

Abstract
Background
Changes in the intestinal microbiome of patients with short bowel syndrome (SBS) are thought to significantly affect clinical outcome. These changes may not only delay enteral diet advancement but may also predispose patients to bacterial translocation, bacteremia, and liver disease. Patients with SBS are thought to be more susceptible to changes in gut microbial communities due to intestinal dysmotility and/or lack of anatomic safeguards such as the ileocecal valve.
Materials and Methods
We analyzed the bacterial composition of 21 fecal specimens from 9 children with SBS and 8 healthy children ages 4 months to 8 years by 16S ribosomal RNA gene sequencing. The sequences were quality filtered and analyzed using QIIME, the Ribosomal Database Project Classifier, and the randomForest supervised learning algorithm.
Results
The fecal microbiome of patients with SBS is different from that of healthy controls. Stool from patients with SBS had a significantly greater abundance of the bacterial classes Gammaproteobacteria and Bacilli. Stool from patients with SBS who experienced increased stool frequency tended to have increased abundance of Lactobacillus (P = .057) and decreased abundance of Ruminococcus.
Conclusion
This study shows that the fecal microbiome of patients with SBS is significantly different from that of healthy controls when analyzed by 16S metagenomics. Differences in the composition and function of gut microbiomes in children with SBS may affect bowel physiology, and these findings may provide new opportunities for intestinal rehabilitation and clinical management.
Keywords: short bowel syndrome, microbiome, gammaproteobacteria, dysbiosis
Introduction
Short bowel syndrome (SBS) is defined as insufficient small intestinal length to maintain protein-energy, fluid, electrolyte, or micronutrient balances when on a conventionally accepted diet.1 In children, SBS can result from congenital causes such as gastroschisis or intestinal atresias as well as postnatal loss of bowel, most commonly from necrotizing enterocolitis (NEC).2,3 The clinical outcome of children with SBS is influenced by their age, etiology of SBS, length of remaining bowel, incidence of sepsis and liver disease, and the ability to increase the proportion of calories taken enterally.2,4–7
Children with SBS often require long-term intravenous (IV) nutrition supplementation while slowly increasing enteral intake.8,9 The progress of diet advancement is often slowed or reversed by recurrent bouts of diarrhea and increased gas production, a process thought to result from changes in the composition of the intestinal microbiome.10–14 Clinically, these symptoms are managed empirically by diet modification and rotating antibiotic regimens.14
Changes in the microbiome may not only delay enteral diet advancement but also predispose patients to bacterial translocation, bacteremia, and liver disease.15–17 Patients with SBS are thought to be more susceptible to changes in gut microbial communities due to intestinal dysmotility and/or lack of anatomic safeguards such as the ileocecal valve.15,18,19 The exact mechanisms by which changes in microbial communities lead to complications are poorly understood, but bacterial by-products and mucosal inflammation may contribute.20–22
In the era of the Human Microbiome Project23–26 and next-generation sequencing technology, we have the ability to identify many uncultured intestinal bacteria.27,28 For example, using high-throughput 16S ribosomal RNA (rRNA) gene sequencing techniques, several recent studies have shown shifts in the communities of intestinal bacteria in a variety of disease states.29–31 Specifically, the class Gammaproteobacteria was present in greater quantities in children who developed NEC,32,33 adults and children with irritable bowel syndrome,34,35 and adults following gastric bypass surgery.36
Prior attempts at identifying the intestinal microbiome of pediatric patients with SBS have relied on culture-based methods.18 In the only published study of adult patients with SBS using next-generation sequencing techniques, an increased abundance of Escherichia coli, a member of the taxonomic class Gammaproteobacteria, and Lactobacillus were found in patients with SBS compared with healthy controls.37 The aim of our study was to characterize the fecal microbiome of the pediatric SBS population using high-throughput sequencing of the bacterial 16S rRNA gene. We also hypothesized that patients with SBS with and without diarrhea would differ with respect to microbial composition.
Materials and Methods
Patients
Children with SBS were recruited from the Intestinal Rehabilitation Clinic at Texas Children's Hospital, Houston, Texas. All recruitment and study procedures were approved by the Baylor College of Medicine Institutional Review Board, and informed consent was obtained from the parents and assent from the children where appropriate.
A total of 9 children with SBS were enrolled in the study (ages 4 months to 4 years), providing 14 fecal specimens. Specimens were considered “diarrhea” if their presence necessitated a change in clinical management, such as dietary modification and/or antibiotic usage. Inclusion criteria specified that children must have a diagnosis of SBS resulting from surgical resection or a congenital anomaly such as gastroschisis or intestinal atresia. Exclusion criteria included congenital malabsorptive disorders or functional short bowel with expected bowel length, known bacterial or viral causes of diarrhea, and bacteremia.
Sequence libraries generated from feces of SBS children were compared with identically extracted and sequenced 16S rRNA gene libraries (V3–V5 region) obtained from feces of 8 healthy control children (ages 7–8 years), using data previously published.35 Exclusion criteria for healthy control children included use of antimicrobial agents, probiotics, or steroids (oral, nasal, or inhaled) within 6 months of fecal sampling. Full inclusion and exclusion criteria for control children, along with subject metadata, are archived at dbGAP under accession phs000265.v3.p1.
