

SUMMARY
Effector T cells and fibroblasts are major components in the tumor microenvironment. The means through which these cellular interactions affect chemoresistance is unclear. Here, we show that fibroblasts diminish nuclear accumulation of platinum in ovarian cancer cells, resulting in resistance to platinum-based chemotherapy. We demonstrate that glutathione and cysteine released by fibroblasts contribute to this resistance. CD8+ T cells abolish the resistance by altering glutathione and cystine metabolism in fibroblasts. CD8+ T-cell-derived interferon (IFN)γ controls fibroblast glutathione and cysteine through upregulation of glutamyltransferases and transcriptional repression of system xc− cystine and glutamate antiporter via the JAK/STAT1 pathway. The presence of stromal fibroblasts and CD8+ T cells is negatively and positively associated with ovarian cancer patient survival, respectively. Thus, our work uncovers a mode of action for effector T cells: they abrogate stromal-mediated chemoresistance. Capitalizing upon the interplay between chemotherapy and immunotherapy holds high potential for cancer treatment.
Graphical abstract
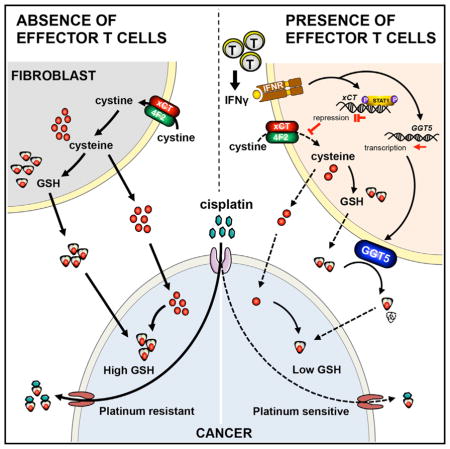
INTRODUCTION
Initial clinical response of ovarian cancer patients to surgical de-bulking and chemotherapy with platinum-based drugs is often excellent. However, relapse and metastasis due to drug resistance is common and patients oftentimes succumb to their disease. Studies regarding chemoresistance in ovarian cancer have focused on cancer genetic alterations, apoptosis, and drug metabolism. However, it is unknown whether and how effector CD8+ T cells play a role in drug resistance.
The human cancer immune microenvironment holds the key to understanding the nature of immunity in response to tumor progression and tumor immunotherapy (Topalian et al., 2015; Zou, 2005; Zou et al., 2016). Interestingly, the efficacy of chemotherapy, radiotherapy, and oncologic antibody targeting therapy also depends upon interferon signaling and CD8+ T cell immunity (Binder et al., 2015; Lee et al., 2009; Zitvogel et al., 2010). Regardless of the type of therapy, the presence of tumor-infiltrating CD8+ T cells is a favorable prognostic factor in many types of cancer, including ovarian cancer (Peng et al., 2015; Sato et al., 2005; Zhang et al., 2003; Zhao et al., 2016). This raises the possibility that CD8+ T cells may play a role in the prevention of chemoresistance in human cancer. In the current work, we have tested this possibility in the context of ovarian cancer.
In addition to T cells and tumor cells, fibroblasts are one of the major cellular components in the tumor microenvironment. Tumor-associated fibroblasts are involved in immune regulation (Kraman et al., 2010) and tumor progression (Özdemir et al., 2014) in tumor bearing mouse models. Thus, in the current work, we examined the potential interaction between CD8+ T cells and fibroblasts in ovarian cancer and found that this interaction shapes the balance between chemotherapeutic resistance and sensitivity in ovarian cancer. We have further dissected the underlying cellular and molecular mechanisms and its pathological relevance in patients with ovarian cancer.
RESULTS
Cancer-Associated Fibroblasts Confer Platinum Resistance to Ovarian Cancer Cells
High grade serous ovarian carcinoma (HGSOC) is the most aggressive subtype of epithelial ovarian cancer. To test if HGSOC-associated fibroblasts play a role in platinum resistance, we isolated fibroblasts from HGSOC tissues for our studies (Table S1). Polychromatic flow cytometry analysis showed that fibroblasts were CD45−, epithelial cell adhesion molecule-negative (EPCAM−), CD24−, CD44+, CD105+, and platelet-derived growth factor receptor-alpha positive (PDGFRA+) (Figure S1A). Immunofluorescence staining showed that isolated fibroblasts expressed alpha-smooth muscle actin (α-SMA) (Figure S1B).
We isolated primary epithelial ovarian cancer cells (OC8) from a patient with HGSOC (Cui et al., 2013). TP53 mutation is a feature of HGSOC (Domcke et al., 2013). We sequenced TP53 gene in OC8 and found that OC8 carried a hotspot TP53 mutation on exon 6 (Figure S1C; Table S2) (Soussi et al., 2010). Next, we inoculated the mixture of GFP-labeled OC8 and primary fibroblasts into female NOD.SCID γc-deficient (NSG) mice and treated the mice with cisplatin. Fibroblasts had minimal effect on tumor volume in the control mice that were not treated with cisplatin (Figure 1A). However, tumor volume was increased in the cisplatin-treated mice injected with both OC8 and fibroblasts, compared with the cisplatin-treated mice injected with OC8 alone (Figure 1A). TUNEL assay demonstrated less cisplatin-induced apoptosis in tumor cells in the presence of fibroblasts (Figure 1B).
Figure 1. Fibroblasts Induce Cancer Platinum Resistance.

(A–D) Effect of fibroblasts on cisplatin resistance to OC8 (A and B) or A2780 (C and D) in vivo. Tumor cells or tumor plus fibroblasts were inoculated subcutaneously into mice and treated with or without cisplatin every 3 days for three cycles. Tumor volume was monitored (A and C) (mean ± SEM; n = 5). Apoptosis was determined by TUNEL staining. Representative images and TUNEL-positive cells (percentage of control) are shown (B and D). Scale bar, 40 μm. Mean ± SD; n = 10 random fields from three sections. *p < 0.05.
(E) Cisplatin-induced apoptosis on GFP+ tumor cells in the mixed co-culture of A2780-GFP and fibroblasts. Representative FACS data are shown (left). Mean ± SD, n = 3. *p < 0.05.
(F–H) Cisplatin-induced apoptosis in A2780 (F), OC8 (G), or NIH: OVCAR3 (H) co-cultured with fibroblasts from different ovarian cancer patients in the Transwell. Mean ± SD; n = 3, *p < 0.05.
(I and J) Cisplatin-induced apoptosis on A2780 (I) or OC8 (J) cultured in fibroblast medium. Mean ± SD; n = 3, *p < 0.05.
A2780 ovarian cancer cells are used in cisplatin resistance studies. Whether A2780 are HGSOC may be controversial (Domcke et al., 2013). We mixed fibroblasts with A2780 and inoculated subcutaneously into nude mice and treated the mice with cisplatin. Similar to the OC8 model, fibroblasts protected A2780 from cisplatin-mediated apoptosis (Figures 1C and 1D). Immunohistochemistry demonstrated the existence of SMA+ fibroblasts in tumors established by injection of both fibroblasts and cancer cells (Figure S1D). Thus, fibroblasts enhance cisplatin resistance in ovarian cancer cells in vivo.
