

Abstract
Background & Aims
Alcoholic liver disease (ALD) ranges from fatty liver to inflammation and cirrhosis. miRNA-155 is an important regulator of inflammation. In this study, we describe the in vivo role of miR-155 in ALD.
Methods
Wild type, WT (C57/BL6J) or miR-155 KO and TLR4 KO mice received Lieber-DeCarli diet for 5 weeks. Some mice received corn oil or CCl4 for 2 or 9 weeks.
Results
We found that miR-155 KO mice are protected from alcohol-induced steatosis and inflammation. The reduction in alcohol-induced fat accumulation in miR-155 KO mice was associated with increased PPRE and PPARα (miR-155 target) binding and decreased MCP1 production. Treatment with a miR-155 inhibitor increased PPARγ expression in naive and alcohol treated RAW macrophages. Alcohol increased lipid metabolism genes (FABP4, LXRα, ACC1 and LDLR) in WT mice and this was prevented in KO mice. Alcohol diet induced increase in the number of CD163+CD206+ infiltrating macrophages and neutrophils in WT was prevented in miR-155 KO mice. Kupffer cells isolated from miR-155 KO mice exhibited predominance of M2 phenotype when exposed to M1 polarized signals and this was due to increased C/EBPβ. Profibrotic genes were attenuated in miR-155 KO mice after alcohol diet or CCl4 treatment. Compared to WT attenuation in CCl4 induced hydroxyproline and α SMA was observed in KO mice. Finally, we show TLR4 signaling regulates miR-155 as TLR4 KO mice showed no induction of miR-155 after alcohol diet.
Conclusions
Collectively our results demonstrated the role of miR-155 in alcohol-induced steatohepatitis and fibrosis in vivo.
Keywords: microRNA, inflammation, alcohol, PPARα, PPARγ, fibrosis
INTRODUCTION
Alcoholic liver disease (ALD) is one of the most common causes of liver diseases in the world [1]. The clinical manifestations of ALD range from simple fatty liver to more severe forms of liver injury, including alcoholic hepatitis, cirrhosis, and hepatocellular carcinoma. Alcoholic hepatitis has high mortality and morbidity with limited treatment options. The pathomechanism of ALD involves complex interactions between the effects of alcohol and its toxic metabolites on various cell types in the liver and gut, induction of reactive oxygen species, and up-regulation of the inflammatory cascade [2–5]. Direct toxic effects of alcohol on hepatocytes, increased intestinal permeability and activation of liver macrophages (Kupffer cells; KCs) by gut-derived lipopolysaccharide (LPS) are major factors contributing to ALD [1–3, 5]. LPS is recognized by the Toll-like receptor (TLR) 4 complex expressed on immune cells as well as parenchymal cells and induces pro-inflammatory cytokine activation [5]. Chronic alcohol exposure also sensitizes liver resident macrophages (KCs) to LPS-induced inflammatory cytokine production [6].
Fatty liver (steatosis) is the first stage of response in the liver to binge drinking or chronic ethanol consumption. Accumulation of lipid products such as triglycerides in hepatocytes leads to lipid superoxidation and oxidative stress, resulting in apoptosis, and hepatic inflammation [7]. microRNAs (miRNAs) are shown to modulated by alcohol in the liver and in hepatocytes to exert inflammatory and fat accumulation effects [6, 8–10]. Among other miRNAs, miR-155 is a major regulator of immune responses [11]. Various TLR ligands are shown to induce miR-155 in macrophages [11]. Previously we identified miR-155 as an alcohol-induced regulator of increased KC activation and TNFα production in macrophages [6]. In recent years, the role of miR-155 in other cellular processes like fatty acid metabolism and fibrogenic events has also been emerging [12–14]. In a methionine choline-deficient (MCD) diet induced steatohepatitis, we showed that miR-155 targets genes involved in lipid metabolism (Fab4, cpt1a) and early fibrosis (C/EBPβ, Smad3) [12].
We earlier demonstrated that miR-155 was increased in both KCs and hepatocytes isolated from alcohol-fed mice. Therefore we hypothesized that miR-155 has a role in alcohol-induced liver injury, inflammation and steatosis. To test this hypothesis, we determined the in vivo role of miR-155 in a mouse model of ALD. Our results revealed that alcohol-induced liver injury, steatosis and inflammation were significantly reduced in miR-155 KO mice compared to wild-type (WT) mice. The reduction in fat accumulation in miR-155 KO mice was associated with increased PPRE and PPARα binding after alcohol diet. The alcohol-induced increase in neutrophil leukocyte and CD163+CD206+ (M2) macrophage (profibrotic) infiltration observed in WT mice was prevented in miR-155 KO mice. miR-155 KO mice also showed attenuation in pro-fibrotic genes after alcohol diet as well as in a CCl4-induced liver fibrosis. Together, these results provide in vivo evidence for the role of miR-155 in ALD and fibrosis.
MATERIALS AND METHODS
Animal studies
Animal studies were approved by the University of Massachusetts Medical School (UMMS) Institutional Animal Use and Care Committee (Worcester, MA). miR-155 KO mice were purchased from Jackson Laboratory (Bar Harbor, Maine, USA) and a breeding colony was maintained in the animal facility of UMMS. Wild type (WT) control mice, C56BL/6 were also obtained from Jackson Laboratory. For chronic ethanol feeding, female mice (n=8–10, 8 weeks old) received 5% (v/v) ethanol (36% ethanol-derived calories) containing Lieber-DeCarli diet or control (pair-fed) diet for 5 weeks. For pair-fed diet, alcohol-derived calories were substituted with dextrin-maltose (Bio-Serv, New Jersey, USA) as described previously [6]. For CCl4 treatment, WT or miR-155 KO male mice (n=6, 8–10 weeks old) were treated either with corn oil (vehicle control) or CCl4 (0.6ml/kg i.p.) for 2 or 9 weeks, as described [15]. Mice were sacrificed 72h after last CCl4 injection. At the end of the experiment, blood was collected by cheek bleeding and mice were sacrificed [6]. Liver tissue was collected and snap frozen for proteins and in RNA later (Qiagen, Germany) for RNA extraction. All samples were stored at −80°C.
Liver cell isolation and Flow cytometery
Primary murine hepatocytes, Kupffer cells (KCs) and liver mononuclear cells (MNCs) were isolated from WT chow fed mice by an enzyme-based tissue digestion method as described [16]. Hepatocytes were plated onto 6 well collagen-coated plates and free-floating cells were removed 3h after plating. Both hepatocytes and MNCs were treated with LPS (100ng/ml) for 6h and processed for RNA extraction. Some hepatocytes were treated with MCP1 (100ng/ml) for 36h and nuclear proteins were used for PPRE binding. KCs isolated from WT or KO mice were treated with or without LPS (100ng/ml), IL-4 (20ng/ml) and IFNγ (20ng/ml) for 18h.
For flow cytometry, MNCs were isolated from perfused livers of WT and miR-155 KO mice after 5 weeks of alcohol diet and processed as described [17]. For cell viability, Live/Dead Fixable Blue Dead staining was used (Life Technologies, NY, USA). Briefly, after blocking for non- specific binding to Fc γRs, MNCs (∼106 cells) were stained with CD163, CD11b-FITC, F4/80 PerCP-Cy5.5, and CD206-Alexa 647 (BioLegend, CA, USA) followed by incubation with PE-conjugated secondary antibody for CD163 as described [17]. For neutrophils, the percentage of CD11b+ PE and Ly6Ghigh − FITC was determined. Respective isotype-matched control antibodies were used (BioLegend). After washing with FACS buffer, cells were fixed with 1% paraformaldehyde and acquired on a BD LSR II instrument (BD Biosciences, CA, USA). Data was analyzed with FlowJo software (OR, USA).
Additional methods are in the supporting material.
Statistical analysis
Statistical significance was determined using the non-parametric Mann-Whitney test. Data is represented as mean±standard error and considered statistically significant at p<0.05.