Fecal Specimen Collection and Microbial DNA Extraction
Fecal specimens were collected directly from a diaper or in a stool “hat” placed in the toilet, immediately transferred into a sterile cup, and stored at −80°C. DNA extraction, bacterial 16S rRNA gene amplification, and 16S metagenomic sequencing were performed as previously described by the Human Microbiome Project Consortium (http://www.hmpdacc.org/doc/HMP_MDG_454_16S_Protocol.pdf).25,26
In brief, DNA was extracted from stool using a commercial manual DNA extraction kit (PowerSoil DNA Isolation Kit; MO-BIO Laboratories, Carlsbad, CA), with minor modifications. The resulting bacterial DNA was quantitated and assessed for quality using the NanoDrop ND-1000 (Thermo Fisher Scientific, Wilmington, DE) and the Qubit 2.0 Fluorometer (Life Technologies, Grand Island, NY).
16S rRNA Gene Sequence Generation and Analysis
Individual libraries were constructed via amplification of the V3–V5 region of the 16S rRNA gene using barcoded universal bacterial primers 357F (5′-CCTACGGGAGGCAGCAG-3′) and 926R (5′-CCGTCAATTCMTTTRAGT-3′). Following emulsion polymerase chain reaction (PCR) and sequence reaction preparation, sequencing was performed on the GS-FLX platform (454 Life Sciences/Roche, Branford, CT), using the 926R amplification primer as the sequencing primer.
Sequence data were parsed by barcode and quality filtered using the QIIME software package v. 1.2.0,38 as implemented in the Genboree Microbiome Toolbench (Bioinformatics Research Laboratory, Baylor College of Medicine, Houston, TX).39 Sequences exceeding 200 bp length, having average quality scores of 20 or greater, and harboring no ambiguous bases or mismatches to their barcode or sequencing primer were retained. The quality-filtered sequence data from both the healthy and SBS cohorts were pooled, and sequences were assigned to operational taxonomic units (OTUs, sequences sharing ≥97% similarity), de novo, using Cd-hit.40 Potentially chimeric sequences were identified using the ChimeraSlayer algorithm,41 and all potential chimeras were excluded from downstream analysis. Identities were assigned to each OTU using the Ribosomal Database Project Classifier42 and RDP release version 10 (Center for Microbial Ecology, Michigan State University, East Lansing, MI).
Prior to the calculation of diversity indices or comparison of sequence libraries across our SBS and healthy cohorts, each of the sequence libraries was randomly subsampled to an even depth across all sequences (1134 sequences). Two of 14 SBS sequence libraries were exceptionally small and failed to meet this threshold; these libraries were excluded from further analysis.
The subsampled sequence libraries were analyzed further, evaluating beta-diversity via principal coordinates analysis of UniFrac distances in QIIME. Using the randomForest supervised learning algorithm43 in conjunction with Boruta feature selection,44 the possible existence of predictive taxa or community “signatures” was evaluated within these microbial communities. The combined randomForest and Boruta feature selection analysis was conducted using the Genboree Microbiome Toolbench. The sequence data associated with this study have been deposited in the NCBI Short Read Archive under accession number SRA054875.
Statistical Analysis
The Mann-Whitney test was applied to the relative abundances of microbial taxa to compare pediatric SBS samples and healthy pediatric samples and to compare the SBS samples with each other based on clinical phenotype. Mann-Whitney and standard error calculations were performed on GraphPad Prism version 5 (GraphPad Software, La Jolla, CA). Data are shown as median or mean ± the standard error of the mean as appropriate to the data.
Results
Characteristics of Healthy Controls and Patients With SBS
The children with SBS (n = 9) were younger than the healthy controls (n = 8): 2.2 ± 0.4 vs 7.6 ± 0.2 years, respectively (P < .0001). Three patients with SBS and 6 healthy controls were female. All patients with SBS had been treated with antibiotics within 6 months, and 7 of 9 patients were treated with metronidazole. None of the patients with SBS were on motility agents or probiotics. They also did not have evidence of liver dysfunction or portal hypertension. Additional clinical, surgical, and dietary history of the 9 children with SBS is included in Table 1. Clinical data on the healthy control children are included in Table 2.
Table 1.
Clinical Characteristics of Patients With SBS.
| Clinical Characteristic | Value |
|---|---|
| Age, mean ± SD (range) | 2.2 ± 0.4 (4 mo to 4 y) |
| Female, No. | 3 |
| Weight % | 17 ± 6.4 |
| Antibiotic use within 6 mo, No. | 9/9 (metronidazole 7/9) |
| Etiology of SBS, No. | |
| Gastroschisis | 4 |
| NEC | 3 |
| Midgut volvulus | 1 |
| Congenital atresia | 1 |
| Presence of ileocecal valve, No. | 3 |
| Bowel length, mean ± SD (range), cm | 50 ± 8 (20–80) |
| Surgical lengthening procedure, No. | 4 |
| Enteral feeding type, No. | |
| Elemental formula | 6 |
| Intact protein formula | 2 |
| Breast milk | 1 |
| Calories from PN, mean ± SD (range), % | 25 ± 11.5 (0–100) |
NEC, necrotizing enterocolitis; PN, parenteral nutrition; SBS, short bowel syndrome.
Table 2.
Clinical Characteristics of 8 Healthy Control Children.
| Clinical Characteristic | Value |
|---|---|
| Age, mean ± SD (range), y | 7.6 ± 0.2 (7–8) |
| Female, No. | 6 |
| Body mass index, mean ± SD, kg/m2 | 16.1 ± 0.7 |
| Antibiotic use within 6 mo, No. | 0/8 |
A total of 20 fecal specimens were collected and analyzed, including 12 specimens from 9 patients with SBS and 8 specimens from 8 different healthy control subjects. Five of 12 SBS specimens were collected when the patients were experiencing diarrhea, necessitating a change in clinical management, such as diet modification and/or antibiotic usage.
