

Abstract
Persistent infection of HCV is one of the leading cause of end stage liver diseases such as decompensated cirrhosis and liver cancer. Of particular note, nearly half of HCV infected people in the United States are reported to be heavy drinkers. This particular group of patients are known to rapidly progress to the end stage liver diseases. Although, the accelerated disease progression among alcohol abusers infected with HCV is clinically well recognized, the molecular pathophysiology behind this manifestation has not been well elucidated. Hepatocytes metabolize ethanol (EtOH) primarily through two steps of oxidative catabolism in which alcohol dehydrogenase (ADH) and aldehyde dehydrogenase (ALDH) play central roles. The ADH-ALDH pathway also governs the metabolism of Retinol (Vitamin A) to its transcriptionally active metabolite, Retinoic Acid (RA). In this study, we defined that the ADH-ALDH pathway serves as a potent antiviral host factor in hepatocytes, which regulates the expression of Interferon Stimulated Genes (ISGs) via biogenesis of RA. ISGs constitute over 300 antiviral effectors, which cooperatively govern intracellular antiviral innate immunity. Our study revealed that intracellular RA levels greatly influence ISGs expression under basal conditions. Moreover, RA augments ISGs induction in response to viral infection or exposure to IFN in a gene-specific manner. Lastly, our results demonstrated that EtOH attenuates the antiviral function of the ADH-ALDH pathway which suggests the possibility that EtOH-Retinol metabolic competition is one of the molecular mechanisms for the synergism between HCV and alcohol abuse in liver disease progression. In conclusion, our study provides novel insights into the critical role of RA in the regulation of intracellular antiviral innate immunity in hepatocytes.
INTRODUCTION
Over 200 million people worldwide are chronically infected with HCV(1). In the US, at least 3.5 million people suffer from chronic HCV infection with recent significant increase observed among IV drug users(2, 3). Of particular note, half of the HCV infected population in the US have been reported to be heavy drinker(4). This group of patients exhibits pronounced HCV replication and refractoriness to antiviral therapy which results in a 30- and 48-fold increase in developing decompensated cirrhosis and hepatocellular carcinoma, respectively(5). This synergism between chronic HCV infection and alcohol abuse is well recognized; however, the underlying molecular pathophysiology has not been well understood.
Hepatocytes play a central role in EtOH metabolism through ADH and to a lesser extent through cytochrome P450-2E1(CYP2E1) oxidation to acetaldehyde(6). Acetaldehyde is subsequently metabolized to acetate by ALDH(6). The relevance of metabolic byproduct(s) of EtOH and/or the cellular response in the pathogenesis of HCV has not been well understood, primarily due to the lack of appropriate research tools. Here, we established Huh7 cell-line based systems that expresses individual enzymes required in EtOH metabolism. Of great interest, our results demonstrate that the ADH-ALDH pathway serves as a potent antiviral element; whereas, CYP2E1 is a proviral host factor. We also found that the antiviral phenotype of the ADH-ALDH pathway is significantly attenuated in the presence of EtOH.
ADH also plays a critical role in the conversion of retinol(ROL) to retinaldehyde(RAL), followed by the oxidation of RAL to RA by ALDH(7). For ADH1,which is abundantly expressed in hepatocytes, ROL is the far preferred substrate as compared to EtOH. However, the ROL concentration in the serum is much lower than the ADH1 Km for ROL. In contrast, the blood EtOH concentration of heavy drinkers often approaches or surpasses the ADH1 Km for EtOH, thus providing a situation in which the biogenesis of RA is impaired(7, 8). This leds us to hypothesize that the EtOH-ROL metabolic competition might be an underlying mechanism for the synergism between HCV and alcoholism.
Numerous studies indicate that RA exhibits antiviral activities against a variety of pathogens(9–11). The antiviral properties of RA have been mainly explained in the context of professional innate immune cells and adaptive immunity(12); however, the role in innate immunity in terminally differentiated non-immune cells such as hepatocytes remains undefined. ISGs, constituting over 300 genes, represent the antiviral innate immune effectors which cooperatively restrict viral lifecycle(13). ISGs expression at basal levels determines cellular susceptibility to viral infection(14). During infection, host cells robustly induce additional ISGs upon Pattern Recognition Receptor(PRR) sensing of Pathogen Associated Molecular Patterns(PAMPs) such as viral genome(15). This event also results in the secretion of endogenous Type-1 IFN such as IFN-β. The IFN then promotes the expression of the grossly redundant ISGs in both infected and neighboring cells via activation of Jak-STAT signaling. The magnitude of the additional ISGs induction is a major predictor of the clinical outcome(16). Although, a few ISGs have been reported as RA-inducible(15, 17, 18), the fundamental role of RA in the regulation of ISGs has not been determined. Our findings revealed that the restoration of the ADH-ALDH pathway in Huh7 cells greatly enhanced ISG expression under both basal and induced condition in a gene-specific manner, which was associated with successful clearance of the pathogen. Moreover, our study revealed that EtOH-inducible enzyme, CYP2E1(19), significantly attenuates RA-mediated gene expression and thus supports viral replication.
In summary, our study demonstrated that the impaired biogenesis of RA leads to decreased expression of ISGs in hepatocytes, thereby providing a molecular explanation for the synergism between HCV infection and alcoholic liver disease.
MATERIALS and METHODS
Cells and Transfection
Huh7 cells were maintained as previously described(20) and transfected with TransIT®-LT1(Mirus) or TransIT®-mRNA(Mirus). Huh7 stable cell lines expressing hADH1B or hCYP2E1 were established via lentiviral transduction(System Bioscience). The lentiviral transduction was carried out at MOI:5 followed by Puromycin selection(2μg/ml) to established polyclonal(pooled) cell lines. Primary Human Hepatocytes(PHH) were obtained from Life Technologies. Primary mouse hepatocytes of C57BL/6 were obtained from Non-Parenchymal Liver Cell Core at SCRC for ALPD and cirrhosis.
Viruses
Lentiviral and HCV(JFH1) particle was propagated and tittered as described previously(20). Huh7 subgenomic replicon cells were established through G418(400ug/ml) selection upon in vitro transcribed subgenome transfection. HCV pseudo-particle system was propagated as previously described(21). Sendai virus(SeV) was obtained from Charles River Laboratories and the infection(10–100 HAU/ml) was conducted in serum-free DMEM for 1 hour at 37C.
Chemical, measurement of EtOH and acetaminophen(APAP)-protein adducts
EtOH treatments were carried out at 10mM, 12.5mM and/or 50mM, which correspond to the physiological blood alcohol concentration(BAC) among alcoholics of 0.046%, 0.057%, and 0.229% respectively(22). EtOH in the cell culture medium was measured via Gas-chromatography similarly to a previous report(23). Concentrations of APAP protein adducts were measured by high performance liquid chromatography with electrochemical detection(HPLC-EC) as described previously(24). Acetaldehyde, Acetate, all trans retinoic acid(ATRA), and all trans ROL, were obtained from Sigma-Aldrich. EC23(ATRA analogue) was purchased from Tocris. RAR antagonist(4310) was described previously(25).
Statistical Analysis
P-values were obtained using Student t-test, and the results were considered significant at p< 0.05.
RESULT
Restoration of the EtOH metabolic pathways in Huh7 cells
The ADH- and ALDH-families are comprised of 8 and 10 family members, respectively(6). We first conducted meta-analysis of NCBI-GEO microarray data to determine the isoforms of ADHs and ALDHs that are predominantly expressed in human hepatocytes. Consistent with previous reports, this analysis demonstrated that hepatocytes express the following isoforms; 1)ADH: ADH1(A, B, and C), 4, 6, and FE1, 2)ALDH:ALDH1A1, ALDH2, and ALDH4A1(Supplemental Figure1A and 1B)(26).
Because in vitro HCV studies are restricted to the use of Huh7 cells, we assessed the expression of liver dominant ADHs and ALDHs in Huh7 cells. The gene- and protein-expression analysis demonstrated that Huh7 cells lack the expression of these ADHs and CYP2E1(Figure1A and 1B). However, they preserved the expression of ALDHs, suggesting that the reconstitution of ADHs should restore their EtOH metabolism capability.
Figure 1. Restoration of functional EtOH metabolic pathways in Huh7 cells.

