literature regarding cardiac amyloidosis focused on the light-chain variant (AL), where a plasma cell dyscrasia produces amyloidogenic kappa or lambda light chains.14 Two types of cardiac amyloidosis arise from TTR, a tetrameric protein synthesized primarily in the liver. Senile systemic amyloidosis arises from wild-type, genetically unaltered TTR and leads to cardiac amyloid deposition in patients after their sixth decade of life.15 A hereditary amyloidosis that is transmitted in an autosomal dominant pattern can arise with mutation of the TTR genome (ATTRmt). More recently it has become evident that ATTRmt in United States disproportionately afflicts Black Americans.
Hereditary Transthyretin Amyloidosis
Transthyretin is a 127 amino-acid plasma protein made of four non–covalently associated, β-sheet enriched subunits that form two thyroxine (T4) binding sites.16 TTR is a homotetramer and individual monomers unfold, misfold and then aggregate to produce amyloid. Point mutations within the TTR protein may alter kinetics for dissociation and thus promote amyloid formation.17
In the United States, ATTRmt disproportionately affects Black Americans and arises from a variant of TTR with a point mutation resulting in a substitution of isoleucine for valine at position 122 (Val122Ile) which was first identified by Gorevic and colleagues.18,19 Similar to the senile amyloidosis, the Val122Ile mutation result in clinically significant amyloid deposition that is predominantly isolated to the heart.
The association of Val122Ile mutation with amyloid heart disease in elderly Black Americans became apparent in a series of autopsy studies. Upon review of 52,000 autopsies performed in the Los Angeles area, the prevalence of non-light chain cardiac amyloidosis in patients over the age of age of 60 years was noted to be higher in Blacks compared to Whites (1.6% vs. 0.42%).20 When comparing Blacks to Whites, there were markedly higher proportions of Blacks with TTR cardiac amyloid in the 7th (8:1) and 8th (13:1) decades of life.20,21 Of the patients with TTR cardiac amyloid on autopsy, 23% of Blacks (vs. 0% of Whites) were heterozygous for the Val122Ile mutation. Furthermore, all Black patients with the Val122Ile mutation had pathological evidence TTR amyloid deposition in the heart (100% penetrance) at the time of death. Evaluation of living patients has revealed that the mutation is highly prevalent in Black Americans, with 3.43% carrying the Val122Ile mutation (allele prevalence of 0.0173).12
Carriers of the mutation are at increased risk of developing congestive heart failure. In an analysis of the Cardiovascular Health Study (CHS), an observational study of community dwelling patients over the age of 65 years, there was an increased prevalence of heart failure in carriers compared to non-carriers (38% vs 15%, RR= 2.62 [95%CI1.28–5.37], P = .04).11 In the Atherosclerosis Risk in Communities Study (ARIC), a prospective cohort study that enrolled followed for a median of 21.5 years to an average age of 74 years, carriers were more likely to develop evidence of systolic or diastolic heart failure (HR = 1.47 [95%CI 1.03–2.10]) and time free from HF was 2.0 years shorter among carriers of the Val122Ile mutation than among non-carriers.10
Overall survival did not statistically differ in either ARIC or CHS, but both analyses were of limited power. However, after adjusting for age and gender in the CHS cohort, the carriers of the Val122Ile mutation were at increased risk of dying at 15 years of follow-up (RR= 2.59 [95%CI 1.01–6.63], P= 0.009).11 Val122Ile carriers in the ARIC cohort did not exhibit increased mortality, even after adjusting for gender and age. However ARIC was a predominantly female cohort (62%) whereas previous data have established that males have a higher penetrance of disease.22 Furthermore, longer follow-up of the ARIC cohort may have revealed an age dependent difference in survival, as was observed in the older CHS cohort. Larger studies with greater age dispersion and gender representation may further clarify mortality risk for mutation carriers.
Findings from ARIC also suggest incomplete echocardiographic penetrance of the Val122Ile mutation. The degree of left ventricular hypertrophy (11%) or overt echocardiographic findings consistent with amyloidosis (7%) were lower than expected. This was unexpected especially considering the higher prevalence of heart failure in the carriers of the mutations. One explanation is that these imaging data were limited by small sample of subjects with Val122Ile and incomplete enrollment in the echocardiography follow-up (data available only for ~1/3 of patients). Furthermore, the number of males with follow-up imaging, who tend to have more aggressive disease at younger age, was quite low (n = 11, 24%). Lastly, these data underscore the limitations of classic echocardiographic measurements as a surveillance tool for TTR cardiac amyloidosis. Considering that prior autopsy data in Val122Ile carriers showed 100% prevalence of TTR amyloid at the time of death, imaging modalities such as 99mTc-PYP or 99mTc-DPD based nuclear imaging, which have been shown to detect cardiac amyloid prior to echocardiographic manifestations, may be more effective to detect early disease.23,24
Nonetheless, the findings suggest that clinical heart failure in Val122Ile carriers is not solely dependent on the presence of the mutation and point to other as yet undefined factors that interact to promote phenotypic expression and subsequent heart failure. Population studies suggest that carriers of the mutation on average develop symptoms of congestive heart failure in the seventh decade of life but phenotypic expression is seen earlier in men than women and those with homozygosity for the Val122Ile mutation.11,25 Factors related to disease penetrance in other TTR mutations that could play a role in Val122Ile include (1) mitochondrial polymorphisms that result in earlier penetrance when the mutation is inherited from the mother, (2) relation to disease endemic foci, (3) modifier gene mutations that stabalize the TTR tetramer and delay disease onset, (4) environmental factors such as inflammation and the gut microbiome, (5) and fibril composition.26–31
When evaluating Black patients with clinical heart failure, the prevalence of the mutation is even higher. The Beta-Blocker Evaluation in Survival Trial (BEST) studied the effect of bucindolol in a clinical trial population with symptomatic systolic heart failure and cardiac amyloidosis was an exclusion. However, of the 207 Black Americans enrolled in the trial, the Val122Ile mutation was detected in 6.3% of patients, and in 10% in those greater than 60 years of age, suggesting that Val122Ile may be under-recognized by heart failure experts in the presence of systolic dysfunction.32
Early Diagnostic Clues
Most studies detailing clinical characteristics of cardiac amyloidosis are limited to tertiary amyloid centers in which the number of subjects with Val122Ile is small and provide limited data on TTR amyloid in Blacks. Such data has assessed a skewed profile of patients presenting with classic clinical features of late-stage disease. Table 1 highlights the both subtle and more advanced clinical clues that should raise concern for the presence of hereditary cardiac amyloidosis.
Table 1.
Clinical clues or “red flags” that may prompt evaluation for hereditary amyloidosis.
| Category | Clinical Clues |
|---|---|
| Clinical History |
|
| Clinical Exam |
|
| Imaging |
|
| Laboratory Data |
|
Clinical Presentation
Compared to Black Americans presenting with AL amyloidosis, ATTRmt patients with the Val122Ile mutation are older, have less severe symptoms despite having greater ventricular hypertrophy, lower left ventricular ejection fractions and greater atrial dilation on echocardiography.33
Early disease with ATTRmt may be minimally symptomatic, especially compared to AL amyloidosis. Patients may complain of mild exercise intolerance or manifest arrhythmias. Clinical and imaging findings may be attributed to other causes of left ventricular hypertrophy and delay diagnosis, while more extensive deposition can mimic hypertrophic cardiomyopathy.34 Atrial arrhythmias are frequent and the risk of left atrial appendage thrombosis is increased.35,36
As the disease progresses, symptoms of right heart failure predominate. Patients complain of fatigue, lightheadedness and breathlessness. On physical examination, patients often present with hypotension, edema, ascites, hepatomegaly and elevated jugular venous pressure.
The survival in ATTRmt is considerably better than AL cardiac amyloidosis (median 27 vs. 5 months).33 In contrast, data on outcomes of Black Americans with TTR cardiac amyloidosis compared to Caucasians with wild type TTR amyloidosis, a phenotypically similar condition, reveal that Black Americans present with more advanced disease despite having a non-invasive method for early identification (genetic testing) and outcomes in Black Americans may be worse. Compared to ATTRwt, Val122Ile patients in a small multicenter prospective study, Transthyretin Cardiac Amyloid Study (TRACS), had higher rates of cardiovascular hospitalization (64% vs 28%) and decreased survival (73% vs 22% died, median survival 26 vs 43 months).37 Other studies have showed similar trends, raising the concern whether this is the result of inadequate access to care or has a biological basis.38,39
Electrocardiography
As amyloid deposits progressively replace conducting myocardium, the patient may develop Q waves on the surface ECG in the absence of epicardial coronary disease.40 The prevalence of this “pseudoinfarction” pattern or poor R wave progression is increased as amyloid deposition becomes more diffuse.41
Amyloid infiltration dampens the QRS complexes on surface electrocardiography, and reduced voltage on the electrocardiogram (ECG) relative to the left ventricular mass should heighten consideration of cardiac amyloidosis. Unique to amyloidosis (low voltage on ECG and hypertrophy on imaging), this feature can be the clue that differentiates it from hypertensive cardiomyopathy, hypertrophic cardiomyopathy or even other infiltrative cardiomyopathies such as Anderson-Fabry disease (high ECG voltage and hypertrophy on imaging).(Figure 1)42 However, the sensitivity of low voltage on the ECG for identifying cardiac amyloid is poor, especially in patients with ATTRmt. Compared to AL amyloidosis patients with ATTRmt have higher voltage on ECG and up to 15% of patients meet ECG criteria for left ventricular hypertrophy.43 Measuring the ratio of ECG voltage to the left ventricular cross sectional area improves the sensitivity and specificity to greater than 80%.44–46
Figure 1.
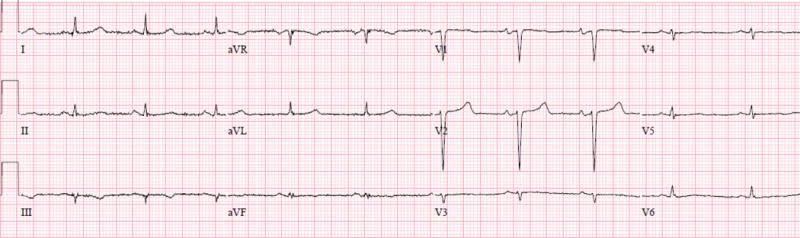
A surface twelve-lead electrocardiograms from a patient with ATTRmt (Val122Ile). The patient had severe left ventricular hypertrophy on two-dimensional echocardiography and no coronary artery disease on coronary angiography. The electrocardiogram displayed lower than expected QRS voltage and suggestion of an anterior wall myocardial infarction (poor R-wave progression).
Key Clinical Points
Electrocardiographic features of cardiac amyloid, especially for the ATTRmt that affects Black Americans, are not sensitive for identifying patients with disease. Thus the absence of low voltage, pseudoinfarction pattern or conduction disease should not exclude the diagnosis of cardiac amyloidosis. Clinicians are encouraged to integrate the voltage on ECG with the wall thickness or mass as determined by echocardiography.