Bacterial Community Composition Within Healthy Children and Patients With SBS
Samples from patients with SBS and healthy controls yielded between 1134 and 13,928 reads a piece, with an average read length of 532 bp. Given the substantial variation in sequence yield, all libraries were randomly subsampled to 1134 sequences per sample. A total of 793 OTUs were detected across all samples with individual sequence libraries containing 37–131 OTUs each (73 ± 5). Bacteria identified in samples from patients with SBS included members of the phyla Actinobacteria, Bacteroidetes, Firmicutes, Fusobacteria, and Proteobacteria, while bacteria identified in samples from control subjects included members of the Actinobacteria, Bacteroidetes, Cyanobacteria, Firmicutes, Proteobacteria, and Verrucomicrobia. As seen in Figure 1, the relative abundance of bacterial phyla differed between study groups. In SBS samples, Firmicutes was the most abundant bacterial phylum (48%), followed by Bacteroidetes (29%). Conversely, the 2 most abundant phyla in control samples were Bacteroidetes (67%) and Firmicutes (30%). Whereas Proteobacteria comprised 22% of the relative abundance of SBS samples, this phylum comprised <1% of the relative abundance of control samples. Ninety-nine percent of SBS samples comprised 4 bacterial classes: Bacteroidetes (29%), Bacilli (26%), Clostridia (22%), and Gammaproteobacteria (22%). In contrast, >95% of the bacterial communities in control samples comprised 2 classes, Bacteroidetes (67%) and Clostridia (27%).
Figure 1.
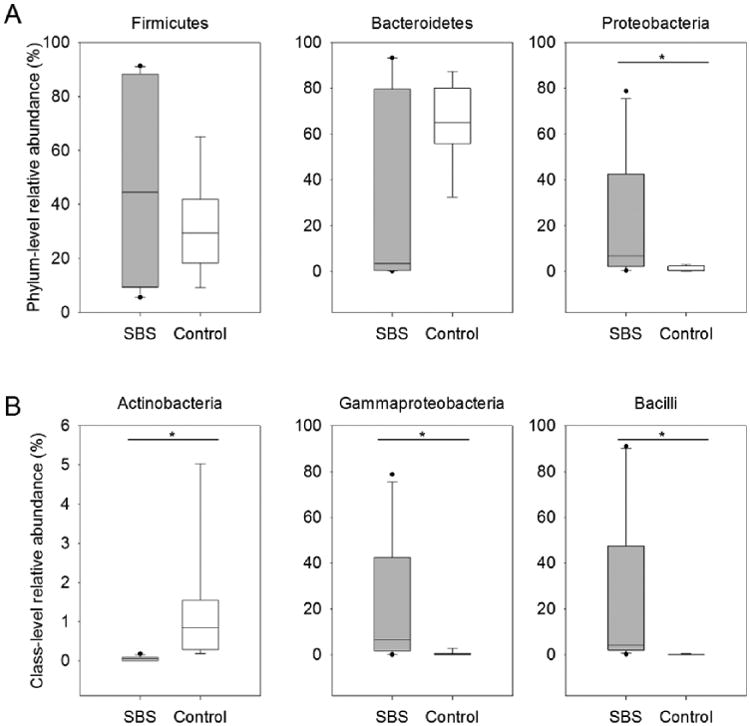
The relative abundances of bacterial phyla (A) and classes (B) in 8 fecal samples from healthy controls and 12 fecal samples from patients with short bowel syndrome (SBS), as detected by sequencing of the 16S ribosomal RNA gene (V3–V5 region). The box plots depict median and interquartile range, their whiskers denote the 10th and 90th percentiles of each group, and dots indicate outlying values. Plots that contain an asterisk (*) are taxa that significantly differ between patients with SBS and controls at P < .05 using a Mann-Whitney test.
We identified multiple taxa that differed significantly between the 2 study groups using the Mann-Whitney test. We found that the microbiomes of patients with SBS were significantly enriched with respect to the relative abundances of taxa belonging to the phylum Proteobacteria (P < .01) and, more specifically, to the class Gammaproteobacteria (P < .001), while healthy control microbiomes were enriched with respect to the relative abundance of taxa belonging to the phylum Actinobacteria (P < .001). The relative abundance of the phylum Firmicutes tended to be greater in fecal samples from patients with SBS relative to those from the healthy controls. Contributing toward this trend, the relative abundance of members of the class Bacilli was significantly greater (P < .001) in patients with SBS than in control subjects, regardless of clinical symptoms.
Genus-Level Differences Detected in the Microbiome Composition of Patients With SBS Relative to Healthy Controls
To obtain a comprehensive perspective of our data, we used principal coordinates analysis (PCoA) to visualize community-level similarity between our sample sets. Using the unweighted UniFrac distance algorithm, distances between the bacterial communities harbored in each specimen were calculated in a phylogenetically informed manner. Our PCoA plot (Figure 2) shows a strong distinction between samples from patients with SBS and healthy controls. While not as uniformly clustered as those from the healthy control group, most SBS samples tended to be more similar to one another than to their healthy counterparts. When the SBS sample communities were further subdivided by clinical criteria such as frequency of stools, bowel length, and etiology of SBS, no further clustering was evident.