Results
miR-155 deficiency attenuates chronic alcohol-induced steatosis, liver injury, and oxidative stress in the liver
Inflammation, a major component in alcoholic liver disease, contributes to the perpetuation of liver disease and amplifies steatosis [1]. In our earlier study, we showed induction of miR-155 in the liver, isolated Kupffer cells and hepatocytes from alcohol fed mice [6], however, the functional role of miR-155 in ALD in vivo is unknown. In this study, we determined the role of miRNA-155 using a miR-155 KO mouse model system. Alcohol feeding for 5 weeks in WT mice resulted in hepatic steatosis as evidenced by H&E (Fig.1A), Oil-O-Red staining (Fig.1B), histological scores (Fig.1C) and liver triglyceride levels (Fig.1D). Compared to WT mice, a significant decrease was observed in fat accumulation in miR-155 KO mice after alcohol feeding (Fig.1A–D). In miR-155 KO mice we found a significant decrease in plasma ALT, a marker of liver injury, compared to WT mice after alcohol feeding (Fig.1E). Alcohol and its metabolites increase the production of reactive oxygen species and enhance peroxidation of lipids, proteins, and DNA [1]. We found that the increased oxidative stress (measured by thiobarbituric acid reactive substances assay) observed in WT mice was prevented in miR-155 KO mice after alcohol diet (Fig.1F), indicating that miR-155 deficiency attenuates alcohol-induced oxidative damage.
Figure 1. miR-155 deficiency attenuates chronic alcohol-induced steatosis and liver injury.
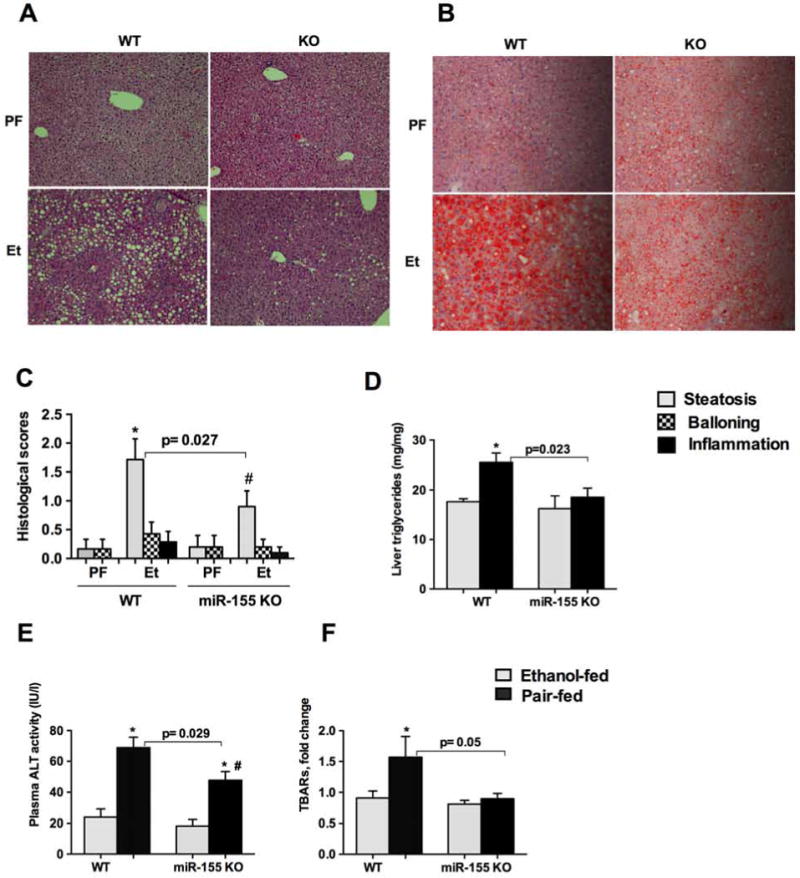
C57BL/6 wild type (WT) or miR-155 KO mice (n=8–10) were fed with Lieber DeCarli diet containing either 5% ethanol or isocaloric control diet for 5 weeks. Formalin fixed liver sections were stained with Hematoxylin and eosin (A) Oil-O-red stain (B). Slides were observed under light microscope (200X) and representative slides are shown. Histological scores were recorded by a pathology expert (C). Triglyceride levels were measured from the livers (D). ALT levels were measured from plasma (E). Oxidative stress was measured from whole liver cell lysates by TBARs assay (F). Data is presented as mean ± SEM. * indicates p<0.05 vs. pair fed (PF) mice and # p<0.05 compared to pair fed KO mice.
Alcohol diet results in increased PPRE and PPARα DNA binding in miR-155 KO mice
Because we found a decrease in liver fat accumulation in miR-155 KO mice after alcohol diet (Fig.1), we sought to evaluate possible mechanisms involved. Lipid and glucose homeostasis are regulated by peroxisome proliferator-activated receptors (PPARs) that belong to the nuclear hormone receptor superfamily [7, 18]. There are three PPAR subtypes: PPARα, PPARγ and PPARδ [7, 18]. PPARα regulates alcoholic steatosis via its effects on genes of lipid metabolism, and a decrease in PPARα has been reported in mouse models of ALD [7, 18, 19]. We extensively performed bioinformatics analysis and found that miR-155 has a seed region at the 3′UTR of PPARγ gene (www.microrna.org) (Supplementary Fig.1A). A very recent study identified PPARα, a direct target of miR-155 [20] and showed that miR-155 regulates inflammation via targeting PPARα after inducing diffuse alveolar hemorrhage. We found a decrease in PPARα mRNA after alcohol diet in WT mice and this decease was prevented in miR-155 KO mice (Fig.2A). We didn’t find a significant change in the PPARγ mRNA after alcohol diet in WT mice, whereas miR-155 KO mice showed increased transcription of PPARγ (Supplementary Fig.1B). Next we examined peroxisome proliferator-activated receptor response element (PPRE) binding using EMSA. We found a decrease in PPRE binding in WT mice after alcohol diet compared to pair-fed mice (Fig.2B–C). In contrast, alcohol feeding resulted in no decrease in PPRE binding in miR-155 KO mice (Fig.2B–C). PPRE binding was similar between genotypes on PF diet. To determine the binding of PPARα in miR-155 KO mice after alcohol diet, we performed a supershift assay. Our results (Fig.2B) indicate a decrease in PPARα binding in alcohol-fed WT mice compared to PF diet fed mice (Fig.2B). Further, we found an increase in PPARα binding in miR-155 KO mice compared to WT mice after alcohol diet (Fig.2B–C).
Figure 2. Alcohol diet results in increased PPRE and PPARα DNA binding in miR-155 KO mice.