A–B. Extracts from either Huh7 or human primary hepatocyte(PHH) were subjected to gene expression(A) or immunoblotting(IB)(B) analysis of the indicated enzymes involved in EtOH metabolism. GAPDH was used for normalization to compare relative expression(A). C. Huh7 cell lysates containing over-expressed the indicated ADH isozyme were incubated with NAD(3mM) in the presence or absence of EtOH(10mM) to assess the NADH conversion rate(μMoles/min/ml). D. Huh7 cells expressing the indicated vectors were treated with EtOH(12.5mM and 50mM). The culture media was used to measure EtOH concentration via gas chromatography. E. Huh7 cells expressing CYP2E1 were treated with the indicated concentrations of acetaminophen. Concentrations of NAPQI-adducts were determined using HPLC-EC.
Among the enzymes responsible for the conversion of EtOH to acetaldehyde in hepatocytes, ADH1 is the most abundant in hepatocytes(7, 26). Thus, ADH1 is known to be the primary ADH involved in liver EtOH metabolism. Next, we tested the enzymatic activity of ADH1B(a delegate of ADH1) on EtOH by determining the reduction rate of Nicotinamide Adenine Dinucleotide(NAD+) conversion to NADH. NAD+ is a co-enzyme required for the oxidation of EtOH, serving as an electron acceptor; and therefore the conversion rate can be used to indirectly monitor ADH activity(26). Huh7 cellular lysates upon ADH1B over-expression exhibited potent activity while ADH4- and ADH6-expressing cells showed negligible activity(Figure1C). Next, we evaluated whether reconstitution of ADH1B or CYP2E1 restores the EtOH metabolic function in Huh7 cells. The result showed that over-expression of ADH1B, and CYP2E1 to a lesser extent, restored the metabolism to physiological concentrations of EtOH (22)(Figure1D).
In addition to CYP1A2, 3A4, and 2D6, CYP2E1 is also known to catabolize xenobiotics such as acetaminophen(APAP). Upon metabolism of APAP, CYPs produces the electrophile N-acetyl-p-benzoquinone imine(NAPQI), which exhibit toxicity through covalent binding to cellular proteins and nucleic acids(27). Thus, we tested whether CYP2E1 in Huh7 cells produces APAP protein adducts using HPLC-EC. Cell lysates from the CYP2E1-expressing cells contained higher NAPQI protein adducts compared to control condition(Figure1E). These results assure that Huh7 cells offer an environment for these enzymes to be functional although these capacities appeared inferior to that of primary hepatocytes likely due to the relative abundance (Supplemental Figure1C).
The ADH-ALDH pathway is a potent antiviral host factor
The restoration of EtOH metabolic capacity in Huh7 cells enabled us to investigate the association between EtOH metabolic pathways and the HCV lifecycle. Surprisingly, transient ADH1B over-expression in Huh7 cells harboring HCV subgenomic replicon(HCV-SGR) dramatically suppressed HCV replication(Figure2A and 2B). In contrast, over-expression of CYP2E1 was associated with an enhancement of HCV replication. Moreover, the antiviral effect of ADH1B was reduced in the presence of EtOH(Figure2B). Next, we tested whether ADH1B affects the efficiency in establishing viral lifecycle via HCV-SGR replicon transduction assay. The transfection of HCV subgenome in Huh7 cells that stably express ADH1B demonstrated a significantly less number of HCV replicating foci formation, however the difference between control- and ADH1B-cells diminished upon EtOH treatment(Figure2C). These results indicate that the ADH-ALDH pathway serves as an antiviral host factor in the absence of EtOH. Furthermore, our results suggest that either 1) The EtOH metabolic byproducts generated by ADH1B may offer a suitable environment for efficient HCV replication, or 2)EtOH prohibits the biogenesis of antiviral molecules produced by the ADH-ALDH pathway. To distinguish between these possibilities, we addressed whether EtOH metabolites increase HCV replication efficiency. The results showed that neither acetaldehyde nor acetate treatment changed viral replication efficiency (Supplemental Figure2A and 2B).
Figure 2. The ADH-ALDH pathway is a potent antiviral host factor.

A. Huh7 cells harboring HCV-SGR were transduced with the indicted vectors for 48 hours. Extracts were subjected to immune-fluorescent analysis. Red: HCV NS3, Green: Flag- ADH1 or CYP2E1, Blue: DAPI. B. The indicated expression vectors were transfected into Huh7 cells containing HCV-SGR for 24 hours followed by EtOH treatment for 48 hours. The extracts were subjected to IB analysis. C. Huh7 cells expressing ADH1B or control vector were transfected with in vitro transcribed HCV-SGR genome followed by G418 selection in the presence or absence of EtOH(12.5mM). Upon completion of G418 selection, the culture dishes were fixed and stained with crystal violet. D. Huh7 cells stably expressing ADH1B or control vector were challenged with HCV pseudo-particles containing a GFP reporter at the indicated titer. 24 hours post infection, cells were subjected to fluorescent microscopic analysis. Green: GFP and Blue: DAPI. The GFP positive cells at each condition was shown in %. E. RT-PCR analysis with primer sets targeting the indicated region of HCV genome. The RNA was extracted from Huh7 cells stably expressing ADH1B or control vector 7 days after SGR-RNA transfection .
To further understand how the ADH-ALDH pathway suppresses HCV, we assessed the effect of ADH1B expression on each stage of the viral lifecycle. First, a HCV pseudo particle containing a GFP reporter was employed to test the effect of ADH1B expression on viral entry. Under these conditions, ADH1B expression in Huh7 cells was found to have a negligible effect on viral entry(Figure2D). Next, we examined the effect of ADH1B expression on genome replication using RT-PCR. The abundance of each region of the viral genome decreased with distance from the replication initiation site(5’UTR) of the viral genome, especially in ADH1B expressing cells(Figure2E). This result may suggest that the ADH-ALDH pathway restrict HCV by promoting premature termination of genome replication. Taken together, these results indicate that the ADH-ALDH pathway restricts the HCV at the genome replication/translation stage.
Shared antiviral properties of liver dominant ADH isoforms
We extended our investigations to the other liver dominant ADHs to determine whether these enzymes also suppress HCV. To accomplish this, we expressed ADH4 and ADH6 in Huh7–SGR cells and assessed their antiviral potency. The result showed that the expression of ADH4 and ADH6 in HCV SGR cells exhibited a comparable degree of viral suppression as to that observed with ADH1B expression(Figure3A). Similar results were observed with immuno-fluorescent microscopic analysis and protein analysis, wherein these ADH isoforms potently inhibited the HCV(Figure3B and 3C), indicating that the antiviral activity of ADH is not unique to ADH1B but is shared among other ADHs.
Figure 3. Shared antiviral properties of liver dominant ADHs.
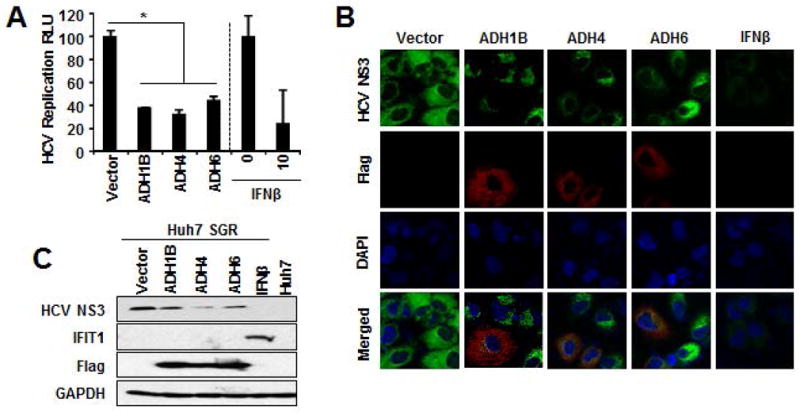
A. HCV-SGR cells containing HCV IRES-dependent firefly luciferase reporter were transduced with the indicated expression vectors along with renilla luciferase plasmid for 48 hours. The firefly luciferase values were normalized with renilla luciferase and presented as relative luciferase unit(RLU). *p<0.01. B–C. HCV-SGR cells were transfected with the indicated expression vectors for 48 hours followed by immuno-fluorescent microscopic analysis(Green:HCV-NS3, Red:Flag, Blue:DAPI)(B) or IB analysis of the indicated proteins(C). IFNβ 10IU/ml served as positive control for HCV suppression(A–C).