Echocardiography
Echocardiography plays an important role in diagnosis and staging of cardiac amyloidosis.(Figure 2) Amyloid infiltration of the myocardium early in the disease process presents as diastolic dysfunction while late disease can be associated with reduced systolic function.47,48 Myocardial infiltration of amyloid impairs cardiac relaxation and reduces compliance. Independent of fibril deposition, the precursor proteins themselves may have a cardiotoxic effect and directly reduce myocyte function. Light chains infused from patients with AL amyloidosis result in impaired myocardial relaxation in mouse models and in culture cells clearly demonstrating toxicity to the myocardium independent of amyloid deposition.49,50 Whether such toxicity also occurs with transthyretin amyloid precursors has not been defined.
Figure 2.
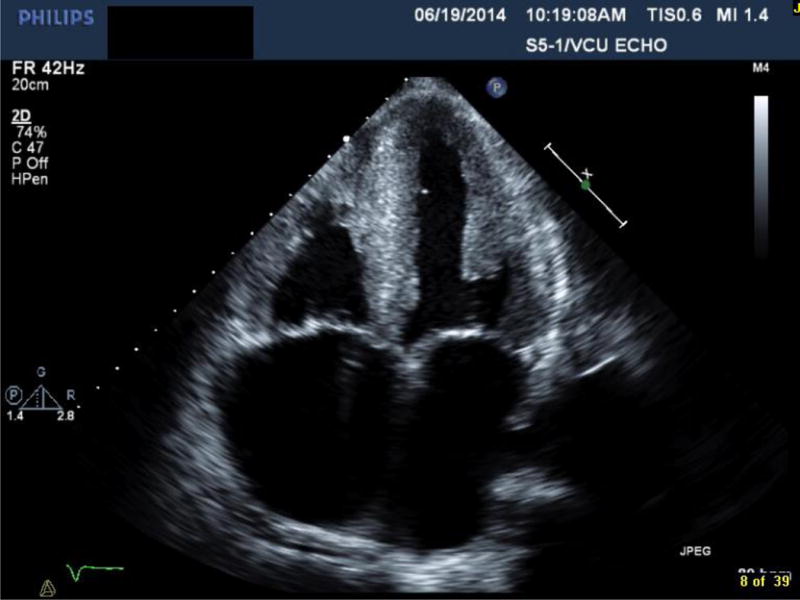
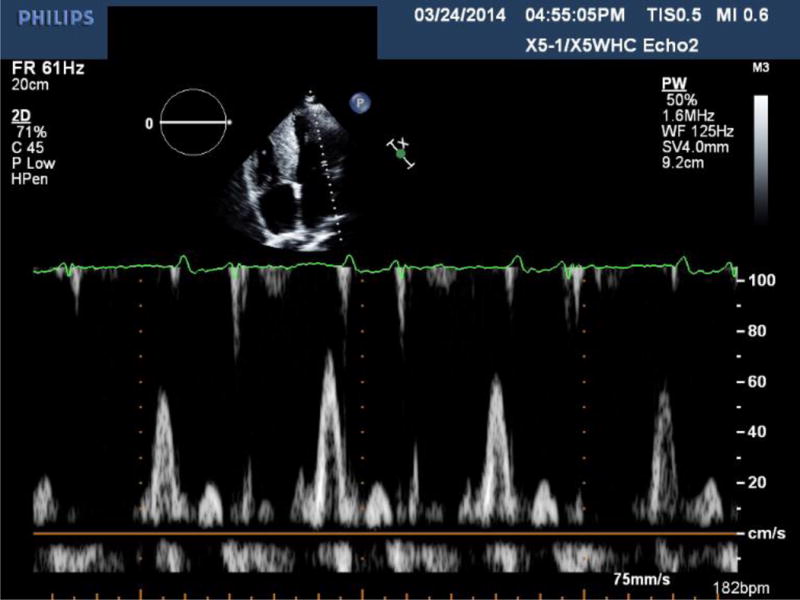
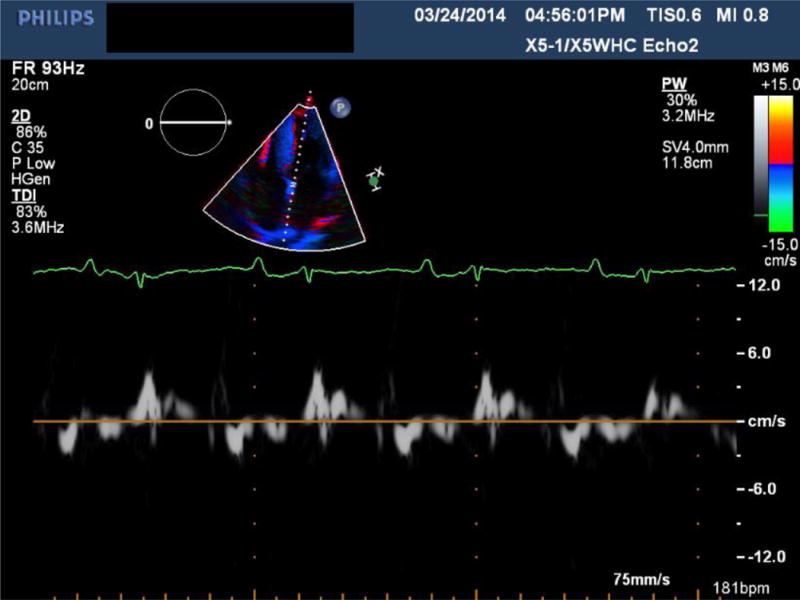
Transthoracic echocardiography of a patient with ATTRmt (Val122Ile) cardiac amyloidosis with classic features of restrictive heart disease. (A) Two-dimensional imaging obtained in the apical four chamber view shows left and right ventricular wall hypertrophy, bi-atrial dilation and increased myocardial echogenicity.(B) Measurements of Doppler inflow velocities across the mitral valve show an increased E/A ratio and decreased deceleration time of early diastolic inflow. (C) Tissue Doppler imaging quantifying the mitral annular velocities are depressed.
Early studies defining the classic echocardiographic findings associated with cardiac amyloid focused on AL amyloidosis. Child et al. in 1976 described a series of patients with symmetrically increased left ventricular wall thickness in the absence of hypertension or aortic valve disease, decreased systolic thickening of the interventricular septum and left ventricular posterior wall, and small to normal size of the left ventricular cavity.51 Also discussed was an associated finding of pericardial effusion. A decade later Falk and colleagues diagnosed the now common reference to a “speckled” appearance to the ventricular wall, due to increased myocardial echogenicity, and atrial septal thickening.45
Contemporary studies have delineated differences on two-dimensional echocardiography among the subtypes of cardiac amyloidosis. In an Italian study that did not include any blacks with Val122Ile, Rapezzi and colleagues studied 233 subjects divided into AL, ATTRwt and ATTRmt.43 The mean left ventricular wall thickness was significantly greater in the ATTRwt (18.8+/−3.8mm) than the AL (15.1+/−2.7mm) or ATTRmt (16+/−3.2mm; p<0.0001) groups, despite having the best survival (2-year survivals ATTRwt 100%, ATTRmt 63%, and AL 63%). Left ventricular ejection fraction was lowest in the ATTRwt group while findings of atrial septal thickening and pericardial effusion were no different among the three groups.
In a case control study evaluating patients that had the Val122Ile mutation without a clinical diagnosis of cardiac amyloidosis, Jacobson et al. compared 23 carriers to 46 non-carriers. Those in the Val122Ile group had significantly greater ventricular septal thickness (1.35+/−0.31mm vs 1.19+/−mm, p=0.04), right ventricular hypertrophy (26% vs 2%, p=0.004), and frequency of granular sparkling (35% vs 2%, p=0.0004) compared to 46 matched controls.52
Early studies of strain rate imaging reported echocardiographic findings in patients with AL amyloidosis. Koyama’s group showed that when compared to subjects with non-cardiac AL amyloid, those with infiltration of the left ventricle (mean wall thickness of greater than 12 mm) have significantly reduced systolic, diastolic longitudinal strain and strain rate at the base and mid-ventricle, but not apex.53 Sun et al. showed that in addition to longitudinal strain, cardiac amyloidosis is associated with severely reduced strain measured in the radial and circumferential aspects, distinguishing it from hypertrophic cardiomyopathy and secondary causes of left ventricular hypertrophy.54 Quarta and colleagues evaluated strain imaging in ATTRmt and confirmed that preservation of apical longitudinal strain was consistent across all subtypes.46
Key Clinical Points
While the classical morphological features of amyloidosis on two-dimensional echocardiography and Doppler studies help identify patients with amyloidosis, preserved apical strain is a unique feature distinguishing amyloid heart disease from other causes of left ventricular hypertrophy such as hypertensive and hypertrophic cardiomyopathy. Other clues include thickened intra-atrial septum, right ventricular hypertrophy, small pericardial effusions and thickening of AV valves.
Cardiovascular Biomarkers
Cardiac troponin, as a marker of myocardial cell death, and B-type natriuretic peptide (BNP) as a sensitive marker of myocardial dysfunction, have been shown to have prognostic significance in AL amyloid.55,56 There are limited data on biomarkers in ATTRmt variant amyloidosis. Suhr and colleagues assessed 29 subjects with Val30Met variant ATTR and found that BNP was elevated above normal in 76% and correlated with ventricular septal wall thickness, left atrial diameter, and basal septal strain.57 As a predictor of echocardiographic involvement of the heart, BNP had a sensitivity of 93% and a specificity of 40% with a negative predictive value of 86%; on par with basal septal strain echocardiographic imaging (sensitivity of 81%, specificity of 36%, and negative predictive value of 50%). Troponin concentrations are not as frequently elevated in ATTR as in AL cardiac amyloidosis, however emerging data suggests a prognostic role for ATTR.58,59
Key Clinical Points
Biomarker concentrations, especially BNP, are frequently abnormal in patients with ATTRmt. The role for cardiovascular biomarkers for diagnosis, prognosis and measuring progression/regression of disease needs further study.
Advanced Diagnostic Tools
Figure 3 summarizes the evolving advanced imaging technologies for diagnosing and monitoring progression of TTR cardiac amyloidosis.
Figure 3.