When we co-cultured A2780 with fibroblasts in vitro, we found that fibroblasts protected tumor cells from cisplatin-induced apoptosis as shown by reduced proportion of Annexin V+ tumor cells (Figure 1E) in a dose-dependent manner (Figure S1E). To further investigate if the protective effects from fibroblasts required cell-to-cell contact, we cultured A2780 with fibroblasts in a Transwell and observed that all three fibroblast samples protected A2780 from cisplatin-induced apoptosis (Figure 1F). We observed similar protective effects of fibroblasts on OC8 (Figure 1G) and NIH:OVCAR3 (Figure 1H), another p53 mutant HGSOC cell (Table S2) (Domcke et al., 2013), and cisplatin-sensitive TOV21G ovarian cancer cells (Figure S1F). We collected serum-free culture medium from fibroblasts (fibroblast medium) and performed identical experiments. Fibroblast medium also protected A2780 and OC8 from cisplatin-induced apoptosis (Figures 1I and 1J). We extended our studies to include carboplatin and oxaliplatin and observed similar results (Figures S1G and S1H). Thus, fibroblasts release soluble factor(s) and promote platinum resistance in ovarian cancer cells.
CD8+ T Cells Abolish Fibroblast-Mediated Chemoresistance by IFNγ
The presence of CD8+ T cells is associated with prolonged survival in cisplatin-treated ovarian cancer patients (Zhang et al., 2003; Zhao et al., 2016). To examine the potential involvement of CD8+ T cells in fibroblast-mediated platinum resistance, we collected the supernatants from activated CD8+ T cells and added it into the fibroblast and ovarian cancer cell co-culture system. We found that CD8+ T cell-derived supernatants abolished the protective effects of fibroblasts on cisplatin-induced apoptosis in tumor cells (Figure 2A). We next pre-treated fibroblasts with CD8+ T cell supernatants and again fibroblasts lost the protective function on cancer cells treated with cisplatin (Figures 2B and S2A). The fibroblast numbers (Figure S2B) and viability (Figure S2C) were not changed by CD8+ T cell supernatants.
Figure 2. CD8+ T Cells Abolish Fibroblast-Induced Platinum Resistance via IFNγ.

(A and B) Effect of CD8+ T cells on fibroblast-induced platinum resistance. A2780 and fibroblast were cultured with the supernatant from activated CD8+ T cells (A). Fibroblasts were primed with CD8+ T cell supernatants and cultured with tumor cells in the presence of cisplatin (B). Tumor cell apoptosis was determined by Annexin V staining. Mean ± SD; n = 3, *p < 0.05.
(C) IFNγ released by activated CD8+ T cells. Mean ± SD; n = 3.
(D–G) Effect of IFNγ on fibroblast-induced platinum resistance. Fibroblasts were primed with IFNγ (D and E) or with CD8+ T cells supernatant in the presence of anti-IFNγ R1 (F) or JAK inhibitor I (G) and subsequently cultured with tumor cells in the Transwell. Cisplatin-induced tumor cell apoptosis was determined. Mean ± SD; n = 3, *p < 0.05.
(H) Effect of IFNγ on fibroblast-induced cisplatin resistance to OC8 in vivo. OC8 and fibroblasts were inoculated subcutaneously into NSG mice and then treated with IFNγ, cisplatin, or IFNγ + cisplatin for three cycles. Tumor volume was monitored (mean ± SEM, n = 5). *p < 0.05.
See also Figure S2.
IFNγ is a major effector cytokine produced by CD8+ T cells. We detected high levels of IFNγ in the activated CD8+ T cell-derived supernatants (Figure 2C). We hypothesized that CD8+ T cell-derived IFNγ abolished the protective effect of fibroblasts. To test this, we pre-treated the fibroblasts with IFNγ and observed more apoptotic A2780 (Figure 2D) or OC8 (Figure 2E) cells in the cultures containing IFNγ-primed fibroblasts compared to control fibroblasts. IFNγ treatment had no effect on fibroblast cell numbers or apoptosis (Figures S2B and S2C). Moreover, we cultured fibroblasts with CD8+ T cell supernatant in the presence of neutralizing anti-IFNγ receptor 1 (IFNGR1) antibody (Figure 2F) or JAK inhibitor I (Figure 2G). IFNγ signaling blockade restored the protective effect of fibroblasts on cisplatin-induced tumor cell apoptosis (Figures 2F and 2G). Thus, effector CD8+ T cells abolish fibroblast-mediated chemoresistance through the IFNγ signaling pathway.
We further tested the effect of IFNγ on fibroblast-mediated cisplatin resistance in the OC8 bearing NSG model in vivo. IFNγ administration had no effect on tumor volume (Figure 2H). However, IFNγ administration increased the therapeutic efficacy of cisplatin treatment as shown by reduced tumor volume (Figure 2H). Thus, IFNγ diminishes fibroblast-mediated platinum resistance in vivo.
Fibroblasts Reduce Platinum Intracellular Content in Cancer Cells
Cisplatin induces cytotoxicity mainly through DNA damage-mediated apoptosis (Wang and Lippard, 2005). To uncover the molecular mechanism of fibroblast-mediated cisplatin resistance, we first examined whether fibroblasts change the cisplatin-induced apoptotic signaling pathway in cancer cells. Cisplatin treatment activated caspase 3 and caspase 9 in A2780, while the presence of fibroblasts reduced their activation (Figure S3A). Cisplatin causes DNA crosslinking and stimulates H2AX phosphorylation at Serine 139 to generate γH2AX as a major marker for DNA damage signaling in response to DNA double-strand break (Revet et al., 2011). When ovarian cancer cells were cultured with fibroblasts, cisplatin-triggered γH2AX in A2780, OC8, and OVCAR3 was reduced (Figure 3A). Immunofluorescence staining confirmed that fibroblast medium also reduced γH2AX in cisplatin-treated A2780 (Figure S3B). Moreover, immunohistochemistry revealed that in the fibroblast and ovarian tumor cell co-injection animal model (Figure 1), fibroblasts led to decreased levels of γH2AX in tumor cells following cisplatin treatment (Figure 3B). Thus, fibroblasts may control cisplatin resistance in cancer cells at the DNA damage induction level.
Figure 3. Fibroblasts Reduce the Accumulation of Cisplatin in Cancer Cells.

(A) Cisplatin-induced γH2AX in cancer cells cultured with fibroblasts was detected by western blotting. Fibroblasts were from four patients. One of three experiments is shown.
(B) Cisplatin-induced γH2AX in vivo was determined in A2780 tissues and representative images are shown. Scale bar, 50 μm.
(C–E) Effect of fibroblasts on cisplatin intracellular content in tumor cells. A2780 (C and D) or OC8 (E) were cultured with fibroblasts in the presence of different concentrations of cisplatin (C) or 10 μg/ml cisplatin (D and E). Platinum content in the whole tumor cells was measured by ICP-MS in triplicate (mean ± SD). *p < 0.05.
(F and G) Effect of fibroblasts on cisplatin DNA content in A2780 (F) or OC8 (G). Platinum content in the genomic DNA was measured by ICP-MS in triplicate (mean ± SD). *p < 0.05.