Analysis of ADH enzymatic activity revealed that the different ADHs possess distinctive EtOH metabolic activity(Figure1C). These ADHs have also been shown to metabolize retinol(ROL)(7, 26), leading to the possibility that the ADH-ALDH pathway suppresses HCV through the metabolism of ROL. To test this possibility, cell lysates containing these ADH isoforms were treated with ROL in the presence of NAD, and the enzymatic activity was assessed by the rate of NAD+ reduction to NADH. All ADHs tested were found to exhibit comparable activity on ROL(Supplemental Figure3A). In addition, the antiviral effect of ADH1B but not ADH4 was antagonized by EtOH (Supplemental Figure3B), likely reflecting the enzymatic activity on EtOH(Figure1C) as well as KiEtOH to ROL(7). These observations collectively suggest that the antiviral activity of ADH may be through the metabolism of ROL to the biogenesis of retinoic acid(RA) and EtOH impairs the antiviral effect of ADHs-ALDH pathway depending on their affinity to EtOH.
The ADH-ALDH pathway regulates gene expression through the biogenesis of RA
To test whether ADH1B expression enables the production of RA from ROL in Huh7 cells, a few well accepted RA-inducible genes expression were assessed via RT-qPCR. The result showed that ADH1B expressing cells robustly induced these genes in response to ROL treatment(Figure4A). RA regulates gene expression by binding to the heterodimer nuclear receptor complex that is comprised of RAR-RXR(28). The RAR-RXR heterodimer binds preferentially to a retinoic acid response element(RARE), which contains two receptor cognate binding sequences typically separated by 5 bp, Direct Repeat 5(DR5). The RA binding to the RAR leads to recruitment of co-activators and activation of gene transcription(28). Therefore, we investigated whether ADH1B expression in Huh7 cells impacts RAR-mediated gene expression using a RARE-containing luciferase reporter. First, a series of RARE luciferase reporters comprising of 1, 3, and 5bp separated RARE(DR1, DR3, or DR5 respectively) were expressed in Huh7 cells and the response to ATRA treatment was monitored. Addition of ATRA selectively induced significant luciferase activity only in the DR5 RARE reporter(Supplemental Figure4A). Moreover, the DR5 reporter was found to exhibit increased and reduced activity in response to an RA agonist(EC23) or an RAR antagonist(4310), respectively(Supplemental Figure 4B and 4C). Given these results, the RARE-DR5 reporter was employed as a surrogate to follow RAR activity. Following co-transfection of the RARE-DR5 into Huh7 cells and treatment with ROL, co-expression of ADH1B induced activation of RARE-DR5(Figure4B). In addition, both gene expression analysis and reporter assay results showed that ADH1B expression enhances the RA-gene regulation even under basal condition. We speculate that this is likely due to the metabolism of up to 50nM ROL in standard 10% FBS DMEM via ADH1B-ALDH pathway. To further test the influence of ADH1B-ALDH pathway in gene regulation, RT-qPCR array comprising RA-regulated genes was employed(Figure4C and 4D). The result demonstrated that ADH1B expression globally up-regulates RA-inducible genes at basal condition, and the degree of the up-regulation was significantly higher than that of exogenous ROL treated control Huh7 cells. Next, we compared the gene expression changes seen in Huh7 cells that express ADH1B to that of PHH treated with ATRA. These results showed identical patterns (Figure4C and 4D), thereby indicating that the reconstitution of ADH1B in Huh7 cells leads to the production of RA from ROL. This notion is also well supported by the fact that the control cells minimally changed the expression of RA-regulated genes even in response to exogenous ROL(Figure4A, 4B and 4C).
Figure 4. Reconstitution of the ADH-ALDH pathway restores biogenesis of RA.
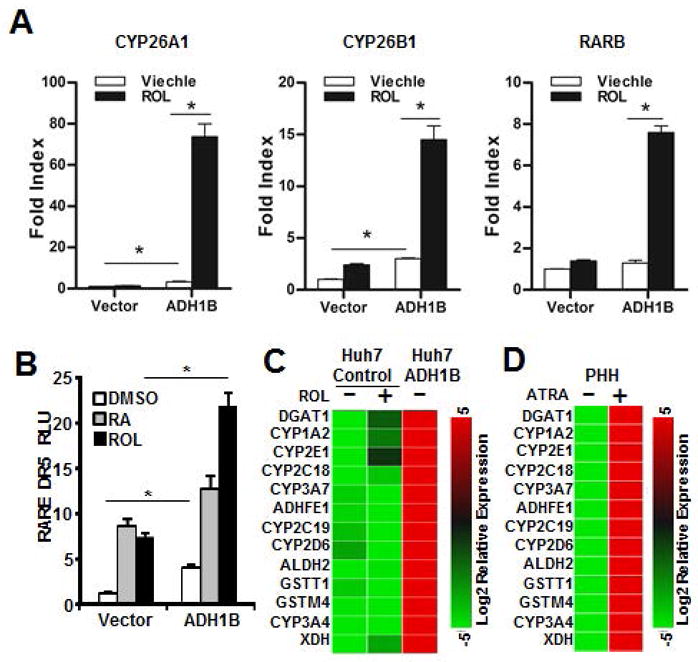
A. Huh7 cells expressing ADH1B or control vector were treated with DMSO(viechle) or ROL(1μM) for 16 hours. Extracts were subjected to RT-qPCR analysis of the indicated RA-inducible gens. B. Huh7 cells were co-transfected with the indicated expression vectors, a firefly luciferase reporter regulated by RARE-DR5, and a renilla luciferase vector. 24 hours after transfection, cells were treated with ROL(1μM), ATRA(0.1μM), or DMSO for 36 hours followed by dual luciferase assay. C–D. Extracted RNA from Huh7 cells stably expressing ADH1B or control vector(C) or primary human hepatocytes(PHH)(D) were subjected to RT-qPCR array of RA-regulated genes. The heat map represents the relative abundance of the indicated genes. Cells were treated with either ROL(1μM)(C), ATRA(0.1μM)(D), or DMSO(control) for 16 hours prior to RNA extraction. *p<0.01
Critical role of the ADH-ALDH pathway for the ISG expression
Next, we directly tested whether RA alone was sufficient to suppress HCV. For this purpose, HCV-SGR cells were treated with RA and this led to dramatic inhibition of viral replication(Figure5A). RA is known to play an important role in a variety of cellular functions including, cell proliferation, death, and differentiation(8). To better understand the mechanisms underlying RA suppression of HCV replication, we initially tested whether the ADH-ALDH pathway influences cell growth and/or susceptibility to cell death, which may nonspecifically influence viral replication as the lifecycle heavily relies on the cellular machinery. Expression of ADH1B neither changed cell growth/proliferation nor cell viability(Supplemental Figure5A and 5B). These results led us to hypothesize that ADH-ALDH pathway plays a role in the regulation of antiviral host factors such as ISGs for the suppression of HCV.
Figure 5. Potent anti-HCV suppression by RA and its enhancement of ISG expression.