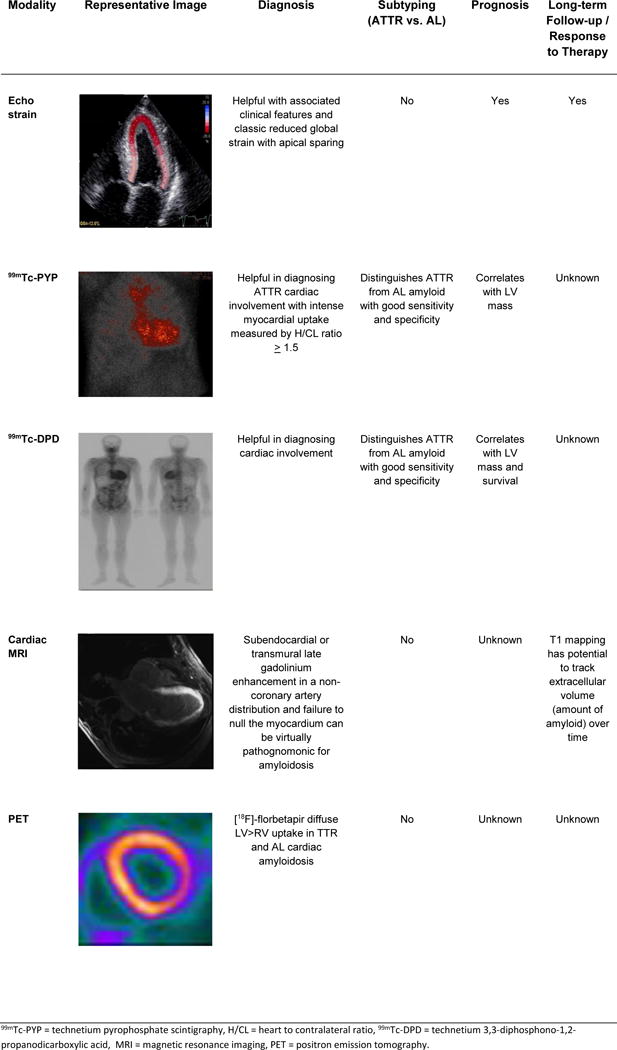
Summary of the advanced imaging modalities for diagnosis and monitoring of cardiac amyloidosis.
Cardiac Magnetic Resonance Imaging
Cardiac magnetic resonance (CMR) imaging has become a valuable tool in assessing for cardiac amyloid infiltration. Cardiac morphologic features can be evaluated using steady-state free precession sequence.(Figure 4) This particular sequence allows assessment of both cardiac structure and function. Images of the heart are taken throughout the cardiac cycle, affording the ability to determine cardiac volumes and ejection fraction. Similarly, myocardial thickness in the atria and ventricles can be quantified. Steady-state free precession sequence is also useful in the identification of pericardial and pleural effusions, which may be present. Although diastolic function indices can be obtained by assessment of mitral inflow parameters using cine phase-contrast pulse sequence, the assessment is limited when compared to echocardiography. Apical sparing in longitudinal strain, a unique finding in cardiac amyloidosis on echocardiography has been reproduced in CMR imaging.60
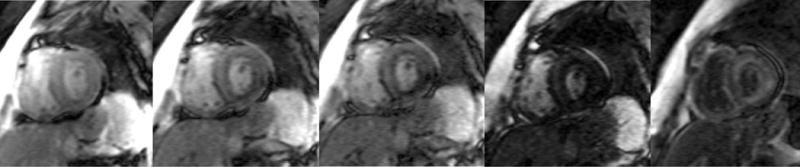
Figure 4.
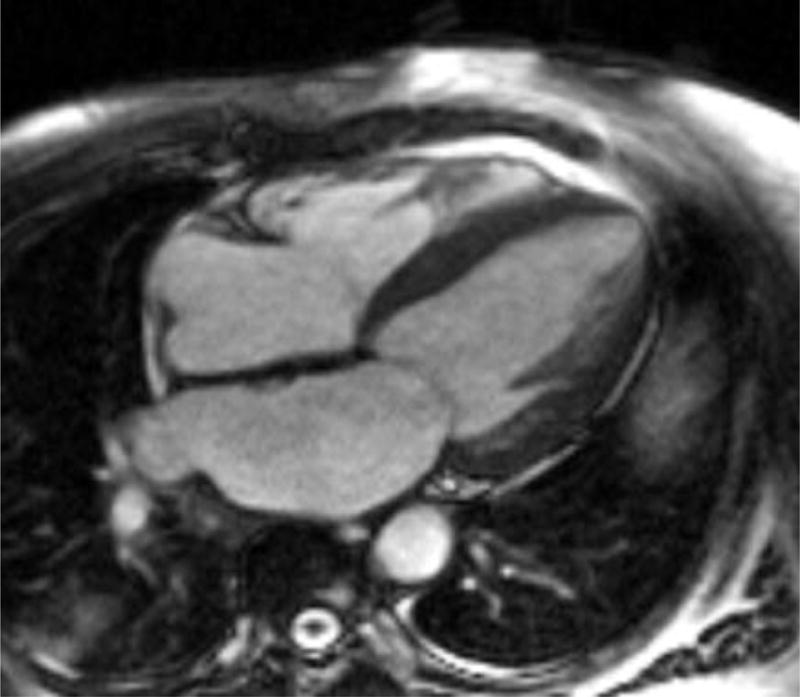
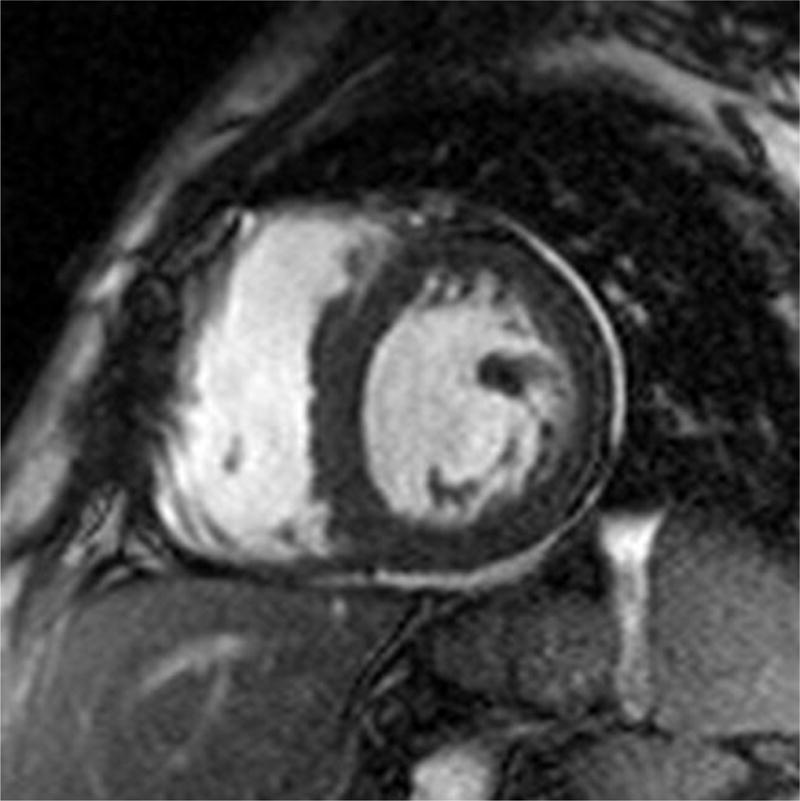
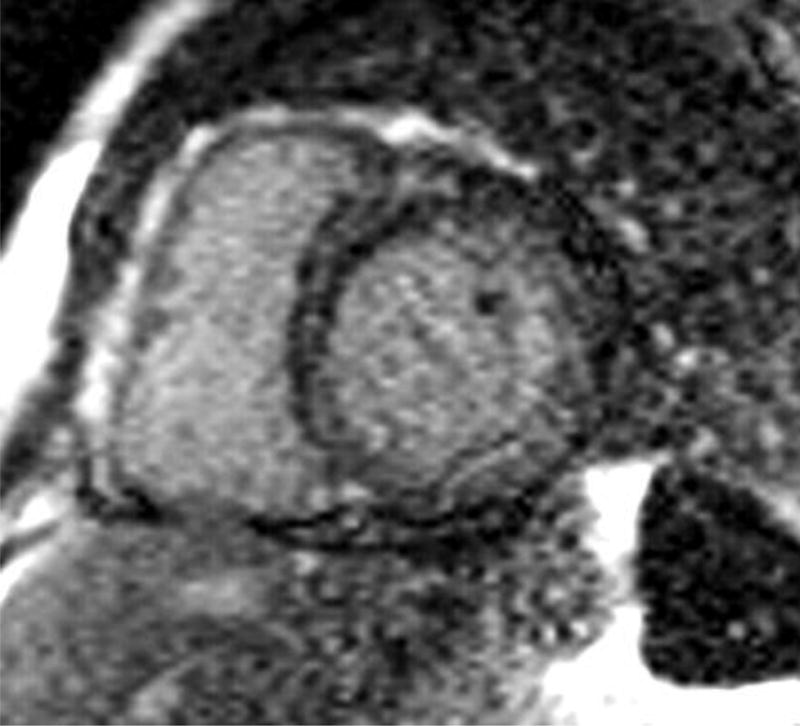
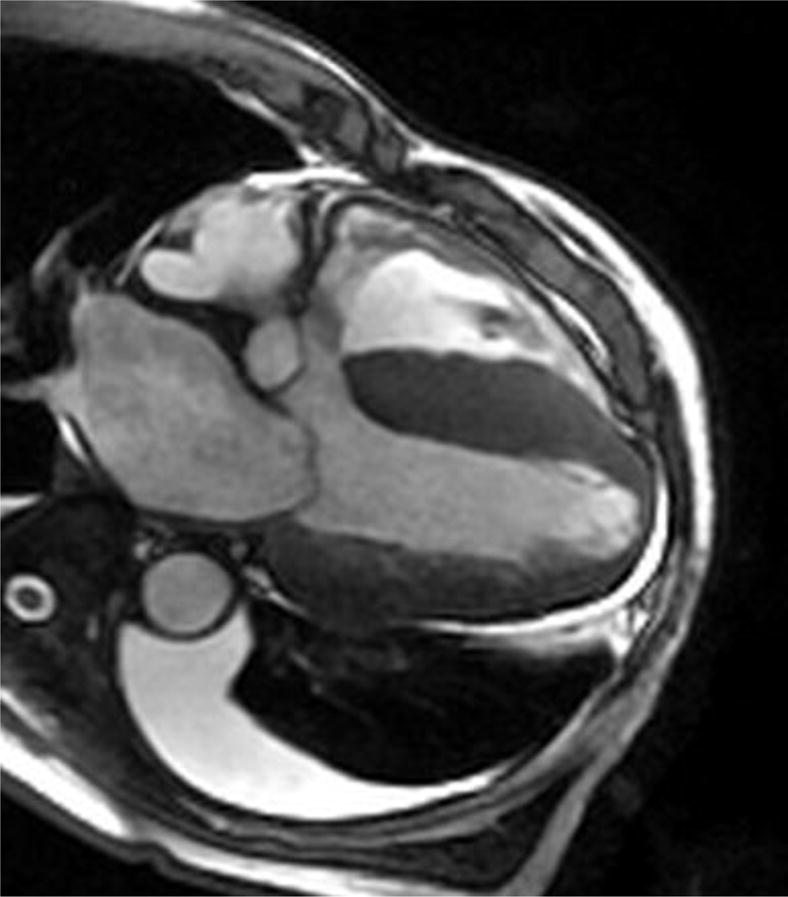
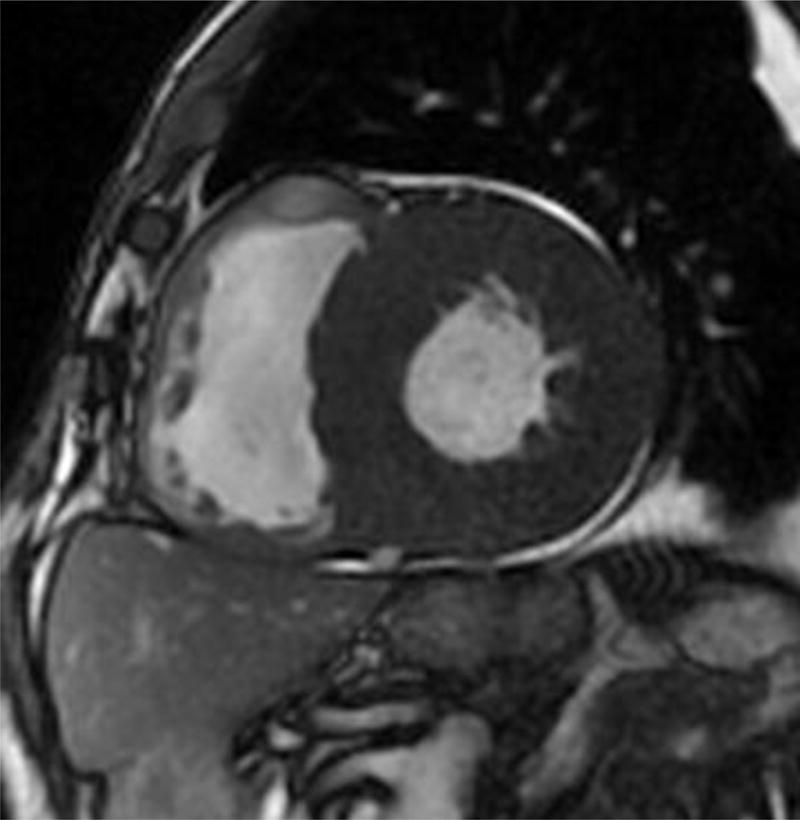
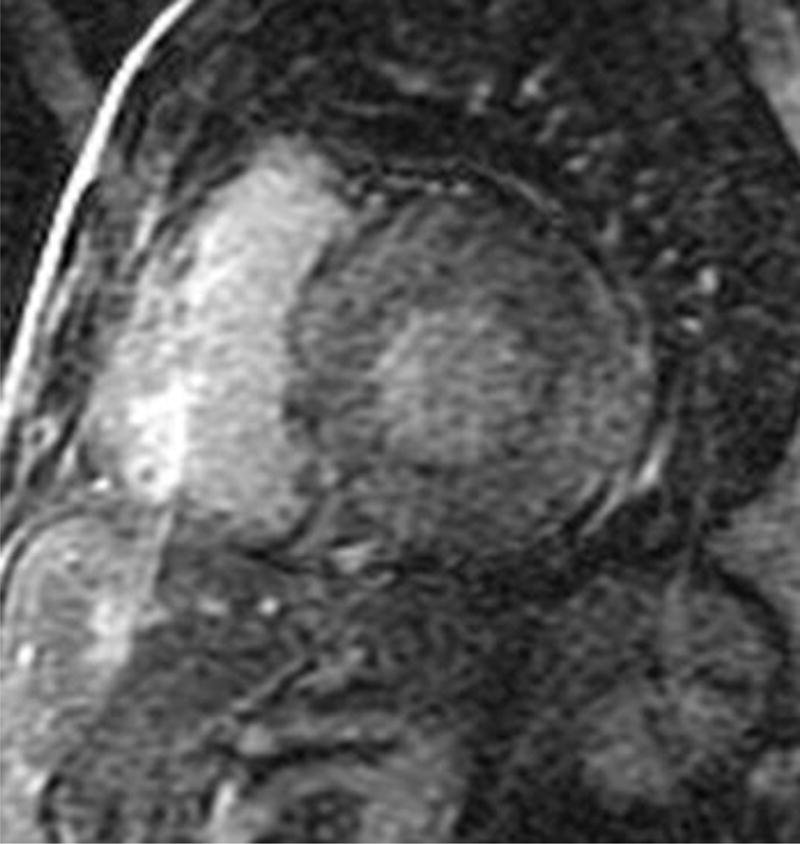
Four chamber and mid-ventricular short axis views of patients with ATTRmt (Val122Ile) and (A,B) mild and (D,E) severe left ventricular hypertrophy. (C) The patient with mild ventricular hypertrophy exhibits only mild late gadolinium enhancement (LGE) in a subendocardial distribution. (F) The patient with severe hypertrophy has a transmural pattern of LGE. Not shown is an example of patchy LGE. (Courtesy of Dr. John D. Grizzard)
Tissue characterization, specifically with late gadolinium enhancement imaging has been reported to be the most accurate predictor of endomyocardial biopsy-positive amyloidosis.61 Enhancement patterns on late gadolinium images may be described as either diffuse, circumferential subendocardial, or patchy focal.(Figure 4)41
A study of 97 patients with cardiac amyloidosis showed that left ventricular mass was significantly larger in ATTR than in AL.62 Late gadolinium enhancement was substantially more extensive in ATTR, with up to 90% of ATTR patients demonstrating transmural enhancement. Subendocardial enhancement was more common in AL patients. Right ventricular gadolinium enhancement was apparent in 100% of ATTR patients compare with 72% of AL.
Early darkening of the blood pool after contrast, due to rapid washout of gadolinium into an enlarged subendocardial interstitial space can be quantitatively assessed by mapping of T1 kinetics of the blood pool and subendocardium early after gadolinium administration. White and colleagues recently published a study in CMR to assess prognosis in cardiac amyloidosis.63 They employed the observation that in contradistinction to normal myocardium which crosses the null point (becomes black) after the blood pool, in cardiac amyloidosis, there is “failure to null the myocardium” represented as greater than 50% of myocardial tissue becoming black at an earlier inversion time as compared to blood.(Figure 5) The inversion time, an accurate indicator of global myocardial hyper-enhancement, was a strong predictor of mortality (HR of 6.0, 95% CI: 3.0–12.1).
Figure 5.
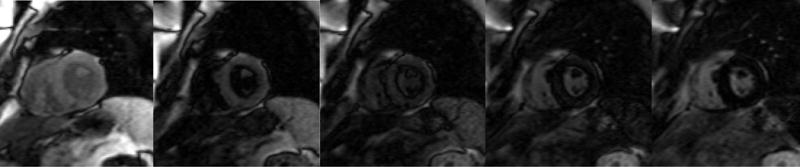
T1 inversion scout sequences of (A) a normal heart and (B) a patient with cardiac amyloidosis. (A) In the normal heart, the blood pool “nulls” or blackens before the surrounding myocardium and nearby spleen. (B) In the patient with amyloid heart disease, the order of “nulling” between the myocardium and blood pool is inversed, where the blood pool “nulls” after the spleen after the myocardium. (Courtesy of Dr. John D. Grizzard)
A new imaging approach of phase-sensitive inversion recovery (PSIR) may further improve the sensitivity for late gadolinium enhancement by automating the nulling of the myocardial signal.64 Fontana and colleagues studied 250 ATTR and AL cardiac amyloidosis patients and found that with PSIR, transmural patterns of late gadolinium enhancement were associated with the worse prognosis (HR of 5.4; 95% CI: 2.1–13.7; P<0.0001), even after adjusting for biomarkers and echocardiographic parameters (HR of 4.1; 95% CI: 1.3–13.1; P<0.05).65