(H) Dot blot assay of cisplatin-DNA adduct in genomic DNA of OC8 cultured with fibroblasts in the presence of cisplatin.
(I) Effect of fibroblasts on cisplatin content in tumor cells in vivo. GFP+ OC8 were enriched from xenograft tumor tissues and platinum content was measured by ICP-MS (n = 5 or 6). *p < 0.05.
(J) Effect of IFNγ blockade on tumor cisplatin content in vivo. GFP+ OC8 and fibroblasts were inoculated into NSG mice with CD8+ T cells. The mice were treated with cisplatin and anti-human IFNγ blocking antibody or control antibody. GFP+ OC8 were enriched and platinum content was measured by ICP-MS (n = 4). *p < 0.05.
See also Figure S3.
Reduced intracellular cisplatin content can be linked to less DNA damage (Galluzzi et al., 2012; Wang and Lippard, 2005). Inductively coupled plasma-high resolution mass spectrometry (ICP-MS) measurement revealed that intracellular cisplatin content was reduced in whole A2780 cells in the presence of fibroblasts with different concentrations of cisplatin (Figure 3C). Similar effects were observed in A2780 (Figure 3D) and OC8 (Figure 3E) using fibroblasts from several clinical specimens. Diminished cisplatin content was also detected in the genomic DNA of cisplatin-treated A2780 (Figure 3F) and OC8 (Figure 3G) cultured with fibroblasts. The presence of fibroblast-conditioned medium from different ovarian cancer patients also resulted in diminished cisplatin accumulation in the genomic DNA of cisplatin-treated tumor cells (Figure S3C). Using an antibody specific for cisplatin-DNA adducts, we found that cisplatin-DNA adduct formation in OC8 was decreased in the presence of fibroblasts (Figure 3H). In support of this observation, fibroblasts led to decreased tumor cell accumulation of cisplatin (Figure 3I) in tumor cells in cisplatin-treated mice (OC8-NSG model) (Figure S3D). Thus, fibroblasts confer cisplatin-resistance in ovarian cancer cells by reducing intracellular cisplatin accumulation.
To further investigate the causal link between CD8+ T cell-derived IFNγ and cisplatin tumor accumulation in vivo, we adoptively transferred CD8+ T cells into the mice bearing tumors established with a mixture of tumor cells and fibroblasts and treated the mice with anti-human IFNγ antibody and cisplatin. Twenty-four hours after the treatment, we enriched GFP-labeled tumor cells (Figure S3D) and found platinum content was reduced by anti-human IFNγ (Figure 3J). Thus, fibroblasts confer cisplatin-resistance in ovarian tumor cells by reducing intracellular cisplatin accumulation and CD8+ T cell-derived IFNγ abolishes this effect.
Fibroblast-Mediated Chemoresistance Is Linked to Intracellular Glutathione Level
We dissected the mechanisms by which fibroblasts reduce cisplatin intracellular accumulation. Intracellular cisplatin content is regulated through cisplatin influx or efflux transporters (Wang and Lippard, 2005). We found that fibroblasts neither decreased the expression of cisplatin influx transporters nor increased the expression of cisplatin efflux transporters in tumor cells (Figure S4A). Cisplatin can be chelated by glutathione and the glutathione (GSH)-platinum complex subsequently effluxes from the cell (Chen and Kuo, 2010). We observed that after co-culture with fibroblasts, the intracellular GSH level was increased in A2780 (Figure 4A), OC8 (Figure 4B), and OVCAR3 (Figure S4B). Similarly, fibroblast medium increased the intracellular GSH level in tumor cells compared to fresh culture medium and tumor cell medium (Figure 4C).
Figure 4. Fibroblasts Elevate Cancer Intracellular GSH Conferring Platinum Resistance.

(A and B) Intracellular GSH level in A2780 (A) or OC8 (B) co-cultured with fibroblasts. Mean ± SD, n = 3, *p < 0.05.
(C) Intracellular GSH level in A2780 cultured in control or fibroblast medium. Mean ± SD, n = 3, *p < 0.05.
(D) Intracellular GSH level in A2780 treated with 200 μM NAC for 6 hr. GSH was measured in triplicate (mean ± SD). *p < 0.05.
(E and F) Effects of NAC on cisplatin-induced γH2AX (E) and cisplatin content (F) in A2780. Pro-caspase 9 and γH2AX were detected by western blotting (E). Platinum content in genomic DNA of A2780 was measured by ICP-MS in triplicates (mean ± SD) (F). *p < 0.05.
(G) Intracellular GSH level in A2780 treated with 6 μM BSO for 6 hr. GSH was measured in triplicates (mean ± SD). *p < 0.05.
(H and I) Platinum content in genomic DNA (H) or cisplatin-induced apoptosis (I) of A2780. A2780 were pretreated with 6 μM BSO for 6 hr. Mean ± SD, n = 3, *p < 0.05.
(J and K) Effects of GCLC knockdown on intracellular GSH level (J) and cisplatin-induced apoptosis (K) in A2780. GSH was measured and normalized with total protein. Apoptosis was determined by Annexin V staining. Mean ± SD; n = 3, *p < 0.05.
(L and M) Effect of GSH monoester on cisplatin resistance in vivo. Mice bearing A2780 ovarian cancer were treated with PBS (control) or GSH monoester for 8 hr and followed with cisplatin treatment for 24 hr. Platinum content in tumor tissue was measured by ICP-MS (L). For tumor volume monitoring, mice were treated with GSH monoester and cisplatin for three cycles (M) (mean ± SEM, n = 5 tumors/group). *p < 0.05.
See also Figure S4.
Next, we investigated whether intracellular GSH level in tumor cells affected their apoptotic response to cisplatin. N-acetyl cysteine (NAC) is a GSH precursor. Buthionine sulfoximine (BSO) is an inhibitor of glutamate-cysteine ligase, the first rate-limiting enzyme of glutathione synthesis. NAC increased intracellular GSH level (Figure 4D), decreased the level of cisplatin-induced γ, de (Figure 4E), reduced platinum content in genomic DNA (Figure 4F), and protected ovarian tumor cells from cisplatin-induced apoptosis (Figure S4C). BSO treatment decreased intracellular GSH level (Figure 4G), increased platinum DNA content (Figure 4H), and promoted cisplatin-induced apoptosis (Figure 4I) in A2780. Similarly, BSO treatment increased cisplatin-induced apoptosis in OC8 (Figure S4D) and OVCAR3 (Figure S4E). Treatment with NAC or BSO in the absence of cisplatin had no effect on the viability of A2780, OC8, and OVCAR3 (Figures 4I and S4C–S4E). Next, we constructed a short hairpin RNA (shRNA) vector to downregulate the expression of glutamate-cysteine ligase catalytic subunit (GCLC) in A2780. shGCLC downregulated the protein expression of GCLC (Figure S4F), decreased intracellular GSH level (Figure 4J), and promoted cisplatin-induced apoptosis (Figure 4K).
GSH monoester is utilized to regulate intracellular GSH levels (Mårtensson and Meister, 1989). We found that GSH monoester reduced tumor accumulation of cisplatin in vivo (Figure 4L). Accordingly, tumor volume was increased in mice treated with GSH monoester plus cisplatin as compared to cisplatin treatment alone (Figure 4M). Thus, GSH monoester diminished the therapeutic efficiency of cisplatin in ovarian tumors in vivo. Altogether, fibroblast-mediated chemoresistance is linked to high intracellular GSH in ovarian cancer cells.