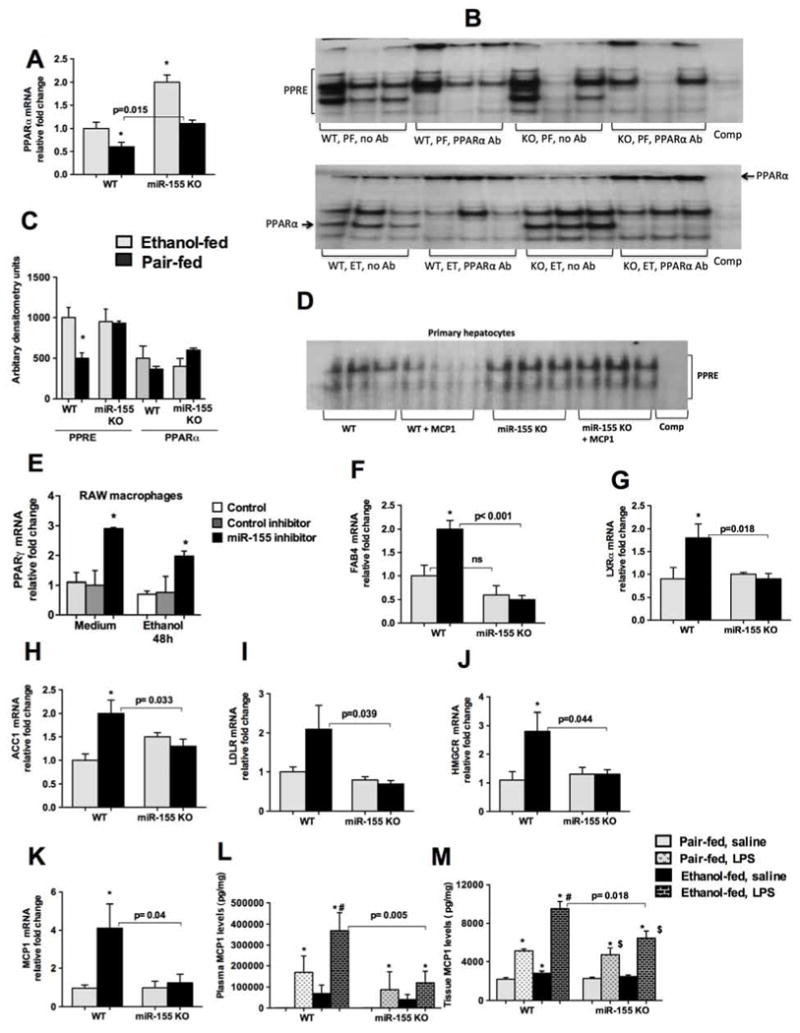
WT or miR-155 KO mice were fed with Lieber DeCarli diet as described in methods. Total RNA was used to determine PPARα levels by qPCR (A). 5ug of nuclear proteins were used to detect PPRE binding by EMSA and PPARα antibody was used for supershift assay and density (B). Primary hepatocytes were isolated from WT and miR-155 KO mice (chow fed) and treated them with or without MCP1 (100ng/ml) for 36h. 10ug of nuclear proteins was used to detect PPRE binding by EMSA (D). RAW 264.7 macrophages were transfected with either miR-155 inhibitor or control inhibitor using lipofectamine siRNA reagent as described in methods. Cells were treated with or without alcohol (50mM), cells were harvested after 48h of transfection and total RNA was used to quantify PPARγ mRNA levels (E). Expression of lipid metabolism and fatty acid uptake gene expression was evaluated by qPCR. FABP4 (F), LXRα (G), ACC1 (H), and LDLR (I), and HMGCR (J).MCP1 mRNA levels were determined by qPCR (K). 18s was used to normalize Cq values. Whole cell liver lysates (L) and plasma (M) were used to detect MCP1 by ELISA. (D). Data is presented as mean ± SEM. * indicates p<0.05 vs. pair-fed mice, # p<0.05 vs PF mice treated with LPS. $ p<0.05 vs PF KO mice.
To further dissect the role of miR-155 in hepatocytes, we isolated hepatocytes from WT and miR-155 KO mice and treated them with or without MCP1 (100ng/ml) to induce steatosis and evaluated for PPRE binding (Fig.2D). MCP1 treatment resulted in a decrease in PPRE binding in hepatocytes from WT mice, however this decrease was prevented in hepatocytes isolated from miR-155 KO mice (Fig.2D), suggesting a role of miR-155 in steatosis via regulation of PPRE signaling. PPARγ is highly expressed in macrophages and low in hepatocytes. Since we found that miR-155 has a seed region at the 3′UTR of PPARγ gene therefore we sought to determine if PPARγ is a direct target of miR-155 using RAW 264.7 macrophages. Inhibition of miR-155 using a specific miR-155 inhibitor resulted in an increase in PPARγ gene both in naïve and alcohol treated cells (Fig.2E), suggesting that miR-155 regulates PPARγ in macrophages.
Since we found that miR-155 regulates PPRE and PPARγ, we checked for PPARγ and PPARα target genes involved in lipid metabolism and fatty acid uptake. Genes such as fatty acid binding protein 4 (FABP4), lipogenesis (LXRα), acetyl-CoA carboxylase 1 [ACC1] and lipid binding transport (LDL receptor) were checked. We found that alcohol induced FABP4 (Fig.2F), LXRα (Fig.2G), ACC1 (Fig.2H), and LDLR (Fig.2I) in the livers of WT mice and not in miR-155 KO mice. Alcohol induced HMGCR mRNA in WT mice and this was prevented in miR-155 KO mice (Fig.2J). No significant changes were observed in genes involved in glucose homeostasis (PCK1 and CEBPα) between genotypes (Supple Fig.1C–D). Other PPARα target genes such as carnitine palmitoyltransferase 1A (CPT1A) and fatty acid synthase (FAS) were also evaluated. We did not any significant differences between genotypes in these genes after alcohol diet (Supple Fig.1E–F).
Recently MCP1 was shown to promote hepatic steatosis [19, 21]. Therefore we assessed MCP1 expression and found that alcohol increased MCP1 mRNA in WT mice but not in miR-155 KO mice (Fig.2K). After an LPS challenge, alcohol fed WT mice showed significant MCP1 production (Fig.2L and M), and a lower MCP1 induction was present in alcohol fed miR-155 KO (Fig.2L and M). Collectively our results suggest that steatogenic, changes observed in miR-155 KO mice after ethanol diet is due to its effect on PPRE signaling.
Chronic alcohol-induced inflammation and macrophage and neutrophil infiltration are attenuated in miR-155 KO mice
miR-155 regulates genes involved in inflammation [11], and alcohol use causes inflammation, therefore, next we evaluated the markers of inflammation. Chronic alcohol feeding increased TNFα (Fig.3A) and IL-1β (Fig.3B) protein production in WT mice compared to mice on PF diet. An LPS challenge further increased these cytokines in alcohol-fed WT mice, however, the LPS induced increase in TNFα and IL-1β proteins was attenuated in alcohol-fed miR-155 KO mice (Fig.3A and B). No significant difference was found in the production of these cytokines between the genotypes that were on PF diet after LPS treatment, supporting a role for miR-155 in the alcohol-induced effects. Macrophage activation markers (CD68 and MIP2) were increased in WT mice, after alcohol feeding, these increases were prevented in miR-155 KO mice (Fig.3C and D).
Figure 3. Chronic alcohol-induced inflammation and macrophage infiltration are attenuated in miR-155 KO mice.