In subjects unable to receive gadolinium, native (pre-contrast) T1 mapping can identify early cardiac amyloid infiltration and can differentiate ATTR or AL amyloid from hypertrophic cardiomyopathy.66
Key Clinical Points
Cardiac MRI offers a non-invasive diagnostic and prognostic tool for cardiac amyloidosis. In addition to defining morphological characteristics, delayed gadolinium enhancement and altered gadolinium kinetics are highly specific findings for cardiac amyloid disease. In TTR amyloid, patterns other than sub-endocardial delayed enhancement predominate. Furthermore, T1 mapping may offer an opportunity to identify early disease and quantitatively follow disease progression and regression without the need for contrast.
Nuclear Imaging
Nuclear imaging modalities can aid in the diagnosis of TTR cardiac amyloidosis. The utility of 99mTc-PYP for detecting ATTR was demonstrated in a recent study of 45 patients (12 AL, 16 ATTRwt, 17 ATTRmt [12 mutants with Val122Ile]) biopsy-proven amyloidosis. Subjects with ATTR had significantly higher semi-quantitative cardiac visual score than the AL cohort as well as a higher quantitative score. A heart to contralateral (H/CL) ratio ≥1.5 was consistent with intensely diffuse myocardial tracer retention with 97% sensitivity and 100% specificity for identifying ATTR cardiac amyloidosis.
Of note, the most studied bone-seeking radiotracer in cardiac amyloidosis to date is 99mtechnetium-3,3-diphosphono-1,2-propanodicarboxylic acid (99mTc-DPD). However, this isotope is not approved by the FDA and therefore not available for clinical use in the United States and has not been used widely to evaluate the Val122Ile mutation. Among 79 patients (28 ATTRmt [0 mutants with Val122Ile], 17 ATTRwt, and 34 AL) who underwent 99mTc-DPD with tracer retention calculated by a heart-to-whole body ratio (H/WB), the test was 100% sensitive and 88% specific for detecting ATTR due to about 1/3 of AL patients having unexpected tracer uptake.67 In a follow-up study in 63 patients with ATTR (23 of which had no echocardiographic features of amyloid involvement), the utility of 99mTc-DPD scintigraphy for identifying myocardial infiltration across a wide spectrum of morphologic and functional phenotypes was evaluated.68 All patients showed moderate to severe myocardial tracer uptake (semi-quantitative score ≥2), H/WB ratio was positively correlated with left ventricular mean wall thickness (Pearson’s r=0.695, p<0.001), negatively correlated with left ventricular ejection fraction (r=−0.368, p=0.004), and was an unfavorable predictor of MACE-free survival at Cox univariate analysis. Smaller studies have also suggested using bone scintigraphy with 99mTechnetium-hydroxymethylene diphosphonate (99mTc-HDP) may allow for early detection of ATTR cardiac disease.23,24
Key Clinical Points
Various nuclear imaging radiotracers are useful to diagnose TTR cardiac amyloidosis. 99mTc-PYP and 99mTc-DPD can distinguish AL from ATTR with excellent sensitivity and specificity in advanced disease. Early data suggests that these modalities detect amyloid deposition prior to echocardiography, thus offering an opportunity for improved pre-clinical diagnosis. Further studies are needed in a broader spectrum of disease including early phase cardiac amyloid and in Val122Ile in particular.
Establishing the Diagnosis
Histopathology
Tissue confirmation of amyloid deposition is considered the gold standard for confirmation of the diagnosis of amyloidosis. Cardiac deposition of amyloid is diffuse and homogenous in its distribution, allowing for a high sensitivity when obtaining a pathological diagnosis. Because amyloidosis is a systemic disease, multiple sources can provide a diagnosis, but the yield can vary greatly from organ to organ. Fine et al. published the largest study in ATTR and fat pad aspiration to date (including 286 subjects) comparing the yield of cardiac and non-cardiac biopsies in patients with ATTRwt and ATTRmt.69 The yield for ATTRmt in abdominal fat pad aspiration (67% of 141 specimens) far exceeded that for ATTRwt (14% of 84 specimens), but only 18 patients (11% of those with genotype data) had the Val122Ile mutation. Fat pad aspiration was less sensitive than endomyocardial biopsy (100% of 42 ATTRmt and 89 ATTRwt specimens). The highest yield for ATTRmt for non-cardiac biopsies came from rectal (81% of 52 specimens) and sural nerve (83% of 54 specimens) biopsies.