Fibroblast-Derived GSH and Cysteine Confer Chemoresistance in Tumor Cells
Fibroblasts release soluble factor(s), elevate intracellular GSH, and reduce intracellular cisplatin content in tumor cells. To identify these fibroblast-released factor(s), we divided the fibroblast medium or tumor cell medium (control) into two fractions based on a 3 kDa molecular weight cutoff filter. We found that the fraction with <3 kDa molecular size, but not the faction with >3 kDa molecular size, inhibited cisplatin-induced apoptosis (Figure 5A) and increased intracellular GSH level (Figure 5B) in A2780. When fibroblast medium was incubated with dextran-coated charcoal for removing hormones and steroids, the protective effect was diminished (Figure S5A). When fibroblast medium was treated with trypsin or proteinase K digestion, the protective effect was not changed (Figure S5B). Thus, the fibroblast factor(s) may be small molecules, rather than protein(s) or large peptide(s).
Figure 5. Fibroblasts Release GSH and Cysteine Conferring to Cisplatin Resistance.

(A and B) Cisplatin-induced apoptosis (A) and intracellular GSH (B) in A2780. Fibroblast medium was filtered into two fractions, >3 kDa and <3 kDa. Tumor cells were cultured with unfiltered or filtered fibroblast medium in the presence of cisplatin. Apoptosis and intracellular GSH in A2780 cells were measured. Mean ± SD; n = 3, *p < 0.05.
(C and D) Total thiol (C) and GSH (D) in fresh medium, A2780 medium, and fibroblast medium. Mean ± SD, n = 3.
(E) Cysteine, Cys-Gly, and γ-Cys-Glu concentrations in fresh medium, A2780 medium, and fibroblast medium. Mean ± SD, n = 3.
(F–K) Effects of exogenous GSH (F–H) and cysteine (I–K) on intracellular GSH level (F and I), cisplatin content (G and J), and cisplatin-induced apoptosis (H and K) in A2780. A2780 were pretreated with 50 μM GSH or 100 μM cysteine for 6 hr and followed with cisplatin treatment. Mean ± SD; n = 3, *p < 0.05.
(L–N) Effects of cystine deficiency on fibroblast-generated thiol (L), GSH (M), and fibroblast-mediated tumor protection (N). Total thiol (L) and GSH (M) were measured in medium that were generated in cystine complete (Cystine+) or cystine free (Cystine−) culture. Mean ± SD; n = 3. A2780 were incubated in Cystine+ or Cystine− fibroblast medium for 6 hr and followed with cisplatin treatment. For Cystine− medium, 100 μM cystine was supplemented before A2780 were exposed to the medium. Tumor apoptosis was determined by Annexin V staining (N). Mean ± SD; n = 3, *p < 0.05.
See also Figure S5.
Given that fibroblast medium increased tumor cell intracellular GSH level, we hypothesized that the fibroblast-derived small molecule(s) were related to GSH metabolism. GSH is the major thiol-containing component synthesized de novo in mammalian cells (Lushchak, 2012). We detected higher levels of thiols in fibroblast medium than in fresh culture medium or in A2780 medium (Figure 5C). Fibroblast medium contained 25–50 μM GSH compared to <1 μM GSH in fresh medium and in A2780 medium (Figure 5D). We confirmed high GSH levels in fibroblast medium from six additional patient samples (Figure S5C). These data suggest that in addition to GSH, there are other thiol component(s) in the fibroblast medium. GSH, cysteine, cysteine-glycine (Cys-Gly), and γ-glutamyl-cysteine (γ-Glu-Cys) are four thiol-containing metabolites. We measured these metabolites by liquid chromatography-mass spectrometry (LC-MS) (Figures S5D and S5E). We detected <3 μM of cysteine-glycine and γ-glutamyl-cysteine in all mediums, <10 μM of cysteine in fresh medium and tumor medium, and 70–100 μM of cysteine in fibroblast medium (Figure 5E). Thus, the major thiol components in fibroblast medium are GSH and cysteine. Fibroblast-derived GSH and cysteine may mediate the protective effect from cisplatin-induced tumor apoptosis. We showed that exogenous GSH (Figures 5F–5H) and cysteine (Figures 5I–5K) increased intracellular levels of GSH (Figures 5F and 5I), decreased platinum DNA accumulation (Figures 5G and 5J), and protected ovarian tumor cells from cisplatin-induced apoptosis (Figures 5H and 5K).
Cystine is a precursor of cysteine, which is the rate-limiting substrate for GSH synthesis (Zhang et al., 2012). Hence, we hypothesized that manipulation of cystine would alter levels of endogenous GSH and cysteine in fibroblasts and consequently impact their protective effect on tumor cells. To this end, we generated fibroblast medium with or without cystine (cystine+ or cystine−) complementation. We showed that cystine deficiency (cystine−) resulted in dramatic reduction of total thiol (Figure 5L) and GSH (Figure 5M) in fibroblast medium. Functionally, cystine+, but not cystine−, fibroblast medium reduced Annexin V+ tumor cells in response to cisplatin (Figure 5N). Therefore, fibroblasts utilize cystine to generate and release cysteine and GSH, which leads to tumor cell uptake of cysteine and GSH to elevate intracellular GSH, ultimately resulting in cisplatin resistance.
CD8+ T Cells Alter GSH/Cystine Metabolism and xCT Expression in Fibroblasts
Fibroblast-mediated cisplatin resistance was abolished by IFNγ and CD8+ T cells (Figure 2). Next, we tested whether IFNγ and CD8+ T cells targeted GSH and cysteine metabolism in fibroblasts. Treatment of fibroblasts with IFNγ or CD8+ T cell supernatants reduced the total thiol (Figures 6A and S6A) and GSH (Figures 6B and S6B) in fibroblast medium. Neutralizing anti-IFNGR1 antibody disabled the effects of CD8+ T cells on fibroblasts-derived thiol and GSH (Figures S6A and S6B).
Figure 6. Effector CD8+ T Cell-Derived IFNγ Reduces GSH and Cysteine in Fibroblasts.

(A and B) Total thiol (A) and GSH (B) concentrations in samples. Fibroblasts were primed with 5 ng/ml IFNγ. Mean ± SD; n = 3, *p < 0.05.
(C) Real-time PCR quantification of GGT mRNAs in IFNγ-treated fibroblasts. Data are presented as fold change relative to control group, n = 3, mean ± SD.
(D) GGT enzymatic activity in fibroblasts after 48 hr of IFNγ treatment. Data are presented as fold change relative to control (mean ± SD), n = 3, *p < 0.05.
(E) Role of GGT5 knockdown in IFNγ-mediated protective effect. Fibroblasts expressing scramble shRNA or shGGT5 were primed with IFNγ. A2780 were cultured with these fibroblasts and cisplatin-induced tumor apoptosis was determined. n = 3, mean ± SD. *p < 0.05.
(F) Quantification of cysteine and cystine concentrations in fibroblast medium by LC-MS. Fibroblasts from three different patients were treated with IFNγ. *p < 0.05.