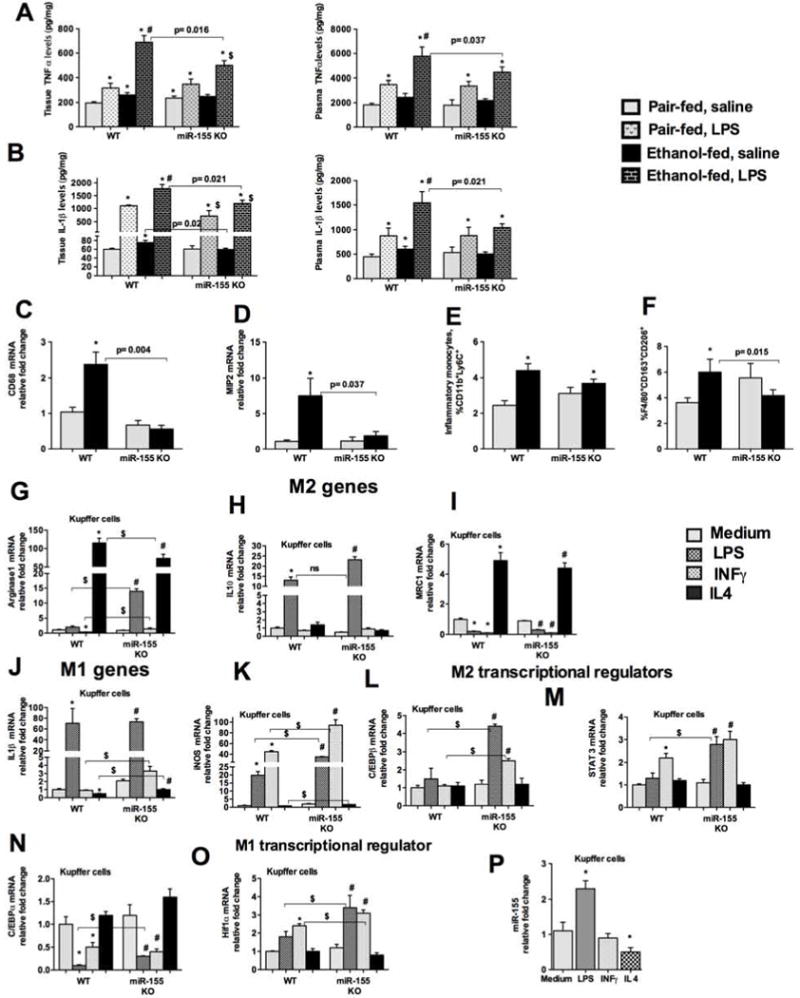
WT or miR-155 KO mice were fed with Lieber DeCarli diet as described in methods. Whole cell liver lysates (A and B, left panel) and plasma (A and B, right panel) were used to detect TNFα (A) and IL-1β (B) by ELISA. RNA was used to determine CD68 (C) and MIP2 (D) mRNA levels by qPCR. MNCs isolated from WT and miR-155 KO mice after alcohol feeding and were stained with combination of antibodies for monocytes, macrophages and neutrophils as described in methods and processed for flow cytometery. % of inflammatory monocytes (E), and M2 macrophages (F) is shown. Kupffer cells were isolated from WT and KO mice and treated with or without LPS (100ng/ml), IL-4 (20ng/ml) and IFNγ (20ng/ml) for 18h. Total RNA was evaluated for M1 and M2 markers and their regulators. Arginase1 (G), IL-10 (H), MRC1 (I), IL-1β (J), iNOS (K), C/EBPβ (L), STAT3 (M), C/EBPα (N), HiF1α (O), and miR-155 levels (P). 18s was used to normalize Cq values. Data is presented as mean ± SEM. * indicates p<0.05 vs. pair fed mice, # p<0.05 vs PF mice treated with LPS. $ p<0.05 vs PF mice KO mice. ns: non significant.
Recently we showed that alcohol induces both M1 (classical activation) and M2 (alternatively activated profibrotic) phenotype of macrophages in mice [17]. In this context, miR-155 was shown to promote the M1 phenotype of bone marrow derived macrophages [22]. To determine if deficiency of miR-155 KO have any effect on macrophage phenotype after alcohol diet, we performed flow cytometry analysis of the liver immune cells. We found that chronic alcohol diet increased the number of inflammatory monocytes Cd11b+ Ly6C+ in WT mice, while in miR-155 KO mice alcohol moderately decreased the number of inflammatory monocytes (Fig.3E). Interestingly, alcohol diet also increased the percentage of F4/80+ CD163+CD206+ M2 macrophages in WT mice, but no increase was found in miR-155 KO mice (Fig.3F). miR-155 KO mice on PF diet showed increased % of M2 macrophages. To evaluate how miR-155 is involved in KCs polarization, we isolated KCs from WT and miR-155 KO mice (on chow fed diet) and polarized to M1 and M2 phenotype. As M2 markers, we evaluated for Arginase1, IL-10 and MRC1 levels. LPS or INFγ treatment decreased and IL-4 treatment increased Arginase1 levels in KCs isolated from WT mice (Fig.3G). Compared to WT, KCs isolated from miR-155 KO mice showed a higher increase in Arginase1 levels after LPS or INFγ treatment (Fig.3G). KCs isolated from miR-155 KO mice showed a significant increase in IL-10 mRNA compared to KCs isolated from WT mice after LPS treatment (Fig.3H). No significant differences in MRC1 levels were found between genotypes (Fig.3I). As M1 markers we tested for IL-1β and iNOS genes. No significant differences were found in IL-1β mRNA between genotypes after LPS treatment (Fig.3J). Surprisingly, KCs isolated from miR-155 KO mice showed a significant increase in iNOS mRNA compared to WT KCs after LPS or INFγ or IL-4 treatment (Fig.3K). To determine the possible explanation for this mixed phenotype/polarization, we checked for transcriptional regulators of macrophage polarization.
miR-155 is involved in modulating macrophage polarization via targeting C/EBPβ. We found induction of C/EBPβ only in KCs isolated from miR-155 KO and not from WT mice after LPS or INFγ treatment (Fig.3L). A higher increase in STAT3 (M2 regulator) was also observed in KCs isolated from miR-155 KO compared to WT mice after LPS or INFγ treatment (Fig.3M). This was specific to C/EBPβ as C/EBPα levels were not so different between genotypes (Fig.3N). These results suggest that miR-155 deficient KCs exhibited predominance of M2 phenotype when treated with M1 polarizing stimuli. Next, we attempted to find the explanation for increased iNOS expression in KCs isolated from miR-155 KO after LPS or INFγ treatment. For this, we checked for Hif1α, a M1 macrophage transcriptional regulator, and found enhanced Hif1α in KCs isolated from miR-155 KO compared to WT mice after LPS or INFγ treatment (Fig.3O). Collectively, our results suggest that miR-155 plays a role in M1 macrophage polarization. Further, we found that LPS induced whereas IL-4 decreased miR-155 levels, however we did not find any increase in miR-155 after INFγ treatment (Fig.3P). It is possible that kinetics of miR-155 induction with INFγ is different than LPS.
In addition to Kupffer cell/macrophage activation, alcoholic hepatitis is also characterized by neutrophil infiltration to the liver [23, 24]. Neutrophil infiltration has been shown to correlate with the severity of acute alcoholic hepatitis [24, 25]. Alcohol diet resulted in an infiltration of neutrophils (CD11b+Ly6Ghi) in the livers of WT mice compared to PF mice (Fig.4A). However, neutrophil infiltration was prevented in miR-155 KO mice after alcohol diet (Fig. 4A). M2 macrophages are known to play roles in tissue remodeling, and fibrogenesis [22, 26, 27]. Chronic alcohol feeding induces pro-fibrotic gene expression [4]. Because our results indicated no increase in M2 macrophages in miR-155 KO mice after alcohol diet, we next evaluated the expression of pro-fibrotic genes. We found that chronic alcohol feeding resulted in an increase in mRNA levels of TGFβ, (Fig.4B), type1 procollagenα (Fig.4C, and vimentin (Fig.4D) in WT mice whereas these increases were either prevented (TGFβ and vimentin) or reduced (procollagen 1α) in miR-155 KO mice (Fig.4 B–D). These results suggested that miR-155 deficiency could be protective from development of liver fibrosis.
Figure 4. Reduction in CCl4-induced liver fibrosis in miR-155 KO mice.