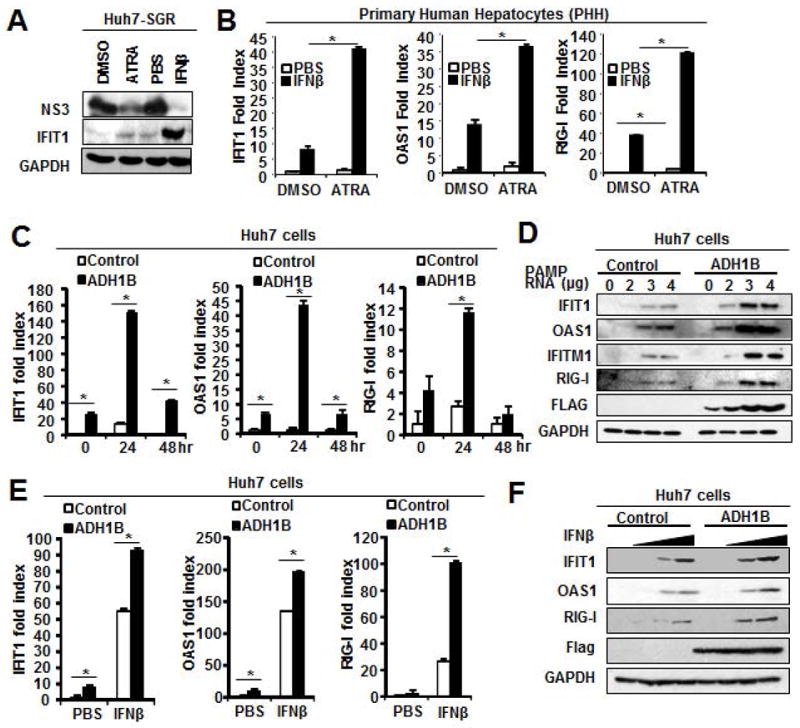
A. Huh7 HCV-SGR cells were treated with ATRA(0.1μM) or IFNβ(10IU/ml) for 72 hours. Extracts were subjected to IB analysis of the indicated proteins. B. RT-qPCR analysis of RNA extracted from PHH pretreated with ATRA(0.1μM) or vehicle for 16 hours followed by IFNβ(100IU/ml) for 8 hours. C and E. RT-PCR for the ISGs expression analysis. Huh7 cells stably expressing ADH1B were transfected with HCV-PAMP-RNA(1μg/5×105cells) for indicated hours(C) or treated with IFNβ(100IU/ml) for 8 hours(E). *p<0.01. D and F. IB analysis of the ISGs expression. Huh7 cells stably expressing ADH1B were transfected with HCV PAMP RNA(0, 2, 3, 4 μg/5×105cells) for 24 hours(D) or treated with IFNβ(0, 1, 10, 100IU/ml)(F).
Clinical studies reported that RA enhances the response to IFN based antiviral therapy(11, 29). These observations suggest that the expression of ISGs could be governed, at least partially, by RA. To further explore this hypothesis, we conducted a bioinformatics search of ISG(446 genes) promoter and found that 88% of the regulatory region of ISGs contain RARE-DR5 sequence(Supplemental Table1). This observation suggests that up-regulation of ISGs via RA may underlie the ADH-ALDH-mediated suppression of HCV.
To further test this idea, we first investigated whether RA augments the expression of ISGs in terminally differentiated primary human hepatocytes(PHH). These experiments showed that ATRA treatment increased the basal expression level of, at least, a few selected ISGs(Figure5B). In addition, the RA treatment significantly enhanced ISG expression in response to Type-1 IFN, well supporting aforementioned clinical observations(Figure5B). Similarly, the synthetic ATRA analogue(EC23) enhanced the IFN mediated ISG induction in PHH(Supplemental Figure5C). These observations with non-cancerous primary cells further indicate the critical roles of RA in ISGs expression in hepatocytes.
Next, we tested whether the ADH-ALDH pathway enhance the expression of ISGs in Huh7 cells. Gene expression analyses showed that ADH1B expression increases ISGs expression under basal conditions(Figure5C and 5E). We also tested whether ADH1B expression augments ISG expression in induced condition with HCV-PAMP-RNA or IFNβ treatment. These are the ligands of two distinct ISG induction pathways: RIG-I or Jak-STAT pathway(15), which leads to robust induction of ISGs during infection in infected or neighboring cells respectively. The result showed that ADH1B expression substantially increased the ISGs expression in response to these ligands at both message level(Figure5C and 5E) and protein level(Figure5D and 5F). Lastly, we also found that ADH1B expression has negligible effect on the signaling activation potency by these ligands(Supplemental Figure5D and 5E), suggesting that the augmentation of ISG expression by RA is likely at the promoter level.
Selective up-regulation of ISGs by the ADH-ALDH pathway
Given chromatin remodeling activity of RAR-RXR, we hypothesized that RA regulates ISG expression at the promoter level(28). Thus, we speculated that the effect of ADH-ALDH pathway on ISGs induction is highly dependent on the accessibility of activated RAR-RXRs to individual gene promoters rather than indiscriminate global gene up-regulation. Thus, we assessed the pattern of ISG expression in ADH1B-expressing cells using a RT-qPCR array. In agreement with our hypothesis, ADH1B-expressing cells enhanced ISG expression in a gene-specific manner under both basal conditions and during acute HCV(JFH-1) infection(Figure6A). Of note, the up-regulated ISGs include well studied anti-HCV genes such as IFIT1 and IFITM1. Furthermore, HCV replication efficiency in ADH1B-expressing cells demonstrated an inverse association with the degree of ISG inducibility(Figure6B and 6C). Lastly, we challenged Huh7 cells expressing ADH1B with Sendai virus, that induces ISGs in a RIG-I signaling dependent manner similar to HCV(15). ADH1B expression was found to attenuate viral replication efficiency, which is correlated with the degree of ISG induction(Supplemental Figure6A and 6B). These observation indicate that the ADH-ALDH pathway restrict viral infection through the augmentation of ISG expression at the gene promoter/enhancer level.
Figure 6. Selective up-regulation of ISGs governed by the ADH-ALDH pathway protect hepatocytes from HCV infection.
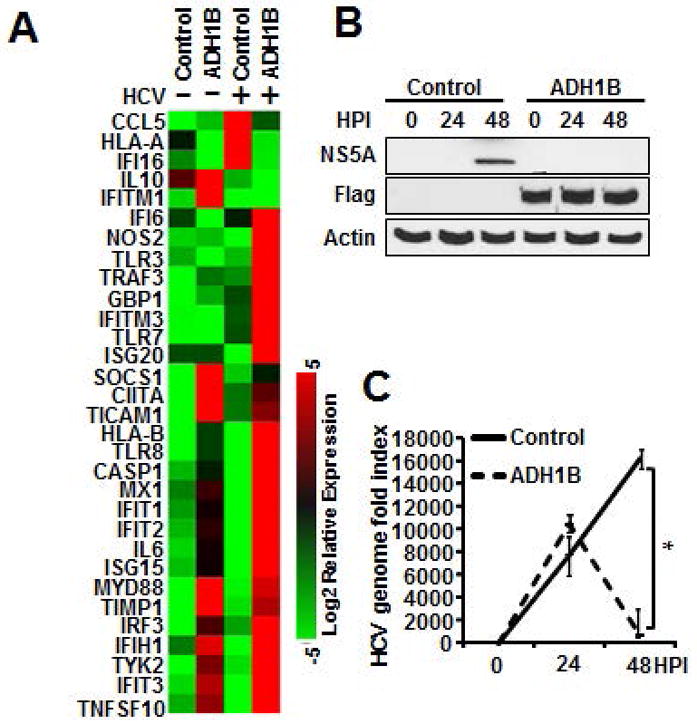
A–C. Huh7 cells stably expressing ADH1B or control vector were infected with HCV(JFH1 MOI:0.5) for 24 and 48 hours. RNA was extracted 24 hours post infection and used for RT-qPCR array for the detection of a panel of ISGs(A), RNA and protein extracts were subjected to IB analysis(B) or RT-qPCR(C) for the detection of viral products. * p<0.01.
Two distinct EtOH metabolizing pathways impair RA regulation of intracellular innate immunity
Our results suggest that the impairment of RA-mediated ISGs regulation in hepatocytes serves as, at least in part, an underlying mechanism for pronounced HCV replication among alcoholics. To further test this idea, we assessed whether EtOH attenuates RAR-mediated gene regulation. Indeed, the analysis with the RARE-DR5 reporter system(Supplemental Figure7A) and RT-qPCR analysis(Figure7A) demonstrated that EtOH impaired RA gene regulation in a dose-dependent manner. Accordingly, the cell lysates from ADH1B-expressing cells demonstrated comparable enzymatic activity on both EtOH and ROL, especially at higher concentrations(Figure7B). Moreover, the effect of ADH1B in enhancing ISGs expression was attenuated in the presence of EtOH(Supplemental Figure7B). These observations suggest that EtOH-ROL competition attenuates RA-mediated ISG expression.
Figure 7. EtOH deregulation of retinoid homeostasis through two distinct mechanisms.