(G) Effect of IFNγ and CD8+ T cell supernatants on 14C-Cystine uptake by fibroblasts. Mean ± SD, n = 3. *p < 0.05.
(H and I) Platinum content (H) and γH2AX level (I) in A2780 cultured with IFNγ-primed fibroblasts in the presence of cisplatin. The intracellular platinum was measured by ICP-MS. Mean ± SD n = 3, *p < 0.05. Tumor γH2AX was detected by western blotting.
(J) Representative immunoblots of xCT and SLC3A2 in fibroblasts treated with IFNγ for 24 hr.
(K) Effect of xCT knockdown on fibroblast-mediated cisplatin resistance. A2780 were cultured with fibroblasts expressing scramble shRNA or shxCT, and followed with cisplatin treatment. Tumor apoptosis was determined by Annexin V staining. n = 3, mean ± SD. *p < 0.05. The knockdown efficiency of shxCT was determined by western blotting.
(L) Real-time PCR assays of mRNAs in fibroblasts treated with IFNγ for different time points. Data are presented as fold change relative to control (mean ± SD), n = 3.
(M) Real-time PCR assays of xCT pre-mRNA in above fibroblasts. xCT pre-mRNA was analyzed with two different primer pairs and normalized to GAPDH pre-mRNA. Mean ± SD, n = 3.
(N) ChIP of RNA Pol II in fibroblasts treated with or without IFNγ. RNA Pol II binding to xCT TSS and intragenic regions (+900 and +6 k) was quantified by qPCR. Results are expressed as the fold changes in site occupancy over control fibroblasts. Mean ± SD from two fibroblasts samples.
(O) IFNγ-induced STAT1 phosphorylation and xCT downregulation in fibroblasts were determined by western blotting.
(P) Graphics map showing the positions of primers used to quantify potential STAT1 binding sites around xCT promoter region. Lower panel shows UCSC genome browser tracks of the xCT promoter region with data for STAT1 ChIP-seq.
(Q) ChIP of STAT1 in IFNγ-treated fibroblasts. The binding of STAT1 to the three GAS regions around xCT promoter was determined. Results are expressed as the fold enrichment over IgG. Mean ± SD from two fibroblasts samples.
(R) Effect of xCT promoter GAS elements on luciferase activity. Fibroblasts were transfected with luciferase reporter constructs containing full-length xCT promoter (GAS1+2+3) or individual GAS elements. Luciferase activity was assessed after IFNγ treatment and presented as the relative activity compared with controls (mean ± SD). n = 3, *p < 0.05.
See also Figure S6.
Gamma-glutamyltransferases (GGT), the only enzyme of the gamma-glutamyl cycle present on the cell membrane, controls GSH homeostasis by breaking down extracellular GSH (Lushchak, 2012). We observed that IFNγ stimulated the mRNA expression of GGT5 (Figure 6C) and increased the GGT enzymatic activity in fibroblasts (Figure 6D), but not in tumor cells (Figures S6C and S6D). Moreover, we found positive correlations between GGT5, CD8 (Figure S6E), and an IFNγ responsive gene, interferon regulatory factor 1 (IRF1) expression (Figure S6F) in the microdissected stroma compartments from HGSOC specimens (GEO: GSE40595) (Yeung et al., 2013). We further explored the importance of IFNγ-stimulated GGT5 expression in fibroblast-mediated cisplatin resistance by knocking down GGT5 expression in fibroblasts. Fibroblasts expressing shGGT5 and scrambled shRNA similarly protected tumor cells from cisplatin-induced apoptosis (Figures 6E and S6G). IFNγ treatment abolished the protective function of fibroblasts expressing scrambled shRNA and partially reversed this protective function of fibroblasts expressing shGGT5 (Figure 6E). These data suggest that IFNγ targets GGT5 in fibroblasts to accelerate GSH degradation and in turn partially reduces the protective activity of fibroblasts on cisplatin-induced tumor cell apoptosis.
Next, we analyzed whether IFNγ and CD8+ T cells could regulate cysteine level in fibroblasts. We measured both cysteine and cystine (Figures S6H and S6I) with LC-MS in the cultured fibroblast medium. IFNγ treatment reduced cysteine and increased cystine in fibroblast medium (Figure 6F). This suggests that IFNγ treatment slows down cysteine synthesis by reducing cystine consumption in fibroblasts. Treatment with IFNγ or CD8+ T cell supernatants resulted in decreased 14C-Cystine uptake in fibroblasts at multiple time points (Figures 6G and S6J), but not in tumor cells (Figure S6K). To further evaluate the effects of CD8+ T cells on fibroblast-mediated chemoresistance, we pretreated fibroblasts with IFNγ and found that these fibroblasts restored intracellular cisplatin accumulation (Figure 6H) and cisplatin-induced γH2AX expression (Figure 6I) in tumor cells.
System xc− cystine/glutamate antiporter is a membrane amino acid transporter that mediates the exchange of extracellular cystine and intracellular glutamate, which is a heterodimer composed of the 4F2 heavy chain (SLC3A2) and the light chain xCT (Ishimoto et al., 2011). We found that the expressions of xCT and SLC3A2 protein were both reduced by IFNγ in fibroblasts (Figure 6J). Knockdown of xCT expression in fibroblasts attenuated fibroblast-mediated cisplatin resistance in A2780 (Figure 6K). The effect of IFNγ on regulation of system xc− is cell-type-specific. IFNγ treatment had no effect on xCT and SLC3A2 expressions in tumor cells (Figure S6L). Cystine transporter xCT expression was higher in fibroblasts than tumor cells (Figure S6M). Thus, IFNγ restricts the production of cysteine and promotes the degradation of GSH in fibroblasts through downregulating system xc− and upregulating GGT expression and consequently abrogates the protective effects of fibroblasts on cisplatin-induced apoptosis in tumor cells.
We dissected the mechanism by which IFNγ regulated system xc− and GGT expression in fibroblasts. We monitored the dynamic expression of xCT, SLC3A2, and GGT5 mRNA in fibroblasts upon IFNγ treatment. Twenty-four hours after IFNγ treatment, we detected a moderate increase in the expression of GGT5 mRNA (Figure 6L). However, the expression of xCT, but not SLC3A2 mRNA, was sharply decreased within 4 hr of IFNγ treatment and continued to decrease during the experimental time period (Figure 6L). In contrast, expression of the classical IFNγ responsive genes IRF1 and CXCL10 rapidly increased within 4 hr (Figure S6N). Given the rapid and potent changes in xCT and high expression of xCT in fibroblasts, we particularly focused on xCT regulation by IFNγ.
Rapid downregulation of xCT mRNA by IFNγ suggests a potential direct transcriptional regulation at the levels of pre-mRNA and/or mature mRNA. We observed that the xCT pre-mRNA level was rapidly decreased in fibroblasts within 30 min of IFNγ treatment and continued to decrease following the experimental time period (Figures 6M and S6O). CXCL10 pre-mRNA was rapidly induced by IFNγ (Figure S6P). We then conducted a RNA polymerase II (Pol II) chromatin immunoprecipitation (ChIP) assay. RNA Pol II occupancy at the transcription start site was comparable in fibroblasts treated with or without IFNγ; however, the occupancy in xCT intragenic regions was attenuated after IFNγ treatment (Figures 6N and S6O). In addition, IFNγ had no effect on the Pol II occupancy at both TSS and intragenic regions of xCT in OC8 (Figure S6Q). Thus, IFNγ treatment had no effect on RNA Pol II loading onto xCT promoter, but potentially decreased elongation of RNA Pol II into xCT gene body, and ultimately resulted in xCT transcriptional repression in fibroblasts.