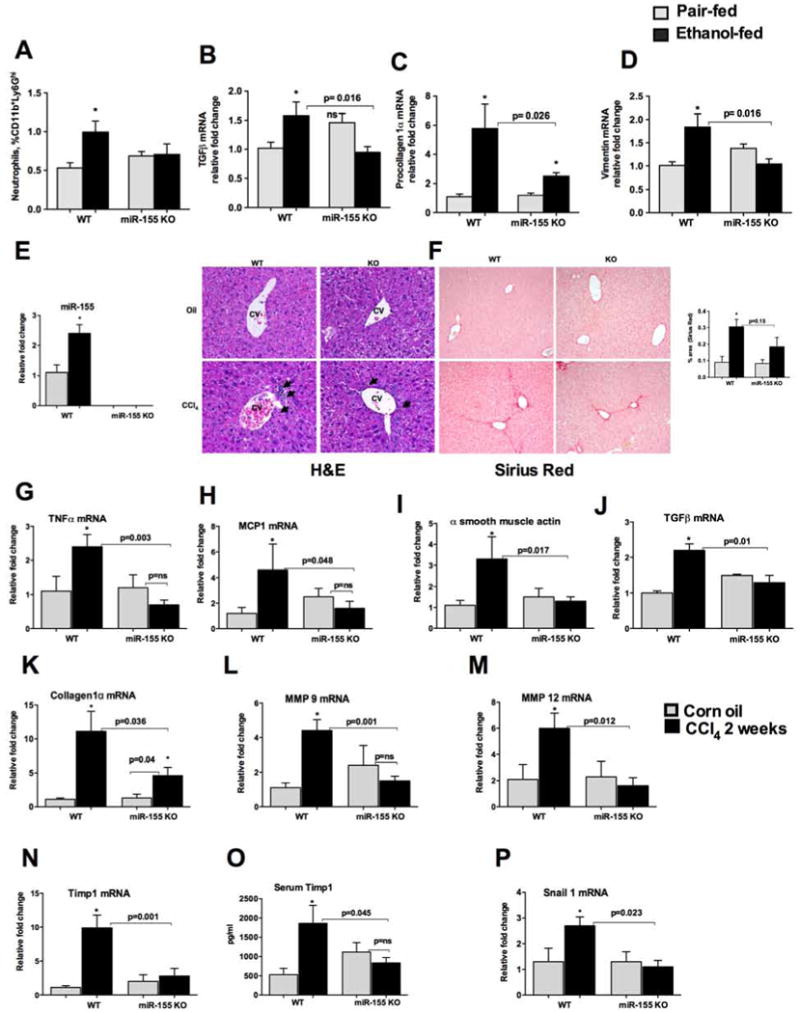
WT or miR-155 KO mice were fed with Lieber DeCarli diet. MNCs were stained for neutrophils (A). Liver RNA was used to determine TGFβ (B) procollagen 1α (C) and vimentin (D) mRNA levels by qPCR. 18s was used to normalize Cq values. Data is presented as mean ± SEM. * indicates p<0.05 vs. pair fed mice, ns: non significant. WT or miR-155 KO mice (n=6) were treated either with corn oil or CCl4 for 2 weeks as described in methods. Total RNA was used to determine miR-155 levels by qPCR (E). Formalin fixed liver sections were stained with Hematoxylin and eosin (F, left panel) and Sirius red stain (F, right panel). Slides were observed under light microscope (100X) and representative slides are shown. RNA was used to determine TNFα (G), MCP1 (H), α smooth muscle actin (I), TGF β (J), collagen 1α (K), MMP9 (L), MMP12 (M), Timp1 (N), Snail1 (P) mRNA levels by qPCR. Timp1 levels were measured from plasma by ELISA (O). 18s or SnoRNA202 was used to normalize Cq values for mRNA, and mouse miR-155 respectively. Data is presented as mean ± SEM. * indicates p<0.05 vs. corn oil treated mice.
Reduction in CCl4-induced liver fibrosis in miR-155 KO mice
Because our results indicated a decrease in pro-fibrotic genes and M2 macrophages in miR-155 KO mice after alcohol diet, we further evaluated the role of miR-155 in liver fibrosis. The Lieber-DeCarli alcohol mouse model results in a modest increase in early markers of fibrosis [4] while the CCl4-induced liver fibrosis mouse model induces significant fibrosis [28]. We found that CCl4 treatment increases hepatic miR-155 expression in WT mice compared to corn oil (Fig.4E). miR-155 levels were not detected in miR-155 KO mice (Fig.4E). CCl4 treatment for 2 weeks resulted in lymphocyte infiltration and initiation of collagen deposition in the livers of WT mice and an attenuation was observed in miR-155 KO mice (H&E and Sirius red staining (morphometery density units) (Fig.4F). Since CCl4 induces inflammation, we next evaluated the markers of inflammation and found an increase in the mRNA levels of pro-inflammatory cytokines (TNFα and MCP1) in WT mice and not in miR-155 KO mice after CCl4 administration (Fig.4G–H). CCl4 treatment caused the induction of pro-fibrotic genes including α smooth muscle actin (Fig.4I), and TGFβ (Fig.4J) and type 1 collagen α (Fig.4K) in WT mice, where as miR-155 KO mice either showed no increases or reduction (Fig.4I–K). Matrix metalloproteinases (MMPs) and metallopeptidase inhibitor (TIMP1) play a crucial role in matrix remodeling [29]. CCl4 treatment resulted in the induction of the mRNA levels of MMP9 and MMP12 (Fig.4L–M) and TIMP1 (Fig.4N) only in WT mice and not in miR-155 KO mice (Fig.4L–N). Consistent with the mRNA data (Fig.4N), we found that the serum levels of TIMP1 were increased in WT mice after CCl4 treatment and this increase was prevented in miR-155 KO mice (Fig.4O). Epithelial-mesenchymal cell transition (EMT) is an important event in the initiation of organ fibrosis. EMT-promoting transcription factor Snail1 has been shown to play a role in liver fibrosis and hepatocyte specific deletion of Snail1 was shown to protect the mice from liver fibrosis [30]. Consistent with previous report [30] we found increase in Snail1 in WT mice after CCl4 treatment, however this increase in Snail1 was prevented in miR-155 KO mice (Fig.4P).
To show that miR-155 is indeed involved in the development of advanced liver fibrosis we carried out 9 weeks of CCl4 treatment. Histological scoring of Sirius red staining performed by pathologist expert revealed less bridging fibrosis and perisinosoidal fibrosis in miR-155 KO compared to WT mice (Fig.5A). In concordance with this, less production of hydroxyproline was found in miR-155 KO compared to WT mice (Fig.5B). α smooth muscle actin protein levels were also significantly decreased in miR-155 KO mice compared to WT mice after 9 weeks of CCl4 treatment (Fig.5C). Collectively, these results suggest a role of miR-155 in liver fibrosis. Finally, to validate observations from the mouse models, we tested the expression of miR-155 levels in the cirrhotic livers of alcoholic patients. We found a significant induction of miR-155 levels in the alcoholic cirrhotic livers compared to livers without cirrhosis (Fig.5D).
Figure 5. miR-155 promotes liver fibrosis.
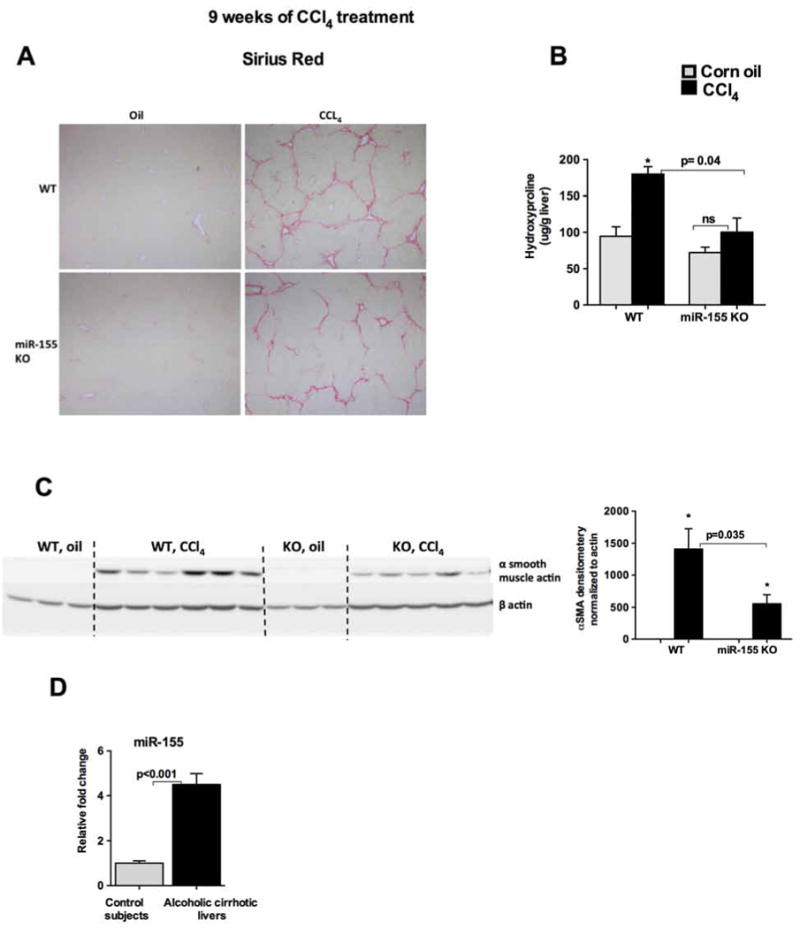
WT or miR-155 KO mice (n=9) were treated either with corn oil or CCl4 for 9 weeks. Formalin fixed liver sections were stained with Sirius red stain (A). Slides were observed under light microscope (100X) and representative slides are shown. The amount of hydroxyproline was measured from the liver (B). Western blot analysis for α SMA and density units (C), miR-155 levels were determined from alcoholic cirrhotic livers by qPCR (D). RNU48 was used to normalize Cq values. Data is presented as mean ± SEM. * indicates p<0.05 vs. corn oil treated mice.