Congo Red stain, which exhibits apple-green birefringence under polarized light, is commonly employed; however, implementing fluorescent microscopy improves sensitivity for amyloid deposits.70 More specific techniques for identifying the subtype of amyloid include immunohistochemical (IF) staining for light chains or TTR. While valid for amyloid typing, there are considerable pitfalls to IF including failure to react to truncated AL light chains and a number of hereditary amyloidoses can be missed by a limited antibody panel.71 It is noteworthy that when assessing for the gene found in ATTRmt of Black Americans, all 10 samples from subjects with Val22Ile ATTRmt studied by Connors et al. positively stained for anti-human TTR antibodies (9 cardiac, 1 fat pad).33
Finally, mass spectrometry can be performed to examine the affected tissue and study the protein constituents of the amyloid fibril.72 Inclusion of laser microdissection of the fibrils followed by tandem mass spectrometry can generate a nearly complete protein composition of the amyloid fibril and has been shown to have a specificity of 100% and sensitivity of 98%.73
Key Clinical Points
Tissue diagnosis is a required step towards making the diagnosis of amyloidosis. For ATTRmt, non-cardiac biopsies may identify disease however the sensitivity is lower than for AL amyloidosis. A negative non-cardiac biopsy does not exclude disease, and an endomyocardial biopsy should be considered. Biopsies revealing amyloid deposition should be further evaluated to confirm protein composition with immunohistochemical staining and mass spectrometry.
Genetic Testing
There are over 100 polymorphisms encoding the variant gene that transcripts the transport protein transthyretin, located on the long (q) arm of chromosome 18 at position 12.1 (18q12.1), with 80 confirmed pathogenic mutations.74 Isoelectric focusing gel electrophoresis, a method of separating molecules by their isoelectric point (pI) and pH, can differentiate ATTRwt from greater than 95% of variants of ATTRmt.40 Among those with ATTRwt, only one protein band is visualized, while heterozygous ATTRmt will show both the wild type TTR at the typical pI and a second mutant TTR (2 distinct protein bands). A single protein band at this pI would suggest homozygousity for the Ile122 mutation. Conclusive genetic distinction between ATTRmt and ATTRwt is obtained from polymerase chain reaction amplification and DNA sequencing of the transthyretin exons 1 to 4, validating isoelectric focusing.40
Key Clinical Points
Genetic testing can provide a method for early identification of at risk or affected individuals and is critical in distinguishing hereditary from senile TTR amyloidosis. Due to the high prevalence of the Val122Ile in Black Americans, one must not attribute the type of cardiac amyloidosis to the mutation until AL amyloid has been comprehensively excluded.
Diagnostic Algorithm
Considering the heterogeneous presentation and insidious progression of this disease, diagnosis is often delayed until the late stages. We propose a diagnostic algorithm in Figure 6 incorporating clinical clues (Table 1) and advanced imaging modalities (Figure 2) for identifying Black Americans with hereditary amyloidosis. Furthermore, common misconceptions and “pit-falls” for diagnosis are presented in Table 2.
Figure 6.
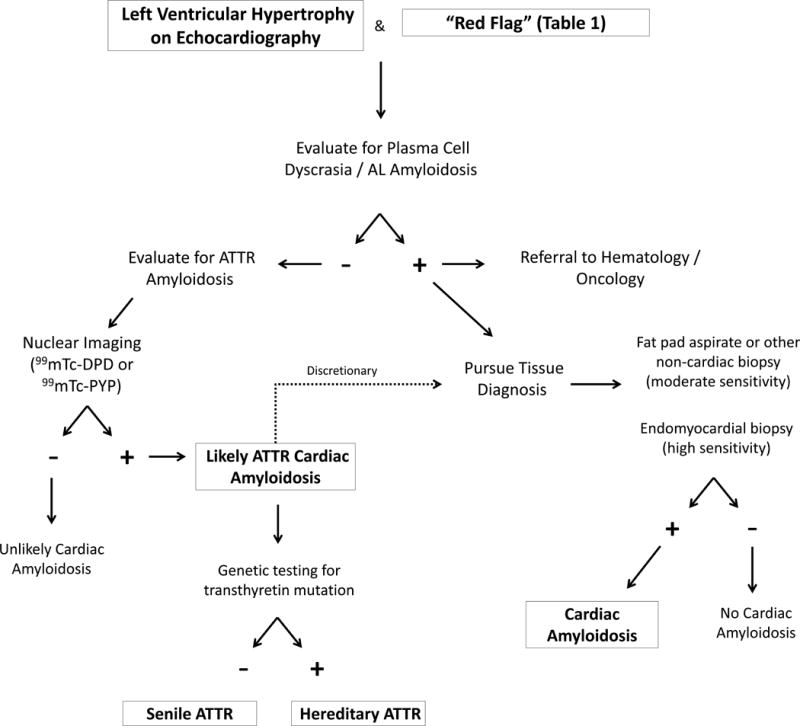
An algorithm for evaluating patients for amyloid heart disease. The first step is to exclude any evidence of a plasma cell disorder.
Table 2.
Common misconceptions about transthyretin amyloid that lead to delayed or missed diagnosis of cardiac amyloidosis.
| Pitfall | Explanation |
|---|---|
| “The voltage on the electrocardiogram was normal” | Low voltage is a relatively late-phase phenomenon in transthyretin related cardiac amyloidosis and many patients with biopsy-proven disease do not meet electrocardiographic criteria for low voltage. In fact, some patients have electrocardiographic evidence of left ventricular hypertrophy. |
| “The fat pad aspiration biopsy didn’t show evidence of amyloidosis” | The sensitivity of a fat pad biopsy for TTR amyloid is low and thus cannot be used to exclude the diagnosis. |
| “The protein electrophoresis showed no evidence of a plasma cell dyscrasia, thus the patient does not have cardiac amyloidosis” | Transthyretin amyloidosis is not caused by a plasma cell dyscrasia, evaluation for a monoclonal gammopathy is expected to be normal. However the first step once a diagnosis of cardiac amyloidosis is considered is to exclude light chain amyloid, given its malignant nature and available therapy |
| “The history of hypertension/diabetes/kidney disease explained the patients ventricular hypertrophy” | Transthyretin cardiac amyloidosis often coexists with these medical conditions that are common in elderly African Americans. |
| “The ejection fraction was low, thus it is unlikely to be a restrictive cardiomyopathy like cardiac amyloidosis” | As cardiac amyloidosis progresses, the systolic function becomes compromised. |
| “Cardiac amyloidosis is a contraindication for heart transplantation” | Unlike AL amyloidosis, outcomes after solid organ transplantation are quite good with transthyretin amyloidosis. Patients with significant cardiac involvement may be eligible for heart or heart + liver transplantation. |
Therapeutic Options
Clinical Pearls for Medical Management
The clinical management of a patient with symptomatic restrictive amyloid from ATTRmt is similar to AL cardiomyopathy, and is centralized around managing fluid balance. Excessive intravascular volume leads to breathlessness and edema, while even modest hypovolemia can cause hypotension and exacerbate renal injury. The mainstays of therapy are loop diuretics, but combination blockade of the renal nephron with thiazide diuretics and aldosterone antagonist may become necessary in the setting of refractory edema or hypokalemia, respectively.
Traditional disease-modifying medications for heart failure have no proven role in the treatment of amyloid heart disease, which is progressive in nature. In fact, most medications have potential to cause harm. Drugs that slow the heart rate (beta-blockers, calcium channel blockers) are poorly tolerated, as patients with restrictive heart disease often have fixed stroke volumes and the cardiac output is highly dependent on the heart rate. In addition to worsening symptoms, there are published case reports of sudden death of patients with amyloidosis after administration such therapies.75,76 Digitalis may bind to amyloid fibrils, leading to unpredictable and possibly toxic circulating concentrations, prohibiting its use in amyloid heart disease. Angiotensin converting enzyme inhibitors, angiotensin receptor blockers, and combination vasodilators may be poorly tolerated due to hypotension and lightheadedness. Inotropes may have a palliative role, but from clinical experience the reduction in symptomatology and improvement in quality of life is only modest. Patients with pacemakers may derive clinical benefit from increased lower-rate limits for pacing.
Atrial arrhythmias are frequent and symptomatic arrhythmias may need to be addressed with pharmacological or ablative therapies. Most importantly, patients with atrial fibrillation or atrial standstill should receive anticoagulation as the risk for intracardiac thrombus and thromboembolism is markedly increased.35,36 The benefit of an implantable defibrillator or cardiac resynchronization is undefined. The efficacy of defibrillation can be limited in an amyloid heart in which extensive protein deposition may limit detection of arrhythmias and increase the defibrillation thresholds for delivery of successful therapy.
Novel therapies for ATTR: stabilizers, silencers, and degraders
No Food and Drug Administration-approved drugs are currently available for treatment of the ATTR cardiac amyloidosis, but several treatment strategies are under clinical investigation.
Agents with transthyretin stabilizing properties are in various stages of clinical development. Diflunisal, a nonsteroidal anti-inflammatory drug, binds and stabilizes TTR variants against acid-mediated fibril formation in vitro and has been tested in animal safety studies and human clinical trials.77 However, the chronic inhibition of cyclooxygenase enzymes including gastrointestinal bleeding, renal dysfunction, fluid retention, and hypertension that may exacerbate heart failure. Currently under clinical trial, tafamidis (Pfizer Inc; New York, NY) binds to the thyroxine-binding sites on the TTR tetramer and inhibits its dissociation into monomers thereby blocking the rate-limiting step in the amyloid fibril cascade.78 A phase 3 trial evaluating the efficacy, safety, and tolerability of tafamidis oral dose of 20mg or 80mg compared with placebo is fully enrolled and expected to conclude in mid-2018.
Ribonucleic acid (RNA) interference has emerged as an endogenous cellular mechanism for controlling gene expression in which small interfering ribonucleic acids (siRNAs) mediate the cleavage of target messenger RNA.79 Lipid nanoparticle formulations and newer N-acetylgalactosamine conjugates have emerged as agents to deliver siRNAs to hepatocytes, the source of mutated Val122Ile TTR production.80 A phase III trial in familial amyloid cardiomyopathy is currently enrolling.
Antisense oligonucleotides are also under clinical investigation as inhibitors of hepatic expression of amyloidogenic TTR and a Phase III trial enrolling both ATTRwt and ATTRmt cardiac amyloid is targeted for enrollment in mid-2016.