Janus kinase (JAK) and STAT1 mediate cellular response to IFNγ stimulation. We observed that IFNγ induced STAT1 phosphorylation in fibroblasts within 5 min and increased IRF1 protein expression within 2 hr, but decreased xCT protein expression within 8 hr (Figure 6O). JAK inhibitor I reversed IFNγ-mediated downregulation of xCT pre-mRNA and mature mRNA in fibroblasts (Figure S6R). STAT1 homodimers bind to DNA at the gamma interferon activation site (GAS) of the consensus sequence. We analyzed the STAT1 ChIP sequencing (ChIP-seq) database of IFNγ-simulated K562 or HeLa cells in the ENCODE project. We found three potential STAT1 binding sites at the xCT promoter region: one at intron1 (GAS1), one near the TSS (GAS2), and one upstream of promoter (GAS3) (Figure 6P). We conducted STAT1 ChIP assay on fibroblasts and OC8. ChIP data demonstrated that IFNγ exclusively enhanced STAT1 binding to the GAS2 region in fibroblasts (Figure 6Q), while IFNγ treatment resulted in increased STAT1 binding to both GAS1 and GAS2 regions in tumor cells (Figure S6S). To assess whether the GAS2 element in xCT promoter is involved in the regulation of xCT expression, we performed xCT promoter luciferase reporter assays. Similar to the full-length xCT promoter, the luciferase activity of reporter construct carrying GAS2, not GAS1 and GAS3, was repressed by IFNγ treatment (Figure 6R). These results suggest that the GAS2 element near the xCT TSS may selectively be linked to IFNγ-mediated xCT transcriptional repression in fibroblasts. Altogether, our data demonstrates that IFNγ represses xCT transcriptional expression in fibroblasts.
Clinical Impact of Fibroblasts and Stromal CD8+ T Cells on Chemoresistance
Finally, we examined the relationship between stromal fibroblasts, chemoresistance, and potential association with stromal CD8+ T cells in HGSOC patients whose clinical and pathological information was available (Table S3). Based on their responses to platinum-based chemotherapy, we divided patients into chemosensitive and chemoresistant groups. As expected, overall survival was much shorter in chemoresistant patients compared to chemosensitive patients (Figure 7A; Table S3).
Figure 7. Fibroblasts and Stromal CD8+ T Cells Clinically Impact Chemoresistance.

(A) Impact of chemotherapeutic responses on patient outcome. Patients were divided into platinum-resistant and platinum-sensitive groups. The Kaplan-Meier curve of overall survival is shown (p < 0.0001, n = 178).
(B) Impact of stromal fibroblasts on patient outcome. Patients were divided into high and low stromal fibroblast groups. The Kaplan-Meier curve of overall survival is shown (p = 0.0026, n = 176).
(C) Association between the levels of stromal fibroblasts and platinum response. The proportion of stromal fibroblasts is shown in chemoresistant and chemosensitive patients (chi-square = 15.02, p = 0.0001).
(D) Impact of stromal CD8+ T cells on patient outcome. Patients were divided into high and low groups. The Kaplan-Meier curve of overall survival is shown (p < 0.0001, n = 176).
(E) Correlation between the levels of stromal CD8+ T cells and platinum response in patients with high stromal fibroblasts. The proportion of stromal CD8+ T cells is shown in chemoresistant and chemosensitive patients (Fisher’s exact test, p = 0.0298).
See also Figure S7 and Tables S3, S4, S5, and S6.
Next, we quantified α-SMA+ fibroblasts in the tumor stroma with immunohistochemistry in ovarian tumor tissues (Figure S7A). Based on the stromal α-SMA staining score, patients were divided into “α-SMAlow” and “α-SMAhigh” groups. We observed that high stromal α-SMA expression was associated with poor overall survival (Figure 7B; Table S3). We further observed that increased tumor stromal fibroblasts strongly correlated with chemoresistance (Table S4), and the proportion of high levels of stromal fibroblasts were elevated in chemoresistant patients compared to chemosensitive patients (Figure 7C).
To determine whether the association between stromal fibroblasts and patient survival (Figure 7C) is linked to platinum response, we adjusted for platinum response and compared the significance of stromal fibroblasts using Cox’s proportional hazard regression (Table S5). We found that stromal fibroblasts were not associated with patient survival when platinum resistance was included as a covariant in regression analysis (Table S5). Altogether, these data indicate that the levels of stromal fibroblasts in ovarian tumors correlate with platinum response and affect patient survival by modulating chemotherapeutic response.
We quantified CD8+ T cells in the tumor stroma (Figure S7B). Based on the stromal CD8+ T cell levels, patients were divided into “CD8low” and “CD8high” groups (Table S3). We observed that high levels of stromal CD8+ T cells were associated with improved overall survival (Figure 7D). As we observed that CD8+ T cells interacted with stromal fibroblasts and abrogated chemoresistance induced by fibroblasts (Figures 2 and 6) and noted that an increased proportion of fibroblasts correlated with diminished platinum response (Figure 7C) and shorter survival in patients (Figure 7B), we analyzed the clinical significance of these findings. We found that the levels of CD8+ T cells were higher in the tumors of chemosensitive patients as compared to chemoresistant patients (Figure 7E; Table S6). Thus, the patients with high SMA who had high stromal CD8+ T cells are more likely to respond to platinum-based chemotherapy as compared to patients with high SMA and low CD8+ T cells (Table S6). Collectively, our results suggest that the density of stromal fibroblasts and CD8+ T cells affect response to platinum-based chemotherapy and impacts patient survival.
DISCUSSION
Effector T cells, fibroblasts, and tumor cells are major cellular components in the tumor microenvironment. Effector T cell tumor trafficking and function shape tumor immunotherapy including PD-L1 blockade and adoptive T cell therapy (Peng et al., 2015). We have now demonstrated that effector T cells and the interaction between effector T cells and fibroblasts play a role in platinum-based chemoresistance and modulate chemotherapeutic response in patients with ovarian cancer.