Alcohol induces miR-155 in the liver at the transcriptional level via the TLR4 pathway
Both chronic alcohol and CCl4 treatments were shown to increase serum LPS levels in mice [31]. Recently, we showed that miR-155 KO mice were protected from alcohol-induced intestinal permeability and inflammation [2]. We found an increase in circulating endotoxin levels in WT mice and not in miR-155 KO mice after alcohol diet (Fig.6A). Upon activation by LPS, KCs release various pro-inflammatory and pro-fibrotic cytokines [6]. The effects of circulating LPS on the kinetics of hepatic miR-155 levels are not known; therefore we next checked the levels of liver miR-155 after a single treatment of LPS in WT mice. Our results indicate that miR-155 levels start to increase 2h after LPS injection and steadily increase over time (Fig.6B). No significant differences were found in miR-155 levels between 6h and 18h after LPS injection (Fig.6B) suggesting that miR-155 levels reached a plateau after 6h of LPS injection. To investigate the cellular source of miR-155 increase after LPS treatment, liver mononuclear cells (MNCs) and hepatocytes isolated from WT mice were treated with or without LPS (100ng/ml). Our results indicate that LPS induces miR-155 only in MNCs but not in hepatocytes (Fig.6C). To corroborate these finding in alcoholic steatohepatitis, we examined the expression of miR-155 in WT mice that were challenged with LPS for 3h after alcohol feeding. We found no significant differences in the induction of miR-155 levels between alcohol and control diet fed mice after LPS challenge (Fig.6D), suggesting that alcohol and LPS may increase miR-155 via the same mechanism.
Figure 6. Alcohol induces miR-155 in the liver at the transcriptional level via the TLR4 pathway.
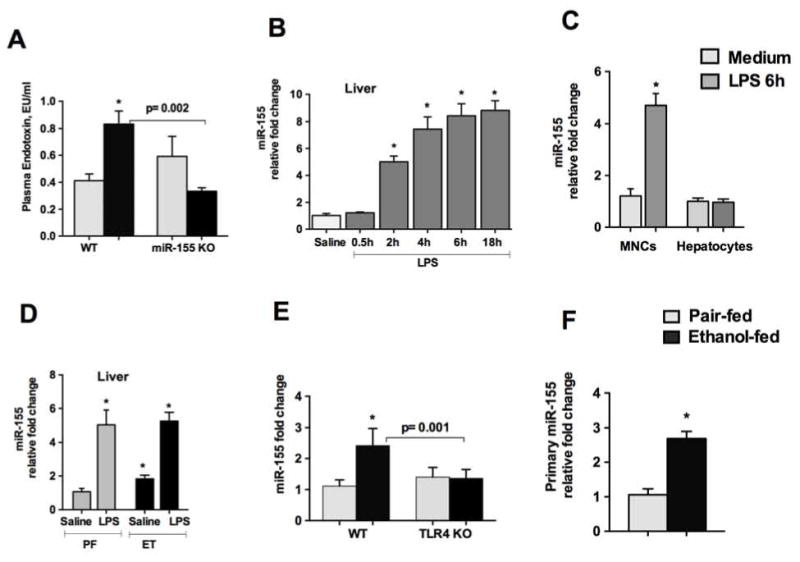
WT or miR-155 KO mice were fed with Lieber DeCarli diet. Endotoxin levels were measured from plasma (A). WT mice were either administered with saline or LPS (2.5mg/kg, ip) for indicated times and miR-155 levels were measured from livers (B). MNCs or hepatocytes were isolated from perfused WT livers as described in methods. Cells were treated with LPS (100ng/ml) for 6h and miR-155 levels were detected (C). WT or miR-155 KO pair- or alcohol- fed mice were injected with LPS (0.5mg/kg, ip) for 3h and livers were isolated and miR-155 levels were detected (D). C57BL/6 WT or TLR4 KO mice (n=6–8) were fed with Lieber DeCarli diet. (E). Primary miR-155 levels were detected using TaqMan gene expression assay. GAPDH was used to normalize Cq values (F). Data is presented as mean ± SEM. * indicates p<0.05 vs. pair fed mice (A, D and F). * indicates p<0.05 vs. saline treated mice (B) or cells in medium only (C).
LPS is recognized by the TLR4 receptor resulting in downstream activation of NF-kB and pro-inflammatory cytokines. TLR4 signaling plays a crucial role in alcohol-induced effects and mice deficient in TLR4 have attenuation in alcohol-mediated cellular response [32]. Our results indicate a role of TLR4 signaling as no induction in mature miR-155 levels were observed in TLR4 KO mice after alcohol feeding compared to WT mice (Fig.6E). To determine if alcohol has any effect on the transcription of miR-155, we checked primary miR-155 (pri-miR-155) levels. Our results demonstrate a significant increase in pri-miR-155 levels in alcohol-fed WT mice compared to controls (Fig.6F). These results suggest that in vivo alcohol increase miR-155 transcription.
Discussion
microRNAs play a key role in various liver diseases including alcoholic liver disease (ALD) and fibrosis. miR-155 is a major regulator of inflammation and immunity. In this study we defined the functional role of miR-155 in ALD and liver fibrosis using miR-155 KO mice. We found that miR-155 KO mice were protected from alcohol-induced liver injury, oxidative stress, steatosis and inflammation. Our data revealed that miR-155 KO mice exhibited reduced liver fibrosis markers in both alcohol and CCl4-induced fibrosis. miR-155 is a major mediator of inflammation, however in recent years the role of miR-155 in steatosis and fibrosis has been emerging [12–14] and our results support the role of miR-155 in these processes. We found that the decrease in PPRE and PPARα binding observed in WT mice was prevented in miR-155 KO mice after alcohol diet. Our results revealed that miR-155 regulates PPRE signaling in both macrophages and hepatocytes. In primary isolated hepatocytes we found that MCP1 treatment decreased PPRE in WT mice where as this decrease was prevented in miR-155 KO mice suggesting a role of miR-155 in regulation of steatosis via PPRE signaling. In consistent with our results MCP1 treatment was shown to decrease PPRE binding in Huh7 cells [19, 21]. We further identified and validated PPARγ a direct target miR-155 in macrophages as inhibition of miR-155 using a specific miR-155 inhibitor resulted in an increase in PPARγ gene in both naïve and alcohol treated cells. Our in vitro findings from hepatocytes and macrophages are in concordance with in vivo findings as we found that alcohol-induced decrease in PPRE binding in WT mice was prevented in miR-155 KO mice.