A. RT-qPCR analysis of RA-inducible genes. Mouse primary hepatocytes were treated with EtOH for 60 hours. *p<0.01. B. Cell lysates expressing ADH1B or control cells were incubated with NAD(3mM) and indicated ADH substrates. The ADH activity was calculated by the rate of NAD+ to NADH conversion(μMoles/min/ml). C. RARE-DR5 dual luciferase assay. Huh7 cells were cotransfected with the indicated cytochrome P450 expression vectors, RARE DR5 luciferase, and renilla reporter followed by RA for 36 hours. *p<0.01. CYP26A1:positive control. CYP27A1 that catabolizes vitamin D was employed as a negative control. D. IB analysis of the HCV protein expression. Huh7 cells were transfected with CYP2E1 or control vector for 24 hours followed by HCV JFH1 infection for 48 hours. E. RT-qPCR of the indicated ISGs expression. PHHs were treated with EtOH for 24 hours followed by IFNβ(50IU/ml) for 8 hours. *p<0.01.
Recent evidence has shown that CYP2E1 potently catabolize RA to transcription inactive polar metabolites(30). CYP2E1 is an EtOH inducible enzyme that plays a role in EtOH metabolism(19)(Supplemental Figure7C and Figure1D). Of note, our results(Figure2B) demonstrated that HCV replication was enhanced upon CYP2E1 expression. Thus, we hypothesized that CYP2E1 expression in hepatocytes can be a secondary mechanism that impairs RA-regulation of ISG expression. An assay utilizing the RARE-DR5 reporter showed that RA-mediated gene regulation was significantly attenuated in the presence of CYP2E1 both under basal and induced conditions(Figure7C and Supplemental Figure7D). Interestingly, our observation suggests that CYP2E1 catabolic activity on ATRA is nearly comparable to that of CYP26A1, which is the most critical RA catabolizing enzyme with the Km/RA~10nM(31). This suggests that CYP2E1 may preferentially catabolizes RA as compared to other substrates such as EtOH or APAP and may play a role in the impairment of ISG expression. Consistent with our findings with HCV-SGR system(Figure2B), the expression of CYP2E1 substantially increased susceptibility to HCV infection(Figure7D).
Lastly, we tested the effect of EtOH in ISGs expression with human primary hepatocytes, in which both EtOH-ROL competition on ADH-ALDH pathway and CYP2E1 induction contribute to the depletion of RA. The results showed EtOH treatment dramatically impaired the expression of ISGs(Figure7E).
Discussion
Alcohol is the most common intoxicating substance in the US. Alcoholics are known to have a significantly higher risk of acquiring HCV, due to the high prevalence of IV drug use in this population(32). The combination of HCV infection and alcohol abuse rapidly progress to end stage liver diseases(5, 33), however, the molecular mechanism underlying this phenomenon has been unclear.
Our study demonstrated that the ADH-ALDH pathway is a potent antiviral pathway. We also found that the antiviral effect of ADH-ALDH pathway is mediated by the metabolism of ROL to the biogenesis of RA. Moreover, the antiviral property of ADH-ALDH pathway was diminished in the presence of physiological concentration of EtOH among alcoholics(34) likely due to EtOH-ROL metabolic competition.
The majority of dietary retinoid is stored as Retinyl Ester(RE) mainly in hepatic stellate cells(HSCs)(35). These stored retinoids are distributed as ROL or RE to maintain serum retinoid concentration. The metabolism of up-taken ROL utilizes a two-step oxidative processes, which medium chain ADHs or retinol dehydrogenases catabolize ROL to retinaldehyde followed by the second oxidation facilitated by ALDH to produce RA(26). In particular to hepatocytes, ADH1 presumably plays a critical role in biogenesis of RA due to its relative abundance(36, 37).
RA governs various aspects of cell biology, especially in the regulation of cell differentiation and proliferation(8). Thus, it is thought that RA has great influence on cells continuously undergoing proliferation and differentiation, such as professional immune cells. Indeed, RA is known to potentiate adaptive immunity by regulating T-cell proliferation, Th1-2 balancing and cytotoxicity, as well as modulating B-cell proliferation and immunoglobulin production(12). However, the role of RA in intracellular innate antiviral immunity, in which ISGs play a central role, has not been determined.
The contribution of RA in ISGs induction appear not unique only to genes which nomenclature contains “RA inducible”(15, 17, 18). Indeed, a number of ISGs were noted to be up-regulated in the gene list obtained from transcriptome analysis of ATRA treated leukemia cells(38). Our results indicated that the effect of RA in ISGs induction is gene-specific rather than global up-regulation(Figure6A), suggesting that RA unlikely induces ISGs via secretion of IFNs nor confer the activation of ISGs induction pathways. In fact, we did not observe the phosphorylation of STATs and IRF3 in ATRA treated cells(data not shown) or ADH1B expression in Huh7 cells(Supplemental Figure5D and 5E). These notions collectively suggest that RA augments ISG expression at the gene promoter level. Our bioinformatics analysis demonstrated the occurrence of RARE-DR5 in promoters of randomly selected genes was 2.11475(-5,000 to +500). Based on this number, we categorize ISGs into: 1)high DR5:>= 4(82 ISG, average 4.904 DR5 occurrence), 2)medium DR5:>=2 and <4(204 ISG, average 2.441 DR5 occurrence), and 3)low DR5:<2(160 ISG, average 0.688 DR5 occurrence)(Table1). These results indicate that RA can influence the expression of up to 88% of ISGs at the promoter level. Moreover, the occurrence of RARE-DR5 is significantly higher in nearly 20% of ISGs promoters, further suggesting the role of RA in ISGs expression. However, we have also noted that the RARE-DR5 frequency is not well correlated to the degree of ISG expression enhanced by the ADH-ALDH pathway(Figure6A and Table1). This can be explained either by the cell type based differential response to RA or the discrepancy between the bioinformatics algorithms and the actual RAR-RXR occupancy. In fact, a study by others has noted differential RA-gene regulation in cell type specific manner and have shown that genome wide RAR binding loci does not correspond to the expression of RA target genes(39). The study attributed this discrepancy to the diversity of RAR-RXR binding sequence and/or chromatin topology.
Table 1.