Figure 2.
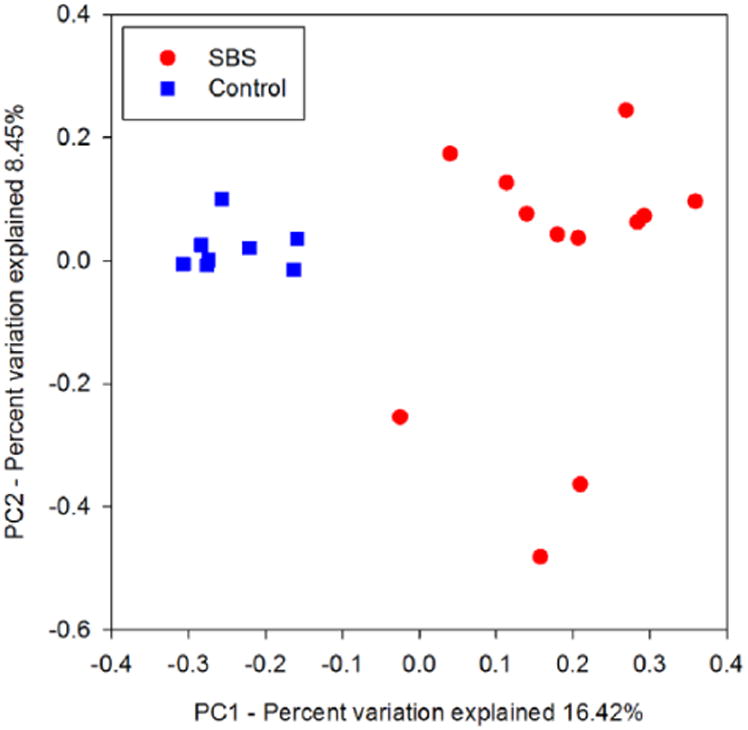
Healthy children and patients with short bowel syndrome (SBS) harbored different gut microbiome structures, as detected by 16S ribosomal RNA gene sequencing. The bacterial communities within the 20 fecal samples of control subjects and patients with SBS clustered by group when analyzed by principal coordinates analysis of unweighted UniFrac distances. Blue square = healthy control subjects. Red circle = patients with SBS.
We used supervised learning to identify a set of bacterial taxa that distinguish different phenotypes. Using randomForest for supervised learning and Boruta for feature selection, the algorithm selected the bacterial taxa that best discriminated between patients with SBS and healthy controls (Figure 3). Our supervised learning approach indicated that increased relative abundances of OTUs resembling Escherichia/Shigella and Streptococcus were indicative of the SBS phenotype. In contrast, increased relative abundances of OTUs belonging to the genera Alistipes and Parabacteroides and the family Lachnospiraceae were indicative of the control state. Similar to our findings with PCoA, when we attempted to further define phenotypes based on SBS clinical data such as bowel length and etiology of SBS, no representative bacterial taxa were identified (data not shown).
Figure 3.
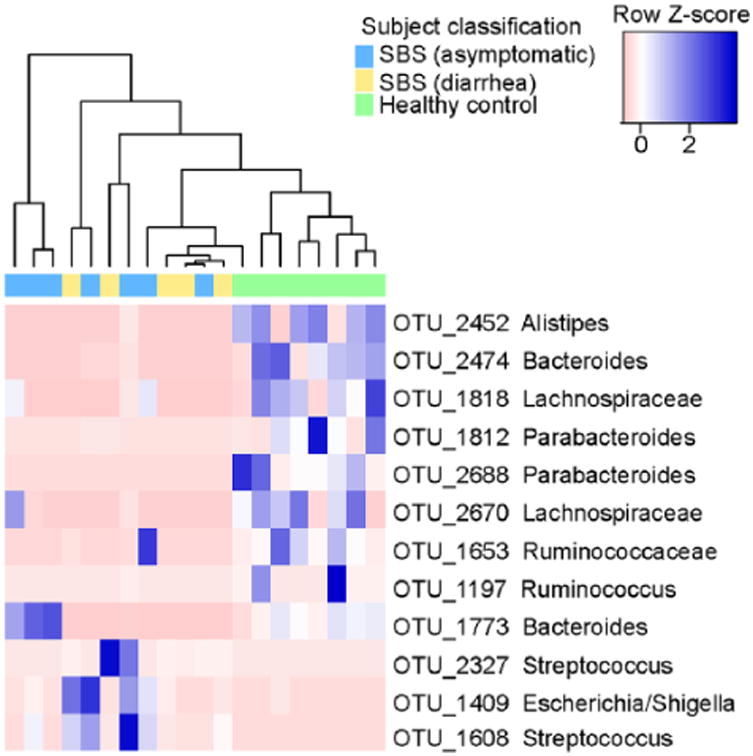
Heatmap of bacterial operational taxonomic units (OTUs) contributing to the differentiation of short bowel syndrome (SBS) stool communities (diarrheal and asymptomatic) from those of healthy controls. Taxa were characterized using 16S ribosomal RNA gene sequencing, and those features that were identified by the randomForest classification algorithm as differentiating the 2 subject groups are highlighted here. The relative abundances of each OTU are scaled by row (blue = enriched relative to the median, pink = depleted relative to the median), and each differed significantly between patients with SBS and healthy controls when analyzed via Mann-Whitney tests. Taxonomic identities associated with each OTU represent the lowest taxonomic depth that could be assigned with >80% confidence by the Ribosomal Database Project Classifier.