Ethanol-induced hepatic steatosis is multifunctional and alcohol affects several pathways, including fatty acid uptake, oxidation, and export [7]. Alcohol is known to reduce PPARα [7], thereby inhibiting fatty acid oxidation in hepatocytes. PPARα and PPARγ not only play a pivotal role in hepatic lipid metabolism but also in oxidative stress, inflammatory response, and fibrogenesis [7]. Collectively, our results indicate that steatogenic, inflammatory and fibrogenic changes observed in miR-155 KO mice after ethanol diet are likely due to the effect of miR-155 on PPAR signaling.
miR-155 plays a key role in macrophage polarization and promotes the M1 phenotype of macrophage [22, 33]. While no significant differences were found in the quantity of immune cell infiltration between WT and miR-155 KO mice before and after alcohol feeding, we found that miR-155 deficiency alter the phenotype of infiltrating immune cells. Macrophage and KC activation are major elements in ALD [1]. Studies suggest that miR-155 favors M1 macrophages via its effect on C/EBP-β, SHIP1, and IL13Rα1[11, 22, 33, 34]. Interestingly in our ALD mouse model, the absence of miR-155 prevented an increase in the number of M2 macrophages after alcohol diet, suggesting that miR-155 has a tissue/cell/microenvironment specific role. Consistent with the role of miR-155 in promoting M1 macrophage phenotype we found a higher baseline of M2 macrophages in miR-155 KO mice on control diet compared to WT. Results from our in vitro studies revealed that KCs isolated from miR-155 KO mice (on chow diet) exhibited predominance of M2 phenotype but also had increased M1 phenotype after M1 polarizing signals. This was somewhat unexpected and could be explained by the fact that we found an increase of both C/EBPβ (M2 transcriptional regulator) and HIf1α (M1 transcriptional regulator) after polarization with LPS or INFγ in isolated KCs from miR-155 KO mice. A recent study revealed that iNOS deficient mice had enhanced M1 macrophage differentiation after polarization but without having major effects on M2 macrophages [35]. Our results support the notion that macrophages likely exist as a mixed population of M1/M2 in vivo or as hybrid phenotypes instead of an entirely polarized M1 or M2 phenotype [35].
It is well known that alcoholic steatohepatitis, if sustained, can lead to fibrosis, cirrhosis and hepatocellular carcinoma [1]. CCl4-induced liver fibrosis is a robust and widely used model of liver fibrosis [28]. Sustained/chronic inflammation leads to fibrosis, therefore, we hypothesized that deficiency of miR-155 will also attenuate CCl4-induced inflammatory response and fibrosis. Consistent with findings from the alcohol diet, we found reduced fibrosis in miR-155 KO mice after CCl4 treatment. Emerging studies support a role of miR-155 in liver fibrosis. Recently, we reported that miR-155 deficiency attenuated MCD diet-induced early liver fibrosis [12]. EMT promoting transcription factor, Snail1 (a putative miR-155 target), plays a role in liver fibrosis and we found that CCl4-induced increase in Snail1 was prevented in miR-155 KO mice. We found that miR-155 KO mice not only were protected from early fibrosis but also showed attenuation from advanced fibrosis after CCl4 treatment (9 weeks), revealing a role of miR-155 in promoting liver fibrosis. PPARγ also acts as an anti-fibrotic gene and we identified PPARγ as a direct target of miR-155. Also miR-155 targets several genes involved in fibrogenesis (such as SMAD2/5, Snail1, STAT3) it is highly likely that the fibrosis phenotype observed in our study is the cumulative effect of miR-155 on various target genes. Our finding of miR-155 being induced in a mouse model of liver fibrosis is relevant to human disease as we found increased miR-155 levels in the cirrhotic livers of alcoholic patients. Given the critical importance of PPAR signaling in normal lipid and glucose metabolism as well as in the regulation of inflammatory responses, it is highly likely that changes in steatosis, inflammation and fibrosis seen in miR-155 KO mice are due to miR-155’s effect on PPAR signaling.
We didn’t find differences in alcohol metabolism between genotypes as serum ethanol levels and Cyp2E1 protein levels were comparable between WT and miR-155 KO mice after alcohol diet (Supple Fig.1G–H). Alcohol intake also increases intestinal permeability, which causes an increase in LPS in the circulation [2]. We found higher endotoxin levels in miR-155 KO mice on pair fed diet after 5 weeks, however this effect was not observed when endotoxin levels were checked after 4 weeks of alcohol diet (Supple Fig.1I). Circulating LPS is recognized by hepatic macrophages via TLR4 receptor resulting is [6]. The role of the LPS/TLR4 axis is very well known in ALD and TLR4 KO mice were protected from ALD [32]. Our results support a role of TLR4 signaling in ALD as we found that alcohol induces miR-155 via the TLR4 pathway. In summary, our data suggest that miR-155 most likely modulates the pro-inflammatory and pro-fibrotic programs via targeting various signaling cascades that impact not only immune cells but also non-immune cells including hepatocytes. Additionally, our results indicate that the absence of miR-155 not only halts alcohol-induced steatohepatitis but also fibrosis in mouse models.
Supplementary Material
Acknowledgments
The authors greatly appreciate Dr. Jin-Kyu Park help in evaluation and scoring of the histology slides. Human liver samples were provided by the NIH-Funded Liver Tissue Procurement and Cell Distribution System (N01-DK-70004/HHSN26700700004C).
Financial support: This work was supported by grant from National Institute on Alcohol Abuse and Alcoholism-AA020744 to G.S.
List of abbreviations
- ALD
alcoholic liver disease
- KC
Kupffer cell
- LPS
lipopolysaccharide
- TLR
toll-like receptor
- miRNA
microRNA
- WT
wild type
- KO
knock out
- PPRE
peroxisome proliferator-activated receptor (PPAR) response element
- M2
macrophage2
- TNF α
tumor necrosis factor α
- MCP1
monocyte chemo attractant protein 1
- IL-1β
Interleukin-1
- TGF β
Transforming growth factor β
- LMNCs
liver mononuclear cells
- EtOH
ethanol
- CCl4
carbon tetrachloride
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest: None
Author’s contributions: Idea conceived and design the experiments: SB, GS. Performed and analyzed the experiments: SB, TC, BS, KK, DC, JZ. Interpreted the data and wrote the paper: SB, and GS. All authors approved the final manuscript.