| A. ISG containing high frequency of RARE DR5 in gene regulatory region (−5000 to +500) | |||
|---|---|---|---|
| High | # | High | # |
| DEFB1 | 8 | ABLIM3 | 4 |
| DUOXA2 | 7 | APOL6 | 4 |
| IGFBP2 | 7 | B2M | 4 |
| LY6E | 7 | B4GALT5 | 4 |
| MX2 | 7 | CMAH | 4 |
| ODC1 | 7 | CRP | 4 |
| SCO2 | 7 | CXCL9 | 4 |
| TREX1 | 7 | CYorf15A | 4 |
| TYMP | 7 | DDX60L | 4 |
| ANKRD22 | 6 | ELF1 | 4 |
| ARG2 | 6 | ENPP1 | 4 |
| C22orf28 | 6 | FUT4 | 4 |
| CCL2 | 6 | GBA | 4 |
| CLEC4D | 6 | GBP2 | 4 |
| FAM26F | 6 | GNB4 | 4 |
| GK | 6 | HLA-A | 4 |
| LGALS9 | 6 | HLA-DOB | 4 |
| RAB4B | 6 | HLA-G | 4 |
| RPL22 | 6 | HMCN2 | 4 |
| ABTB2 | 5 | IDO1 | 4 |
| ANGPTL1 | 5 | IL17RB | 4 |
| ARNTL | 5 | ISG20 | 4 |
| BCL2L14 | 5 | KIAA0040 | 4 |
| BMI1 | 5 | LGALS3 | 4 |
| BUB1 | 5 | MT1M | 4 |
| C1S | 5 | P2RY6 | 4 |
| CCR1 | 5 | PARP14 | 4 |
| DDO | 5 | PDK1 | 4 |
| FAM134B | 5 | PLA2G2A | 4 |
| FAM46C | 5 | RARB | 4 |
| HEG1 | 5 | RARRES3 | 4 |
| IL1R1 | 5 | RASSF5 | 4 |
| IL28RA | 5 | RNF19B | 4 |
| IMPA2 | 5 | SAMHD1 | 4 |
| LAMP3 | 5 | SP110 | 4 |
| MIA | 5 | ST3GAL4 | 4 |
| PIM3 | 5 | TCF7L2 | 4 |
| PLA1A | 5 | TMEM140 | 4 |
| PLEK | 5 | TRAFD1 | 4 |
| PUS1 | 5 | WHAMM | 4 |
| SERPINE1 | 5 | ||
| UPP2 | 5 | ||
| B. ISG containing average frequency of RARE DR5 in gene regulatory region (−5000 to +500) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Average | # | Average | # | Average | # | Average | # | Average | # |
| APOBEC3A | 3 | MAB21L2 | 3 | ADAMDEC1 | 2 | IFI16 | 2 | S100A8 | 2 |
| AQP9 | 3 | MCL1 | 3 | ALDH1A1 | 2 | IFI35 | 2 | SAMD4A | 2 |
| ARHGEF3 | 3 | MKX | 3 | AMPH | 2 | IFI44 | 2 | SAMD9L | 2 |
| ATF3 | 3 | MOV10 | 3 | ANG | 2 | IFI6 | 2 | SERPING1 | 2 |
| BAG1 | 3 | MSR1 | 3 | ANKFY1 | 2 | IFIT3 | 2 | SLC15A3 | 2 |
| C15orf48 | 3 | MT1G | 3 | APOL1 | 2 | IFITM1 | 2 | SLC1A1 | 2 |
| C3AR1 | 3 | MT1H | 3 | APOL2 | 2 | IFITM3 | 2 | SMAD3 | 2 |
| C4orf33 | 3 | MT1X | 3 | BATF2 | 2 | IFNGR1 | 2 | SOCS1 | 2 |
| CA7 | 3 | MX1 | 3 | BCL3 | 2 | IL15RA | 2 | SPATS2L | 2 |
| CARHSP1 | 3 | MYOF | 3 | BLVRA | 2 | IL6ST | 2 | SPSB1 | 2 |
| CCL4 | 3 | NAPA | 3 | BTN3A3 | 2 | ISG15 | 2 | SPTLC2 | 2 |
| CD38 | 3 | NMI | 3 | C10orf10 | 2 | KLHL17 | 2 | SRGN | 2 |
| CD53 | 3 | NUP50 | 3 | C1orf38 | 2 | LAP3 | 2 | STAT2 | 2 |
| CDKN1A | 3 | OAS2 | 3 | C4orf32 | 2 | LIPA | 2 | STEAP4 | 2 |
| CLEC2B | 3 | OPTN | 3 | C5orf27 | 2 | LRG1 | 2 | TLR2 | 2 |
| CLEC4E | 3 | OTUD4 | 3 | CAPN8 | 2 | MAFB | 2 | TMEM192 | 2 |
| CSF2RB | 3 | PARP12 | 3 | CCDC92 | 2 | MAP3K5 | 2 | TMEM50A | 2 |
| CSRNP1 | 3 | PMAIP1 | 3 | CCL19 | 2 | MARCKS | 2 | TMEM51 | 2 |
| CTSO | 3 | PNRC1 | 3 | CD163 | 2 | MCOLN2 | 2 | TMEM86A | 2 |
| CYTH1 | 3 | PSMB10 | 3 | CD40 | 2 | MEG3 | 2 | TNFAIP3 | 2 |
| DCP1A | 3 | RABGAP1L | 3 | CD80 | 2 | MICB | 2 | TNFRSF10A | 2 |
| DDX58 | 3 | RASGRP3 | 3 | CEBPD | 2 | MYC | 2 | TRIM14 | 2 |
| DNAL4 | 3 | RBCK1 | 3 | CES1 | 2 | MYD88 | 2 | TRIM25 | 2 |
| DTX3L | 3 | RBP2 | 3 | CHUK | 2 | NEURL3 | 2 | TRIM46 | 2 |
| DUSP5 | 3 | RNF34 | 3 | CPT1A | 2 | NPAS2 | 2 | TXNIP | 2 |
| FCGR1A | 3 | SAA1 | 3 | CTCFL | 2 | NRN1 | 2 | VCAM1 | 2 |
| FNDC3B | 3 | SLC16A1 | 3 | CTSS | 2 | NT5C3 | 2 | VEGFC | 2 |
| FNDC4 | 3 | SLC25A28 | 3 | CXCL10 | 2 | OAS1 | 2 | WARS | 2 |
| G6PC | 3 | SNN | 3 | DUOXA1 | 2 | OAS3 | 2 | WNT10B | 2 |
| GABBR1 | 3 | SSBP3 | 3 | EIF2AK2 | 2 | PHF11 | 2 | XAF1 | 2 |
| GAK | 3 | STARD5 | 3 | ETV6 | 2 | PI4K2B | 2 | ZNF263 | 2 |
| GCA | 3 | STAT1 | 3 | FAM70A | 2 | PLIN2 | 2 | ZNFX1 | 2 |
| GLRX | 3 | SUN2 | 3 | FFAR2 | 2 | PLSCR1 | 2 | ||
| GPX2 | 3 | TAGAP | 3 | FTSJD1 | 2 | PPM1K | 2 | ||
| GZMB | 3 | TBX3 | 3 | GBP1 | 2 | PRIC285 | 2 | ||
| HLA-DPA1 | 3 | THBD | 3 | GBP4 | 2 | PSMB8 | 2 | ||
| IFI30 | 3 | THOC4 | 3 | GEM | 2 | PSMB9 | 2 | ||
| IFI44L | 3 | TLR3 | 3 | GJA4 | 2 | RNASE4 | 2 | ||
| IFIT1 | 3 | TMEM49 | 3 | GTPBP1 | 2 | ||||
| IFIT5 | 3 | TNFSF10 | 3 | HERC6 | 2 | ||||
| IRF1 | 3 | UBD | 3 | HESX1 | 2 | ||||
| JUNB | 3 | UBE2L6 | 3 | HLA-DRB1 | 2 | ||||
| LMO2 | 3 | UNC93B1 | 3 | HLA-F | 2 | ||||
| LOC285194 | 3 | VAMP5 | 3 | HPSE | 2 | ||||
| XRN1 | 3 | ||||||||
| ZBP1 | 3 | ||||||||
| C. ISG containing low frequency of RARE DR5 in gene regulatory region (−5000 to +500) | |||||||
|---|---|---|---|---|---|---|---|
| Low | # | Low | # | Low | # | Low | # |
| ADAR | 1 | IFIH1 | 1 | TAP1 | 1 | JAK2 | 0 |
| AGPAT9 | 1 | IFIT2 | 1 | TAP2 | 1 | LGMN | 0 |
| AHNAK2 | 1 | IFITM2 | 1 | TDRD7 | 1 | MAFF | 0 |
| AIM2 | 1 | IL15 | 1 | TIMP1 | 1 | MXD1 | 0 |
| AKT3 | 1 | IL1RN | 1 | TLK2 | 1 | NCOA3 | 0 |
| APOBEC3B | 1 | IRF2 | 1 | TNFSF13B | 1 | NDC80 | 0 |
| ATP10D | 1 | IRF7 | 1 | TRIM22 | 1 | NFIL3 | 0 |
| BAZ1A | 1 | ITIH4 | 1 | TRIM31 | 1 | PADI2 | 0 |
| BLZF1 | 1 | LRRIQ3 | 1 | TRIM38 | 1 | PHF15 | 0 |
| CASP1 | 1 | MAP2K5 | 1 | TRIM5 | 1 | PTMA | 0 |
| CASP7 | 1 | MAP3K14 | 1 | UBE2D4 | 1 | RGS1 | 0 |
| CCDC75 | 1 | MASTL | 1 | ULK4 | 1 | RSAD2 | 0 |
| CCL5 | 1 | MAX | 1 | USP18 | 1 | SELL | 0 |
| CCL8 | 1 | MB21D1 | 1 | ZCCHC2 | 1 | STAP1 | 0 |
| CD274 | 1 | MS4A4A | 1 | ZNF385B | 1 | TNFAIP6 | 0 |
| CD74 | 1 | MS4A6A | 1 | ACSL1 | 0 | TP53 | 0 |
| CD9 | 1 | MT1F | 1 | ADM | 0 | TRIM21 | 0 |
| CDC25B | 1 | MTHFD2L | 1 | ARL5B | 0 | ||
| CDK17 | 1 | NAMPT | 1 | BST2 | 0 | ||
| CMPK2 | 1 | NLRC5 | 1 | C5orf39 | 0 | ||
| COMMD3 | 1 | NOD2 | 1 | C9orf91 | 0 | ||
| CSDA | 1 | OASL | 1 | CCDC109B | 0 | ||
| CXCL11 | 1 | OGFR | 1 | CCNA1 | 0 | ||
| CYP1B1 | 1 | PDGFRL | 1 | CCND3 | 0 | ||
| DUSP6 | 1 | PFKFB3 | 1 | CCRL1 | 0 | ||
| DYNLT1 | 1 | PLAUR | 1 | CD69 | 0 | ||
| EHD4 | 1 | PLEKHA4 | 1 | CFB | 0 | ||
| EPSTI1 | 1 | PML | 1 | CHMP5 | 0 | ||
| ETV7 | 1 | PMM2 | 1 | CREB3L3 | 0 | ||
| EXT1 | 1 | PNPT1 | 1 | CRY1 | 0 | ||
| FAM125B | 1 | PRAME | 1 | CX3CL1 | 0 | ||
| FAM46A | 1 | PRKD2 | 1 | DDIT4 | 0 | ||
| FAM72B | 1 | PXK | 1 | DDX3X | 0 | ||
| FBXO6 | 1 | RAB8B | 1 | DHX58 | 0 | ||
| FST | 1 | RASSF4 | 1 | EIF3L | 0 | ||
| FTSJD2 | 1 | RIPK2 | 1 | EPAS1 | 0 | ||
| FZD5 | 1 | RNF114 | 1 | ERLIN1 | 0 | ||
| GALNT2 | 1 | RNF213 | 1 | FAM111A | 0 | ||
| GBP5 | 1 | RTP4 | 1 | FKBP5 | 0 | ||
| GCH1 | 1 | SCARB2 | 1 | FLJ39639 | 0 | ||
| GLB1 | 1 | SDS | 1 | GBP3 | 0 | ||
| GMPR | 1 | SECTM1 | 1 | GCNT1 | 0 | ||
| GTPBP2 | 1 | SERPINB9 | 1 | HES4 | 0 | ||
| HIST2H2AA | 1 | SIRPA | 1 | HK2 | 0 | ||
| HLA-C | 1 | SLC22A23 | 1 | HSH2D | 0 | ||
| HLA-DQB1 | 1 | SLC25A30 | 1 | IFI27 | 0 | ||
| HLA-E | 1 | SLFN5 | 1 | IL18BP | 0 | ||
| SOCS2 | 1 | IRF8 | 0 | ||||