Treatment approaches to amyloidosis have focused on reducing amyloid precursor protein production, but the ability to remove already deposited amyloid is another important therapeutic strategy. The utility of combined doxycycline and tauroursodeoxycholic aid (TUDCA) has been studied for TTR degradation and clinical trial are under development for ATTRmt cardiac amyloidosis. 81 The role of normal, nonfibrillar glycoprotein present in all human amyloid deposits, serum amyloid P component (SAP), is currently under intense investigation to address the unmet need for therapy that safely promotes clearance of established amyloid deposits.82
Conclusion/Future Directions
In summary, the Val122Ile mutation in the TTR gene is very common in Blacks (prevalent in 1 in 25) and results in an age dependent, late onset restrictive/hypertrophic cardiomyopathy that can mimic other forms of heart failure leading to misdiagnosis and under-diagnosis. Penetrance of the phenotype is low according to recent data and the biologic, genetic and environmental factors that lead to phenotypic penetrance remain unknown but if defined could provide insights into other forms of cardiovascular disease that disproportionately afflict Black Americans. This form of cardiac amyloidosis could be more easily identified than ATTRwt (senile cardiac amyloidosis) if widespread genetic testing were implemented. The available data suggests that despite the ability for early diagnosis, those with the Val122Ile mutation present initially with a more advanced phenotype than ATTRwt. Whether this is due to a more malignant phenotype in the presence of the Val122Ile or is the result of racial differences in access to care is unknown. With the advent of non-invasive nuclear medicine techniques for identifying TTR cardiac amyloid due to either mutant or wild type disease, there is an opportunity to identify Blacks with both Val122Ile and wild type TTR and follow their progression and outcomes in order to address these issues. Ongoing investigation of disease modifying therapies including TTR stabilizers and TTR silencers will determine if these agents can alter the natural history of Val122Ile cardiac amyloid which is a relentlessly progressive form of heart failure.
Supplementary Material
Acknowledgments
Sources of Funding: Dr. Castano is supported by a dual grant awarded by the American College of Cardiology and Merck & Co.,Inc. Dr. Maurer is supported by a K24 from NIA AG036778 entitled Midcareer Mentoring Award for Patient Oriented Research in Geriatric Cardiology.
Disclosures: KBS, OOA and MSM have received research grants for clinical trials from Alnylam Pharmaceuticals and Pfizer, Inc. KBS is on the speaker’s bureau for Amgen Pharmaceuticals. KBS has received research grants from Thoratec Corp. AKM has received research grants for clinical trials from Alnylam Pharmaceuticals. AC has received a research grant from the American College of Cardiology and Merck & Co., Inc. PBD is on the speaker’s bureau for Arbor Pharmaceuticals and serves on the medical advisory boards for Alnylam Pharmaceuticals, Novartis Pharmaceuticals Amgen Pharmaceuticals. IVF is on the speaker’s bureau for Novartis Pharmaceuticals.
References
- 1.Ayanian JZ, Weissman JS, Chasan-Taber S, Epstein AM. Quality of care by race and gender for congestive heart failure and pneumonia. Med Care. 1999;37:1260–1269. doi: 10.1097/00005650-199912000-00009. [DOI] [PubMed] [Google Scholar]
- 2.Dickson VV, McCarthy MM, Howe A, Schipper J, Katz SM. Sociocultural influences on heart failure self-care among an ethnic minority black population. J Cardiovasc Nurs. 2013;28:111–118. doi: 10.1097/JCN.0b013e31823db328. [DOI] [PubMed] [Google Scholar]
- 3.Chaudhry SI, Herrin J, Phillips C, Butler J, Mukerjhee S, Murillo J, Onwuanyi A, Seto TB, Spertus J, Krumholz HM. Racial disparities in health literacy and access to care among patients with heart failure. J Card Fail. 2011;17:122–127. doi: 10.1016/j.cardfail.2010.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yancy CW, Abraham WT, Albert NM, Clare R, Stough WG, Gheorghiade M, Greenberg BH, O’Connor CM, She L, Sun JL, Young JB, Fonarow GC. Quality of care of and outcomes for African Americans hospitalized with heart failure: findings from the OPTIMIZE-HF (Organized Program to Initiate Lifesaving Treatment in Hospitalized Patients With Heart Failure) registry. J Am Coll Cardiol. 2008;51:1675–1684. doi: 10.1016/j.jacc.2008.01.028. [DOI] [PubMed] [Google Scholar]
- 5.Kamath SA, Drazner MH, Wynne J, Fonarow GC, Yancy CW. Characteristics and outcomes in African American patients with decompensated heart failure. Arch Intern Med. 2008;168:1152–1158. doi: 10.1001/archinte.168.11.1152. [DOI] [PubMed] [Google Scholar]
- 6.Wright JT, Dunn JK, Cutler JA, Davis BR, Cushman WC, Ford CE, Haywood LJ, Leenen FHH, Margolis KL, Papademetriou V, Probstfield JL, Whelton PK, Habib GB, ALLHAT Collaborative Research Group Outcomes in hypertensive black and nonblack patients treated with chlorthalidone, amlodipine, and lisinopril. JAMA. 2005;293:1595–1608. doi: 10.1001/jama.293.13.1595. [DOI] [PubMed] [Google Scholar]
- 7.Carson P, Ziesche S, Johnson G, Cohn JN. Racial differences in response to therapy for heart failure: analysis of the vasodilator-heart failure trials. Vasodilator-Heart Failure Trial Study Group. J Card Fail. 1999;5:178–187. doi: 10.1016/s1071-9164(99)90001-5. [DOI] [PubMed] [Google Scholar]
- 8.Gillmore JD, Hawkins PN. V122I transthyretin variant in elderly black Americans. N Engl J Med. 2015;372:1769. doi: 10.1056/NEJMc1503222. [DOI] [PubMed] [Google Scholar]
- 9.Connors LH, Sam F, Skinner M, Salinaro F, Sun F, Ruberg FL, Berk JL, Seldin DC. Heart Failure Resulting From Age-Related Cardiac Amyloid Disease Associated With Wild-Type Transthyretin: A Prospective, Observational Cohort Study. Circulation. 2016;133:282–290. doi: 10.1161/CIRCULATIONAHA.115.018852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Quarta CC, Buxbaum JN, Shah AM, Falk RH, Claggett B, Kitzman DW, Mosley TH, Butler KR, Boerwinkle E, Solomon SD. The amyloidogenic V122I transthyretin variant in elderly black Americans. N Engl J Med. 2015;372:21–29. doi: 10.1056/NEJMoa1404852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buxbaum J, Alexander A, Koziol J, Tagoe C, Fox E, Kitzman D. Significance of the amyloidogenic transthyretin Val 122 Ile allele in African Americans in the Arteriosclerosis Risk in Communities (ARIC) and Cardiovascular Health (CHS) Studies. Am Heart J. 2010;159:864–870. doi: 10.1016/j.ahj.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jacobson DR, Alexander AA, Tagoe C, Buxbaum JN. Prevalence of the amyloidogenic transthyretin (TTR) V122I allele in 14 333 African-Americans. Amyloid. 2015;22:171–174. doi: 10.3109/13506129.2015.1051219. [DOI] [PubMed] [Google Scholar]
- 13.Sipe JD, Benson MD, Buxbaum JN, Ikeda S, Merlini G, Saraiva MJM, Westermark P. Nomenclature 2014: Amyloid fibril proteins and clinical classification of the amyloidosis. Amyloid. 2014;21:221–224. doi: 10.3109/13506129.2014.964858. [DOI] [PubMed] [Google Scholar]
- 14.Dubrey SW, Cha K, Anderson J, Chamarthi B, Reisinger J, Skinner M, Falk RH. The clinical features of immunoglobulin light-chain (AL) amyloidosis with heart involvement. QJM. 1998;91:141–157. doi: 10.1093/qjmed/91.2.141. [DOI] [PubMed] [Google Scholar]
- 15.Cornwell GG, Murdoch WL, Kyle RA, Westermark P, Pitkänen P. Frequency and distribution of senile cardiovascular amyloid. A clinicopathologic correlation. Am J Med. 1983;75:618–623. doi: 10.1016/0002-9343(83)90443-6. [DOI] [PubMed] [Google Scholar]
- 16.Blake CC, Geisow MJ, Oatley SJ, Rérat B, Rérat C. Structure of prealbumin: secondary, tertiary and quaternary interactions determined by Fourier refinement at 1.8 A. J Mol Biol. 1978;121:339–356. doi: 10.1016/0022-2836(78)90368-6. [DOI] [PubMed] [Google Scholar]
- 17.Hammarström P, Jiang X, Hurshman AR, Powers ET, Kelly JW. Sequence-dependent denaturation energetics: A major determinant in amyloid disease diversity. Proc Natl Acad Sci USA. 2002;99(Suppl 4):16427–16432. doi: 10.1073/pnas.202495199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gorevic PD, Prelli FC, Wright J, Pras M, Frangione B. Systemic senile amyloidosis. Identification of a new prealbumin (transthyretin) variant in cardiac tissue: immunologic and biochemical similarity to one form of familial amyloidotic polyneuropathy. J Clin Invest. 1989;83:836–843. doi: 10.1172/JCI113966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jacobson DR, Gorevic PD, Buxbaum JN. A homozygous transthyretin variant associated with senile systemic amyloidosis: evidence for a late-onset disease of genetic etiology. Am J Hum Genet. 1990;47:127–136. [PMC free article] [PubMed] [Google Scholar]
- 20.Jacobson DR, Pastore RD, Yaghoubian R, Kane I, Gallo G, Buck FS, Buxbaum JN. Variant-sequence transthyretin (isoleucine 122) in late-onset cardiac amyloidosis in black Americans. N Engl J Med. 1997;336:466–473. doi: 10.1056/NEJM199702133360703. [DOI] [PubMed] [Google Scholar]
- 21.Buck FS, Koss MN, Sherrod AE, Wu A, Takahashi M. Ethnic distribution of amyloidosis: an autopsy study. Mod Pathol. 1989;2:372–377. [PubMed] [Google Scholar]