Fibroblasts promote tumor progression through multiple mechanisms, including increased tumor cell proliferation, angio-genesis, invasion, sustain of stemness, and inhibition of tumor cell death (Kalluri and Zeisberg, 2006; Loeffler et al., 2006). We have found that primary cancer-associated fibroblasts confer resistance to platinum-based treatment in ovarian cancer cells. It has been reported that fibroblasts mediate carboplatin resistance to pancreatic cancer cell lines (Straussman et al., 2012). We found that fibroblasts diminish cisplatin accumulation in ovarian cancer cells by releasing GSH and cysteine. Biological thiols have been suggested to regulate platinum sensitivity by decreasing DNA platinum accumulation in cultured tumor cells (Chen and Kuo, 2010; Kröning et al., 2000). GSH and cysteine are both involved in maintaining intracellular GSH hemostasis (Lushchak, 2012). Previous studies have suggested that intracellular GSH could react with cisplatin to form a Pt(GS)2 conjugate and this Pt(GS)2 may be eliminated across the membrane through the ATP-dependent glutathione S-conjugate export pump (Ishikawa and Ali-Osman, 1993). Moreover, biological thiols could directly react with platinum atoms to compete with DNA-platinum binding in both intracellular and extracellular reaction systems (Dabrowiak et al., 2002). In our study, we found that fibroblasts released thiols, elevated intracellular GSH levels, and reduced platinum DNA accumulation in tumor cells. Thus, these findings establish a surprising non-genetic paradigm in shaping drug resistance: stromal fibroblasts alter thiol metabolism and control chemotherapeutic response in tumor cells. Clinically, stromal fibroblasts predict platinum-based therapeutic response and patient survival. Moreover, the association between stromal fibroblasts and patient survival depends upon platinum response. Thus, we provide mechanistic and clinical insight into platinum-based resistance.
Intraepithelial (Peng et al., 2015; Sato et al., 2005; Zhao et al., 2016) and stromal CD8+ T cells correlate with improved survival in ovarian cancer. Tumor stromal CD8+ T cells may not directly participate in tumor cell killing activities. We have found that stromal CD8+ T cells modulate stroma function and in turn regulate tumor progression and therapeutic response. For example, CD8+ T cells can abolish fibroblast-mediated platinum resistance via IFNγ. CD8+ T cells alter the metabolism of cystine and GSH in fibroblasts. CD8+ T cell-derived IFNγ activates GGT and promotes extracellular GSH degradation and diminishes fibroblast cysteine generation through downregulation of system xc− cysteine and glutamate antiporter. Furthermore, IFNγ-activated STAT1 binds to specific promoter region of xCT and rapidly inhibits xCT gene transcription through JAK-STAT1 signaling in fibroblasts but not in tumor cells. Hence, our work identifies an unexpected mode of action for effector T cells and IFNγ in the tumor: CD8+ T cells and IFNγ selectively target stromal fibroblasts, shape their thiol metabolism, and impact tumor chemotherapeutic response. Therefore, effective immunotherapy may subvert chemoresistance and revitalize tumor sensitivity to chemotherapy. Platinum-based drugs remain the first line of chemotherapy for ovarian cancer patients. Clinical responses to PD-L1 and PD-1 blockade are observed in a fraction of patients (Zou et al., 2016). Capitalizing upon the interplay between chemotherapy and immunotherapy holds promising clinical potential for ovarian cancer treatment.
EXPERIMENTAL PROCEDURES
Ovarian Cancer Patients, Cancer Tissue Samples, and Cells
Patients diagnosed with HGSOC were recruited for this study. All patients received standardized platinum-based chemotherapy that was administered every 3 weeks for six cycles. Platinum resistance was defined as a progression within 6 months of the platinum therapy. A total of 178 formalin-fixed, paraffin-embedded ovarian tumor tissue blocks and nine fresh ovarian cancer tissues were collected for this study (Curiel et al., 2003, 2004; Peng et al., 2015). A tissue microarray (TMA) was constructed, and immunohistochemical staining was performed as previously described (Cui et al., 2013; Peng et al., 2015). Single cell suspension was prepared from fresh ovarian cancer tissues and fibroblasts were isolated from the adherent cells. CD8+ T cells were isolated from peripheral blood mononuclear cells using the EasySep Human CD8+ T Cell Isolation Kit (Stemcell) and then stimulated with immobilized anti-CD3 and anti-CD28.
Tumor and Fibroblast Co-culture
For mixed co-culture, fibroblasts and A2780-GFP cells were co-cultured and Annexin V+ cells were analyzed with GFP gating. For Transwell co-culture, fibroblasts were seeded in the plates and tumor cells were in the inserts.
Animal Studies
Female nude mice or NSG mice (6- to 8-week-old) were used for in vivo experiments. Cisplatin or recombinant human IFNγ was administered intraperitoneally every 3 days for total three treatments or as indicated.
Detection of GSH-Related Metabolites
Fresh medium was analyzed with LC-triple quadrupole mass spectrometer (Agilent 6490 series) with jet stream electrospray ionization source (Agilent). Chromatography was carried out on a Waters C18 High Strength Silica T3 column (50 mm × 2 mm) on Agilent 1200 Series liquid chromatography.
ChIP
ChIP assay was described previously (Peng et al., 2015) (Table S7).
Detection of Cisplatin
Cell pellets or total DNA were digested and platinum concentration was measured using ICP-HRMS Element 2 (Thermo Scientific).
Radiolabeled Cystine Uptake Assay
Cells were cultured in different conditions in medium containing L-14C-cystine (0.2 μCi/ml) and lysed in NaOH (100 mM). Radioactivity was measured by a Beckman liquid scintillation counter.
Statistical Analysis
Data were shown as mean ± SEM or mean ± SD. Statistical analysis was performed using two sample t test, Mann-Whitney test, or one-way or two-way ANOVA. For all tests, p < 0.05 was considered significant.
Supplementary Material
Highlights.
Fibroblasts diminish platinum content in cancer cells, resulting in drug resistance
GSH and cysteine released by fibroblasts contribute to platinum resistance
T cells alter fibroblast GSH and cystine metabolism and abolish the resistance
Fibroblasts and CD8+ T cells associate with patient chemotherapy response
Acknowledgments
This work is supported (in part) by the Department of Defense (W81XWH-10-1-0865), the NIH grants (CA123088, CA099985, CA156685, CA171306, CA190176, CA193136, and 5P30CA46592), the Ovarian Cancer Research Fund, and Marsha Rivkin Center for Ovarian Cancer Research. This work utilized Metabolomics Core Services supported by grant U24 DK097153 of NIH Common Funds Project to the University of Michigan. We thank Kathleen Cho for her intellectual support. We thank D. Postiff, M. Vinco, R. Craig, and J. Barikdar for their assistance. We thank Chunhai Ruan and Charles F. Burant at the Metabolomics Core, Ted J. Huston at the Department of Earth and Environmental Sciences, Peng Huang at the University of Texas, and Stephen B. Howell at the University of California San Diego for their support. We appreciate the support from Barbara and Don Leclair.
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and seven tables and can be found with this article online at http://dx.doi.org/10.1016/j.cell.2016.04.009.
AUTHOR CONTRIBUTIONS
W.W., I.K., J.R.L., and W.Z. designed the experiments and wrote the paper. S.W., W.W., L.T., and G.H. performed the xenograft tumor experiments. W.W., I.K., and L.T. processed clinical specimens. W.W. and F.L. performed the DNA damage experiments. W.W., I.K., D.P., and J.R.L. performed immunohistochemical and pathological analysis. L.D. performed platinum quantification on ICP-MS. H.L. performed ChIP experiments. J.K., R.T., and J.R.L. provided clinical samples and information. R.K., W.W., I.K., L.Z., T.M., and W.Z. analyzed data. L.V. and W.S. provided technical support.