Our study found that ADH-ALDH pathway restricts HCV lifecycle at either protein translation and/or genome replication stage. As a similar pattern of HCV suppression were observed in IFN treated HCV-SGR cells(data not shown), it is possible to speculate that the ISGs up-regulated by the ADH-ALDH pathway cooperatively drives this phenomenon. Because these two stages of viral lifecycle have great influence on each other, further investigation is required in order to precisely define the mechanism of how RA-regulated ISGs exhibit antiviral activities.
Our results also imply the possibility that the catabolism of RA by CYP2E1 may contribute to the impairments of ISGs expression. In addition, others has reported that the oxidative stress triggered by CYP2E1 impairs the IFN-mediated ISGs expression through inhibition of STAT1 tyrosine phosphorylation(40), indicating that CYP2E1 impairs ISGs expression through multiple mechanisms. Furthermore, chronic liver diseases such as ALD and HCV infection is known to cause hypovitaminosis A due to the myo-fibroblastic transformation of HSC(29, 41). This mechanism likely further aggravates the impairment of RA-regulated ISG expression, and thus explain the refractoriness to IFN-based antiviral therapy among advanced liver disease patients.
In summary, our study provided novel insights on the pathophysiology behind the synergistic liver disease progression between HCV infection and ALD. The insights obtained from our study are well applied to the high-risk demographics of HCV infection, as nearly half of infected populations are heavy drinkers.
Supplementary Material
Acknowledgments
We would like to thank: T. Tellinghuisen(The Scripps Research Institute) and M. Gale(University of Washington) for reagents, Dr. Jessica Y. Rathbun and EeLyn Ooi for technical assistance.
Financial support: This work was supported by funds from AASLD/ALF Liver Scholar Award, Baxter Foundation Award, SCRC for ALPD & Cirrhosis Pilot Project Grant(5P50AA011999) and USC RCLD pilot grant(5P30DK048522), NIAAA(R21AA022751), NIDDK(RO1DK101773), ACS IRG(to T.S), NIGMS (P20GM103408)(to J.R.C).
Abbreviations
- ATRA
all trans retinoic acid
- ALD
alcoholic liver disease
- IFN
interferon
- ISG
Interferon Stimulated Genes
- HCV
hepatitis C virus
References
- 1.Gravitz L. Introduction: a smouldering public-health crisis. Nature. 2011;474:S2–4. doi: 10.1038/474S2a. [DOI] [PubMed] [Google Scholar]
- 2.Edlin BR, Carden MR. Injection drug users: the overlooked core of the hepatitis C epidemic. Clin Infect Dis. 2006;42:673–676. doi: 10.1086/499960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Edlin BR, Eckhardt BJ, Shu MA, Holmberg SD, Swan T. Toward a more accurate estimate of the prevalence of hepatitis C in the United States. Hepatology. 2015;62:1353–1363. doi: 10.1002/hep.27978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Russell M, Pauly MP, Moore CD, Chia C, Dorrell J, Cunanan RJ, Witt G, et al. The impact of lifetime alcohol use on hepatitis C treatment outcomes in privately insured members of an integrated health care plan. Hepatology. 2012;56:1223–1230. doi: 10.1002/hep.25755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peters MG, Terrault NA. Alcohol use and hepatitis C. Hepatology. 2002;36:S220–S225. doi: 10.1053/jhep.2002.36811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Crabb DW, Matsumoto M, Chang D, You M. Overview of the role of alcohol dehydrogenase and aldehyde dehydrogenase and their variants in the genesis of alcohol-related pathology. Proc Nutr Soc. 2004;63:49–63. doi: 10.1079/pns2003327. [DOI] [PubMed] [Google Scholar]
- 7.Chase JR, Poolman MG, Fell DA. Contribution of NADH increases to ethanol's inhibition of retinol oxidation by human ADH isoforms. Alcohol Clin Exp Res. 2009;33:571–580. doi: 10.1111/j.1530-0277.2008.00871.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Balmer JE, Blomhoff R. Gene expression regulation by retinoic acid. J Lipid Res. 2002;43:1773–1808. doi: 10.1194/jlr.r100015-jlr200. [DOI] [PubMed] [Google Scholar]
- 9.Villamor E, Koulinska IN, Aboud S, Murrin C, Bosch RJ, Manji KP, Fawzi WW. Effect of vitamin supplements on HIV shedding in breast milk. Am J Clin Nutr. 2010;92:881–886. doi: 10.3945/ajcn.2010.29339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Neuzil KM, Gruber WC, Chytil F, Stahlman MT, Graham BS. Safety and pharmacokinetics of vitamin A therapy for infants with respiratory syncytial virus infections. Antimicrob Agents Chemother. 1995;39:1191–1193. doi: 10.1128/aac.39.5.1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bocher WO, Wallasch C, Hohler T, Galle PR. All-trans retinoic acid for treatment of chronic hepatitis C. Liver Int. 2008;28:347–354. doi: 10.1111/j.1478-3231.2007.01666.x. [DOI] [PubMed] [Google Scholar]
- 12.Mora JR, Iwata M, von Andrian UH. Vitamin effects on the immune system: vitamins A and D take centre stage. Nat Rev Immunol. 2008;8:685–698. doi: 10.1038/nri2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schoggins JW, Rice CM. Interferon-stimulated genes and their antiviral effector functions. Curr Opin Virol. 2011;1:519–525. doi: 10.1016/j.coviro.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cho H, Proll SC, Szretter KJ, Katze MG, Gale M, Jr, Diamond MS. Differential innate immune response programs in neuronal subtypes determine susceptibility to infection in the brain by positive-stranded RNA viruses. Nat Med. 2013;19:458–464. doi: 10.1038/nm.3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saito T, Owen DM, Jiang FG, Marcotrigiano J, Gale M. Innate immunity induced by composition-dependent RIG-I recognition of hepatitis C virus RNA. Nature. 2008;454:523–527. doi: 10.1038/nature07106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sen GC. Viruses and interferons. Annual Review of Microbiology. 2001;55:255–281. doi: 10.1146/annurev.micro.55.1.255. [DOI] [PubMed] [Google Scholar]
- 17.Mao M, Yu M, Tong JH, Ye J, Zhu J, Huang QH, Fu G, et al. RIG-E, a human homolog of the murine Ly-6 family, is induced by retinoic acid during the differentiation of acute promyelocytic leukemia cell. Proc Natl Acad Sci U S A. 1996;93:5910–5914. doi: 10.1073/pnas.93.12.5910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu M, Tong JH, Mao M, Kan LX, Liu MM, Sun YW, Fu G, et al. Cloning of a gene (RIG-G) associated with retinoic acid-induced differentiation of acute promyelocytic leukemia cells and representing a new member of a family of interferon-stimulated genes. Proc Natl Acad Sci U S A. 1997;94:7406–7411. doi: 10.1073/pnas.94.14.7406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takahashi T, Lasker JM, Rosman AS, Lieber CS. Induction of cytochrome P-4502E1 in the human liver by ethanol is caused by a corresponding increase in encoding messenger RNA. Hepatology. 1993;17:236–245. [PubMed] [Google Scholar]