Microbiomes of SBS Phenotypes Differ Based on Clinical Symptoms
Fecal samples from patients with SBS were further analyzed to explore differences in the relative abundance of microbial taxa in the context of additional clinical metadata. Samples obtained while patients with SBS had diarrhea tended to harbor a greater relative abundance of Lactobacillus species compared with patients with SBS without diarrhea (1.3% ± 0.6% vs 0.3% ± 0.2%, P = .057). Interestingly, the relative abundance of Lactobacillus in the samples from patients with diarrhea was significantly different from those in healthy controls (0.00617 vs 0.00000, respectively; 95% confidence interval [CI], 0.00177–0.03263; P = .0034), while that from samples without diarrhea was not. In addition, while patients with SBS with diarrhea had no measurable component of the genus Ruminococcus in their sequence libraries, those without diarrhea yielded this genus at lower relative abundances than in healthy controls (0.00176 vs 0.00705; 95% CI, −0.06702 to 0.00001; P = .057).
Discussion
Our data suggest that the fecal microbiomes of children with SBS are significantly different from those found in healthy control children. The SBS fecal microbiomes harbored greater relative abundances of species from the phylum Proteobacteria and the class Gammaproteobacteria. The class Bacilli was also observed at greater relative abundances in SBS samples. Furthermore, supervised learning was able to demonstrate that specific taxa (OTUs) from the genera Escherichia/Shigella and Streptococcus, members of the classes Gammaproteobacteria and Bacilli, respectively, were indicative of the SBS phenotype.
The findings of this study are consistent with a study published by Joly et al37 on the intestinal microbiome of adults with SBS. In that study, analysis of the fecal microbiome with genus-specific primers showed increased relative abundances of Lactobacillus, a member of the class Bacilli, in fecal samples of patients with SBS compared with healthy controls. The authors also analyzed colonic mucosal biopsy specimens and found increased relative abundances of both Lactobacillus as well as Escherichia coli, a member of the class Gammaproteobacteria, in patients with SBS. Since members of the genus Lactobacillus are more commonly found in the small intestine of healthy subjects,31 Lactobacillus may thrive in the SBS colonic milieu or these bacteria may contribute to disease symptoms in patients with SBS.
Several recent studies using next-generation sequencing techniques have described increased relative abundances of the class Gammaproteobacteria in the intestinal microbiomes of various disease states. Studies by Wang et al32 and Mai et al33 found increased quantities of Gammaproteobacteria in fecal samples of premature infants who developed NEC. Another study by Zhang et al36 discovered that obese patients who underwent gastric bypass surgery had an increased relative abundance of Gammaproteobacteria in their fecal samples. Two studies on irritable bowel syndrome (IBS) found similar changes in the abundance of Gammaproteobacteria. A study by Rajiliċ-Stojanoviċ et al34 showed that increased relative abundances of Gammaproteobacteria in adults were associated with worse IBS symptoms. Similarly, Saulnier et al35 showed that children with IBS had an increased relative abundance of Gammaproteobacteria in their fecal samples.
As a class, Gammaproteobacteria contain the pathogenic genera Haemophilus, Proteus, Klebsiella, Escherichia, and Shigella that have been associated with patients in the intensive care unit (ICU) setting.45 While present in small amounts in healthy intestines as shown in our study and others,25 they may contribute directly to chronic disease states involving the human intestine. Alternatively, these microbes may thrive in specific pathological conditions relative to other bacterial taxa. In the case of SBS, increased amounts of these specific bacterial groups or taxa may result in chronic intestinal inflammation and possibly translocation, leading to sepsis and liver disease.15–17 In addition, specific bacterial taxa may fill a void left in the microbiome by the application of antibiotics. Alternatively, it is possible that these microorganisms are increased in patients with SBS because they contribute to physiologic adaptation, maintenance of basic functions, and growth of the intestine. The identification of roles for different bacterial species in SBS pathogenesis would be desirable before physicians begin to change clinical practice and antibiotic selection.
Although our comparative numbers are small, it is interesting to note that the relative abundance of Lactobacillus species tended to be greater in the SBS diarrheal vs nondiarrheal samples. The relative abundance of Lactobacillus was also greater in the SBS diarrheal samples compared with control samples. As noted above, the colonic environment of patients with SBS, particularly those with diarrhea, may promote the growth of Lactobacillus or simply affect less adherent or persistent bacterial taxa to a greater extent by promoting the loss of other bacterial taxa. Alternatively, the acid-producing Lactobacillus may play a role in causing diarrheal symptoms in the context of a short bowel by lowering the intestinal pH, thereby promoting a microbial community that produces deleterious microbial factors.