- 22.Coelho T, Maurer MS, Suhr OB. THAOS – The Transthyretin Amyloidosis Outcomes Survey: initial report on clinical manifestations in patients with hereditary and wild-type transthyretin amyloidosis. Curr Med Res Opin. 2013;29:63–76. doi: 10.1185/03007995.2012.754348. [DOI] [PubMed] [Google Scholar]
- 23.Glaudemans AWJM, van Rheenen RWJ, van den Berg MP, Noordzij W, Koole M, Blokzijl H, Dierckx RAJO, Slart RHJA, Hazenberg BPC. Bone scintigraphy with (99m)technetium-hydroxymethylene diphosphonate allows early diagnosis of cardiac involvement in patients with transthyretin-derived systemic amyloidosis. Amyloid. 2014;21:35–44. doi: 10.3109/13506129.2013.871250. [DOI] [PubMed] [Google Scholar]
- 24.Galat A, Rosso J, Guellich A, Van Der Gucht A, Rappeneau S, Bodez D, Guendouz S, Tissot C-M, Hittinger L, Dubois-Randé J-L, Plante-Bordeneuve V, Itti E, Meignan M, Damy T. Usefulness of (99m)Tc-HMDP scintigraphy for the etiologic diagnosis and prognosis of cardiac amyloidosis. Amyloid. 2015:1–11. doi: 10.3109/13506129.2015.1072089. [DOI] [PubMed] [Google Scholar]
- 25.Reddi HV, Jenkins S, Theis J, Thomas BC, Connors LH, Van Rhee F, Highsmith WE. Homozygosity for the V122I mutation in transthyretin is associated with earlier onset of cardiac amyloidosis in the African American population in the seventh decade of life. J Mol Diagn. 2014;16:68–74. doi: 10.1016/j.jmoldx.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 26.Bonaïti B, Olsson M, Hellman U, Suhr O, Bonaïti-Pellié C, Planté-Bordeneuve V. TTR familial amyloid polyneuropathy: does a mitochondrial polymorphism entirely explain the parent-of-origin difference in penetrance? Eur J Hum Genet. 2010;18:948–952. doi: 10.1038/ejhg.2010.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hellman U, Alarcon F, Lundgren H-E, Suhr OB, Bonaiti-Pellié C, Planté-Bordeneuve V. Heterogeneity of penetrance in familial amyloid polyneuropathy, ATTR Val30Met, in the Swedish population. Amyloid. 2008;15:181–186. doi: 10.1080/13506120802193720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koike H, Ando Y, Ueda M, Kawagashira Y, Iijima M, Fujitake J, Hayashi M, Yamamoto M, Mukai E, Nakamura T, Katsuno M, Hattori N, Sobue G. Distinct characteristics of amyloid deposits in early- and late-onset transthyretin Val30Met familial amyloid polyneuropathy. J Neurol Sci. 2009;287:178–184. doi: 10.1016/j.jns.2009.07.028. [DOI] [PubMed] [Google Scholar]
- 29.Noguchi H, Ohta M, Wakasugi S, Noguchi K, Nakamura N, Nakamura O, Miyakawa K, Takeya M, Suzuki M, Nakagata N, Urano T, Ono T, Yamamura K. Effect of the intestinal flora on amyloid deposition in a transgenic mouse model of familial amyloidotic polyneuropathy. Exp Anim. 2002;51:309–316. doi: 10.1538/expanim.51.309. [DOI] [PubMed] [Google Scholar]
- 30.Soares ML, Coelho T, Sousa A, Batalov S, Conceição I, Sales-Luís ML, Ritchie MD, Williams SM, Nievergelt CM, Schork NJ, Saraiva MJ, Buxbaum JN. Susceptibility and modifier genes in Portuguese transthyretin V30M amyloid polyneuropathy: complexity in a single-gene disease. Hum Mol Genet. 2005;14:543–553. doi: 10.1093/hmg/ddi051. [DOI] [PubMed] [Google Scholar]
- 31.Ihse E, Rapezzi C, Merlini G, Benson MD, Ando Y, Suhr OB, Ikeda S, Lavatelli F, Obici L, Quarta CC, Leone O, Jono H, Ueda M, Lorenzini M, Liepnieks J, Ohshima T, Tasaki M, Yamashita T, Westermark P. Amyloid fibrils containing fragmented ATTR may be the standard fibril composition in ATTR amyloidosis. Amyloid. 2013;20:142–150. doi: 10.3109/13506129.2013.797890. [DOI] [PubMed] [Google Scholar]
- 32.Buxbaum J, Jacobson DR, Tagoe C, Alexander A, Kitzman DW, Greenberg B, Thaneemit-Chen S, Lavori P. Transthyretin V122I in African Americans with congestive heart failure. J Am Coll Cardiol. 2006;47:1724–1725. doi: 10.1016/j.jacc.2006.01.042. [DOI] [PubMed] [Google Scholar]
- 33.Connors LH, Prokaeva T, Lim A, Théberge R, Falk RH, Doros G, Berg A, Costello CE, O’Hara C, Seldin DC, Skinner M. Cardiac amyloidosis in African Americans: comparison of clinical and laboratory features of transthyretin V122I amyloidosis and immunoglobulin light chain amyloidosis. Am Heart J. 2009;158:607–614. doi: 10.1016/j.ahj.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 34.Damy T, Costes B, Hagège AA, Donal E, Eicher J-C, Slama M, Guellich A, Rappeneau S, Gueffet J-P, Logeart D, Planté-Bordeneuve V, Bouvaist H, Huttin O, Mulak G, Dubois-Randé J-L, Goossens M, Canoui-Poitrine F, Buxbaum JN. Prevalence and clinical phenotype of hereditary transthyretin amyloid cardiomyopathy in patients with increased left ventricular wall thickness. Eur Heart J. 2015 doi: 10.1093/eurheartj/ehv583. [DOI] [PubMed] [Google Scholar]
- 35.Santarone M, Corrado G, Tagliagambe LM, Manzillo GF, Tadeo G, Spata M, Longhi M. Atrial thrombosis in cardiac amyloidosis: diagnostic contribution of transesophageal echocardiography. J Am Soc Echocardiogr. 1999;12:533–536. doi: 10.1016/s0894-7317(99)70091-x. [DOI] [PubMed] [Google Scholar]
- 36.Dubrey S, Pollak A, Skinner M, Falk RH. Atrial thrombi occurring during sinus rhythm in cardiac amyloidosis: evidence for atrial electromechanical dissociation. Br Heart J. 1995;74:541–544. doi: 10.1136/hrt.74.5.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ruberg FL, Maurer MS, Judge DP, Zeldenrust S, Skinner M, Kim AY, Falk RH, Cheung KN, Patel AR, Pano A, Packman J, Grogan DR. Prospective evaluation of the morbidity and mortality of wild-type and V122I mutant transthyretin amyloid cardiomyopathy: the Transthyretin Amyloidosis Cardiac Study (TRACS) Am Heart J. 2012;164:222–228.e1. doi: 10.1016/j.ahj.2012.04.015. [DOI] [PubMed] [Google Scholar]
- 38.Givens RC, Russo C, Green P, Maurer MS. Comparison of cardiac amyloidosis due to wild-type and V122I transthyretin in older adults referred to an academic medical center. Aging health. 2013;9:229–235. doi: 10.2217/ahe.13.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Connors LH, Doros G, Sam F, Badiee A, Seldin DC, Skinner M. Clinical features and survival in senile systemic amyloidosis: comparison to familial transthyretin cardiomyopathy. Amyloid. 2011;18:157–159. doi: 10.3109/13506129.2011.574354059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ruberg FL, Berk JL. Transthyretin (TTR) Cardiac Amyloidosis. Circulation. 2012;126:1286–1300. doi: 10.1161/CIRCULATIONAHA.111.078915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Syed IS, Glockner JF, Feng D, Araoz PA, Martinez MW, Edwards WD, Gertz MA, Dispenzieri A, Oh JK, Bellavia D, Tajik AJ, Grogan M. Role of cardiac magnetic resonance imaging in the detection of cardiac amyloidosis. JACC Cardiovasc Imaging. 2010;3:155–164. doi: 10.1016/j.jcmg.2009.09.023. [DOI] [PubMed] [Google Scholar]
- 42.Kampmann C, Wiethoff CM, Martin C, Wenzel A, Kampmann R, Whybra C, Miebach E, Beck M. Electrocardiographic signs of hypertrophy in fabry disease-associated hypertrophic cardiomyopathy. Acta Paediatr Suppl. 2002;91:21–27. doi: 10.1111/j.1651-2227.2002.tb03105.x. [DOI] [PubMed] [Google Scholar]
- 43.Rapezzi C, Merlini G, Quarta CC, Riva L, Longhi S, Leone O, Salvi F, Ciliberti P, Pastorelli F, Biagini E, Coccolo F, Cooke RMT, Bacchi-Reggiani L, Sangiorgi D, Ferlini A, Cavo M, Zamagni E, Fonte ML, Palladini G, Salinaro F, Musca F, Obici L, Branzi A, Perlini S. Systemic cardiac amyloidoses: disease profiles and clinical courses of the 3 main types. Circulation. 2009;120:1203–1212. doi: 10.1161/CIRCULATIONAHA.108.843334. [DOI] [PubMed] [Google Scholar]
- 44.Carroll JD, Gaasch WH, McAdam KP. Amyloid cardiomyopathy: characterization by a distinctive voltage/mass relation. Am J Cardiol. 1982;49:9–13. doi: 10.1016/0002-9149(82)90270-3. [DOI] [PubMed] [Google Scholar]
- 45.Falk RH, Plehn JF, Deering T, Schick EC, Boinay P, Rubinow A, Skinner M, Cohen AS. Sensitivity and specificity of the echocardiographic features of cardiac amyloidosis. Am J Cardiol. 1987;59:418–422. doi: 10.1016/0002-9149(87)90948-9. [DOI] [PubMed] [Google Scholar]
- 46.Quarta CC, Solomon SD, Uraizee I, Kruger J, Longhi S, Ferlito M, Gagliardi C, Milandri A, Rapezzi C, Falk RH. Left ventricular structure and function in transthyretin-related versus light-chain cardiac amyloidosis. Circulation. 2014;129:1840–1849. doi: 10.1161/CIRCULATIONAHA.113.006242. [DOI] [PubMed] [Google Scholar]
- 47.Cueto-Garcia L, Reeder GS, Kyle RA, Wood DL, Seward JB, Naessens J, Offord KP, Greipp PR, Edwards WD, Tajik AJ. Echocardiographic findings in systemic amyloidosis: spectrum of cardiac involvement and relation to survival. J Am Coll Cardiol. 1985;6:737–743. doi: 10.1016/s0735-1097(85)80475-7. [DOI] [PubMed] [Google Scholar]
- 48.Klein AL, Hatle LK, Burstow DJ, Seward JB, Kyle RA, Bailey KR, Luscher TF, Gertz MA, Tajik AJ. Doppler characterization of left ventricular diastolic function in cardiac amyloidosis. J Am Coll Cardiol. 1989;13:1017–1026. doi: 10.1016/0735-1097(89)90254-4. [DOI] [PubMed] [Google Scholar]
- 49.Liao R, Jain M, Teller P, Connors LH, Ngoy S, Skinner M, Falk RH, Apstein CS. Infusion of light chains from patients with cardiac amyloidosis causes diastolic dysfunction in isolated mouse hearts. Circulation. 2001;104:1594–1597. [PubMed] [Google Scholar]
- 50.Brenner DA, Jain M, Pimentel DR, Wang B, Connors LH, Skinner M, Apstein CS, Liao R. Human amyloidogenic light chains directly impair cardiomyocyte function through an increase in cellular oxidant stress. Circ Res. 2004;94:1008–1010. doi: 10.1161/01.RES.0000126569.75419.74. [DOI] [PubMed] [Google Scholar]