- 20.Ooi EL, Chan ST, Cho NE, Wilkins C, Woodward J, Li M, Kikkawa U, et al. Novel antiviral host factor, TNK1, regulates IFN signaling through serine phosphorylation of STAT1. Proc Natl Acad Sci U S A. 2014;111:1909–1914. doi: 10.1073/pnas.1314268111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bartosch B, Dubuisson J, Cosset FL. Infectious hepatitis C virus pseudo-particles containing functional E1–E2 envelope protein complexes. J Exp Med. 2003;197:633–642. doi: 10.1084/jem.20021756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brick J. Standardization of alcohol calculations in research. Alcohol Clin Exp Res. 2006;30:1276–1287. doi: 10.1111/j.1530-0277.2006.00155.x. [DOI] [PubMed] [Google Scholar]
- 23.Clemens DL, Halgard CM, Miles RR, Sorrell MF, Tuma DJ. Establishment of a recombinant hepatic cell line stably expressing alcohol dehydrogenase. Arch Biochem Biophys. 1995;321:311–318. doi: 10.1006/abbi.1995.1400. [DOI] [PubMed] [Google Scholar]
- 24.Muldrew KL, James LP, Coop L, McCullough SS, Hendrickson HP, Hinson JA, Mayeux PR. Determination of acetaminophen-protein adducts in mouse liver and serum and human serum after hepatotoxic doses of acetaminophen using high-performance liquid chromatography with electrochemical detection. Drug Metab Dispos. 2002;30:446–451. doi: 10.1124/dmd.30.4.446. [DOI] [PubMed] [Google Scholar]
- 25.Dranse HJ, Sampaio AV, Petkovich M, Underhill TM. Genetic deletion of Cyp26b1 negatively impacts limb skeletogenesis by inhibiting chondrogenesis. J Cell Sci. 2011;124:2723–2734. doi: 10.1242/jcs.084699. [DOI] [PubMed] [Google Scholar]
- 26.Duester G. Families of retinoid dehydrogenases regulating vitamin A function: production of visual pigment and retinoic acid. Eur J Biochem. 2000;267:4315–4324. doi: 10.1046/j.1432-1327.2000.01497.x. [DOI] [PubMed] [Google Scholar]
- 27.James LP, Capparelli EV, Simpson PM, Letzig L, Roberts D, Hinson JA, Kearns GL, et al. Acetaminophen-associated hepatic injury: evaluation of acetaminophen protein adducts in children and adolescents with acetaminophen overdose. Clin Pharmacol Ther. 2008;84:684–690. doi: 10.1038/clpt.2008.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bastien J, Rochette-Egly C. Nuclear retinoid receptors and the transcription of retinoid-target genes. Gene. 2004;328:1–16. doi: 10.1016/j.gene.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 29.Bitetto D, Bortolotti N, Falleti E, Vescovo S, Fabris C, Fattovich G, Cussigh A, et al. Vitamin A deficiency is associated with hepatitis C virus chronic infection and with unresponsiveness to interferon-based antiviral therapy. Hepatology. 2013;57:925–933. doi: 10.1002/hep.26186. [DOI] [PubMed] [Google Scholar]
- 30.Liu C, Russell RM, Seitz HK, Wang XD. Ethanol enhances retinoic acid metabolism into polar metabolites in rat liver via induction of cytochrome P4502E1. Gastroenterology. 2001;120:179–189. doi: 10.1053/gast.2001.20877. [DOI] [PubMed] [Google Scholar]
- 31.Lutz JD, Dixit V, Yeung CK, Dickmann LJ, Zelter A, Thatcher JE, Nelson WL, et al. Expression and functional characterization of cytochrome P450 26A1, a retinoic acid hydroxylase. Biochem Pharmacol. 2009;77:258–268. doi: 10.1016/j.bcp.2008.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Falk DPD, Yi HYPD, Hiller-Sturmhofel SPD. An Epidemiologic Analysis of Co-Occurring Alcohol and Drug Use and Disorders: Findings From the National Epidemiologic Survey of Alcohol and Related Conditions (NESARC) Alcohol Res Health. 2008;31:100–110. [PMC free article] [PubMed] [Google Scholar]
- 33.Bhattacharya R, Shuhart MC. Hepatitis C and alcohol: interactions, outcomes, and implications. J Clin Gastroenterol. 2003;36:242–252. doi: 10.1097/00004836-200303000-00012. [DOI] [PubMed] [Google Scholar]
- 34.Vonghia L, Leggio L, Ferrulli A, Bertini M, Gasbarrini G, Addolorato G. Acute alcohol intoxication. Eur J Intern Med. 2008;19:561–567. doi: 10.1016/j.ejim.2007.06.033. [DOI] [PubMed] [Google Scholar]
- 35.Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008;88:125–172. doi: 10.1152/physrev.00013.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Deltour L, Foglio MH, Duester G. Metabolic deficiencies in alcohol dehydrogenase Adh1, Adh3, and Adh4 null mutant mice. Overlapping roles of Adh1 and Adh4 in ethanol clearance and metabolism of retinol to retinoic acid. J Biol Chem. 1999;274:16796–16801. doi: 10.1074/jbc.274.24.16796. [DOI] [PubMed] [Google Scholar]
- 37.Duester G. Involvement of alcohol dehydrogenase, short-chain dehydrogenase/reductase, aldehyde dehydrogenase, and cytochrome P450 in the control of retinoid signaling by activation of retinoic acid synthesis. Biochemistry. 1996;35:12221–12227. doi: 10.1021/bi961176+. [DOI] [PubMed] [Google Scholar]
- 38.Zheng PZ, Wang KK, Zhang QY, Huang QH, Du YZ, Zhang QH, Xiao DK, et al. Systems analysis of transcriptome and proteome in retinoic acid/arsenic trioxide-induced cell differentiation/apoptosis of promyelocytic leukemia. Proc Natl Acad Sci U S A. 2005;102:7653–7658. doi: 10.1073/pnas.0502825102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Delacroix L, Moutier E, Altobelli G, Legras S, Poch O, Choukrallah MA, Bertin I, et al. Cell-specific interaction of retinoic acid receptors with target genes in mouse embryonic fibroblasts and embryonic stem cells. Mol Cell Biol. 2010;30:231–244. doi: 10.1128/MCB.00756-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McCartney EM, Semendric L, Helbig KJ, Hinze S, Jones B, Weinman SA, Beard MR. Alcohol metabolism increases the replication of hepatitis C virus and attenuates the antiviral action of interferon. J Infect Dis. 2008;198:1766–1775. doi: 10.1086/593216. [DOI] [PubMed] [Google Scholar]
- 41.Blaner WS, O'Byrne SM, Wongsiriroj N, Kluwe J, D'Ambrosio DM, Jiang H, Schwabe RF, et al. Hepatic stellate cell lipid droplets: a specialized lipid droplet for retinoid storage. Biochim Biophys Acta. 2009;1791:467–473. doi: 10.1016/j.bbalip.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.