One possible limitation of this study is that the children with SBS were younger than the healthy children. Although the human gut microbiome is known to change as we age,46,47 studies examining the gut microbial communities of healthy, young children (eg, 2–6 years of age) are limited in number.48,49 Given the limited availability of healthy reference data, we have no evidence that the taxa reported here are differentially abundant between healthy children in these age groups. In addition, despite the difference in age, the healthy control specimens were well matched with SBS patient specimens in terms of limiting potential sources of technical bias. Identical DNA extraction, PCR, and sequencing protocols were used for both specimen groups, and many of these steps were performed by the same group of technicians. Although previous publications have demonstrated that sources of technical variation, including extraction method, primer choice, and run-to-run variation, can be detected in microbiome data sets,50,51 they are often outweighed by the effects of interindividual variation.52,53
Another potential limitation is that unlike the healthy children, all of the children with SBS had received antibiotics within the 6 months prior to sample collection, with 7 of 9 children receiving metronidazole. Several studies have shown how antibiotics alter the intestinal microbiome.54,55 A study in 2010 by Jakobsson et al56 showed that an adult fecal microbiome can be altered for 4 years following a 1-week course of metronidazole and clarithromycin. Similar to our study, the authors found decreased quantities of the phylum Actinobacteria in fecal samples from the group treated with metronidazole and clarithromycin. In addition, they observed an enrichment in the phylum Proteobacteria following antibiotic treatment. Given the nature of SBS, however, exposures to antibiotics represent the rule rather than the exception and reflect the population typically encountered in medical practice. Future studies should attempt to examine the effects of antibiotic treatment longitudinally in this patient group. Another potential confounder is that some of children with SBS were receiving parenteral nutrition (PN). Several rodent studies have shown that PN can potentially alter the intestinal microbiome.57
In summary, the fecal microbiomes of children with SBS appear to be different from those in healthy children. Differences in the composition and function of the microbiomes in children with SBS may affect bowel physiology, and these findings may provide new prospects for intestinal rehabilitation and clinical management. Future studies, including whole-genome shotgun metagenomics of fecal samples and longitudinal sampling of children with SBS, are likely to provide additional insights into the potential role of the microbiome in gut adaptation and barrier function (eg, bacterial translocation) in children with SBS.
Clinical Relevancy Statement.
Changes in the intestinal microbiome of patients with short bowel syndrome (SBS) are thought to significantly affect clinical outcome. This study shows that the fecal microbiome of patients with SBS is significantly different from that of healthy controls when analyzed by 16S metagenomics. Differences in the composition and function of gut microbiomes in children with SBS may affect bowel physiology, and these findings may provide new opportunities for intestinal rehabilitation and clinical management.
Acknowledgments
Financial disclosure: Supported by the National Institute of Diabetes, Digestive, and Kidney Diseases (UH2 DK093990 and UH3 DK083990, and R01 DK065075 to JV); the National Center for Complementary and Integrative Health (RO1 AT004326 to JV); and the National Cancer Institute (U01 CA170930 to JV). This research also was supported by the USDA/ARS under Cooperative Agreement No. 6250-51000-043 to RJS and P30-DK56338, which funds the Texas Medical Center Digestive Disease Center (to JV). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. This work is a publication of the USDA/ARS Children's Nutrition Research Center, Department of Pediatrics, Baylor College of Medicine and Texas Children's Hospital, Houston, Texas. The contents of this publication do not necessarily reflect the views or policies of the USDA, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. government.
References
- 1.O'Keefe SJ, Buchman AL, Fishbein TM, Jeejeebhoy KN, Jeppesen PB, Shaffer J. Short bowel syndrome and intestinal failure: consensus definitions and overview. Clin Gastroenterol Hepatol. 2006;4(1):6–10. doi: 10.1016/j.cgh.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 2.Buchman AL. Etiology and initial management of short bowel syndrome. Gastroenterology. 2006;130((2)(suppl 1)):S5–S15. doi: 10.1053/j.gastro.2005.07.063. [DOI] [PubMed] [Google Scholar]
- 3.Duro D, Kamin D, Duggan C. Overview of pediatric short bowel syndrome. J Pediatr Gastroenterol Nutr. 2008;47(suppl 1):S33–S36. doi: 10.1097/MPG.0b013e3181819007. [DOI] [PubMed] [Google Scholar]
- 4.Vanderhoof JA, Langnas AN, Pinch LW, Thompson JS, Kaufman SS. Short bowel syndrome. J Pediatr Gastroenterol Nutr. 1992;14(4):359–370. doi: 10.1097/00005176-199205000-00001. [DOI] [PubMed] [Google Scholar]
- 5.Weaver LT, Austin S, Cole TJ. Small intestinal length: a factor essential for gut adaptation. Gut. 1991;32(11):1321–1323. doi: 10.1136/gut.32.11.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goulet O, Ruemmele F. Causes and management of intestinal failure in children. Gastroenterology. 2006;130((2)(suppl 1)):S16–S28. doi: 10.1053/j.gastro.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 7.Vanderhoof JA, Langnas AN. Short-bowel syndrome in children and adults. Gastroenterology. 1997;113(5):1767–1778. doi: 10.1053/gast.1997.v113.pm9352883. [DOI] [PubMed] [Google Scholar]