References
- Binder DC, Fu YX, Weichselbaum RR. Radiotherapy and immune checkpoint blockade: potential interactions and future directions. Trends Mol Med. 2015;21:463–465. doi: 10.1016/j.molmed.2015.05.007. [DOI] [PubMed] [Google Scholar]
- Chen HH, Kuo MT. Role of glutathione in the regulation of Cisplatin resistance in cancer chemotherapy. Met Based Drugs. 2010;2010:430939. doi: 10.1155/2010/430939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui TX, Kryczek I, Zhao L, Zhao E, Kuick R, Roh MH, Vatan L, Szeliga W, Mao Y, Thomas DG, et al. Myeloid-derived suppressor cells enhance stemness of cancer cells by inducing microRNA101 and suppressing the corepressor CtBP2. Immunity. 2013;39:611–621. doi: 10.1016/j.immuni.2013.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curiel TJ, Wei S, Dong H, Alvarez X, Cheng P, Mottram P, Krzysiek R, Knutson KL, Daniel B, Zimmermann MC, et al. Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat Med. 2003;9:562–567. doi: 10.1038/nm863. [DOI] [PubMed] [Google Scholar]
- Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- Dabrowiak JC, Goodisman J, Souid AK. Kinetic study of the reaction of cisplatin with thiols. Drug Metab Dispos. 2002;30:1378–1384. doi: 10.1124/dmd.30.12.1378. [DOI] [PubMed] [Google Scholar]
- Domcke S, Sinha R, Levine DA, Sander C, Schultz N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat Commun. 2013;4:2126. doi: 10.1038/ncomms3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O, Castedo M, Kroemer G. Molecular mechanisms of cisplatin resistance. Oncogene. 2012;31:1869–1883. doi: 10.1038/onc.2011.384. [DOI] [PubMed] [Google Scholar]
- Ishikawa T, Ali-Osman F. Glutathione-associated cis-diammine-dichloroplatinum(II) metabolism and ATP-dependent efflux from leukemia cells. Molecular characterization of glutathione-platinum complex and its biological significance. J Biol Chem. 1993;268:20116–20125. [PubMed] [Google Scholar]
- Ishimoto T, Nagano O, Yae T, Tamada M, Motohara T, Oshima H, Oshima M, Ikeda T, Asaba R, Yagi H, et al. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(−) and thereby promotes tumor growth. Cancer Cell. 2011;19:387–400. doi: 10.1016/j.ccr.2011.01.038. [DOI] [PubMed] [Google Scholar]
- Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6:392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- Kraman M, Bambrough PJ, Arnold JN, Roberts EW, Magiera L, Jones JO, Gopinathan A, Tuveson DA, Fearon DT. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science. 2010;330:827–830. doi: 10.1126/science.1195300. [DOI] [PubMed] [Google Scholar]
- Kröning R, Lichtenstein AK, Nagami GT. Sulfur-containing amino acids decrease cisplatin cytotoxicity and uptake in renal tubule epithelial cell lines. Cancer Chemother Pharmacol. 2000;45:43–49. doi: 10.1007/PL00006741. [DOI] [PubMed] [Google Scholar]
- Lee Y, Auh SL, Wang Y, Burnette B, Wang Y, Meng Y, Beckett M, Sharma R, Chin R, Tu T, et al. Therapeutic effects of ablative radiation on local tumor require CD8+ T cells: changing strategies for cancer treatment. Blood. 2009;114:589–595. doi: 10.1182/blood-2009-02-206870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeffler M, Krüger JA, Niethammer AG, Reisfeld RA. Targeting tumor-associated fibroblasts improves cancer chemotherapy by increasing intratumoral drug uptake. J Clin Invest. 2006;116:1955–1962. doi: 10.1172/JCI26532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lushchak VI. Glutathione homeostasis and functions: potential targets for medical interventions. J Amino Acids. 2012;2012:736837. doi: 10.1155/2012/736837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mårtensson J, Meister A. Mitochondrial damage in muscle occurs after marked depletion of glutathione and is prevented by giving glutathione monoester. Proc Natl Acad Sci USA. 1989;86:471–475. doi: 10.1073/pnas.86.2.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Özdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, Laklai H, Sugimoto H, Kahlert C, Novitskiy SV, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 2014;25:719–734. doi: 10.1016/j.ccr.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W, Sun Y, Zhao E, Vatan L, Szeliga W, et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature. 2015;527:249–253. doi: 10.1038/nature15520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revet I, Feeney L, Bruguera S, Wilson W, Dong TK, Oh DH, Dankort D, Cleaver JE. Functional relevance of the histone gammaH2Ax in the response to DNA damaging agents. Proc Natl Acad Sci USA. 2011;108:8663–8667. doi: 10.1073/pnas.1105866108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F, Jungbluth AA, Frosina D, Gnjatic S, Ambrosone C, et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci USA. 2005;102:18538–18543. doi: 10.1073/pnas.0509182102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soussi T, Hamroun D, Hjortsberg L, Rubio-Nevado JM, Fournier JL, Béroud C. MUT-TP53 2.0: a novel versatile matrix for statistical analysis of TP53 mutations in human cancer. Hum Mutat. 2010;31:1020–1025. doi: 10.1002/humu.21313. [DOI] [PubMed] [Google Scholar]
- Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, Davis A, Mongare MM, Gould J, Frederick DT, et al. Tumour microenvironment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012;487:500–504. doi: 10.1038/nature11183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27:450–461. doi: 10.1016/j.ccell.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Lippard SJ. Cellular processing of platinum anticancer drugs. Nat Rev Drug Discov. 2005;4:307–320. doi: 10.1038/nrd1691. [DOI] [PubMed] [Google Scholar]
- Yeung TL, Leung CS, Wong KK, Samimi G, Thompson MS, Liu J, Zaid TM, Ghosh S, Birrer MJ, Mok SC. TGF-β modulates ovarian cancer invasion by upregulating CAF-derived versican in the tumor microenvironment. Cancer Res. 2013;73:5016–5028. doi: 10.1158/0008-5472.CAN-13-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, Makrigiannakis A, Gray H, Schlienger K, Liebman MN, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med. 2003;348:203–213. doi: 10.1056/NEJMoa020177. [DOI] [PubMed] [Google Scholar]
- Zhang W, Trachootham D, Liu J, Chen G, Pelicano H, Garcia-Prieto C, Lu W, Burger JA, Croce CM, Plunkett W, et al. Stromal control of cystine metabolism promotes cancer cell survival in chronic lymphocytic leukaemia. Nat Cell Biol. 2012;14:276–286. doi: 10.1038/ncb2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao E, Maj T, Kryczek I, Li W, Wu K, Zhao L, Wei S, Crespo J, Wan S, Vatan L, et al. Cancer mediates effector T cell dysfunction by targeting microRNAs and EZH2 via glycolysis restriction. Nat Immunol. 2016;17:95–103. doi: 10.1038/ni.3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zitvogel L, Kepp O, Kroemer G. Decoding cell death signals in inflammation and immunity. Cell. 2010;140:798–804. doi: 10.1016/j.cell.2010.02.015. [DOI] [PubMed] [Google Scholar]
- Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer. 2005;5:263–274. doi: 10.1038/nrc1586. [DOI] [PubMed] [Google Scholar]
- Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016;8:328rv4. doi: 10.1126/scitranslmed.aad7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.