- 51.Child JS, L JA. Echocardiographic manifestations of infiltrative cardiomyopathy. A report of seven cases due to amyloid. Chest. 1977;70:726–31. doi: 10.1378/chest.70.6.726. [DOI] [PubMed] [Google Scholar]
- 52.Jacobson D, Tagoe C, Schwartzbard A, Shah A, Koziol J, Buxbaum J. Relation of clinical, echocardiographic and electrocardiographic features of cardiac amyloidosis to the presence of the transthyretin V122I allele in older African-American men. Am J Cardiol. 2011;108:440–444. doi: 10.1016/j.amjcard.2011.03.069. [DOI] [PubMed] [Google Scholar]
- 53.Koyama J, Ray-Sequin PA, Falk RH. Longitudinal myocardial function assessed by tissue velocity, strain, and strain rate tissue Doppler echocardiography in patients with AL (primary) cardiac amyloidosis. Circulation. 2003;107:2446–2452. doi: 10.1161/01.CIR.0000068313.67758.4F. [DOI] [PubMed] [Google Scholar]
- 54.Sun JP, Stewart WJ, Yang XS, Donnell RO, Leon AR, Felner JM, Thomas JD, Merlino JD. Differentiation of hypertrophic cardiomyopathy and cardiac amyloidosis from other causes of ventricular wall thickening by two-dimensional strain imaging echocardiography. Am J Cardiol. 2009;103:411–415. doi: 10.1016/j.amjcard.2008.09.102. [DOI] [PubMed] [Google Scholar]
- 55.Dispenzieri A, Kyle RA, Gertz MA, Therneau TM, Miller WL, Chandrasekaran K, McConnell JP, Burritt MF, Jaffe AS. Survival in patients with primary systemic amyloidosis and raised serum cardiac troponins. The Lancet. 2003;361:1787–1789. doi: 10.1016/S0140-6736(03)13396-X. [DOI] [PubMed] [Google Scholar]
- 56.Palladini G, Campana C, Klersy C, Balduini A, Vadacca G, Perfetti V, Perlini S, Obici L, Ascari E, d’Eril GM, Moratti R, Merlini G. Serum N-terminal pro-brain natriuretic peptide is a sensitive marker of myocardial dysfunction in AL amyloidosis. Circulation. 2003;107:2440–2445. doi: 10.1161/01.CIR.0000068314.02595.B2. [DOI] [PubMed] [Google Scholar]
- 57.Suhr OB, Anan I, Backman C, Karlsson A, Lindqvist P, Mörner S, Waldenström A. Do troponin and B-natriuretic peptide detect cardiomyopathy in transthyretin amyloidosis? J Intern Med. 2008;263:294–301. doi: 10.1111/j.1365-2796.2007.01888.x. [DOI] [PubMed] [Google Scholar]
- 58.Pinney JH, Whelan CJ, Petrie A, Dungu J, Banypersad SM, Sattianayagam P, Wechalekar A, Gibbs SDJ, Venner CP, Wassef N, McCarthy CA, Gilbertson JA, Rowczenio D, Hawkins PN, Gillmore JD, Lachmann HJ. Senile systemic amyloidosis: clinical features at presentation and outcome. J Am Heart Assoc. 2013;2:e000098. doi: 10.1161/JAHA.113.000098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Maurer MS, Kristen AV, Rapezzi C, Suhr OB, Kolluri S, Thibaud D. Cardiac biomarkers in patients with transthyretin amyloidosis as documented in THAOS: the transthyretin amyloidosis survey. J Am Coll Cardiol. 2013;61(10_S) doi: 10.1016/S0735-1097(13)61244-9. [DOI] [Google Scholar]
- 60.Phelan D, Collier P, Thavendiranathan P, Popović ZB, Hanna M, Plana JC, Marwick TH, Thomas JD. Relative apical sparing of longitudinal strain using two-dimensional speckle-tracking echocardiography is both sensitive and specific for the diagnosis of cardiac amyloidosis. Heart. 2012;98:1442–1448. doi: 10.1136/heartjnl-2012-302353. [DOI] [PubMed] [Google Scholar]
- 61.Austin BA, Tang WHW, Rodriguez ER, Tan C, Flamm SD, Taylor DO, Starling RC, Desai MY. Delayed hyper-enhancement magnetic resonance imaging provides incremental diagnostic and prognostic utility in suspected cardiac amyloidosis. JACC Cardiovasc Imaging. 2009;2:1369–1377. doi: 10.1016/j.jcmg.2009.08.008. [DOI] [PubMed] [Google Scholar]
- 62.Dungu JN, Valencia O, Pinney JH, Gibbs SDJ, Rowczenio D, Gilbertson JA, Lachmann HJ, Wechalekar A, Gillmore JD, Whelan CJ, Hawkins PN, Anderson LJ. CMR-based differentiation of AL and ATTR cardiac amyloidosis. JACC Cardiovasc Imaging. 2014;7:133–142. doi: 10.1016/j.jcmg.2013.08.015. [DOI] [PubMed] [Google Scholar]
- 63.White JA, Kim HW, Shah D, Fine N, Kim K-Y, Wendell DC, Al-Jaroudi W, Parker M, Patel M, Gwadry-Sridhar F, Judd RM, Kim RJ. CMR imaging with rapid visual T1 assessment predicts mortality in patients suspected of cardiac amyloidosis. JACC Cardiovasc Imaging. 2014;7:143–156. doi: 10.1016/j.jcmg.2013.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gerbaud É, Lederlin M, Laurent F. Value of phase-sensitive inversion recovery sequence to perform and analyse late gadolinium enhancement in cardiac amyloidosis. Arch Cardiovasc Dis. 2009;102:859–860. doi: 10.1016/j.acvd.2009.06.006. [DOI] [PubMed] [Google Scholar]
- 65.Fontana M, Pica S, Reant P, Abdel-Gadir A, Treibel TA, Banypersad SM, Maestrini V, Barcella W, Rosmini S, Bulluck H, Sayed RH, Patel K, Mamhood S, Bucciarelli-Ducci C, Whelan CJ, Herrey AS, Lachmann HJ, Wechalekar AD, Manisty CH, Schelbert EB, Kellman P, Gillmore JD, Hawkins PN, Moon JC. Prognostic Value of Late Gadolinium Enhancement Cardiovascular Magnetic Resonance in Cardiac Amyloidosis. Circulation. 2015;132:1570–1579. doi: 10.1161/CIRCULATIONAHA.115.016567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fontana M, Banypersad SM, Treibel TA, Maestrini V, Sado DM, White SK, Pica S, Castelletti S, Piechnik SK, Robson MD, Gilbertson JA, Rowczenio D, Hutt DF, Lachmann HJ, Wechalekar AD, Whelan CJ, Gillmore JD, Hawkins PN, Moon JC. Native T1 mapping in transthyretin amyloidosis. JACC Cardiovasc Imaging. 2014;7:157–165. doi: 10.1016/j.jcmg.2013.10.008. [DOI] [PubMed] [Google Scholar]
- 67.Rapezzi C, Quarta CC, Guidalotti PL, Longhi S, Pettinato C, Leone O, Ferlini A, Salvi F, Gallo P, Gagliardi C, Branzi A. Usefulness and limitations of 99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid scintigraphy in the aetiological diagnosis of amyloidotic cardiomyopathy. Eur J Nucl Med Mol Imaging. 2011;38:470–478. doi: 10.1007/s00259-010-1642-7. [DOI] [PubMed] [Google Scholar]
- 68.Rapezzi C, Quarta CC, Guidalotti PL, Pettinato C, Fanti S, Leone O, Ferlini A, Longhi S, Lorenzini M, Reggiani LB, Gagliardi C, Gallo P, Villani C, Salvi F. Role of (99m)Tc-DPD scintigraphy in diagnosis and prognosis of hereditary transthyretin-related cardiac amyloidosis. JACC Cardiovasc Imaging. 2011;4:659–670. doi: 10.1016/j.jcmg.2011.03.016. [DOI] [PubMed] [Google Scholar]
- 69.Fine NM, Arruda-Olson AM, Dispenzieri A, Zeldenrust SR, Gertz MA, Kyle RA, Swiecicki PL, Scott CG, Grogan M. Yield of noncardiac biopsy for the diagnosis of transthyretin cardiac amyloidosis. Am J Cardiol. 2014;113:1723–1727. doi: 10.1016/j.amjcard.2014.02.030. [DOI] [PubMed] [Google Scholar]
- 70.Linke RP. Highly sensitive diagnosis of amyloid and various amyloid syndromes using Congo red fluorescence. Virchows Arch. 2000;436:439–448. doi: 10.1007/s004280050471. [DOI] [PubMed] [Google Scholar]
- 71.Picken MM. Amyloidosis-where are we now and where are we heading? Arch Pathol Lab Med. 2010;134:545–551. doi: 10.5858/134.4.545. [DOI] [PubMed] [Google Scholar]
- 72.Lavatelli F, Vrana JA. Proteomic typing of amyloid deposits in systemic amyloidoses. Amyloid. 2011;18:177–182. doi: 10.3109/13506129.2011.630762. [DOI] [PubMed] [Google Scholar]
- 73.Vrana JA, Gamez JD, Madden BJ, Theis JD, Bergen HR, Dogan A. Classification of amyloidosis by laser microdissection and mass spectrometry-based proteomic analysis in clinical biopsy specimens. Blood. 2009;114:4957–4959. doi: 10.1182/blood-2009-07-230722. [DOI] [PubMed] [Google Scholar]
- 74.Benson MD, Kincaid JC. The molecular biology and clinical features of amyloid neuropathy. Muscle Nerve. 2007;36:411–423. doi: 10.1002/mus.20821. [DOI] [PubMed] [Google Scholar]
- 75.Pollak A, Falk RH. Left ventricular systolic dysfunction precipitated by verapamil in cardiac amyloidosis. Chest. 1993;104:618–620. doi: 10.1378/chest.104.2.618. [DOI] [PubMed] [Google Scholar]
- 76.Gertz MA, Falk RH, Skinner M, Cohen AS, Kyle RA. Worsening of congestive heart failure in amyloid heart disease treated by calcium channel-blocking agents. Am J Cardiol. 1985;55:1645. doi: 10.1016/0002-9149(85)90995-6. [DOI] [PubMed] [Google Scholar]
- 77.Berk JL, Suhr OB, Obici L, Sekijima Y, Zeldenrust SR, Yamashita T, Heneghan MA, Gorevic PD, Litchy WJ, Wiesman JF, Nordh E, Corato M, Lozza A, Cortese A, Robinson-Papp J, Colton T, Rybin DV, Bisbee AB, Ando Y, Ikeda S, Seldin DC, Merlini G, Skinner M, Kelly JW, Dyck PJ, Diflunisal Trial Consortium Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA. 2013;310:2658–2667. doi: 10.1001/jama.2013.283815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bulawa CE, Connelly S, Devit M, Wang L, Weigel C, Fleming JA, Packman J, Powers ET, Wiseman RL, Foss TR, Wilson IA, Kelly JW, Labaudinière R. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc Natl Acad Sci USA. 2012;109:9629–9634. doi: 10.1073/pnas.1121005109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 80.Kanasty R, Dorkin JR, Vegas A, Anderson D. Delivery materials for siRNA therapeutics. Nat Mater. 2013;12:967–977. doi: 10.1038/nmat3765. [DOI] [PubMed] [Google Scholar]
- 81.Cardoso I, Saraiva MJ. Doxycycline disrupts transthyretin amyloid: evidence from studies in a FAP transgenic mice model. FASEB J. 2006;20:234–239. doi: 10.1096/fj.05-4509com. [DOI] [PubMed] [Google Scholar]
- 82.Pepys MB, Dash AC. Isolation of amyloid P component (protein AP) from normal serum as a calcium-dependent binding protein. Lancet. 1977;1:1029–1031. doi: 10.1016/s0140-6736(77)91260-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.