- 8.Sala D, Chomto S, Hill S. Long-term outcomes of short bowel syndrome requiring long-term/home intravenous nutrition compared in children with gastroschisis and those with volvulus. Transplant Proc. 2010;42(1):5–8. doi: 10.1016/j.transproceed.2009.12.033. [DOI] [PubMed] [Google Scholar]
- 9.Quiros-Tejeira RE, Ament ME, Reyen L, et al. Long-term parenteral nutritional support and intestinal adaptation in children with short bowel syndrome: a 25-year experience. J Pediatr. 2004;145(2):157–163. doi: 10.1016/j.jpeds.2004.02.030. [DOI] [PubMed] [Google Scholar]
- 10.Cole CR, Frem JC, Schmotzer B, et al. The rate of bloodstream infection is high in infants with short bowel syndrome: relationship with small bowel bacterial overgrowth, enteral feeding, and inflammatory and immune responses. J Pediatr. 2010;156(6):941–947.e1. doi: 10.1016/j.jpeds.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goulet O, Joly F, Corriol O, Colomb-Jung V. Some new insights in intestinal failure-associated liver disease. Curr Opin Organ Transplant. 2009;14(3):256–261. doi: 10.1097/MOT.0b013e32832ac06f. [DOI] [PubMed] [Google Scholar]
- 12.Kaufman SS, Loseke CA, Lupo JV, et al. Influence of bacterial overgrowth and intestinal inflammation on duration of parenteral nutrition in children with short bowel syndrome. J Pediatr. 1997;131(3):356–361. doi: 10.1016/s0022-3476(97)80058-3. [DOI] [PubMed] [Google Scholar]
- 13.Lappinga PJ, Abraham SC, Murray JA, Vetter EA, Patel R, Wu TT. Small intestinal bacterial overgrowth: histopathologic features and clinical correlates in an underrecognized entity. Arch Pathol Lab Med. 2010;134(2):264–270. doi: 10.5858/134.2.264. [DOI] [PubMed] [Google Scholar]
- 14.Quigley EM, Quera R. Small intestinal bacterial overgrowth: roles of antibiotics, prebiotics, and probiotics. Gastroenterology. 2006;130((2)(suppl 1)):S78–S90. doi: 10.1053/j.gastro.2005.11.046. [DOI] [PubMed] [Google Scholar]
- 15.Sondheimer JM, Asturias E, Cadnapaphornchai M. Infection and cholestasis in neonates with intestinal resection and long-term parenteral nutrition. J Pediatr Gastroenterol Nutr. 1998;27(2):131–137. doi: 10.1097/00005176-199808000-00001. [DOI] [PubMed] [Google Scholar]
- 16.Sondheimer JM, Cadnapaphornchai M, Sontag M, Zerbe GO. Predicting the duration of dependence on parenteral nutrition after neonatal intestinal resection. J Pediatr. 1998;132(1):80–84. doi: 10.1016/s0022-3476(98)70489-5. [DOI] [PubMed] [Google Scholar]
- 17.Meehan JJ, Georgeson KE. Prevention of liver failure in parenteral nutrition–dependent children with short bowel syndrome. J Pediatr Surg. 1997;32(3):473–475. doi: 10.1016/s0022-3468(97)90609-6. [DOI] [PubMed] [Google Scholar]
- 18.Dibaise JK, Young RJ, Vanderhoof JA. Enteric microbial flora, bacterial overgrowth, and short-bowel syndrome. Clin Gastroenterol Hepatol. 2006;4(1):11–20. doi: 10.1016/j.cgh.2005.10.020. [DOI] [PubMed] [Google Scholar]
- 19.Ricotta J, Zuidema GD, Gadacz TR, Sadri D. Construction of an ileocecal valve and its role in massive resection of the small intestine. Surg Gynecol Obstet. 1981;152(3):310–314. [PubMed] [Google Scholar]
- 20.Haller D, Bode C, Hammes WP, Pfeifer AM, Schiffrin EJ, Blum S. Non-pathogenic bacteria elicit a differential cytokine response by intestinal epithelial cell/leucocyte co-cultures. Gut. 2000;47(1):79–87. doi: 10.1136/gut.47.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Preidis GA, Versalovic J. Targeting the human microbiome with antibiotics, probiotics, and prebiotics: gastroenterology enters the metagenomics era. Gastroenterology. 2009;136(6):2015–2031. doi: 10.1053/j.gastro.2009.01.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.von der Weid T, Bulliard C, Schiffrin EJ. Induction by a lactic acid bacterium of a population of CD4(+) T cells with low proliferative capacity that produce transforming growth factor beta and interleukin-10. Clin Diagn Lab Immunol. 2001;8(4):695–701. doi: 10.1128/CDLI.8.4.695-701.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. The human microbiome project. Nature. 2007;449(7164):804–810. doi: 10.1038/nature06244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pennisi E. Metagenomics: massive microbial sequence project proposed. Science. 2007;315(5820):1781. doi: 10.1126/science.315.5820.1781a. [DOI] [PubMed] [Google Scholar]
- 25.Structure function and diversity of the healthy human microbiome. Nature. 2012;486(7402):207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.A framework for human microbiome research. Nature. 2012;486(7402):215–221. doi: 10.1038/nature11209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zoetendal EG, Collier CT, Koike S, Mackie RI, Gaskins HR. Molecular ecological analysis of the gastrointestinal microbiota: a review. J Nutr. 2004;134(2):465–472. doi: 10.1093/jn/134.2.465. [DOI] [PubMed] [Google Scholar]
- 28.Petrosino JF, Highlander S, Luna RA, Gibbs RA, Versalovic J. Metagenomic pyrosequencing and microbial identification. Clin Chem. 2009;55(5):856–866. doi: 10.1373/clinchem.2008.107565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sokol H, Seksik P, Furet JP, et al. Low counts of Faecalibacterium prausnitzii in colitis microbiota. Inflamm Bowel Dis. 2009;15(8):1183–1189. doi: 10.1002/ibd.20903. [DOI] [PubMed] [Google Scholar]
- 30.Dicksved J, Halfvarson J, Rosenquist M, et al. Molecular analysis of the gut microbiota of identical twins with Crohn's disease. ISME J. 2008;2(7):716–727. doi: 10.1038/ismej.2008.37. [DOI] [PubMed] [Google Scholar]