References
- 1.Wang HJ, Gao B, Zakhari S, Nagy LE. Inflammation in alcoholic liver disease. Annu Rev Nutr. 2012:343–368. doi: 10.1146/annurev-nutr-072610-145138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lippai D, Bala S, Catalano D, Kodys K, Szabo G. Micro-RNA-155 Deficiency Prevents Alcohol-Induced Serum Endotoxin Increase and Small Bowel Inflammation in Mice. Alcohol Clin Exp Res. 2014;8:2217–2224. doi: 10.1111/acer.12483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Petrasek J, Iracheta-Vellve A, Csak T, Satishchandran A, Kodys K, Kurt-Jones EA, et al. STING-IRF3 pathway links endoplasmic reticulum stress with hepatocyte apoptosis in early alcoholic liver disease. Proc Natl Acad Sci U S A. 2013;41:16544–16549. doi: 10.1073/pnas.1308331110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Petrasek J, Bala S, Csak T, Lippai D, Kodys K, Menashy V, et al. IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J Clin Invest. 2012;10:3476–3489. doi: 10.1172/JCI60777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Szabo G, Bala S. Alcoholic liver disease and the gut-liver axis. World J Gastroenterol. 2010;11:1321–1329. doi: 10.3748/wjg.v16.i11.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bala S, Marcos M, Kodys K, Csak T, Catalano D, Mandrekar P, et al. Up-regulation of microRNA-155 in macrophages contributes to increased tumor necrosis factor {alpha} (TNF{alpha}) production via increased mRNA half-life in alcoholic liver disease. J Biol Chem. 2011;2:1436–1444. doi: 10.1074/jbc.M110.145870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nan YM, Wang RQ, Fu N. Peroxisome proliferator-activated receptor alpha, a potential therapeutic target for alcoholic liver disease. World J Gastroenterol. 2014;25:8055–8060. doi: 10.3748/wjg.v20.i25.8055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dong X, Liu H, Chen F, Li D, Zhao Y. MiR-214 promotes the alcohol-induced oxidative stress via down-regulation of glutathione reductase and cytochrome P450 oxidoreductase in liver cells. Alcohol Clin Exp Res. 2014;1:68–77. doi: 10.1111/acer.12209. [DOI] [PubMed] [Google Scholar]
- 9.Meng F, Glaser SS, Francis H, Yang F, Han Y, Stokes A, et al. Epigenetic Regulation of miR-34a Expression in Alcoholic Liver Injury. Am J Pathol. 2012 doi: 10.1016/j.ajpath.2012.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yin H, Hu M, Zhang R, Shen Z, Flatow L, You M. MicroRNA-217 promotes ethanol-induced fat accumulation in hepatocytes by down-regulating SIRT1. J Biol Chem. 2012;13:9817–9826. doi: 10.1074/jbc.M111.333534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O’Connell RM, Chaudhuri AA, Rao DS, Baltimore D. Inositol phosphatase SHIP1 is a primary target of miR-155. Proc Natl Acad Sci U S A. 2009;17:7113–7118. doi: 10.1073/pnas.0902636106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Csak T, Bala S, Lippai D, Kodys K, Catalano D, Iracheta-Vellve A, et al. MicroRNA-155 Deficiency Attenuates Liver Steatosis and Fibrosis without Reducing Inflammation in a Mouse Model of Steatohepatitis. PLoS One. 2015;6:e0129251. doi: 10.1371/journal.pone.0129251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miller AM, Gilchrist DS, Nijjar J, Araldi E, Ramirez CM, Lavery CA, et al. MiR-155 has a protective role in the development of non-alcoholic hepatosteatosis in mice. PLoS One. 2013;8:e72324. doi: 10.1371/journal.pone.0072324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dai W, Zhao J, Tang N, Zeng X, Wu K, Ye C, et al. MicroRNA-155 attenuates activation of hepatic stellate cell by simultaneously preventing EMT process and ERK1 signalling pathway. Liver Int. 2015;4:1234–1243. doi: 10.1111/liv.12660. [DOI] [PubMed] [Google Scholar]
- 15.Bala S, Petrasek J, Mundkur S, Catalano D, Levin I, Ward J, et al. Circulating microRNAs in exosomes indicate hepatocyte injury and inflammation in alcoholic, drug-induced and inflammatory liver diseases. Hepatology. 2012 doi: 10.1002/hep.25873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bala S, Csak T, Momen-Heravi F, Lippai D, Kodys K, Catalano D, et al. Biodistribution and function of extracellular miRNA-155 in mice. Sci Rep. 2015:10721. doi: 10.1038/srep10721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saha B, Bala S, Hosseini N, Kodys K, Szabo G. Kruppel-like factor 4 is a transcriptional regulator of M1/M2 macrophage polarization in alcoholic liver disease. J Leukoc Biol. 2015 doi: 10.1189/jlb.4A1014-485R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pawlak M, Lefebvre P, Staels B. Molecular mechanism of PPARalpha action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J Hepatol. 2015;3:720–733. doi: 10.1016/j.jhep.2014.10.039. [DOI] [PubMed] [Google Scholar]
- 19.Mandrekar P, Ambade A, Lim A, Szabo G, Catalano D. An essential role for MCP-1 in alcoholic liver injury: Regulation of pro-inflammatory cytokines and hepatic steatosis. Hepatology. 2011 doi: 10.1002/hep.24599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou S, Wang Y, Meng Y, Xiao C, Liu Z, Brohawn P, et al. In Vivo Therapeutic Success of MicroRNA-155 (miR-155) Antagomir in a Mouse Model of Lupus Alveolar Hemorrhage. Arthritis Rheumatol. 2015 doi: 10.1002/art.39485. [DOI] [PubMed] [Google Scholar]
- 21.Nath B, Levin I, Csak T, Petrasek J, Mueller C, Kodys K, et al. Hepatocyte-specific hypoxia-inducible factor-1alpha is a determinant of lipid accumulation and liver injury in alcohol-induced steatosis in mice. Hepatology. 2011;5:1526–1537. doi: 10.1002/hep.24256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cai X, Yin Y, Li N, Zhu D, Zhang J, Zhang CY, et al. Re-polarization of tumor-associated macrophages to pro-inflammatory M1 macrophages by microRNA-155. J Mol Cell Biol. 2012;5:341–343. doi: 10.1093/jmcb/mjs044. [DOI] [PubMed] [Google Scholar]
- 23.Lemmers A, Moreno C, Gustot T, Marechal R, Degre D, Demetter P, et al. The interleukin-17 pathway is involved in human alcoholic liver disease. Hepatology. 2009;2:646–657. doi: 10.1002/hep.22680. [DOI] [PubMed] [Google Scholar]
- 24.Ramaiah SK, Jaeschke H. Role of neutrophils in the pathogenesis of acute inflammatory liver injury. Toxicol Pathol. 2007;6:757–766. doi: 10.1080/01926230701584163. [DOI] [PubMed] [Google Scholar]
- 25.Bertola A, Park O, Gao B. Chronic plus binge ethanol feeding synergistically induces neutrophil infiltration and liver injury in mice: a critical role for E-selectin. Hepatology. 2013;5:1814–1823. doi: 10.1002/hep.26419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gratchev A, Guillot P, Hakiy N, Politz O, Orfanos CE, Schledzewski K, et al. Alternatively activated macrophages differentially express fibronectin and its splice variants and the extracellular matrix protein betaIG-H3. Scand J Immunol. 2001;4:386–392. doi: 10.1046/j.1365-3083.2001.00885.x. [DOI] [PubMed] [Google Scholar]
- 27.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;12:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chiang DJ, Roychowdhury S, Bush K, McMullen MR, Pisano S, Niese K, et al. Adenosine 2A receptor antagonist prevented and reversed liver fibrosis in a mouse model of ethanol-exacerbated liver fibrosis. PLoS One. 2013;7:e69114. doi: 10.1371/journal.pone.0069114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hemmann S, Graf J, Roderfeld M, Roeb E. Expression of MMPs and TIMPs in liver fibrosis – a systematic review with special emphasis on anti-fibrotic strategies. J Hepatol. 2007;5:955–975. doi: 10.1016/j.jhep.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 30.Rowe RG, Lin Y, Shimizu-Hirota R, Hanada S, Neilson EG, Greenson JK, et al. Hepatocyte-derived Snail1 propagates liver fibrosis progression. Mol Cell Biol. 2011;12:2392–2403. doi: 10.1128/MCB.01218-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seki E, Schnabl B. Role of innate immunity and the microbiota in liver fibrosis: crosstalk between the liver and gut. J Physiol. 2012;(Pt 3):447–458. doi: 10.1113/jphysiol.2011.219691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hritz I, Mandrekar P, Velayudham A, Catalano D, Dolganiuc A, Kodys K, et al. The critical role of toll-like receptor (TLR) 4 in alcoholic liver disease is independent of the common TLR adapter MyD88. Hepatology (Baltimore, Md) 2008;4:1224–31. doi: 10.1002/hep.22470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.He M, Xu Z, Ding T, Kuang DM, Zheng L. MicroRNA-155 regulates inflammatory cytokine production in tumor-associated macrophages via targeting C/EBPbeta. Cell Mol Immunol. 2009;5:343–352. doi: 10.1038/cmi.2009.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martinez-Nunez RT, Louafi F, Sanchez-Elsner T. The interleukin 13 (IL-13) pathway in human macrophages is modulated by microrna-155 via direct targeting of interleukin 13 receptor alpha1 (IL13R{alpha}1) J Biol Chem. 2010 doi: 10.1074/jbc.M110.169367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lu G, Zhang R, Geng S, Peng L, Jayaraman P, Chen C, et al. Myeloid cell-derived inducible nitric oxide synthase suppresses M1 macrophage polarization. Nat Commun. 2015:6676. doi: 10.1038/ncomms7676. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.