- 31.Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A. 2007;104(34):13780–13785. doi: 10.1073/pnas.0706625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Y, Hoenig JD, Malin KJ, et al. 16S rRNA gene-based analysis of fecal microbiota from preterm infants with and without necrotizing enterocolitis. ISME J. 2009;3(8):944–954. doi: 10.1038/ismej.2009.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mai V, Young CM, Ukhanova M, et al. Fecal microbiota in premature infants prior to necrotizing enterocolitis. PLoS One. 2011;6(6):e20647. doi: 10.1371/journal.pone.0020647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rajiliċ-Stojanoviċ M, Biagi E, Heilig HG, et al. Global and deep molecular analysis of microbiota signatures in fecal samples from patients with irritable bowel syndrome. Gastroenterology. 2011;141(5):1792–1801. doi: 10.1053/j.gastro.2011.07.043. [DOI] [PubMed] [Google Scholar]
- 35.Saulnier DM, Riehle K, Mistretta TA, et al. Gastrointestinal microbiome signatures of pediatric patients with irritable bowel syndrome. Gastroenterology. 2011;141(5):1782–1791. doi: 10.1053/j.gastro.2011.06.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang H, DiBaise JK, Zuccolo A, et al. Human gut microbiota in obesity and after gastric bypass. Proc Natl Acad Sci U S A. 2009;106(7):2365–2370. doi: 10.1073/pnas.0812600106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Joly F, Mayeur C, Bruneau A, et al. Drastic changes in fecal and mucosa-associated microbiota in adult patients with short bowel syndrome. Biochimie. 2010;92(7):753–761. doi: 10.1016/j.biochi.2010.02.015. [DOI] [PubMed] [Google Scholar]
- 38.Riehle K, Coarfa C, Jackson A, et al. The Genboree Microbiome Toolset and the analysis of 16S rRNA microbial sequences. BMC Bioinfomatics. 2012;13(suppl 13):S11. doi: 10.1186/1471-2105-13-S13-S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Caporaso JG, Kuczynski J, Stombaugh J, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7(5):335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22(13):1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 41.Haas BJ, Gevers D, Earl AM, et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011;21(3):494–504. doi: 10.1101/gr.112730.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73(16):5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liaw A, Wiener M. Classification and regression by randomForest. R News. 2002;2(3):18–22. [Google Scholar]
- 44.Kursa MB, Rudnicki WR. Feature selection with the Boruta package. J Stat Software. 2010;36(11):13. [Google Scholar]
- 45.Bousbia S, Papazian L, Saux P, et al. Repertoire of intensive care unit pneumonia microbiota. PLoS One. 2012;7(2):e32486. doi: 10.1371/journal.pone.0032486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yatsunenko T, Rey FE, Manary MJ, et al. Human gut microbiome viewed across age and geography. Nature. 2012;486(7402):222–227. doi: 10.1038/nature11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hollister EB, Gao C, Versalovic J. Compositional and functional features of the gastrointestinal microbiome and their effects on human health. Gastroenterology. 2014;146(6):1449–1458. doi: 10.1053/j.gastro.2014.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schloss PD, Iverson KD, Petrosino JF, Schloss SJ. The dynamics of a family's gut microbiota reveal variations on a theme. Microbiome. 2014;2(1):25. doi: 10.1186/2049-2618-2-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ringel-Kulka T, Cheng J, Ringel Y, et al. Intestinal microbiota in healthy US young children and adults—a high throughput microarray analysis. PLoS One. 2013;8(5):e64315. doi: 10.1371/journal.pone.0064315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schloss PD, Gevers D, Westcott SL. Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS One. 2011;6(12):e27310. doi: 10.1371/journal.pone.0027310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lozupone CA, Stombaugh J, Gonzalez A, et al. Meta-analyses of studies of the human microbiota. Genome Res. 2013;23(10):1704–1714. doi: 10.1101/gr.151803.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wesolowska-Andersen A, Bahl MI, Carvalho V, et al. Choice of bacterial DNA extraction method from fecal material influences community structure as evaluated by metagenomic analysis. Microbiome. 2014;2(1):19. doi: 10.1186/2049-2618-2-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Voigt AY, Costea PI, Kultima JR, et al. Temporal and technical variability of human gut metagenomes. Genome Biol. 2015;16(1):73. doi: 10.1186/s13059-015-0639-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dethlefsen L, Relman DA. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc Natl Acad Sci U S A. 2011;108(suppl 1):4554–4561. doi: 10.1073/pnas.1000087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jernberg C, Lofmark S, Edlund C, Jansson JK. Long-term impacts of antibiotic exposure on the human intestinal microbiota. Microbiology. 2010;156(pt 11):3216–3223. doi: 10.1099/mic.0.040618-0. [DOI] [PubMed] [Google Scholar]
- 56.Jakobsson HE, Jernberg C, Andersson AF, Sjolund-Karlsson M, Jansson JK, Engstrand L. Short-term antibiotic treatment has differing long-term impacts on the human throat and gut microbiome. PLoS One. 2010;5(3):e9836. doi: 10.1371/journal.pone.0009836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Harris JK, El Kasmi KC, Anderson AL, et al. Specific microbiome changes in a mouse model of parenteral nutrition associated liver injury and intestinal inflammation. PLoS One. 2014;9(10):e110396. doi: 10.1371/journal.pone.0110396. [DOI] [PMC free article] [PubMed] [Google Scholar]